 Simona Giglio1*
Simona Giglio1* Viviana Annibali2†Roberto Cirombella1
Viviana Annibali2†Roberto Cirombella1 Omar Faruq1Stefano Volinia3
Omar Faruq1Stefano Volinia3 Claudia De Vitis1Margherita Pesce1Donatella Caserta4
Claudia De Vitis1Margherita Pesce1Donatella Caserta4 Angela Pettinato5Filippo Fraggetta5
Angela Pettinato5Filippo Fraggetta5 Andrea Vecchione1*
Andrea Vecchione1*- 1Department of Clinical and Molecular Medicine, “La Sapienza” University, Sant'Andrea Hospital, Rome, Italy
- 2Department of Neurosciences, Mental Health and Sensory Organs, Centre for Experimental Neurological Therapies (CENTERS), “La Sapienza” University, Sant'Andrea Hospital, Rome, Italy
- 3Department of Internal Medicine, Biosystems Analysis, LTTA, Department of Morphology, Surgery and Experimental Medicine, Università Degli Studi, Ferrara, Italy
- 4Department of Medical-Surgical Sciences and Translational Medicine, “La Sapienza” University, Sant'Andrea Hospital, Rome, Italy
- 5Pathology Unit, Cannizzaro Hospital, Catania, Italy
Endometrial cancer is the most common gynecologic malignancy in developed countries. Estrogen-dependent tumors (type I, endometrioid) account for 80% of cases and non-estrogen-dependent (type II, non-endometrioid) account for the rest. Endometrial cancer type I is generally thought to develop via precursor lesions along with the increasing accumulation of molecular genetic alterations. Endometrial hyperplasia with atypia/Endometrial Intraepithelial Neoplasia is the least common type of hyperplasia but it is the type most likely to progress to type I cancer, whereas endometrial hyperplasia without atypia rarely progresses to carcinoma. MicroRNAs are a class of small, non-coding, single-stranded RNAs that negatively regulate gene expression mainly binding to 3′-untranslated region of target mRNAs. In the current study, we identified a microRNAs signature (miR-205, miR-146a, miR-1260b) able to discriminate between atypical and typical endometrial hyperplasia in two independent cohorts of patients. The identification of molecular markers that can distinguish between these two distinct pathological conditions is considered to be highly useful for the clinical management of patients because hyperplasia with an atypical change is associated with a higher risk of developing cancer. We show that the combination of miR-205, −146a, and −1260b has the best predictive power in discriminating these two conditions (>90%). With the aim to find a biological role for these three microRNAs, we focused our attention on a common putative target involved in endometrial carcinogenesis: the oncosuppressor gene SMAD4. We showed that miRs-146a,−205, and−1260b directly target SMAD4 and their enforced expression induced proliferation and migration of Endometrioid Cancer derived cell lines, Hec1a cells. These data suggest that microRNAs-mediated impairment of the TGF-β pathway, due to inhibition of its effector molecule SMAD4, is a relevant molecular alteration in endometrial carcinoma development. Our findings show a potential diagnostic role of this microRNAs signature for the accurate diagnosis of Endometrial hyperplasia with atypia/Endometrial Intraepithelial Neoplasia and improve the understanding of their pivotal role in SMAD4 regulation.
Introduction
Worldwide, endometrial cancer (EC) represents 4% of all cancers in women and is the most common malignant tumor of the female genital tract in industrialized countries (1). The etiology of EC is not yet fully understood, although there is some evidence that molecular modifications and hormonal influences contribute to its initiation and progression (2).
Endometrial cancer is divided into two major classes: estrogen-dependent tumors (type I, endometrioid endometrial carcinomas) that represent 80% of cases and non-estrogen-dependent (type II, non-endometrioid endometrial carcinomas) that account for the rest. EC type I is thought to develop via precursor lesions along with the increasing accumulation of molecular genetic aberration (3, 4).
Recently, the World Health Organization classification of tumors (WHO) (5) divided endometrial hyperplasias into two categories: hyperplasia without atypia (Benign Hyperplasia, BH) and atypical hyperplasia/endometrioid intraepithelial neoplasia: (AH/EIN) (5).
Indeed, AH/EIN is most likely to progress to type I endometrial carcinoma (~30%), and has been reported to be associated with invasive EC in 62% of endometrial biopsy (6), whereas BH rarely progresses to EC (<5%) (7). Therefore, discerning between these two entities has significant clinical implications (8).
Unfortunately, recognition of atypia in endometrial hyperplasia is subjective among pathologists with a low inter observer reproducibility (<50% in almost all studies) (9, 10).
Although the recent two-tier classification of these entities by WHO (5, 11) improved reproducibility, management of endometrial pre-cancers is compromised by a longstanding debate.
MicroRNAs (miRs) are a class of small, non-coding, single-stranded RNAs that negatively regulate gene expression mainly binding to 3′-untranslated region (UTR) of target mRNAs at the post-transcriptional level (12). Several studies showed that they are important in many biological processes, thus their aberrant expressions are closely associated with the development, invasion, metastasis, and prognosis of various cancers, including EC (13–17).
Up until now, several miR signatures have been documented in either normal or neoplastic endometrium, but the role of miRs in endometrial hyperplasia with or without atypia remains poorly understood (18–22). In the present study, we investigate the hypothesis that changes in miRs may represent useful biomarkers for the diagnosis of AH/EIN.
Materials and Methods
Endometrial Tissue Samples and Patients
Eighty-five archived formalin-fixed, paraffin embedded (FFPE) tissue blocks of BH (41 cases), and AH/EIN (44 cases), were obtained from the Pathology Department of Sant'Andrea Hospital and Ospedale Cannizzaro, Catania from 2004 to 2013. Patient's age ranged from 37 to 84 years, with a median of 56 years. This study was authorized by the institutional ethics committee board at S. Andrea Hospital Rome, Italy (Aut. #168/03). Written informed consent was obtained from all patients enrolled.
The selected cases were randomly divided into a training set (23 BH, 19 AH/EIN) and into a validation set (21 BH, 22 AH/EIN).
Hyperplasia was macro or laser-microdissected, were appropriate, for this study.
RNA Extraction
Total RNA, including miRs fraction, was extracted from FFPE tissues using the High Pure miRNA isolation kit (Roche) according to the manufacturer's instructions. RNAs concentration were assessed using Nanodrop (ThermoScientific).
Affymetrix Gene Chip miRNA Array
RNA quality and purity were assessed with the use of the RNA 6000 Nano assay on Agilent 2100 Bioanalyzer (Agilent). Briefly, 500 ng of total RNA was labeled using FlashTag Biotin HSR (Genisphere LLC) and hybridized to GeneChip® miRNA 2.0 Arrays. The arrays were stained in the Fluidics Station 450 and then scanned on the GeneChip® Scanner 3000 (Affymetrix, USA).
Microarray Data Analysis
The statistical analysis was performed by Transcriptome Analysis Console (TAC) software (Thermo Fisher Scientific).
To survey outliers that could disturb the dataset, a Principal Component Analysis (implemented by means of R statistical software) was performed and its visualization, which led to the knowledge of which subjects needed to be excluded from the dataset. MicroRNA probe outliers were defined from the manufacturer's instructions (Affymetrix, USA), and further analysis included data summarization, normalization, and quality control using the web-based miRNA QC Tool software (Affymetrix).
The microarray data has been submitted and assigned a GEO omnibus accession number GSE85105.
Reverse Transcription and Quantitative Real-Time PCRs
Each sample was reverse-transcribed using miRNA miRCURY LNA Universal RT kit (Exiqon) according to the manufacturer's protocol.
Reverse transcription and quantitative real-time PCRs were performed for miRNAs using miRCURY LNA Universal RT microRNA PCR LNA primers set with miRCURY LNA cDNA Synthesis Kit II and ExiLENT Syber Green master mix, in triplicate (Exiqon). RNU48 (U48) was used to normalize input total small RNA.
Expression of each miR was presented as the ratio between miR and RNU48 (RQ). The relative miRs expression was calculated using the ΔΔCt method. At least three separate experiments were performed, and each sample was assayed in triplicate.
Cells Transfections
Pre-designed Pre-miR (miR Precursors) for each miR was obtained from Ambion (ThermoFisher Scientific). A negative-control miRNA mimic [Pre-miR miRNA negative #1 (Ambion, ThermoFisher Scientific)] was used to address the specificity of the observed effect to the specific miR sequence. Cells were transfected using Lipofectamine RNAimax (ThermoFisher Scientific).
Protein Extraction, Western Blotting, and Antibodies
Hec1a cell lines were obtained from ATCC (Atcc, HTB-112) and cultured according to the manufacturer's protocol. Total cell extracts with RIPA buffer (Sigma Aldrich) were collected at 24 h and analyzed by western blot to assess proteins expression levels. Briefly, Hec1a cells were rinsed in ice-cold PBS and subsequently lysed in ice-cold RIPA lysis buffer (Sigma Aldrich) and Complete inhibitor (Roche). Proteins were analyzed on pre-cast polyacrylamide gel (Bio-Rad), transferred onto nitrocellulose membranes (Bio-Rad) and blocked with 5% BSA (Sigma Aldrich), and incubated with specific primary antibodies.
Polyclonal antibody against SMAD4 (sc-7966, Santa Cruz Biotechnology) diluted 1:200. Monoclonal antibody against PAX2 (aJ1589a, Abgent) diluted 1:1,000. Monoclonal antibody against Pten (560002, BD Biosciences) diluted 1:1,000.
To normalize protein loading, membranes were probed for 1 h at room temperature with an anti-vinculin antibody (sc- 25336, Santa Cruz Biotechnology).
Secondary antibodies (labeled HRP anti-rabbit or anti-mouse, Bio-Rad) were incubated for 45 min at room temperature and revealed with chemiluminescent ECL method (Bio-Rad).
Digital images of autoradiography were acquired with ChemiDOC XRS (Bio-Rad).
Plasmids and Constructs
The 3′UTR of the SMAD4 gene was obtained from GeneArt Gene Synthesis (Invitrogen, Thermo Fisher Scientific) by cloning 900bp of SMAD4 3′UTR into pMir-vector (Promega) giving rise to the pMir-3′UTRSMAD4 construct.
Site-direct mutagenesis into the miR-205, miR146a, and miR-1260b binding sites of the SMAD4 gene 3′UTR were introduced using GeneArt Site-Directed Mutagenesis PLUS System kit (Thermo Fisher Scientific) according to manufacturer's instructions. The primers used were:
FP 5′CTTCACCTGTTATGTAcctgccAATCATTCCAGTGC3′
RP 5′GCACTGGAATGATTggcaggTACATAACAGGTGAAG3′
FP 5′GCTGATTTTAAAGGCAGAGAAccgtcgAAAGTTAATTCACC3′
RP 5′GGTGAATTAACTTTcgacggTTCTCTGCCTTTAAAATCAGC3′
FP 5′GTTATTCCTAGTGacccgtTGTTGATGAAGTATACTTTTCCCC3′
RP 5′GGGGAAAAGTATACTTCATCAACAacgggtCACTAGGAATAAC3
Luciferase Activity Assays
Hec1a cells were cultured in 12-well-plates and transfected with 500 ng of pMir-3′UTRSMAD4 wt or mutated plasmid or pMir control vector together with 50 ng of β-GAL vector and 50 pmoles of pre-miR-205, pre-miR-146a, pre-miR-1260b, or pre-miR-negative control#1 (Thermo Fisher Scientific). Transfections were carried out using Lipofectamine 2000 and OPTI-MEM as recommended by the manufacturer (Thermo Fisher Scientific). At 48 h after transfection, luciferase activity was measured using the Luciferase Reporter Assay (Promega). Each transfection was repeated twice in triplicate. Transfection efficiency was corrected to β-GAL expression in all cases.
Cell Proliferation Assay
Cell proliferation was measured using Muse® Count & Viability Assay Kit and Muse® Cell Analyzer as recommended by the manufacturer (Merck Millipore). Cells were transfected with each pre-miR and the pre-miR-negative control#1 into a 35 mm dish as described below and incubated for 72 h. Three independent experiments were performed in duplicate.
Transwell Migration Assay
The migration ability of Hec1A cells was determined in a Boyden Chamber. Twenty-four hours after transfection, the cells were seeded into 8 μm Transwells (6.5 mm diameter, Corning) at 5 ×104 cells well with serum-free culture medium. Medium containing 10% FBS was added into the lower chamber and served as the chemoattractant. After incubation for 24 h, the cells remaining on the upper surface of the filter were removed by gently wiping with a cotton swab. The cells migrated through the filter were fixed with methanol, stained with MGG quick staining (Bio Optica), and visualized by an inverted fluorescence microscope.
Statistical Analysis
Data were expressed as the mean ± SD from at least three independent experiments. Statistical analysis between two samples was performed using Student's t-test. Statistical comparisons of more than two groups were performed using one-way analysis of variance (ANOVA).
The diagnostic ability of miRs-205, −146a, and −1260b in diagnosing atypical hyperplasia was examined via the area under the corresponding receiver operating characteristic curve (AUC). All statistical analyses were performed using Graph Pad Prism software (GraphPad software). p < 0.05 was considered statistically significant.
Results
Dysregulated miRs in BH vs. AH/EIN
To investigate whether miRs could discriminate BH from AH/EIN, we analyzed the expression of 1,105 human miRs (miRbase version 15) in the training set (23 BH, 19 AH/EIN). Examples of BH and AH are presented in Figures 1A,B.
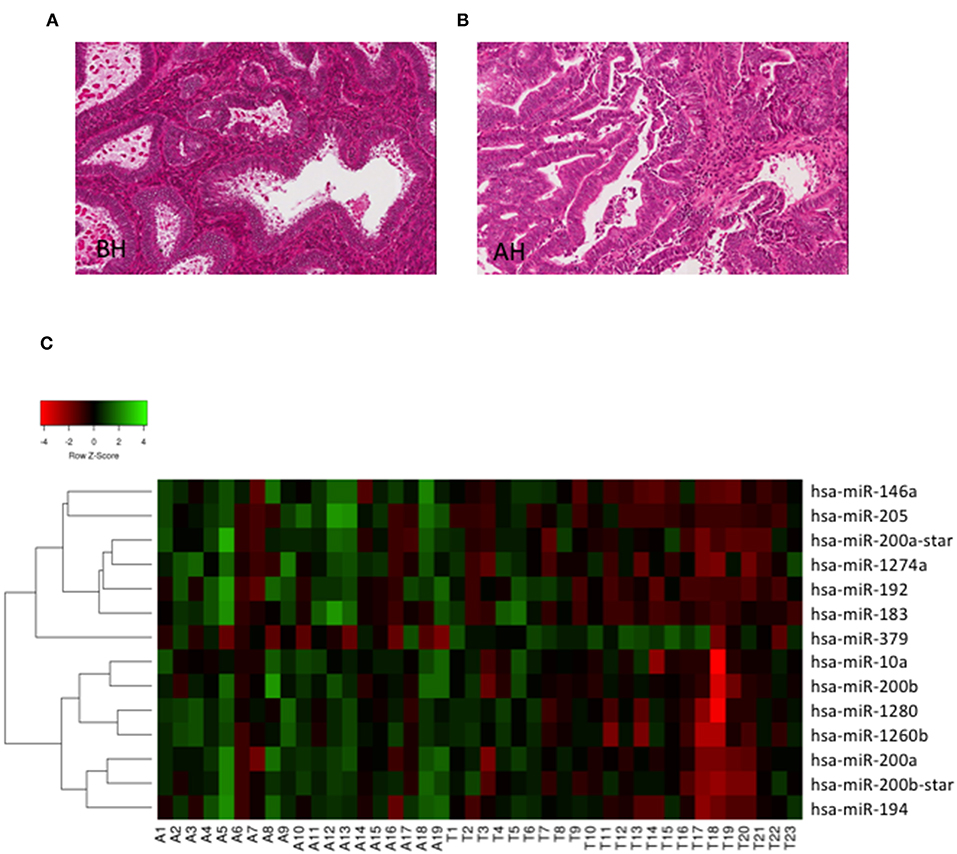
Figure 1. Hierarchical clustering. (A,B) Representative H/E stain images of BH and AH (Magnification 20X). (C) Heat Map depicting hierarchical cluster analysis of the 14 miRs differentially expressed between AH/EIN (A) and BH (T) identified by microarrays analysis.
As shown in Figure 1C, we could identify 14 differentially expressed miRs capable of discriminating BH from AH/EIN (FC ≥ 1.5, p ≤ 0.05). In particular, 13 miRs were upregulated (miRs-205,-146a, −200b_star, −1274a, −1260b, −200b, −200a, −192, −183, −10, −194, and −200a_star) and 1 (miR-379) was downregulated in AH/EIN compared to BH samples.
Using multiple logistic regression, the statistical significant variables (Age, BMI, Parity, miR values) were assessed in univariate analysis and investigated comparing BH to AH/EIN. No significant correlation was observed (data not shown).
Differentially expressed miRs were then validated in an independent validation set (21 BH, 22 AH/EIN). Out of the 14 miRs initially identified, we could confirm three miRs all up-regulated (miR-205, −146a, and −1260b) (Figure 2A), suggesting that these miRs could discriminate between the two groups.
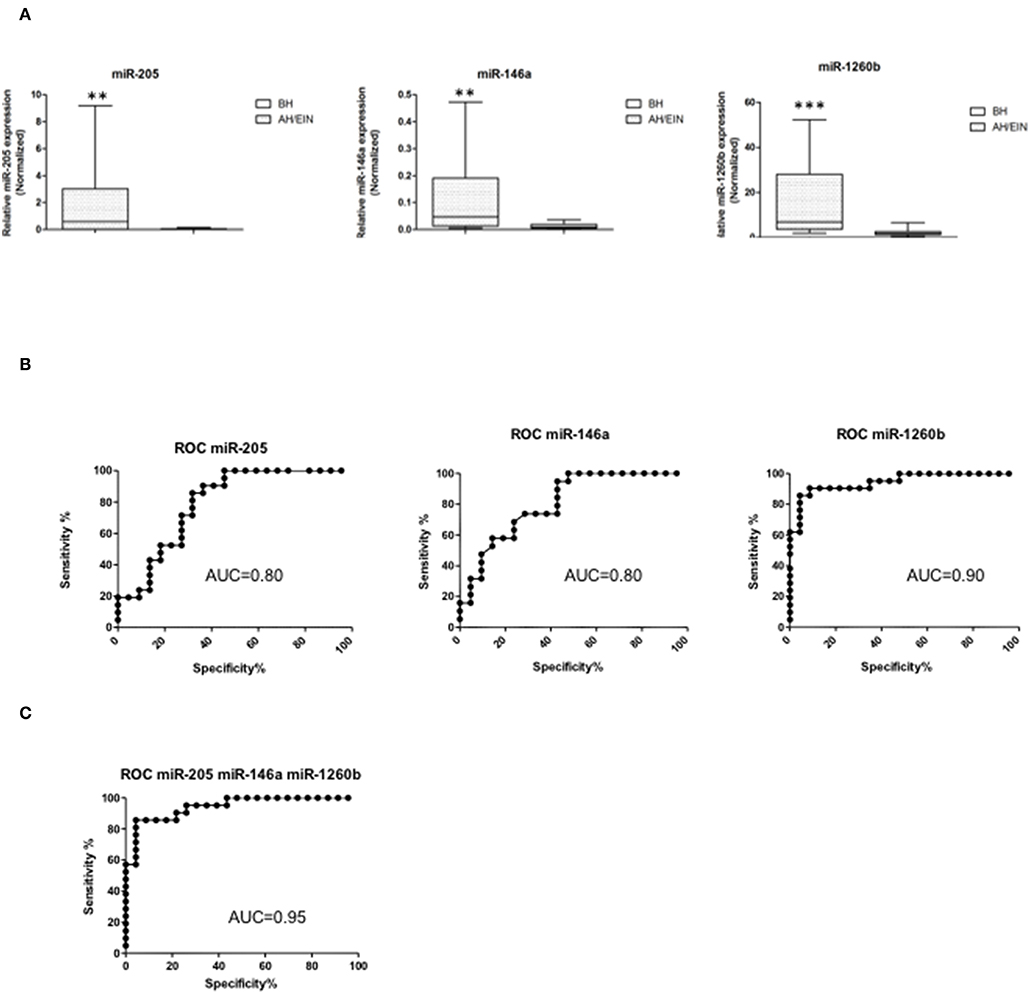
Figure 2. Validation and ROC curve analysis of identified miRs. (A) Validated differentially expressed miRs are shown. Expression levels of miRs-205,−146a, and−1260b were significantly higher in AH/EIN compared with BH. The horizontal lines indicate the median value. **p ≤ 0.01, ***p ≤ 0.001. (B,C) Accuracy for each and for the combination of the three miRs in differentiating AH/EIN from BH, respectively. AUC is shown. All p ≤ 0.05.
To assess the ability of each miR to differentiate between AH/EIN and BH, receiver-operating characteristic curves (ROC) were constructed and the area under the curve (AUC) was calculated. Univariate analysis for each individual miR showed an AUC of 0.8 [95% confidence interval (CI) = 0.66–0.93 p = 0.0009] for miR-205, an AUC of 0.8 (95% CI = 0.68–0.94 p = 0.0008) for miR-146a, and an AUC of 0.9 (95% CI = 0.88–1.01 p < 0.0001) for miR-1260b, respectively (Figure 2B). Performing a multivariate analysis for the combination of the three miRs, we observed an AUC of 0.95 (95% CI 0.88–1.01 p < 0.0001) showing that these three miRs have a high predictive power in discriminating AH/EIN from BH (Figure 2C).
SMAD4 Is a Target of miRs-205, 146a, and 1260b
To investigate a biological role for these miRs in AH/EIN, we searched different prediction algorithms. We found that highly conserved binding sites for each of these miRs were present in the mRNA of the oncosuppressor gene SMAD4, which has been shown to be down-modulated in EC (23).
We used endometrial cancer-derived cell lines Hec1a and tested the endogenous expression of these miRs.
QRT-PCR analysis showed that Hec1a cells expressed detectable amounts of each miRs (Figure 3A). To investigate the effects of these miRs on SMAD4 expression, we transiently transfected pre-miRs-146a, −205, and −1260b or control into Hec1a cells.
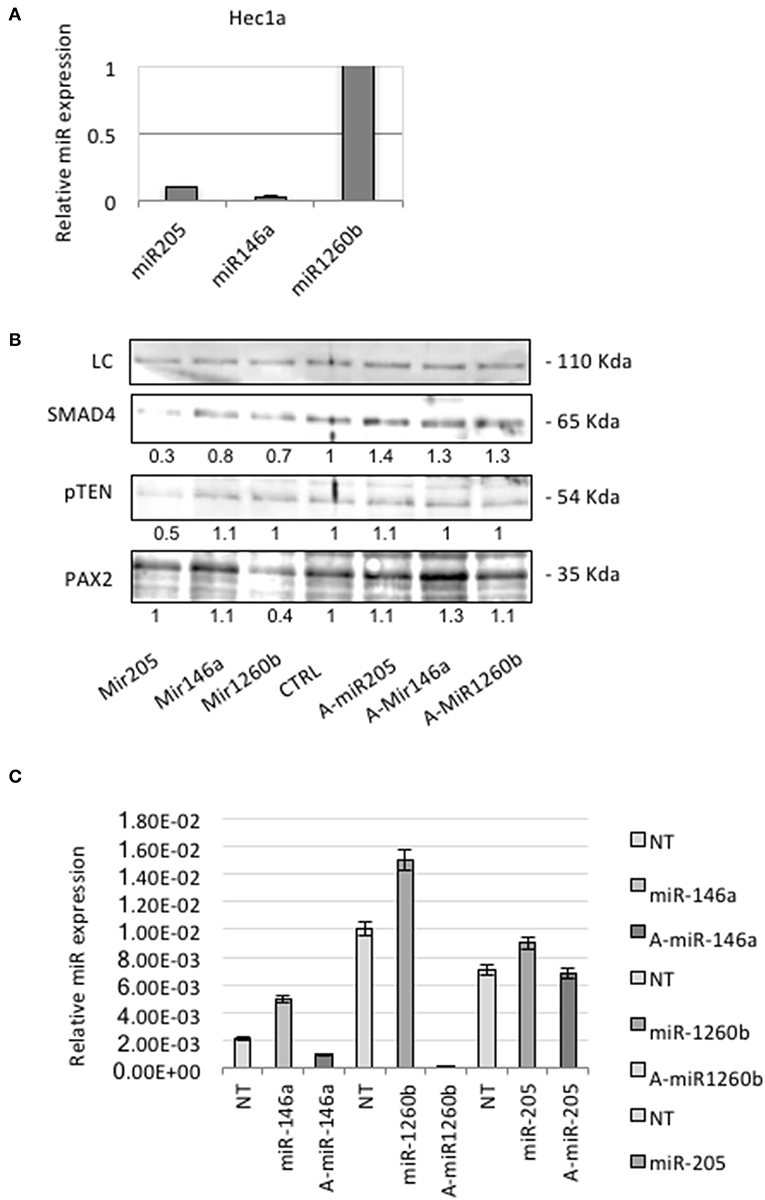
Figure 3. miRs-205,−146a, and−1260b regulate SMAD4 levels. (A) Expression levels of each miR were assessed in Hec1a cell line. Relative expression of miRs-205,−146a, and−1260b is reported. Each Ct value is normalized to RNU48. (B) Hec1a cells transfected as showed. The Smad4 protein appears as a band at approximately 60 kDa. Actin (~40 kDa) was used as a loading control LC. Densitometry value is reported under each line. (C) RT-PCR verification of the transfection efficiency of Hec1a cells transfected in B. Each bar shows miR expression normalized to RNU48 ±SD of three independent experiments.
In miRs transfected cells, we observed a significant suppression of SMAD4 compared to control (Figure 3B). To further confirm this observation, we transiently transfected Hec1a cells with antagomirs. This resulted in an increase of SMAD4 levels, compared to control (Figures 3B,C), as expected.
Co-loss of both PTEN and PAX2 has been reported in AH/EIN and it has been regarded as the reference markers for its diagnosis (24).
Therefore, we analyzed Pax2 levels by Western blot and we observed a strong protein reduction in miR-1260b transfected cells. There is no evidence of interaction between these two players, but miR binding sites algorithm prediction showed that other Pax family members are the putative target of miR-1260b. We also found that pTEN was downregulated in miR-205 transfected cells confirming previous evidence showing the potential role of miR-205 in regulating PTEN in endometrial tissue (25).
Although by different means, Smad4 could be a target of these miRs, one can argue that miR-146a, miR-205, and miR-1260b interact with other unknown targets that down-regulate Smad4 protein levels (18–22) (Figure 4A). To address this concern, we performed a luciferase reporter assay, cloning a 900 bp 3′UTR of human SMAD4 into a pMIR vector (p3′UTRSmad4pMir). Therefore, Hec1a cells were transfected either with miR-146a, miR-205, mir-1260b, or the pre-miR-control, and p3′UTRSmad4pMir vector. As shown in Figure 4B, all miRs decrease luciferase activity of the p3′UTRSmad4pMir compared to control, showing that each miR has a direct effect on their target in this cell lines (Figure 4B). To determine this direct miR-target interaction, we constructed a plasmid with mutagenesis of the three seed sequences (Figure 4C). As expected, we observed only a slight effect on luciferase activity when we compared the wild-type vector with the p3′UTRSmad4pMir mutants in the presence of each miRs overexpressed, showing that the modification of the seed sequence is enough to block the function of each miR (Figure 4C).
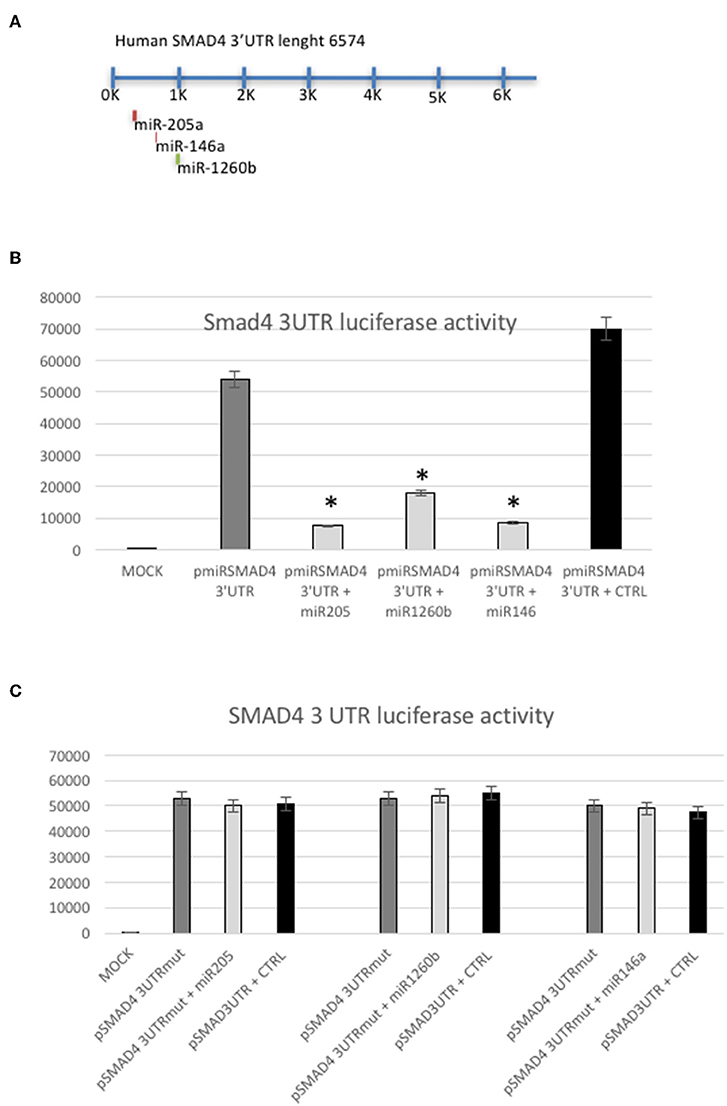
Figure 4. SMAD4 is a target of miRs-205, −146a, and −1260b. (A) Schematic representation of predicted miRs binding sites in the SMAD4 3′UTR. (B) Luciferase reporter assay in Hec1a cells co-transfected with the reporter gene containing the SMAD4 3′UTR alone (pmiRSMAD4 3′UTR) (gray bar), the SMAD4 3′UTR and miRs-205, −1260b, and −146a (light gray bars), respectively and the negative control (CTRL) (black bar). Each reporter plasmid was transfected three times, and each sample was assayed in triplicate. (C) Luciferase reporter assay performed in Hec1a cells co-transfected with the reporter gene containing the SMAD4 3′UTR mutated (pmiRSMAD4 3′UTR-Mut) in the miRs-205, −146a, or −1260b seed sequences (gray bar) alone, the pmiRSMAD4 3′UTR-Mut in each mir seed sequence and miRs-205, −1260b, and −146a (light gray bars), respectively and the negative control (CTRL) (black bar). Bars indicate Firefly Luciferase activity normalized to β-Gal activity ±SD. *p ≤ 0.05 compared to control (CTRL) transfected cells.
SMAD4 Repression by miRs-205, 146a, and 1260b Induces Proliferation and Migration in Hec1a Cell Lines
Smad4 is involved in the signal transduction pathway of the transforming growth factor ß (TGF-ß) that acts as a tumor suppressor gene in several cancers (26, 27). To gain further insights into how dysregulation of these miRs may play a role in endometrial cancer cells, we performed different assays to study the biological effects of the interaction between these miRs and their target Smad4 into endometrial adenocarcinoma-derived cell lines Hec1a.
First, we tested cells proliferation. Cells transfected with pre-miRs-146a, −205, or −1260b showed a higher rate of proliferation compared with non-treated or control transfected cells (Figure 5A). Cells transfected with anti-miRs-146a, −205, or −1260b showed a reduction of proliferation compared to control (Figure 5B).
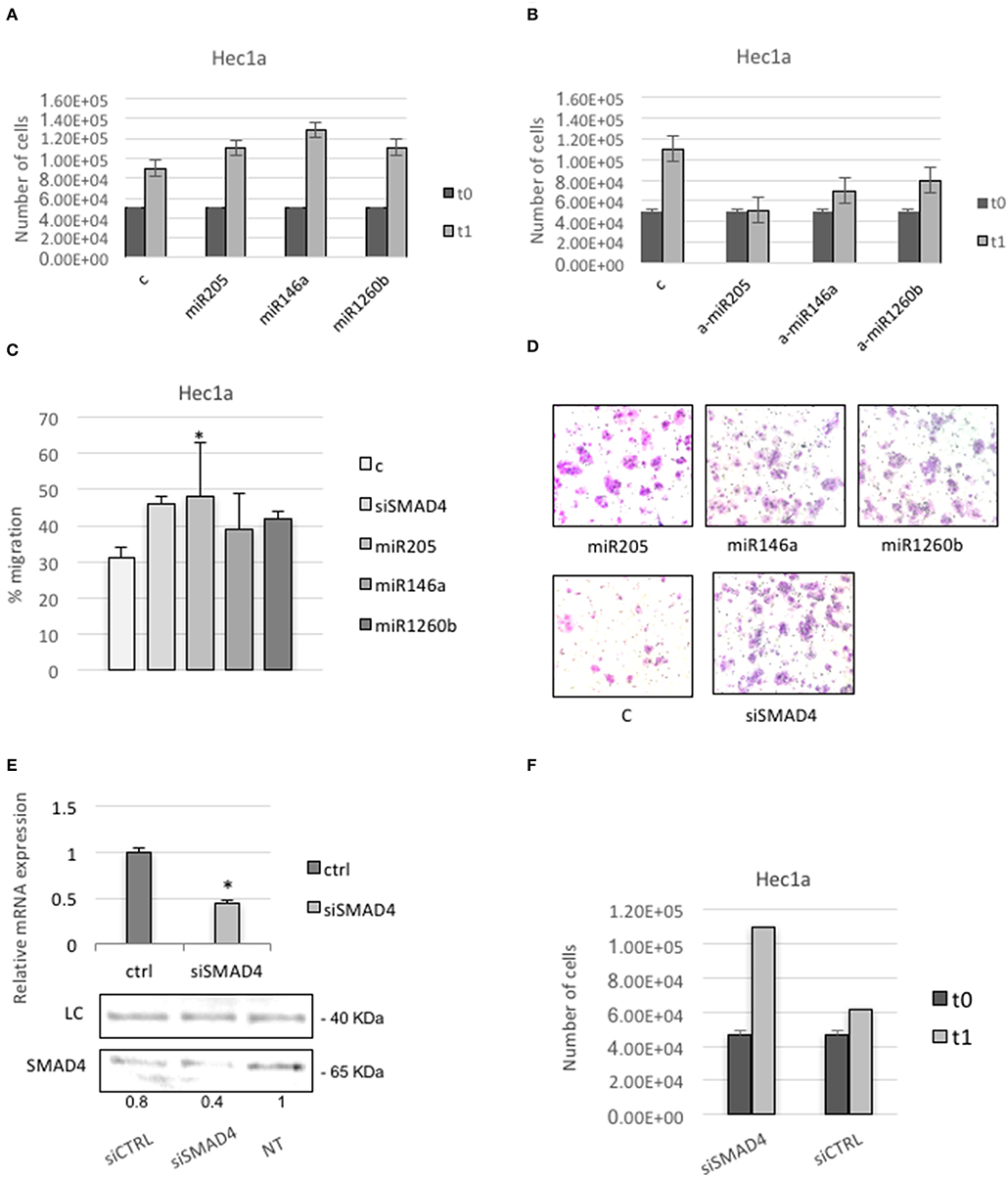
Figure 5. SMAD4 regulation by miRs-205, −146a, and −1260b induces proliferation and migration in Hec1a cells. (A,B) Hec1a cells proliferation was measured at 72 h. Cells were transiently transfected with negative control #1 (C) and miRs-205, −146a, −1260b in A or anti-miRs-205, −146a, and −1260b (a-miRs) in B. (C) Transwell migration assay shows that overexpression of miRs-205, −146a, −1260b, and siSMAD4 enhance cell migration ability of Hec1a cells. (D) Representative photographs of the Transwell migration assay is shown. All data are presented as mean ± SD, *p < 0.05. (E,F) Effects of siSMAD4 expression in Hec1a cells. (E) RT-PCR (upper panel) and western blot analysis (lower panel) are shown. LC (loading control, β-actin). PCR bars depict SMAD4 expression in control (ctrl) and si transfected cells normalized to β-actin. (F) Measure of Hec1a cells proliferation in siSMAD4 and SiCTRL transfected cells is reported. Data represent the mean (from three independent experiments) ±SD. *P < 0.05.
To better understand how the dysregulation of these miRs may change the behavior of endometrial cancer-derived cell line, we examined the influence of Smad4 knockdown on Hec1a cell migration. We found a significant increase in the migration capability in pre-miR-205, −146a, or −1260b transfected cells compared to control, showing that these miRs positively regulate the migration of cultured endometrial cancer cells (Figures 5C,D).
Since miRs may have multiple targets, to ensure that Smad4 mediated the observed effects, we repeated the assay using a specific Smad4 siRNA.
We confirm significant down-regulation of Smad4 through qRT-PCR (Figure 5E). Next, we observed that Hec1a cells transfected with Smad4 siRNA displayed a higher proliferation and migration rate respect to controls (Figures 5C,F).
Discussion
Identification of molecular markers that can differentiate between AH/EIN and BH are considered to be highly useful for clinical management of patients because hyperplasia with atypical change and/or Endometrial Intraepithelial Neoplasia are associated with a higher risk to progress to cancer (4).
Since there aren't reference markers, the diagnosis is based only on histological features, such as the presence of nucleoli and other atypical characteristics, which are not consistently associated with that diagnosis (9).
Although the new WHO classification is more likely to successfully identify premalignant lesions, the low interobserver reproducibility among gynecological pathologist in diagnosing atypical hyperplasia/EIN should be improved (6, 10, 28–30).
Atypical endometrial hyperplasia/EIN and EC shares several molecular alterations with each other, including microsatellite instability, PAX2 inactivation, mutation of PTEN, KRAS, and CTNNB1 (β-catenin), but there is not a linear accumulation of mutational events leading to cancer (31). Identifying the disease-related miRs will improve the diagnosis and understanding of pathogenesis of these lesions.
Since over 50% of miRs reside in cancer-associated genomic regions, they have been indicated to play an important role as diagnostic biomarkers (13, 32, 33).
Most of the miRs studies on endometrium have been focused on the identification of their implications in EC development, almost neglecting their possible diagnostic role in precursor lesions (34). In fact, several authors showed an altered expression of miRs that may discriminate EC from non-atypical or atypical hyperplasia (9, 14, 21, 35–40).
In particular, expression of five miRs (miRs-182, 183, 200a, 200c, and 205) was significantly higher in EC when compared with complex atypical hyperplasia, simple hyperplasia (SH) and normal endometrial tissue (P < 0.05, respectively) (41).
To our knowledge, our study is the first to identify a miRs signature able to discriminate between atypical hyperplasia/EIN and benign endometrial hyperplasia with the capability to better distinguish between low- and high- risk lesions. Identification of miR-target genes and pathways to understand the molecular basis of endometrial cancer pathogenesis is a major challenge, as there are numerous pathways that drive cancer. Accordingly, in this study, we proposed a novel miR-based classification method to categorize the high risk pre-cancerous endometrial lesions.
In fact, we showed a high predictive power, above 90%, using a three miRs-signature (miRs-146a, −205, and −1260b) in distinguishing between non-atypical and atypical hyperplasia/EIN provides a supplementary diagnostic tool when required.
Interestingly, a previous study conducted by Snowdon and colleagues examined a miRs profile in atypical hyperplasia compared to normal proliferative controls. The microarray expression profile shares some important similarities with our data. MiRs-146a, miR-200a, miR-200b, miR-200b-star, and miR-205 resulted up-regulated and miR-542-5p down-regulated in atypical hyperplasia vs. normal endometrium, adding further emphasis to our results (22). The up-regulation of miR-200 family members and miR-205 in EC is observed among different studies, indicating that these miRs may play a role in driving oncogenesis in the endometrium (21, 35, 37, 42, 43).
Some authors showed that miR-205 is a negative prognostic marker for EC and its levels were significantly increased in endometrial cancer cell lines and endometrial tumors compared to normal tissues (25, 26). Mir-205 is directly involved in PTEN regulation that represents one of the most commonly investigated markers implicated in endometrial tumorigenesis (44). Our results have enforced this effect indicating a remarkable influence of miR-205 on regulating essential target genes involved in different signal pathways in endometrial cells. On the other hand, Lacey et al. have shown that a loss of expression of PTEN status was not associated with progression risk of endometrial hyperplasia (45).
Even if EC seems to be characterized by elevated expression of miR-205, a recent study conducted by Wilczynski showed that higher levels of miR-205 may be a marker of an early stage disease and is associated with a more favorable prognosis, whereas patients with lower levels of miR-205 had worse survival (42).
No evidence has revealed the dysregulation of miRs-1260b and −146a in EC, suggesting that up-regulation of these miRs may be specific of AH/EIN.
Interestingly, authors found that a single nucleotide polymorphism (SNP) rs2910164 G>C within miR-146a is associated with the increased risk of gastric cancer and papillary thyroid carcinoma (46, 47). A recent publication showed that overexpression of miR-146a inhibited cell proliferation, enhanced apoptosis, and increased sensitivity to chemotherapy drugs in epithelial ovarian cancers cells showing, therefore, that the role of miR-146a is still to be elucidated (48).
MiR-1260b has been found to be highly expressed in the prostate, renal cell, and in colorectal carcinomas (49, 52). Recently, it was demonstrated that in Hepatocellular carcinoma, MiR-1260b promotes cell migration and invasion through the G-protein signaling 22 (50).
All of this evidence confirmed that our identified miRs regulate genes involved in different signal pathways that may trigger the endometrial cellular transformation.
Thus, we investigated a possible common pathway that could be regulated by these miRs and could be implicated in cellular transformation, and we found that they target SMAD4. Also noteworthy are several other reports, which demonstrated a direct interaction of these miRs and this transcript (51–53).
Smad4 is a gene implicated in several cancers, including EC, albeit its role in endometrial carcinogenesis is yet not clear (54–56).
Impairment of the Smad pathway results in escape from growth inhibition and leads to the promotion of cell proliferation, contributing to carcinogenesis (57).
The disturbances in Smad proteins expression and/or differences in their intracellular distribution, that trigger a TGF-β signaling pathway deregulation, it was reported in endometrial carcinomas, but it is still not well-understood (58). The region within 18q21 where Smad4 is located is frequently deleted in endometrial carcinomas, showing its involvement in EC, however, an immunohistochemical study showed that inactivation of this gene occurs infrequently in this tumor.
Changes in the expression of the TGF-β signaling cascade in type I ECs seem to be associated mainly with deregulation of TGF-β receptors and SMAD expression at the protein level, indicating SMAD4 as a central molecule of this pathway (59, 60).
Thus, the potential pathogenic role of SMAD4 in endometrial hyperplasia is supported by our finding, albeit further studies are required to understand its biological and diagnostic role in this environment.
Our results clearly demonstrated that overexpression of miRs-146a, −205, and −1260b induced Hec1a proliferation and migration through SMAD4 inhibition, providing an insight into the possible mechanisms underlying the function of these miRs in endometrial hyperplasia.
Thus, our work highlights the relevance of miRs in regulating cellular processes that may ultimately lead to tumorigenesis.
Taken together these results strongly show that miRs-205, −146a, and −1260b contribute to enhancing proliferation and migration properties of endometrial cancer cells through Smad4 inhibition.
In conclusion, distinguishing between hyperplasia and true pre-cancerous lesions has significant clinical implications because distinct endometrial pre-cancerous conditions require intervention. Thus, we proposed a three miR-signature (146a −205, −1260b) as a potential biomarker for diagnosis of atypical endometrial hyperplasia/EIN that could have a significant impact on treatment decisions. Furthermore, the regulatory capability of these three miRs on cell proliferation and migration, possibly through impairment of TGF-β signaling Smad4-mediated, highlights their crucial role in endometrial hyperplasia outcome.
Although we believe that this study represents a step forward in investigating the molecular relationship between miR deregulation and EIN lesions, we analyzed a relatively small group of patients, and therefore, a prospective analysis is needed to strengthen the accuracy of our results.
Ethics Statement
This study was carried out in accordance with the recommendations of name of guidelines, name of committee with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the IRB committee.
Author Contributions
SG and AV: study conception and design and drafting the manuscript. SG, VA, RC, OF, CD, and MP: methodology. SG, SV, and AV: analysis and interpretation of data. DC, AP, FF, and AV: access to clinical data. SG, DC, AP, FF, and AV: critical revision.
Funding
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC IG 16862 to AV) and La Sapienza University Fondi Ateneo 2017 to AV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. (2015) 65:5–29. doi: 10.3322/caac.21254
2. Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol. (2007) 2:57–87. doi: 10.1146/annurev.pathol.2.010506.091905
3. Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol. (2014) 15:268–78. doi: 10.1016/S1470-2045(13)70591-6
4. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. (1983) 15:10–7. doi: 10.1016/0090-8258(83)90111-7
5. Kurman RJ, Carcangiu ML, Herrington CS, Young RH. WHO Classification of Tumours of Female Reproductive Organs. Lyon: IARC (2014).
6. Trimble CL, Kauderer J, Zaino R, Silverberg S, Lim PC, Burke JJ, et al. Concurrent endometrial carcinoma in women with a biopsy diagnosis of atypical endometrial hyperplasia: a gynecologic oncology group study. Cancer. (2006) 106:812–9. doi: 10.1002/cncr.21650
7. Kurman RJ, Kaminski PF, Norris HJ. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer. (1985) 56:403–12. doi: 10.1002/1097-0142(19850715)56:2<403::AID-CNCR2820560233>3.0.CO;2-X
8. Emons G, Beckmann MW, Schmidt D, Mallmann P. New WHO classification of endometrial hyperplasias. Geburtsh Frauenheilk. (2015) 75:135–6. doi: 10.1055/s-0034-1396256
9. Kendall BS, Ronnett BM, Isacson C, Cho KR, Hedrick L, Diener-West M, et al. Reproducibility of the diagnosis of endometrial hyperplasia, atypical hyperplasia and well-differentiated carcinoma. Am J Surg Pathol. (1998) 22:1012–9. doi: 10.1097/00000478-199808000-00012
10. Bergeron C, Nogales FF, Masseroli M, Abeler V, Duvillard P, Müller-Holzner E, et al. A multicentric European study testing the reproducibility of the WHO classification of endometrial hyperplasia with a proposal of a simplified working classification for biopsy and curettage specimens. Am J Surg Pathol. (1999) 23:1102–8. doi: 10.1097/00000478-199909000-00014
11. Ordi J, Bergeron C, Hardisson D, McCluggage WG, Hollema H, Felix A, et al. Reproducibility of current classifications of endometrial endometrioid glandular proliferations: further evidence supporting a simplified classification. Histopathology. (2014) 64:284–92. doi: 10.1111/his.12249
13. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. (2006) 6:857–66. doi: 10.1038/nrc1997
14. Boren T, Xiong Y, Hakam A, Wenham R, Apte S, Wei Z, et al. MicroRNAs and their messenger RNAs associated with endometrial carcinogenesis. Gynecol Oncol. (2008) 110:206–15. doi: 10.1016/j.ygyno.2008.03.023
15. Giglio S, Vecchione A. Role of microRNAs in the molecular diagnosis of cancer. JNAI. (2010) 1:e4. doi: 10.4081/jnai.2010.1659
16. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med. (2014) 20:460–9. doi: 10.1016/j.molmed.2014.06.005
17. Jayaraman M, Radhakrishnan R, Mathews CA, Yan M, Husain S, Moxley KM, et al. Identification of novel diagnostic and prognostic miRNA signatures in endometrial cancer. Genes Cancer. (2017) 8:566–76. doi: 10.18632/genesandcancer.144
18. Kaur S, Lotsari JE, Al-Sohaily S, Warusavitarne J, Kohonen-Corish MR, Peltomäki P. Identification of subgroup-specific miRNA, patterns by epigenetic profiling of sporadic and Lynch syndrome-associated colorectal and endometrial carcinoma. Clin Epigene. (2015) 7:20. doi: 10.1186/s13148-015-0059-3
19. Xu YY, Wu HJ, Ma HD. microRNA-503 suppresses proliferation and cell-cycle progression of endometrioid endometrial cancer by negatively regulating cyclin D1. FEBS J. (2013) 280:3768–79. doi: 10.1111/febs.12365
20. Lee TS, Jeon HW, Kim YB. Aberrant microRNA expression in endometrial carcinoma using formalin-fixed paraffin-embedded (FFPE) tissues. PLoS ONE. (2013) 8:e81421. doi: 10.1371/journal.pone.0081421
21. Lee H, Choi HJ, Kang CS, Lee HJ, Lee WS, Park CS. Expression of miRNAs and PTEN in endometrial specimens ranging from histologically normal to hyperplasia and endometrial adenocarcinoma. Mod Pathol. (2012) 25:1508–15. doi: 10.1038/modpathol.2012.111
22. Snowdon J, Zhang X, Childs T, Tron VA, Feilotter H. The microRNA-200 family is upregulated in endometrial carcinoma. PLoS ONE. (2011) 6:e22828. doi: 10.1371/journal.pone.0022828
23. Piestrzeniewicz-Ulanska D, Brys M, Semczuk A, Jakowicki JA, Krajewska WM. Expression and intracellular localization of Smad proteins in human endometrial cancer. Oncol Rep. (2003) 10:1539–44. doi: 10.3892/or.10.5.1539
24. Monte NM, Webster KA, Neuberg D, Dressler GR, Mutter GL. Joint loss of PAX2 and PTEN expression in endometrial precancers and cancer. Cancer Res. (2010) 70:6225–32. doi: 10.1158/0008-5472.CAN-10-0149
25. Karaayvaz M, Zhang C, Liang S, Shroyer KR, Ju J. Prognostic significance of miR-205 in endometrial cancer. PLoS ONE. (2012) 7:e35158. doi: 10.1371/journal.pone.0035158
26. Tabruyn SP, Hansen S, Ojeda-Fernández ML, Bovy N, Zarrabeitia R, Recio-Poveda L, et al. MiR-205 is downregulated in hereditary hemorrhagic telangiectasia and impairs TGF-beta signaling pathways in endothelial cells. Angiogenesis. (2013) 16:877–87. doi: 10.1007/s10456-013-9362-9
27. Zhong H, Wang HR, Yang S, Zhong JH, Wang T, Wang C, et al. Targeting Smad4 links microRNA-146a to the TGF-beta pathway during retinoid acid induction in the acute promyelocytic leukemia cell line. Int J Hematol. (2010) 92:129–35. doi: 10.1007/s12185-010-0626-5
28. Skov BG, Broholm H, Engel U, Franzmann MB, Nielsen AL, Lauritzen AF, et al. Comparison of the reproducibility of the WHO classifications of 1975 and 1994 of endometrial hyperplasia. Int J Gynecol Pathol. (1997) 16:33–7. doi: 10.1097/00004347-199701000-00006
29. Zaino RJ, Kauderer J, Trimble CL. Reproducibility of the diagnosis of atypical endometrial hyperplasia a gynecologic oncology group study. Cancer. (2006) 106:804–11. doi: 10.1002/cncr.21649
30. Raffone A, Travaglino A, Saccone G. Endometrial hyperplasia and progression to cancer: which classification system stratifies the risk better? A systematic review and meta-analysis. Arch Gynecol Obstet. (2019) 299:1233–42. doi: 10.1007/s00404-019-05103-1
31. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. (2013) 497:67–73. doi: 10.1038/nature12113
32. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring, therapeutics. A comprehensive review. EMBO Mol Med. (2012) 4:143–59. doi: 10.1002/emmm.201100209
33. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. (2009) 10:704–14. doi: 10.1038/nrg2634
34. Ratner ES, Tuck D, Richter C, Nallur S, Patel RM, Schultz V, et al. MicroRNA signatures differentiate uterine cancer tumor subtypes. Gynecol Oncol. (2010) 118:251–7. doi: 10.1016/j.ygyno.2010.05.010
35. Pan Q, Chegini N. MicroRNA signature and regulatory functions in the endometrium during normal and disease states. Semin Reprod Med. (2008) 26:479–93. doi: 10.1055/s-0028-1096128
36. Cohn DE, Fabbri M, Valeri N, Alder H, Ivanov I, Liu CG, et al. Comprehensive miRNA profiling of surgically staged endometrial cancer. Am J Obstet Gynecol. (2010) 202:651–8. doi: 10.1016/j.ajog.2010.02.051
37. Hiroki E, Akahira J, Suzuki F, Nagase S, Ito K, Suzuki T, et al. Changes in microRNA expression levels correlate with clinicopathological features and prognoses in endometrial serous adenocarcinomas. Cancer Sci. (2010) 101:241–9. doi: 10.1111/j.1349-7006.2009.01385.x
38. Wu W, Lin Z, Zhuang Z, Liang X. Expression profile of mammalian microRNAs in endometrioid adenocarcinoma. Eur J Cancer Prev. (2009) 18:50–5. doi: 10.1097/CEJ.0b013e328305a07a
39. Pan Q, Luo X, Toloubeydokhti T, Chegini N. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol Hum Reprod. (2007) 13:797–806. doi: 10.1093/molehr/gam063
40. Chung TK, Cheung TH, Huen NY, Wong KW, Lo KW, Yim SF, et al. Dysregulated microRNAs and their predicted targets associated with endometrioid endometrial adenocarcinoma in Hong Kong women. Int J Cancer. (2009) 124:1358–65. doi: 10.1002/ijc.24071
41. Lee JW, Park YA, Choi JJ, Lee YY, Kim CJ, Choi C, et al. The expression of the miRNA-200 family in endometrial endometrioid carcinoma. Gynecol Oncol. (2011) 120:56–62. doi: 10.1016/j.ygyno.2010.09.022
42. Wilczynski M, Danielska J, Dzieniecka M, Szymanska B, Wojciechowski M, Malinowski A. Prognostic and clinical significance of miRNA-205 in endometrioid endometrial cancer. PLoS ONE. (2016) 11:e0164687. doi: 10.1371/journal.pone.0164687
43. Zhang G, Hou X, Li Y, Zhao M. MiR-205 inhibits cell apoptosis by targeting phosphatase and tensin homolog deleted on chromosome ten in endometrial cancer Ishikawa cells. BMC Cancer. (2014) 14:440. doi: 10.1186/1471-2407-14-440
44. Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP, Eng C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. (2000) 92:924–30. doi: 10.1093/jnci/92.11.924
45. Lacey JV Jr, Mutter GL, Ronnett BM. PTEN expression in endometrial biopsies as a marker of progression to endometrial carcinoma. Cancer Res. (2008) 68:6014–20. doi: 10.1158/0008-5472.CAN-08-1154
46. Xia ZG, Yin HF, Long Y, Cheng L, Yu LJ, Guo WJ, et al. Genetic variant of miR-146a rs2910164 C>G and gastric cancer susceptibility. Oncotarget. (2016) 7:34316–21. doi: 10.18632/oncotarget.8814
47. Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci USA. (2008) 105:7269–74. doi: 10.1073/pnas.0802682105
48. Cui Y, She K, Tian D, Zhang P, Xin X. MiR-146a inhibits proliferation and enhances chemosensitivity in epithelial ovarian cancer via reduction of SOD2. Oncol Res. (2016) 23:275–82. doi: 10.3727/096504016X14562725373798
49. Liu DR, Guan QL, Gao MT, Jiang L, Kang HX. MiR-1260b is a potential prognostic biomarker in colorectal cancer. Med Sci Monit. (2016) 22:2417–23. doi: 10.12659/MSM.898733
50. Li X, Song H, Liu Z, Bi Y. miR-1260b promotes cell migration and invasion of hepatocellular carcinoma by targeting the regulator of G-protein signaling. Biotechnol Lett. (2018) 40:57–62. doi: 10.1007/s10529-017-2455-6
51. Xiao B, Zhu ED, Li N, Lu DS, Li W, Li BS, et al. Increased miR-146a in gastric cancer directly targets SMAD4 and is involved in modulating cell proliferation and apoptosis. Oncol Rep. (2012) 27:559–66. doi: 10.3892/or.2011.1514
52. Hirata H, Hinoda Y, Shahryari V, Deng G, Tanaka Y, Tabatabai ZL, et al. Genistein downregulates onco-miR-1260b and upregulates sFRP1 and Smad4 via demethylation and histone modification in prostate cancer cells. Br J Cancer. (2014) 110:1645–54. doi: 10.1038/bjc.2014.48
53. Li J, Hu K, Gong G. Upregulation of MiR-205 transcriptionally suppresses SMAD4 and PTEN and contributes to human ovarian cancer progression. Sci Rep. (2017) 7:41330. doi: 10.1038/srep41330
54. Hahn SA, Schmiegel WH. Recent discoveries in cancer genetics of exocrine pancreatic neoplasia. Digestion. (1998) 59:493–501. doi: 10.1159/000007526
55. Kang YK, Kim WH, Jang JJ. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum Pathol. (2002) 33:877–83. doi: 10.1053/hupa.2002.127444
56. Liu FS, Chen JT, Hsieh YT, Ho ES, Hung MJ, Lu CH, et al. Loss of Smad4 protein expression occurs infrequently in endometrial carcinomas. Int J Gynecol Pathol. (2003) 22:347–52. doi: 10.1097/01.pgp.0000092131.88121.0a
57. Principe Daniel R, Doll Jennifer A, Jessica B, Barbara J, Munshi Hidayatullah G, Laurent B, et al. TGF-β: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. (2014) 106:djt369. doi: 10.1093/jnci/djt369
58. Piestrzeniewicz-Ulanska D, Brys M, Semczuk A, Rechberger T, Jakowicki JA, Krajewska WM. TGF-beta signaling is disrupted in endometrioid-type endometrial carcinomas. Gynecol Oncol. (2004) 95:173–80. doi: 10.1016/j.ygyno.2004.06.032
59. Ijichi H, Otsuka M, Tateishi K, Ikenoue T, Kawakami T, Kanai F, et al. Smad4-independent regulation of p21/WAF1 by transforming growth factor-beta. Oncogene. (2004) 23:1043–51. doi: 10.1038/sj.onc.1207222
Keywords: microRNAs, endometrial hyperplasia, endometrial cancer, biomarkers, SMAD4, TGF-β pathway
Citation: Giglio S, Annibali V, Cirombella R, Faruq O, Volinia S, De Vitis C, Pesce M, Caserta D, Pettinato A, Fraggetta F and Vecchione A (2019) miRNAs as Candidate Biomarker for the Accurate Detection of Atypical Endometrial Hyperplasia/Endometrial Intraepithelial Neoplasia. Front. Oncol. 9:526. doi: 10.3389/fonc.2019.00526
Received: 03 December 2018; Accepted: 30 May 2019;
Published: 21 June 2019.
Edited by:
Jian-Jun Wei, Northwestern University, United StatesReviewed by:
Tamara Louise Kalir, Icahn School of Medicine at Mount Sinai, United StatesMassimo Nabissi, University of Camerino, Italy
Copyright © 2019 Giglio, Annibali, Cirombella, Faruq, Volinia, De Vitis, Pesce, Caserta, Pettinato, Fraggetta and Vecchione. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Giglio, c2ltb25hLmdpZ2xpb0B1bmlyb21hMS5pdA==; Andrea Vecchione, YW5kcmVhLnZlY2NoaW9uZUB1bmlyb21hMS5pdA==
†Present Address: Viviana Annibali, Merck, Rome, Italy