 Philipp Wolf1,2*
Philipp Wolf1,2*- 1Department of Urology, Medical Center - University of Freiburg, Freiburg, Germany
- 2Faculty of Medicine, University of Freiburg, Freiburg, Germany
Introduction
Prostate cancer (PC) remains the second-most frequently diagnosed cancer among men worldwide and the fifth leading cause of cancer deaths (1). Despite great efforts to optimize radiotherapy, androgen deprivation, and chemotherapy, no curative treatment exists for advanced PC to date. This is due to the fact that during tumor progression and treatment, many changes occur in signaling pathways that lead to therapy resistance and treatment failure (2). Therefore, the search has to concentrate on signaling pathways that can be influenced even in advanced stages.
The Intrinsic Apoptotic Pathway in Prostate Cancer
Cells have the ability to commit suicide initiated by a finely tuned signal network after the influence of stimuli that require the cell to die. This is why we are talking about “programmed” cell death or apoptosis. In multicellular organisms apoptosis serves to eliminate surplus or damaged cells to preserve tissue and organ homeostasis (3).
Apoptosis can be triggered by two different pathways. The “extrinsic” apoptotic pathway is initiated through the stimulation of transmembrane death receptors. The “intrinsic” apoptotic pathway is initiated after internal cell damage and marked by the release of cytochrome C from mitochondria (3). Key regulators of the intrinsic apoptotic pathway are pro- and anti-apoptotic Bcl-2 (B-cell lymphoma 2) family proteins (4). In non-apoptotic cells the main anti-apoptotic members, Bcl-2, Bcl-xl, and Mcl-1, bind the pro-apoptotic effectors Bax and Bak. Upon induction of apoptosis, the pro-apoptotic activators (BID, BIM, and PUMA) and sensitizers (BAD and NOXA) are transcriptionally or post-transcriptionally activated and bind via their so called BH3 domain to the anti-apoptotic proteins to free Bax and Bak. Bax and Bak can oligomerize and form pores in the outer membrane of mitochondria. This event is termed mitochondrial outer membrane permeabilization (MOMP) and marks the point of no return in apoptosis. MOMP is followed by cytochrome c release from the mitochondria, activation of caspases, and finally cell death (3).
PC therapies like radiation, androgen deprivation, and chemotherapy aim to activate the intrinsic apoptotic pathway by causing cellular stress (5–7). This cellular stress generally leads to an activation of different signaling pathways and expression of activators to free Bax and Bak for the induction of cell death (8–11). However, two essential conditions must be met for the therapies to be effective. First, the signaling pathways that induce apoptosis must be intact; second, Bax and Bak must not be inhibited by an excess of anti-apoptotic proteins for successful release. In fact, a deregulation of signaling pathways can be observed in PC during tumor progression and therapy, so that the threshold for the induction of apoptosis cannot be reached (2, 12, 13). Moreover, an upregulation of Bcl-2, Bcl-xl, and Mcl-1 was associated with resistance to apoptosis, radiation, androgen deprivation, and chemotherapy (14–19). Immunohistological studies have shown that the anti-apoptotic proteins Bcl-xl and Mcl-1 are continuously present at a high percentage in PC cells (81–100%) independently of grade or metastasis (Table 1). The number of Bcl-2-positive cells varied from study to study between 24 and 70% (17, 20–26). Interestingly, the pro-apoptotic effectors Bax and Bak were also detected at high percentages in all tumors (77.5–100%) and mutations of the Bak and Bax genes were shown to be rare events in PC (17, 20, 26). This means that PC cells may be characterized by a high apoptosis resistance due to their high expression of anti-apoptotic Bcl-2 proteins, but that they are at the same time capable for an induction of apoptosis due to their ubiquitous expression of the pro-apoptotic effector proteins.
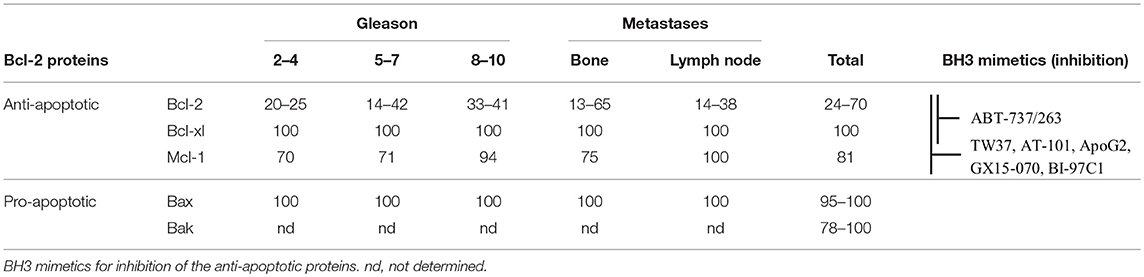
Table 1. Percentage of Bcl-2 protein positive cells in prostate cancer, as determined by immunohistology in different studies.
Direct Activation of the Intrinsic Apoptotic Pathway in Prostate Cancer
The fact that the activation of the effectors Bax and Bak is prevented by Bcl-2, Bcl-xl, and Mcl-1 leads to an alternative therapeutic strategy to induce the programmed cell death by direct inhibition of the anti-apoptotic proteins. In principle, this can be done without previous cell damage and without consideration of upstream signaling pathway elements, which could be altered in advanced stages of the disease.
DNA-antisense and RNA interference methods were used to down-regulate the anti-apoptotic proteins in PC cells. In combination with chemotherapeutic agents or irradiation, synergistic cytotoxicity, and increased antitumor activity in in vivo tumor models were seen (27–30). However, in a phase II study combination of docetaxel with the Bcl-2 antisense oligonucleotide oblimersen did not enhance the clinical outcome of patients with castration-resistant PC (CRPC). Major toxic events were >grade 3 fatigue, mucositis, and thrombocytopenia and primary endpoints (PSA response > 30% and major toxicity event rate < 45%) were not reached (31).
A second focus for the direct activation of the intrinsic apoptotic pathway was on the development of Bcl-2 antagonists, such as BH3 mimetics. Similar to the Bcl-2 activator proteins, these small molecules can bind to the anti-apoptotic proteins Bcl-2, Bcl-xl, or Mcl-1 followed by a release of Bax and Bak (32).
First generation BH3 mimetics included small molecule inhibitors such as TW37, (-)-Gossypol (AT-101), Apogossypolone (ApoG2), BI-97C1 (Sabutoclax), or GX15-070 (Obatoclax Mesylate). They are known as pan-BH3 mimetics, because they can bind Bcl-2, Bcl-xl, and Mcl1. In preclinical experiments, they successfully induced apoptosis in PC cells (33–35). Due to the different structure of the Mcl-1 molecule compared to Bcl-2 and Bcl-xl, however, the pan-BH3 mimetics only show moderate affinity and low specificity against all these proteins and elicit apoptosis-independent off-target effects (36). AT-101 failed in a Phase I/II clinical trial, in which only two of 23 patients with PC experienced a decrease in PSA without objective responses. The main adverse side effect was gastrointestinal toxicity, which was dose-limiting (37). Moreover, combination therapy with AT-101 plus docetaxel/prednisone of patients with metastatic CRPC cancer did not extend overall survival (38).
Second generation BH3 mimetics were therefore created by structural-based design to bind with high affinity and specificity to individual members of the anti-apoptotic proteins (39, 40). The specific BH3 mimetic ABT-737 and its orally administrable analog ABT-263 (Navitoclax) can specifically inhibit Bcl-2 and Bcl-xl, but not Mcl-1. In preclinical studies, they were successfully combined with agents targeting Mcl-1 (e.g., chemotherapeutic agents or kinase inhibitors) to exceed the threshold for the induction of apoptosis in PC cells (35). A phase II study with Navitoclax and abiraterone acetate with or without hydroxychloroquine in patients with metastatic CRPC has been terminated, but outcome has not been published yet (ClinicalTrials.gov, Identifier: NCT01828476).
Making the Induction of Apoptosis Tumor-Specific
Despite the recognition that a combination of specific BH3 mimetics with other drugs is necessary to achieve complete inhibition of the anti-apoptotic proteins Bcl-2, Bcl-xl, and Mcl-1 for the induction of apoptosis, these combinations have only hardly been used in clinical trials against PC to date. The main reason for this is that the combinations tested so far are not tumor-specific, but also can affect healthy cells. Second, most drugs, which showed synergistic effects with BH3 mimetics, target signaling pathways, e.g., androgen receptor (AR) or receptor tyrosine kinase (RTK) pathways, that are altered in advanced tumor stages (2, 13). It is therefore to be feared that the expected side effects of such combination therapies could outweigh the clinical benefits.
In recent years, new strategies were therefore developed to make the targeting of apoptosis more tumor-specific for enhanced efficacy and reduction of adverse side-effects. One such approach is the “BH3 profiling” of tumor cells, in which BH3 peptides interacting with Bcl-2, Bcl-xl, or Mcl-1 are used to identify the dependence of tumor cells on the respective anti-apoptotic Bcl-2 proteins. As a consequence, specific inhibitors could be selectively used to inhibit these proteins in the context of a personalized therapy. Specific Bcl-2 signatures for hematological tumors could be identified [reviewed in (41)]. It remains, however, questionable whether such profiling will also be successful in future for PC patients, because prostate tumors are known to be very heterogeneous and because it is difficult to isolate tumor cells for profiling in advanced stages.
A tumor specific therapy can generally be performed by using antibodies, peptides, or inhibitors against markers on the surface of tumor cells. With regard to a targeted induction of apoptosis, a direct coupling of Bcl-2 antagonists to tumor specific antibodies or a combination of Bcl-2 antagonists with immunotoxins represent promising, new therapeutic approaches. Berguig and colleagues generated an antibody-peptide drug conjugate consisting of an anti-CD22 antibody and a BIM peptide, targeting all anti-apoptotic proteins, to treat B cell lymphoma. The conjugate was applied via a multifunctional polymeric delivery system containing ethylene glycol segments to optimize safety and tumor biodistribution and butyl methacrylate (BMA) and diethylaminoethyl methacrylate (DEAEMA) for enhanced endosomal release. An inhibition of tumor growth marked by an increased apoptosis rate and an enhanced overall survival was reached with this construct in a B cell lymphoma mouse xenograft model (42). In another study combination of low dose EU-5346, a BH3 mimetic targeting Mcl-1, with trastuzumab induced significant cytotoxicity in Her-2 positive breast cancer cells (43).
In various tumor entities it was shown that the toxicity of Pseudomonas Exotoxin A (PEA) based immunotoxins can be significantly increased by the combination with the BH3 mimetic ABT-737/ABT-263 (44–46). PEA is a virulence factor of the bacterium Pseudomonas aeruginosa with ADP-ribosyltransferase activity. It ADP-ribosylates diphtamide, a modified histidine residue of the eukaryotic elongation factor 2 (eEF-2), in human ribosome complexes followed by inhibition of protein biosynthesis and induction of apoptosis (47). Initially, the inhibition of protein biosynthesis affects proteins with a short half-life due to rapid degradation, such as Mcl-1 (half-life < 1 h) (48). For the production of recombinant immunotoxins, the enzymatic domain of PEA is fused to a tumor-specific, internalizing antibody fragment and thus PEA is directed into tumor cells where it triggers apoptosis (49). In our group, we produced a PEA-based immunotoxin using an antibody fragment that specifically binds to the prostate specific membrane antigen (PSMA) on the surface of PC cells. With the anti-PSMA immunotoxin a downregulation of Mcl-1 was detected. When low doses of the immunotoxin and ABT-737 were combined, a synergistic cytotoxicity could be reached in PC cells, representing advanced androgen-dependent and independent stages (50). To our knowledge, this represents the first approach to make the induction of apoptosis specific for PC cells.
Conclusions
The direct induction of the intrinsic apoptotic pathway is a promising new therapeutic option for advanced PC. In recent years, various drugs have been developed that can directly inhibit anti-apoptotic Bcl-2 proteins and induce apoptosis, independently of potentially altered upstream signaling pathways. Since these drugs could also affect healthy cells and lead to severe adverse side effects, future research must focus on strategies to make the induction of the intrinsic apoptotic pathway tumor-specific.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by the Wilhelm Sander Foundation (Grant No. 2016.089.1).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AR, androgen receptor; BAD, Bcl-2 antagonist of cell death; Bak, Bcl-2 antagonist/killer; Bax, Bcl-2 associated X protein; Bcl-2, B-cell lymphoma 2; Bcl-xl, B-cell lymphoma extra-large; BID, BH3 interacting domain death agonist; BIM, Bcl-2 interacting mediator of cell death; BMA, butyl methacrylate; CRPC, castration-resistant PC; DEAEMA, diethylaminoethyl methacrylate; eEF-2, eukaryotic elongation factor 2; Her2, human epidermal growth factor receptor 2; Mcl-1, myeloid cell leukemia sequence; MOMP, mitochondrial outer membrane permeabilization; NOXA, phorbol-12-myristate-13-acetate-induced protein 1; PEA, Pseudomonas Exotoxin A; PC, prostate cancer; PSMA, prostate specific membrane antigen; PUMA, p53 upregulated modulator of apoptosis; RTK, receptor tyrosine kinase.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Katzenwadel A, Wolf P. Androgen deprivation of prostate cancer: leading to a therapeutic dead end. Cancer Lett. (2015) 367:12–7. doi: 10.1016/j.canlet.2015.06.021
3. Kiraz Y, Adan A, Kartal Yandim M, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. (2016) 37:8471–86. doi: 10.1007/s13277-016-5035-9
4. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. (2014) 15:49–63. doi: 10.1038/nrm3722
5. Szostak MJ, Kyprianou N. Radiation-induced apoptosis: predictive and therapeutic significance in radiotherapy of prostate cancer (review). Oncol Rep. (2000) 7:699–706. doi: 10.3892/or.7.4.699
6. Kramer G, Schwarz S, Hagg M, Havelka AM, Linder S. Docetaxel induces apoptosis in hormone refractory prostate carcinomas during multiple treatment cycles. Br J Cancer. (2006) 94:1592–8. doi: 10.1038/sj.bjc.6603129
7. Zhang KX, Firus J, Prieur B, Jia W, Rennie PS. To die or to survive, a fatal question for the destiny of prostate cancer cells after androgen deprivation therapy. Cancers (Basel). (2011) 3:1498–512. doi: 10.3390/cancers3021498
8. Mediavilla-Varela M, Pacheco FJ, Almaguel F, Perez J, Sahakian E, Daniels TR, et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol Cancer. (2009) 8:68. doi: 10.1186/1476-4598-8-68
9. Shiota M, Yokomizo A, Naito S. Pro-survival and anti-apoptotic properties of androgen receptor signaling by oxidative stress promote treatment resistance in prostate cancer. Endocr Relat Cancer. (2012) 19:R243–53. doi: 10.1530/ERC-12-0232
10. Fiandalo MV, Wu W, Mohler JL. The role of intracrine androgen metabolism, androgen receptor and apoptosis in the survival and recurrence of prostate cancer during androgen deprivation therapy. Curr Drug Targets. (2013) 14:420–40. doi: 10.2174/1389450111314040004
11. Yu H, Sun SQ, Gu XB, Wang W, Gao XS. Atorvastatin prolongs the lifespan of radiationinduced reactive oxygen species in PC-3 prostate cancer cells to enhance the cell killing effect. Oncol Rep. (2017) 37:2049–56. doi: 10.3892/or.2017.5447
12. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. (2007) 26:1324–37. doi: 10.1038/sj.onc.1210220
13. Kim JH, Lee H, Shin EA, Kim DH, Choi JB, Kim SH. Implications of Bcl-2 and its interplay with other molecules and signaling pathways in prostate cancer progression. Expert Opin Ther Targets. (2017) 21:911–20. doi: 10.1080/14728222.2017.1369044
14. Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. (1995) 55:4438–45.
15. Reiner T, de Las Pozas A, Parrondo R, Palenzuela D, Cayuso W, Rai P, et al. Mcl-1 protects prostate cancer cells from cell death mediated by chemotherapy-induced DNA damage. Oncoscience. (2015) 2:703–15. doi: 10.18632/oncoscience.231
16. Lebedeva I, Rando R, Ojwang J, Cossum P, Stein CA. Bcl-xL in prostate cancer cells: effects of overexpression and down-regulation on chemosensitivity. Cancer Res. (2000) 60:6052–60.
17. Yoshino T, Shiina H, Urakami S, Kikuno N, Yoneda T, Shigeno K, et al. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin Cancer Res. (2006) 12(20 Pt 1):6116–24. doi: 10.1158/1078-0432.CCR-06-0147
18. Lin Y, Fukuchi J, Hiipakka RA, Kokontis JM, Xiang J. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. (2007) 17:531–6. doi: 10.1038/cr.2007.12
19. Karnak D, Xu L. Chemosensitization of prostate cancer by modulating Bcl-2 family proteins. Curr Drug Targets. (2010) 11:699–707. doi: 10.2174/138945010791170888
20. Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, et al. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol. (1996) 148:1567–76.
21. Matsushima H, Hosaka Y, Suzuki M, Mizutani T, Ishizuka H, Kawabe K. bl-2 [corrected] Expression on prostate cancer and its relationship to cell cycle and prognosis. Int J Urol. (1996) 3:113–7. doi: 10.1111/j.1442-2042.1996.tb00494.x
22. Borre M, Stausbol-Gron B, Nerstrom B, Overgaard J. Immunohistochemical BCL-2 and Ki-67 expression predict survival in prostate cancer patients followed expectantly. Prostate Cancer Prostatic Dis. (1998) 1:268–75. doi: 10.1038/sj.pcan.4500252
23. Kaur P, Kallakury BS, Sheehan CE, Fisher HA, Kaufman RP Jr, Ross JS. Survivin and Bcl-2 expression in prostatic adenocarcinomas. Arch Pathol Lab Med. (2004) 128:39–43. doi: 10.1043/1543-2165(2004)128<39:SABEIP>2.0.CO;2
24. Castilla C, Congregado B, Chinchon D, Torrubia FJ, Japon MA, Saez C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology. (2006) 147:4960–7. doi: 10.1210/en.2006-0502
25. Cho IC, Chung HS, Cho KS, Kim JE, Joung JY, Seo HK, et al. Bcl-2 as a predictive factor for biochemical recurrence after radical prostatectomy: an interim analysis. Cancer Res Treat. (2010) 42:157–62. doi: 10.4143/crt.2010.42.3.157
26. Anvari K, Seilanian Toussi M, Kalantari M, Naseri S, Karimi Shahri M, Ahmadnia H, et al. Expression of Bcl-2 and Bax in advanced or metastatic prostate carcinoma. Urol J. (2012) 9:381–8.
27. Miayake H, Tolcher A, Gleave ME. Chemosensitization and delayed androgen-independent recurrence of prostate cancer with the use of antisense Bcl-2 oligodeoxynucleotides. J Natl Cancer Inst. (2000) 92:34–41. doi: 10.1093/jnci/92.1.34
28. Leung S, Miyake H, Zellweger T, Tolcher A, Gleave ME. Synergistic chemosensitization and inhibition of progression to androgen independence by antisense Bcl-2 oligodeoxynucleotide and paclitaxel in the LNCaP prostate tumor model. Int J Cancer. (2001) 91:846–50. doi: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1131>3.0.CO;2-Y
29. Mu Z, Hachem P, Pollack A. Antisense Bcl-2 sensitizes prostate cancer cells to radiation. Prostate. (2005) 65:331–40. doi: 10.1002/pros.20303
30. Anai S, Brown BD, Nakamura K, Goodison S, Hirao Y, Rosser CJ. Irradiation of human prostate cancer cells increases uptake of antisense oligodeoxynucleotide. Int J Radiat Oncol Biol Phys. (2007) 68:1161–8. doi: 10.1016/j.ijrobp.2007.03.058
31. Sternberg CN, Dumez H, Van Poppel H, Skoneczna I, Sella A, Daugaard G, et al. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): an EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann Oncol. (2009) 20:1264–9. doi: 10.1093/annonc/mdn784
32. Opydo-Chanek M, Gonzalo O, Marzo I. Multifaceted anticancer activity of BH3 mimetics: current evidence and future prospects. Biochem Pharmacol. (2017) 136:12–23. doi: 10.1016/j.bcp.2017.03.006
33. Zeitlin BD, Joo E, Dong Z, Warner K, Wang G, Nikolovska-Coleska Z, et al. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2. Cancer Res. (2006) 66:8698–706. doi: 10.1158/0008-5472.CAN-05-3691
34. Zhang XQ, Huang XF, Hu XB, Zhan YH, An QX, Yang SM, et al. Apogossypolone, a novel inhibitor of antiapoptotic Bcl-2 family proteins, induces autophagy of PC-3 and LNCaP prostate cancer cells in vitro. Asian J Androl. (2010) 12:697–708. doi: 10.1038/aja.2010.57
35. Wolf P. BH3 Mimetics for the treatment of prostate cancer. Front Pharmacol. (2017) 8:557. doi: 10.3389/fphar.2017.00557
36. Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. (2013) 12:1691–700. doi: 10.1158/1535-7163.MCT-13-0058
37. Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res. (2009) 15:3172–6. doi: 10.1158/1078-0432.CCR-08-2985
38. Sonpavde G, Matveev V, Burke JM, Caton JR, Fleming MT, Hutson TE, et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann Oncol. (2012) 23:1803–8. doi: 10.1093/annonc/mdr555
39. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. (2005) 435:677–81. doi: 10.1038/nature03579
40. Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. (2006) 13:1419–21. doi: 10.1038/sj.cdd.4401937
41. Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. (2018) 25:56–64. doi: 10.1038/cdd.2017.183
42. Berguig GY, Convertine AJ, Frayo S, Kern HB, Procko E, Roy D, et al. Intracellular delivery system for antibody-Peptide drug conjugates. Mol Ther. (2015) 23:907–17. doi: 10.1038/mt.2015.22
43. Vallet S, Fan F, Malvestiti S, Pecherstorfer M, Sattler M, Schneeweiss A, et al. Rationally derived drug combinations with the novel Mcl-1 inhibitor EU-5346 in breast cancer. Breast Cancer Res Treat. (2019) 173:585–96. doi: 10.1007/s10549-018-5022-5
44. Mattoo AR, FitzGerald DJ. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int J Cancer. (2013) 132:978–87. doi: 10.1002/ijc.27732
45. Antignani A, Sarnovsky R, FitzGerald DJ. ABT-737 promotes the dislocation of ER luminal proteins to the cytosol, including pseudomonas exotoxin. Mol Cancer Ther. (2014) 13:1655–63. doi: 10.1158/1535-7163.MCT-13-0998
46. Hollevoet K, Antignani A, Fitzgerald DJ, Pastan I. Combining the antimesothelin immunotoxin SS1P with the BH3-mimetic ABT-737 induces cell death in SS1P-resistant pancreatic cancer cells. J Immunother. (2014) 37:8–15. doi: 10.1097/CJI.0000000000000010
47. Michalska M, Wolf P. Pseudomonas exotoxin A: optimized by evolution for effective killing. Front Microbiol. (2015) 6:963. doi: 10.3389/fmicb.2015.00963
48. Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol Cell Biol. (2010) 30:3099–110. doi: 10.1128/MCB.01266-09
49. Wolf P, Elsasser-Beile U. Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol. (2009) 299:161–76. doi: 10.1016/j.ijmm.2008.08.003
Keywords: prostate cancer, apoptosis, Bcl-2 proteins, targeted therapy, tumor specificity
Citation: Wolf P (2019) Tumor-Specific Induction of the Intrinsic Apoptotic Pathway—A New Therapeutic Option for Advanced Prostate Cancer? Front. Oncol. 9:590. doi: 10.3389/fonc.2019.00590
Received: 08 March 2019; Accepted: 17 June 2019;
Published: 02 July 2019.
Edited by:
Suzie Chen, The State University of New Jersey, United StatesReviewed by:
Jian Pan, Sun Yat-Sen University, ChinaCopyright © 2019 Wolf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philipp Wolf, cGhpbGlwcC53b2xmQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==