 Kelsey Sokol1*
Kelsey Sokol1* Saritha Kartan1
Saritha Kartan1 William T. Johnson1
William T. Johnson1 Onder Alpdogan1
Onder Alpdogan1 Neda Nikbakht2
Neda Nikbakht2 Bradley M. Haverkos3
Bradley M. Haverkos3 Jerald Gong4
Jerald Gong4 Pierluigi Porcu1*
Pierluigi Porcu1*- 1Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA, United States
- 2Department of Dermatology and Cutaneous Biology, Thomas Jefferson University, Philadelphia, PA, United States
- 3Blood Cancer and BMT, University of Colorado, Aurora, CO, United States
- 4Department of Pathology, Thomas Jefferson University, Philadelphia, PA, United States
Angioimmunoblastic T-cell lymphoma (AITL) is one of four major subtypes of nodal peripheral T cell lymphoma, characterized by its cell of origin, the follicular helper T-cell (TFH). Patients typically present with prominent constitutional (B) symptoms, generalized lymphadenopathy, hepatosplenomegaly, cytopenias, and rash. Here we present a case of a 62-year-old male with progressive cervical adenopathy, fevers and weight loss presenting with extreme polyclonal plasmacytosis and high plasma EBV viral load. While the initial presentation appeared to mimic plasma cell leukemia or severe infection, lymph node biopsy and bone marrow biopsy confirmed a diagnosis of AITL. This case highlights the heterogeneity of the clinical presentation of AITL to enable physicians to more promptly recognize, diagnose and initiate treatment.
Introduction
Angioimmunoblastic T cell lymphoma (AITL) is recognized in the 2016 World Health Organization (WHO) Classification as one of the four major subtypes of nodal peripheral T cell lymphoma (PTCL), accounting for 15–25% of all PTCL, depending on geographical region, and approximately 1–2% of all non-Hodgkin lymphomas (NHL) (1). AITL is a clinically distinct lymphoid neoplasm, with a well-characterized cell of origin, the follicular helper T-cell (TFH), and with discrete immunophenotypical and genetic features. Most patients with AITL present with very prominent constitutional (B) symptoms, generalized lymphadenopathy, hepatosplenomegaly, multilineage cytopenias (in particular thrombocytopenia), and rash (2). These symptoms may precede the diagnosis by several months, rarely by years. Laboratory signs of B-cell activation and autoimmunity are a hallmark of AITL, including hypergammaglobulinemia, autoimmune hemolytic anemia (AIHA), immune mediated thrombocytopenia (ITP), and presence of autoantibodies (rheumatoid factor, anti-phospholipid, anti-DNA, and anti-smooth muscle antibodies). The majority of AITL patients have detectable Epstein Bar Virus (EBV)-encoded RNA (EBER)-positive cells by in situ hybridization (ISH) (3). in the diagnostic lymph node biopsy and about 50% have measurable plasma EBV DNA (pEBVd) (4), even before initiation of chemotherapy which may contribute to polyclonal B-cell activation and lead to the development of EBV-positive B-cell lymphomas in a subset of patients. While in most cases the hypergammaglobulinemia is polyclonal, monoclonal immunoglobulins (Ig) can be detected in the serum of some patients, mimicking multiple myeloma (5).
There are several reported cases of marked peripheral blood and/or bone marrow plasmacytosis in AITL, mimicking plasma cell leukemia, especially when associated with elevated Ig levels. The polyclonal nature of the plasmacytosis, however, ultimately suggests a reactive etiology, such a viral infection (EBV, parvovirus B19, hepatitis), autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, Sjögren's syndrome), or serum sickness (6, 7). EBV has an established association with numerous B-cell and T cell lymphoproliferative neoplasms, including AITL (8, 9). Here we report the case of a patient with AITL who presented with marked peripheral blood polyclonal plasmacytosis, polyclonal hypergammaglobulinemia, and high levels of pEBVd, suggestive of EBV reactivation. The case highlights the heterogeneity of the clinical manifestations of AITL, to enable physicians to more readily recognize the disease at presentation and initiate treatment promptly.
Case Presentation
A 62-year-old male, with a history of non-ischemic cardiomyopathy presented with new onset atrial flutter. He reported shortness of breath, diaphoresis and lightheadedness for ~ 1 week as well as progressive cervical adenopathy, fevers, and unintentional weight loss. Laboratory evaluation demonstrated white blood cell (WBC) count of 17.3 ×109/L with 37% (6.4 ×109/L) plasma cells, hemoglobin of 11.6 g/dL, and platelet count of 53 ×109/L. Contrast-enhanced computed tomography (CT) scans of the chest, abdomen, and pelvis revealed non-bulky cervical, axillary, mediastinal, retroperitoneal and inguinal lymphadenopathy, ranging in size between 1.4 cm and 2.5 cm, as well as splenomegaly of 15.5 cm, without discrete lesions. Shortly after admission, the patient developed acute renal failure and serum electrolyte abnormalities consistent with tumor lysis syndrome (TLS) (creatinine of 4.2 mg/dL, potassium of 5.4 mmol/L, phosphate of 5.3 mg/dL, urate of 11.9 mg/dL, lactate dehydrogenase of 368 IU/L). Peripheral blood flow cytometry revealed that 46% of the leukocytes were represented by polyclonal plasma cells (CD19+, CD20−, CD22−, CD45+(moderate), CD38+(bright), CD56−, CD117−, CD138+, HLA-DR+(heterogeneous), surface κ or λ−, cytoplasmic κ +(subset), cytoplasmic λ+(subset)). Serum protein electrophoresis (SPEP) and immunofixation electrophoresis (IFE) showed abnormally high gamma globulin levels (IgA 1200 mg/dL and IgG 4200 mg/dL) without a monoclonal paraprotein. Serum kappa and lambda light chain levels were modestly elevated (92.6 mg/dL and 73 mg/dL, respectively) but the ratio was normal (1.27).
There was initial concern for an infectious etiology for the patient's presentation given the signs of systemic inflammatory response syndrome (tachycardia, leukocytosis, fever) and circulating polyclonal plasma cells. Blood cultures and viral studies were performed and were notable for a detectable plasma EBV viral load, which was quantified by qRT-PCR as 71,000 copies/mL of pEBVd, and approximately 24 h later increased to 1.05 ×106 copies/mL. Human immunodeficiency virus (HIV) and hepatitis C serologies were negative, hepatitis B serology was consistent with prior immunization, and plasma cytomegalovirus DNA (pCMVd) was negative. Serum antinuclear antibody (ANA) titer for initial workup of autoimmune disease was also negative. After bone marrow and lymph node biopsies were obtained, dexamethasone 40 mg daily was initiated, as concerns for an EBV associated lymphoproliferative disorder (EBV-LPD) were high. In addition, the patient was started on antiviral therapy with ganciclovir 2.5 mg/kg IV every 12 h for possible active EBV infection. The drug was stopped after 5 days as the treating physicians concluded that the risk of drug toxicity outweighed the benefit, and the data to support its use in this setting were insufficient. Despite these measures and aggressive supportive care, within 1 week of admission, his clinical course progressed to multiorgan failure requiring mechanical ventilation and ultimately cardiac arrest requiring extracorporeal membrane oxygenation (ECMO).
Bone marrow exam revealed hypercellularity (90%) with 30–40% plasma cells (which was deemed to be reactive because of polyclonal kappa/lambda expression), large lymphoid aggregates with predominance of T cells, and scattered EBV+ B cells. Molecular studies for T-cell receptor (TCR) gamma gene and immunoglobulin heavy chain (IGH) gene rearrangements were performed on the blood sample. A monoclonal TCR gamma gene rearrangement was detected; clonal IGH gene rearrangement was not detected. The left inguinal node showed completely effaced normal lymph node architecture with a diffuse infiltration of atypical small to medium sized lymphocytes in a background of vascular proliferation, plasma cells, and scattered immunoblasts. By immunohistochemistry and flow cytometry, there was a diffusely increased T cells infiltrate admixed with scattered B immunoblasts. The T cells expressed CD2, CD3, CD5, CD7, PD-1, and were negative for CD5, CD10, BCL-6, and all B-cell antigens analyzed. CD4 to CD8 ratio was within normal range. Scattered large immunoblasts were positive for CD20 and EBV (by EBER in situ hybridization). CD21 revealed several expanded clusters of follicular dendritic cells. In situ hybridization for immunoglobulin kappa and lambda light chains revealed numerous polyclonal plasma cells. Molecular studies for TCR gamma gene and IGH gene rearrangements were repeated on the lymph node. Identical clonal TCR gamma gene rearrangement was detected in the lymph node. Similarly, clonal IGH gene rearrangement was not detected. Representative images are shown in Figure 1.
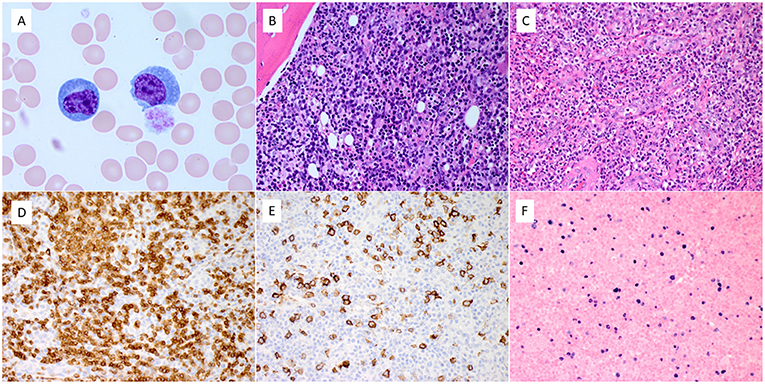
Figure 1. Pathologic features of blood, bone marrow, and lymph node. (A) The blood smear showed a marked increase of reactive circulating plasma cells. (B) Bone marrow biopsy showed diffuse infiltration of atypical lymphocytes admixed with numerous reactive plasma cells. (C) The left inguinal lymph node revealed diffuse infiltration of small to medium-sized atypical lymphocytes and scattered large immunoblasts, with exuberant vascular proliferation and numerous reactive plasma cells. (D–F). The atypical lymphocytes expressed CD3 (D). Scattered immunoblasts expressed CD20 (E) and EBER by in situ hybridization (F) (Magnification: A. x100, B–F. x40).
In aggregate, these findings were thought to be most consistent with a diagnosis of AITL. Pathologically, the complete effacement of nodal architecture, diffuse involvement of bone marrow, and the presence of clonal T-cell gene rearrangement rule out conditions of reactive EBV infection such as infectious mononucleosis and chronic active EBV infection. Unfortunately, the patient failed to improve with dexamethasone and the family decided to withdraw care. A request for autopsy was declined.
Discussion
In this report we present a case of AITL with profound polyclonal peripheral plasmacytosis and rapidly rising pEBVd, reaching extremely high levels (>1 million copies/mL). This presentation offered a significant challenge as plasma cell leukemia and a severe systemic infection were the initial diagnostic considerations.
AITL is an aggressive subtype of nodal peripheral T cell lymphomas. It is derived from follicular helper T-cells (TFH) that normally reside at the border between the mantle zone and the germinal center of the normal physiologic lymph node (10). TFH are a distinct subset of T helper cells necessary for the formation of germinal centers (GC) and B-cell differentiation. They are formed through naïve CD4+ T-cell's interaction with dendritic cells, leading to the activation of the inducible T-cell costimulator (ICOS) and phosphatidylinositol 3-kinase (PI3K) pathways in the CD4+ T-cells. This induces the expression of B-cell lymphoma 6 protein (BCL6), along with other transcription factors which guides the differentiation of the naïve CD4+ T-cells into TFH (10, 11) TFH then help in directing the differentiation of B-cell centroblasts to centrocytes, which ultimately form plasma cells or memory B cells (10). TFH begin to express high levels of C-X-C chemokine receptor type 5 (CXCR5), PD-1, BCL6, myelin-associated glycoprotein (MAG), and serum amyloid P component (SAP), the expression of which can be detected by immunohistochemistry (IHC). The genetic alterations thought to be driving the development of AITL include ten eleven translocation methylcytosine dioxygenase 2 (TET2), DNA (cytosine-5)-methyltransferase 3A (DNMT3A), isocitrate dehydrogenase (NADP(+)) 2 (IDH2), ras homolog gene family member A (RHOA), and components of the TCR pathways, all of which are potentials targets for therapy (11).
Histologic examination of involved lymph nodes in AITL shows small-medium sized tumor cells in small clusters with clear cytoplasm, that can be identified as TFH by immunohistochemistry, based on the expression of PD-1 and CXCL13 (CXCR5 ligand) and pan T-cell markers, such as CD3 and CD4 (12). The associated FDC are typically identified by expression of CD21, CD23, and CD35. These two cell types are present among a population of other small reactive lymphocytes, histiocytes, and plasma cells, with high endothelial venules (13). The B-cells are often EBV+ (EBER+), while the associated T cells and plasma cells are typically not (14). This morphologic appearance can initially be mistaken for a hyperplastic reactive lymph node with EBV reactivation.
In a retrospective subset analysis of 157 patients with AITL enrolled on the GELA (Groupe d'Etude des Lymphomes de l'Adulte) LNH87 and LNH93 randomized clinical trials, bone marrow involvement was seen in 60% of patients (14). The most commonly seen features were paratrabecular and interstitial infiltration of atypical lymphocytes, histiocytes and eosinophils, with infiltration of small to large B cells. Vascular proliferation and reticulin fibrosis have also been reported. Secondary changes include trilineage hyperplasia and polyclonal plasmacytosis. Together, these findings can lead to misdiagnoses, including benign lymphoid hyperplasia, T cell rich large B cell lymphoma, chronic myeloproliferative disease, or plasma cell dyscrasia (15–17). The consensus is that CD10, CXCL13, and PD-1 are expressed in only a fraction of cases, although double labeling with CD3 and BCL6 may specifically highlight the neoplastic T cells (16). Additionally, depending on the methodology used, T-cell receptor (TCR) gene rearrangements are seen in 70–90% of cases and Ig gene rearrangements in approximately 10-20% of cases (18).
The association between polyclonal plasma cell proliferations and AITL has not yet been fully elucidated. While it may in part be secondary to cytokine release such as IL-6, in this case we hypothesize that the EBV likely was a contributing factor (6). EBV reactivation is known to occur when latently infected B cells are exposed to a variety of stimuli, including physiologic signals able to induce proliferation and differentiation (19). The specific mechanisms leading to the occurrence of high levels of pEBVd in AITL merit further investigation.
We report this clinical case study to emphasize the need for consideration of AITL in patients presenting with peripheral blood polyclonal plasmacytosis, high levels of pEBVd, and features highly suggestive of lymphoma, such as extensive lymphadenopathy and TLS. While the initial presentation and tissue morphology appears consistent with a reactive process, closer examination as above reveals histopathology consistent with AITL. In previous reports of AITL with plasmacytosis, patients who received rapid treatment showed a good response to cytotoxic chemotherapy (20). The efficacy of treatment in patients with AITL and significant EBV viral load remains less well known.
Data Availability
No datasets were generated or analyzed for this study.
Patient Consent
Written consent for publication of this case report and any potentially identifying information was obtained by the patient's wife.
Author Contributions
KS and PP wrote the first draft of the manuscript. SK, WJ, OA, NN, BH, JG, and PP wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bal M, Gujral S, Gandhi J, Shet T, Epari S, Subramanian PG. Angioimmunoblastic T-Cell lymphoma: A critical analysis of clinical, morphologic and immunophenotypic features. Indian J Pathol Microbiol. (2010). 53:640–5. doi: 10.4103/0377-4929.72010
2. Dogan A, Attygalle A, Kyriakou C. Angioimmunoblastic T-cell lymphoma. Br J Haematol. (2003) 121:681–91. doi: 10.1046/j.1365-2141.2003.04335.x
3. Dogan A, Gaulard P, Jaffe ES, Ralfkiaer E, Müller-Hermelink HK. (2008). Angioimmunoblastic T-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon: IARC. p. 309–11.
4. Haverkos BM, Huang Y, Gru A, Pancholi P, Freud AG, Mishra A, et al. Frequency and clinical correlates of elevated plasma Epstein-Barr virus DNA at diagnosis in peripheral T-cell lymphomas. Int J Cancer. (2017) 15140:1899–906. doi: 10.1002/ijc.30566
5. Xu J, Tang Y, Zhao S, Zhang W, Xiu Y, Liu T, et al. Angioimmunoblastic T-cell lymphoma with coexisting plasma cell myeloma: a case report and review of the literature. Tohoku J Exp Med. (2015) 235:283–8. doi: 10.1620/tjem.235.283
6. Singh N, Sharma A, Pasricha S, Agrawal N, Bhurani D, Gupta G, et al. Florid plasmacytosis in angioimmunoblastic T cell lymphoma: A diagnostic conundrum. Indian J Hematol Blood Trans. (2017) 34:1–3. doi: 10.1007/s12288-017-0824-x
7. Ahsanuddin AN, Brynes RK, Li S. Peripheral blood polyclonal plasmacytosis mimicking plasma cell leukemia in patients with angioimmunoblastic T-cell lymphoma: report of 3 cases and review of the literature. Int J Clin Exp Pathol. (2011) 4:416–20.
8. Kim HJ, Ko YH, Kim JE, Lee SS, Lee H, Park G, et al. Epstein-barr virus–associated lymphoproliferative disorders: review and update on 2016 WHO Classification. J Pathol Transl Med. (2017) 51:352–8. doi: 10.4132/jptm.2017.03.15
9. Okuyama S, Terada T, Kumagai H, Tsumanuma R, Omoto E, Ueki T, et al. Epstein-Barr virus clonality and plasmacytosis in a patient with atypical angioimmunoblastic T cell lymphoma. Ann Hematol. (2018) 97:537. doi: 10.1007/s00277-017-3189-1
10. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody response and autoimmunity. Nat Rev Immunol. (2005) 5:853–65. doi: 10.1038/nri1714
11. Rodríguez-Cortés J, Palomero T. The curious origins of Angioimmunoblastic T-Cell Lymphoma. Curr Opin Hematol. (2016) 23:434–43. doi: 10.1097/MOH.0000000000000261
12. Hongbo Yu, Aliakbar Shahsafaei, David M. Dorfman. Germinal-center T-helper-cell markers PD-1 and CXCL13 are both expressed by neoplastic cells in angioimmunoblastic T-cell lymphoma. Am J Clin Pathol. (2009) 131:33–41. doi: 10.1309/AJCP62WRKERPXDRT
13. Jaffe ES, Harris NL, Stein H, Vardiman J. World Health Organization Classification: Tumours of Hematopoietic and Lymphoid Tissues. Jaffe ES, Harris H, Stein H, Vardiman JW, editors. Lyon: IARC Press (2007).
14. Mourad N, Mounier N, Brière J, Raffoux E, Delmer A, Feller A, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte (GELA) trials. Blood. (2008) 111:4463–4470. doi: 10.1182/blood-2007-08-105759
15. Grogg KL, Morice WG, Macon WR. Spectrum of bone marrow findings in patients with angioimmunoblastic T-cell lymphoma. Br J Haematol. (2007) 137:416–422 doi: 10.1111/j.1365-2141.2007.06577.x
16. Khokhar FA, Payne WD, Talwalkar SS, Jorgensen JL, Bueso-Ramos CE, Medeiros LJ, et al. Angioimmunoblastic T-cell lymphoma in bone marrow: a morphologic and immunophenotypic study. Hum Pathol. (2010) 41:79–87 doi: 10.1016/j.humpath.2009.06.016
17. Cho YU, Chi HS, Park CJ, Jang S, Seo EJ, Huh J. Distinct features of angioimmunoblastic T-cell lymphoma with bone marrow involvement. Am J Clin Pathol. (2009) 131:640–646 doi: 10.1309/AJCPQXKCHQH4VAJ5
18. Rai MP, Bedi PS, Marinas EB, Khan NNS. Angioimmunoblastic T-cell lymphoma: a rare subtype of peripheral T-cell lymphoma. Clin Case Rep. (2018) 6:750–52. doi: 10.1002/ccr3.1388
19. Odumade OA, Hogquist KA, Balfour HH. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev. (2011) 24:193–209. doi: 10.1128/CMR.00044-10
Keywords: angioimmunoblastic T-cell lymphoma, plasmacytosis, EBV, plasma cell leukemia, follicular helper T-cell
Citation: Sokol K, Kartan S, Johnson WT, Alpdogan O, Nikbakht N, Haverkos BM, Gong J and Porcu P (2019) Extreme Peripheral Blood Plasmacytosis Mimicking Plasma Cell Leukemia as a Presenting Feature of Angioimmunoblastic T-Cell Lymphoma (AITL). Front. Oncol. 9:509. doi: 10.3389/fonc.2019.00509
Received: 05 March 2019; Accepted: 28 May 2019;
Published: 13 June 2019.
Edited by:
Alejandro Gru, University of Virginia Health System, United StatesReviewed by:
Roberto Nicolas Miranda, University of Texas MD Anderson Cancer Center, United StatesCandida Vitale, University of Turin, Italy
Copyright © 2019 Sokol, Kartan, Johnson, Alpdogan, Nikbakht, Haverkos, Gong and Porcu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelsey Sokol, a2Vsc2V5LnNva29sQGplZmZlcnNvbi5lZHU=; Pierluigi Porcu, cGllcmx1aWdpLnBvcmN1QGplZmZlcnNvbi5lZHU=