 Mengjie Guo1,2†
Mengjie Guo1,2† Dongdong Sun3†Zhimin Fan1†Yuxia Yuan2Miaomiao Shao2Jianhao Hou2Yuqi Zhu2Rongfang Wei2Yan Zhu2Jinjun Qian2Fei Li4*
Dongdong Sun3†Zhimin Fan1†Yuxia Yuan2Miaomiao Shao2Jianhao Hou2Yuqi Zhu2Rongfang Wei2Yan Zhu2Jinjun Qian2Fei Li4* Ye Yang2,5*
Ye Yang2,5* Chunyan Gu1,2*
Chunyan Gu1,2*- 1The Third Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, China
- 2School of Medicine and Life Sciences, Nanjing University of Chinese Medicine, Nanjing, China
- 3The First Clinical Medical College, Nanjing University of Chinese Medicine, Nanjing, China
- 4Department of Hematology, The First Affiliated Hospital of Nanchang University, Nanchang, China
- 5School of Holistic Integrative Medicine, Nanjing University of Chinese Medicine, Nanjing, China
MAPKAPK2 (MK2), the direct substrate of p38 MAPK, has been well-acknowledged as an attractive drug target for cancer therapy. However, few studies have assessed the functions of it in multiple myeloma (MM). In the present study, MK2 expression of MM patients was analyzed by gene expression profiling (GEP) and array-based comparative genomic hybridization (aCGH). Several experiments in vitro including MTT assay, Western blot and flow cytometry analysis were performed to identify the function of MK2 in MM. In addition, we conducted mouse survival experiments to explain the effects of MK2 on MM in vivo. mRNA level of MK2 and chromosomal gain of MK2 locus in MM cells significantly increased compared to normal samples. Furthermore, MM patients with high expression of MK2 were associated with a poor outcome. Follow-up studies showed that MK2 exerted a remarkably positive effect on MM cell proliferation and drug-resistance. Further exploration focusing on MK2 inhibitor IV revealed its inhibitory action on MM growth and drug-resistance, as well as improving survival in mouse models. In addition, a combination of MK2 inhibitor IV and the key MM therapeutic agents including bortezomib, doxorubicin, or dexamethasone facilitated curative effects on inhibiting MM cell proliferation. Taken together, our study reveals the clinical relevance of MK2 inhibition in MM and demonstrates that targeting MK2 may afford a new therapeutic approach to MM.
Introduction
Multiple myeloma (MM) is the second most prevalent hematologic malignancy derived from neoplastic growth of bone marrow plasma cells. Chemotherapy has been reported to interfere with the function of cellular machineries (like DNA replication and cell division), and is regarded as one of the useful therapeutic methods for MM; thus more and more novel agents are developed and tested (1). The National Comprehensive Cancer Network (NCCN) delineated the treatment guideline that lenalidomide and bortezomib regimens involving combination therapy with doxorubicin and/or dexamethasone are currently recommended in front-line treatment paradigm for MM (2). Although the survival outcomes of MM patients have been improved by new therapeutic approaches, it is still far away from having satisfactory effects on chemotherapy and relapse remains the major obstacle in MM management, which makes MM incurable (3, 4).
MAPK-activated protein kinase 2 (MK2), a serine/threonine-protein kinase known as the best-understood downstream partner of p38 MAP kinase, is involved in many cellular processes such as stress and inflammatory responses, nuclear export, cell proliferation and invasion (5). A meta-analysis demonstrated that there is a relatively higher expression of MK2 in MM compared with normal plasma cells by the means of ONCOMINE microarray database (6). In particular, overexpression of MK2 could be linked with MM drug resistance and relapse, due to the fact that p38-MAPKAPK2-Hsp27 signaling preserves the survival of cancer stem cells (6). Consequently, it is conceivable that MK2 may act as a powerful hallmark of cancer cells and inhibiting MK2 could disrupt tumor growth (7), which provides new avenues to explore the response of cancers to their therapeutic agents.
As reported, MK2 inhibitors exhibited desirable efficacy without toxic side effects, indicating that they are promising candidates for improving cancer treatment clinically. To this end, the research focused on this issue has been developed (8). MK2 inhibitor IV is a non-ATP competitive MK2 inhibitor and an excellent pharmacologic tool for specifically testing MK2 biology, because of its high degree of selectivity (9). However, little attention has been paid to MK2 inhibitor IV on MM. In addition, as inactivation of MK2 could enhance bortezomib cytotoxicity (10), it is an interesting investigation to explore whether the combination of MK2 inhibitor IV and current chemotherapeutics could achieve synergistic anti-tumoral effects on MM. Several studies have assessed the role of MK2 in chemotherapy, however seemingly conflicting results occur (11, 12). Thus, it is of great importance to choose the suitable chemotherapeutic drug combined with MK2 inhibitors for cancer treatment.
In the present study, we characterized MK2 expression in MM cells compared to normal control cells to reveal the relationship among MK2 expression, clinical characteristics and overall survival in total therapy 2 (TT2). Then the functions of MK2 in MM cell proliferation and drug-resistance were explored. Furthermore, we provided in vitro data declaring the anti-proliferation efficacy of MK2 inhibitor IV in MM cells and gained further insights to determine whether MK2 inhibitor is more effective than monotherapeutic agent (such as bortezomib, doxorubicin, or dexamethasone). Additionally, combination of MK2 inhibitor IV and bortezomib, doxorubicin or dexamethasone presented better inhibition effect on MM cell proliferation.
Materials and Methods
Cell Lines and Cell Culture
Human MM cell lines, APR1, and OCI-MY5 cells were maintained in RPMI 1640 (Gibco, Grand Island, NY) and supplemented with 10% fetal bovine serum (FCS) (Gibco, Grand Island, NY), penicillin and streptomycin solution (100 μg/mL, Sigma, St. Louis, MO) under the condition of 37°C in a humidified atmosphere of 5% CO2.
Reagents
MK2 antibody, MK2 Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) lentiviral activation particles, and MK2-shRNA lentiviral particle were purchased from Santa Cruz Biotechnology (Dallas, Texas). Caspase-3, PARP, β-actin, and pAKT antibodies were obtained from Cell Signaling Technology (Danvers, MA). Trypan Blue, dexamethasone and doxorubicin were from Sigma (St. Louis, MO). Bortezomib was purchased from Selleck Chemicals (Houston, TX). MK2 inhibitor IV was purchased from Cayman Chemical (Ann Arbor, MI).
Cell Growth Assays
Following cell transfection or 48 h after different drug treatment, MK2 inhibitor IV IC50 15 μM; bortezomib IC50 80 nM; dexamethasone IC50 25 μM; doxorubicin IC50 1.5 μM; MK2 inhibitor IV IC25 4.75 μM + bortezomib IC25 46.2 nM; MK2 inhibitor IV IC25 4.75 μM + dexamethasone IC25 6.6 μM; MK2 inhibitor IV IC25 4.75 μM + doxorubicin IC25 0.3 μM. Cell growth was evaluated using MTT assay according to the method described in the literature (13). In short, cells were seeded at 4,000~5,000 cells/well in 96-well plates, 10 μL of MTT and 90 μL of complete medium were added to each well according to the manufacturer's instructions, and the culture plate was incubated at 37°C for 4 h. Then, the culture medium was removed and 150 μL dimethyl sulfoxide was added to each well. After vigorous shaking for 10 min, the absorbance was read by a fluorometer (CytoFluor; Applied Biosystems, Foster City, CA, USA) at 570 nm.
Cell Proliferation
Cells were enumerated using a hemocytometer. The fraction of dead cells was determined by trypan blue staining.
Flow Cytometry
A flow cytometry assay was applied to analyze the cell cycle distribution as previously reported (14). Briefly, samples were washed with PBS and stained with PI solution (Yeasen, Shanghai, China) for 30 min. All samples were analyzed using FlowSight (Merck Millipore, Darmstadt, Germany).
Western Blot Analysis
Western blots were utilized to measure the protein levels of MK2, pAKT and apoptotic markers in MM cells. In brief, around 20 μg protein per sample was extracted and whole-cell lysate were electrophorezed by 4–12% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose. After incubation with primary and second antibodies, blots were subsequently stripped and re-probed for β-actin as protein controls. The immunostaining band was quantified using Image J software.
Soft Agar Clonogenic Assay
Clonogenic formation was monitored by plating 10,000 MM cells in 0.5 mL 0.33% agar RPMI 1640 medium (Invitrogen, Carlsbad, CA) with 10% FBS in 12-well plate. The cells were incubated at 37°C with 5% CO2 and fed by the same medium for 1–2 week, twice per week. The colonies were imaged and colony numbers were counted by Image J.
Apoptosis and Dye-Efflux Multidrug-Resistance Assays
Drug-affected apoptosis following exposure to bortezomib or doxorubicin was detected in MM cells using flow-cytometric Annexin V apoptosis detection kit APC (catalog number 88–8007) from eBioscience (San Diego, CA). In brief, one million cells were washed and suspended in 1 mL binding buffer, followed by adding the Annexin V-APC (5 μL) to the cells suspension (100 μL). Then, cells were incubated for 15 min at room temperature, washed and re-suspended in binding buffer. The labeled cells were analyzed via flow cytometry.
The eFluxx-ID™ Multidrug resistance assay was applied to measure drug resistance as previously described (15). Shortly, 500,000 cells were incubated for 30 min under the circumstance of 37°C in water bath with detection reagent, Golden dye. Afterwards, cells were washed and re-suspended in ice-cold PBS for flow analysis, and MCF7 cells were used as reference.
5TMM Mouse Model
5TMM3VT murine myeloma cells (1 × 106) were injected subcutaneously into the abdomen of 6 weeks old C57BL/KaLwrij mice (Jackson laboratory, Bar Harbor, Maine) (n = 20). After 2 days, MK2 inhibitor IV treatment used on 10 mice and intraperitoneally injected at the dose of 20 mg/kg three times a week lasting for 78 days until all the mice were dead (Tuesday, Thursday, and Saturday). The time of death caused by paralysis in the control group and the MK2i group was recorded in turn, and the survival curve was drawn at the end (16). All experimental procedures were in compliance with the National research council guidance's for the care and use of laboratory animals and approved by Animal Ethical and Experimental Committee of Nanjing University of Chinese Medicine (ACU170501).
Statistical Considerations
All data was shown as means ± SD. Two experimental groups were analyzed by Student's t-test, while multiple (n ≥ 3) groups were analyzed with one-way ANOVA. The correlation of MK2 with disease progression, event-free and overall survivals were measured using the Kaplan-Meier method. Significance was set at p < 0.05.
Results
Amplification of MK2 Is Relevant for Poor Survival in MM Patients
Using the gene expression profiling (GEP) database collected from NIH Gene Expression Omnibus GSE2658 as a discovery tool, we found that MK2 expression levels exhibited a dramatic upward trend from normal plasma cells (NP, n = 22), monoclonal gammopathy of undetermined significance cells (MGUS, n = 44), to newly diagnosed myeloma patient plasma cells (n = 351) (Figure 1A). The analysis of array-based comparative genomic hybridization (aCGH) data showed that MK2 locus was amplified in MM patient samples to a major extent (Figure 1B). To correlate with clinical parameters (Table 1), MK2 expression was impressively associated with clinical parameters like CRP at least 4.0 mg/l (p < 0.05), chromosomal abnormalities (by G-banding) (p < 0.05), and MRI focal bone lesions (p < 0.05). Furthermore, Kaplan-Meier survival curves showed that increment of MK2 expression was linked with an apparently shorter response duration of both event free (EF) and overall survival (OS) in TT2 (Total Therapy 2) cohort, respectively (Figures 1C,D). Continuing our previous study focusing on the relationship between MK2 and outcomes of MM patients in the APEX cohort, which was an independent cohort of MM patients (17), we further confirmed that MK2 acts as a valuable diagnostic and prognostic marker in MM.
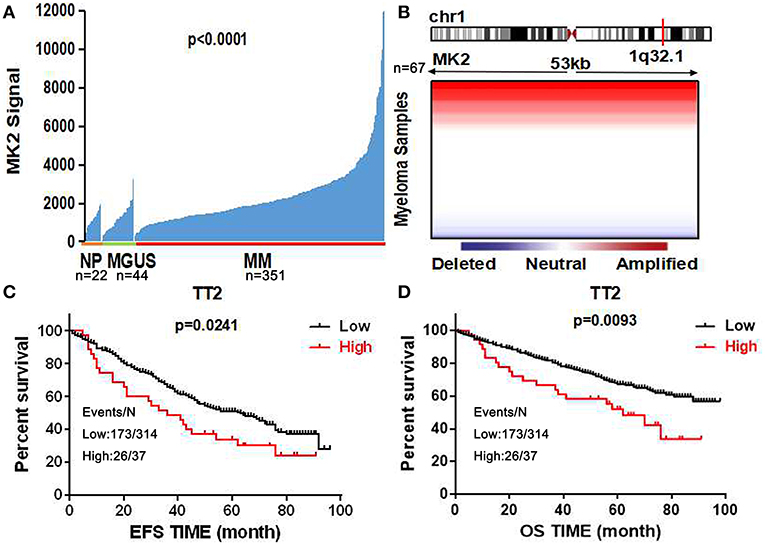
Figure 1. MK2 acts as a poor prognostic marker in MM. (A) MK2 expression of normal plasma cells (NP, n = 22), monoclonal gammopathy of undetermined significance cells (MGUS, n = 44), and myeloma patient plasma cells (n = 351) in GEP dataset. (B) Heatmap illustrating MK2 copy number variation in 67 primary MM samples. (C,D) Kaplan-Meier analysis on the event free survival (C) and overall survival (D) of MM patients according to the MK2 expression in TT2. Data are expressed as mean ± SD.
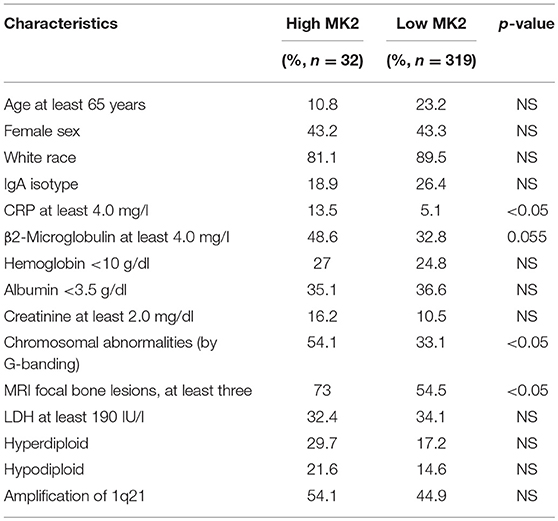
Table 1. The Correlation of MK2 Expression and Clinical Characteristics in TT2.
Alteration in MK2 Expression Is Crucial for MM Cell Growth and Drug-Resistance
To further characterize the carcinogenic role of MK2 in MM, lentiviral shRNA transfection technology was utilized to knock down the endogenous expression of the MK2 gene in ARP1 and OCI-MY5 cells. The results of the western blot showed the remarkable impairment of MK2 expression levels in MK2-shRNA transfected MM cells (KD) relative to the controls (Ctrl) (p < 0.05) (Figure 2A). As demonstrated in Figure 2B, a significant drop in cell growth rate of OCI-MY5 cells was provoked by silencing MK2 (p < 0.05), along with the slight reduction in ARP1 cells. Next, we transfected MM with CRISPR lentiviral activation particles to significantly upregulate the expression of MK2 (p < 0.05) (Figure 2A). Cell proliferation rate of MK2-OE cells was obviously increased relative to the control cells in both ARP1 and OCI-MY5 cells (p < 0.05) (Figure 2B). These findings proposed that MK2 facilitated MM cell proliferation. Flow cytometry analysis for cell cycle distribution found that the proportions of cells in the G2-M phase were increased after MK2 overexpression (ARP1 WT and ARP1 OE: 8.12 vs. 22.9%; OCI-MY5 WT and OCI-MY5 OE: 2.46 vs. 33.3%) (Figure 2C). Conversely, knockdown of MK2 decreased the proportions of cells in the G2-M phase (ARP1 WT and ARP1 KD: 26.6 vs. 6.51%; OCI-MY5 WT and OCI-MY5 KD: 22.0 vs. 4.13%) (Figure 2C). Furthermore, MK2-OE cells generated significant reduction of the mean fluorescence intensity, revealing a marked increment of ABC transporter substrates efflux (Figure 2D). Meanwhile, the data illustrated that treatment of cells with bortezomib (8 nM) or doxorubicin (100 nM) triggered less apoptotic death in the MK2-OE cells compared with its corresponding control cells in the assay (Figure 2E). These results support the point that MK2 is a powerful factor in promoting MM progression and drug resistance, which could be applied as a novel therapeutic target for cancer treatment.
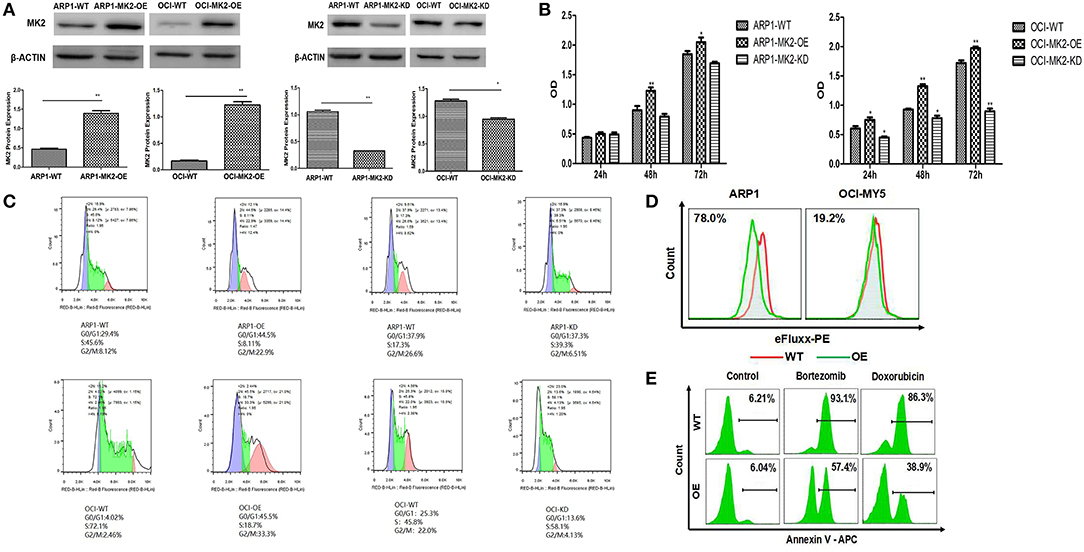
Figure 2. MK2 overexpression promotes proliferation and drug-resistance in MM. (A) Validation of MK2 expression levels in MK2-overexpression (OE) and MK2-knockdown (KD) MM cells compared with untransfected cells (WT). (B) MTT assay on MK2-overexpression (OE) and MK2-knockdown (KD) ARP1 and OCI-MY5 cells compared with untransfected cells (WT). (C) The distribution of MK2 OE and MK2 KD cells in different phases of the cell cycle was determined by flow cytometry. (D) Evaluation of ABC transporter activity in ARP1 and OCI-MY5 WT and OE cells by the means of flow cytometry. (E) The cellular apoptosis of ARP1 and OCI-MY5 MK2-WT and OE cells treated with or without bortezomib or doxorubicin. *p < 0.05, **p < 0.01. Data are expressed as mean ± SD.
MK2 Inhibitor Executes Potential Suppressive Function in MM
To extend our findings into pre-clinics study, we evaluated the effects of MK2 inhibitor IV on MM growth and drug-resistance. MK2 promoted MM progression via activating AKT in our previous study (17). Here we first examined the effect of the MK2 inhibitor on reducing AKT activation. A sharp abolishment on pAKT (the phosphorylation-activated form of AKT) expression was attained by MK2 inhibitor IV treatment, when compared with non-treated control in both ARP1 and OCI-MY5 cells (p < 0.05) (Figure 3A), partly suggesting that MK2 plays a positive role in AKT phosphorylation. Subsequently, the growth rate and cell viability of ARP1 and OCI-MY5 cells were significantly hindered by MK2 inhibitor IV compared with their non-treated counterparts in a time-dependent manner (Figures 3B,C). Alternatively, the inhibitor also diminished long-term self-renewal of MK2-OE cells, which was evidenced by the remarkable declined clonogenicity (p < 0.05) (Figure 3D). Similarly, ABC transporter drug pump in MK2-OE cells was deactivated by MK2 inhibitor IV treatment, indicating the reverse on MK2 induced drug-resistance (Figure 3E). Combined with the western blot data that MK2 inhibitor IV induced remarkable elevation of cleaved both Caspase-3 and PARP (p < 0.05) (Figure 3F), we could speculate that the decreased growth rate of MK2 inhibitor IV treated cells was attributed to the increased apoptotic cell death. Summarily, the data above indicates that MK2 inhibitor IV may act as a potent pre-clinic candidate for targeting MK2 in MM.
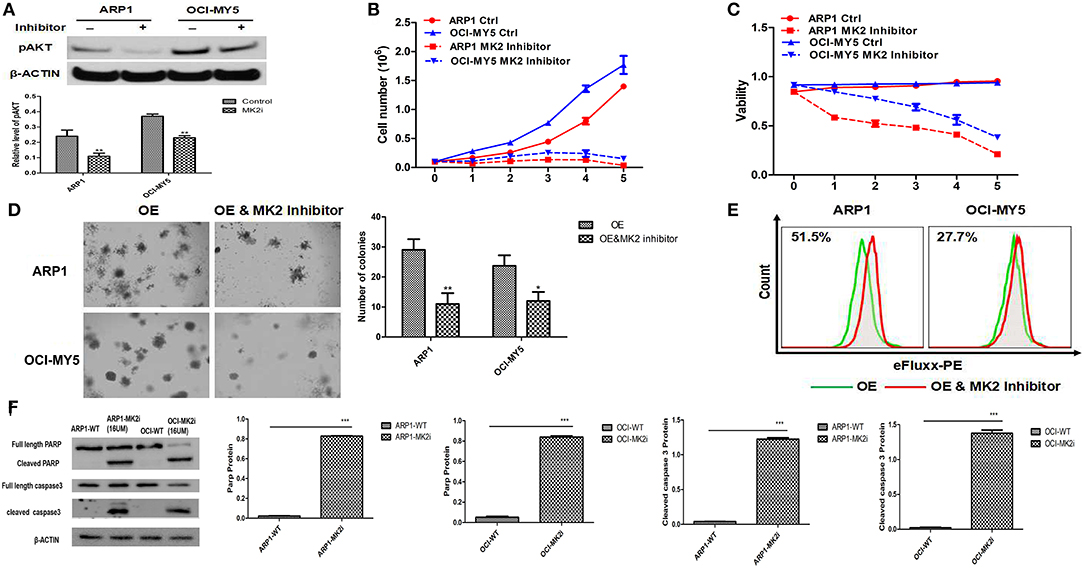
Figure 3. MK2 inhibitor plays a potential suppressive role in MM. (A) pAKT expression in ARP1 and OCI-MY5 cells were detected by western blot after MK2 inhibitor IV (15 μM) treatment for 24 h. (B) Growth curve of ARP1 and OCI-MY5 cells treated with or without MK2 inhibitor IV for 5 days. (C) Cell viability of ARP1 and OCI-MY5 cells treated with or without MK2 inhibitor IV for 5 days. (D) The effect of MK2 inhibitor IV treatment on the MK2-OE MM cells compared with control cells was measured by colony formation assay. (E) The detection of ABC transport pump activity when treated with MK2 inhibitor IV. (F) Western blot of ARP1 and OCI-MY5 cells following MK2 inhibitor IV treatment on the PARP and Caspase-3 expression. Data are expressed as mean ± SD.
MK2 Inhibitor Synergistically Enhances the Repressive Effect of Bortezomib/Dexamethasone/Doxorubicin on MM Cell Proliferation
Bortezomib (BTZ), dexamethasone (DEX), and doxorubicin (DOX) have been well-recognized as key therapeutic agents for MM treatment; hence, MTT assays were performed to examine the effects of the drugs, alone or combined with MK2 inhibitor IV. As shown in Figure 4, when compared with control cells MK2 inhibitor IV and each agent (dexamethasone/doxorubicin/bortezomib) all yielded significant reductions in the growth rates of ARP1 and OCI-MY5 cells (p < 0.05). The cell growth inhibitory function of MK2 inhibitor IV was significantly and slightly superior to doxorubicin and bortezomib in OCI-MY5 cells and ARP1 cells, respectively. However, dexamethasone-exposed cells exhibited lower proliferation rate than MK2 inhibitor IV-exposed cells to some extent. Then we explored combined effects of MK2 inhibitor IV and dexamethasone/doxorubicin/bortezomib and found the combinations remarkably suppressed cell proliferation in comparison with the single drug in ARP1 and OCI-MY5 cells (p < 0.05). Above all, the data unveiled that MK2 inhibitor is capable of improving bortezomib, dexamethasone, or doxorubicin activity in vitro.
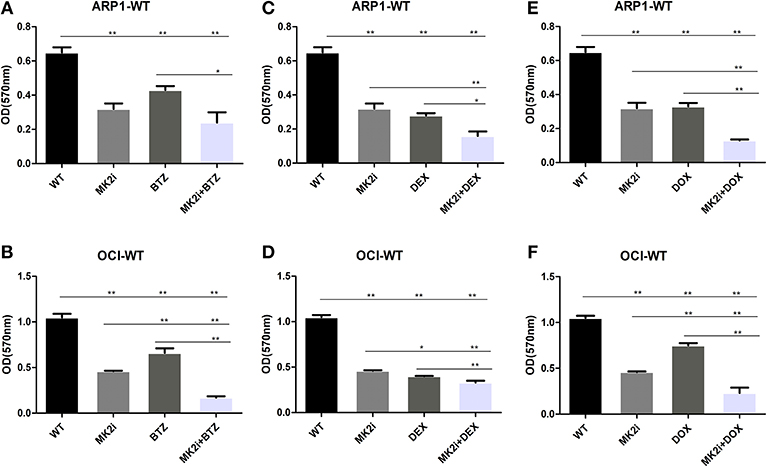
Figure 4. MK2 inhibitor IV enhances the inhibitory effect of bortezomib, dexamethasone, or doxorubicin on cellar proliferation in ARP1 and OCI-MY5. (A,B) ARP1 cells (A) and OCI-MY5 cells (B) were treated with MK2 inhibitor IV with/without BTZ or BTZ alone. (C,D) ARP1 cells (C) and OCI-MY5cells (D) were treated with MK2 inhibitor IV with/without DEX or DEX alone. (E,F) ARP1 cells (E) and OCI-MY5 cells (F) were treated with MK2 inhibitor IV with/without DOX or DOX alone. All the drug cytostatic effect was analyzed by MTT proliferation assay after 48 h. Drug concentrations: MK2 inhibitor IV: 15 μM, bortezomib: 80 nM, dexamethasone: 25 μM, and doxorubicin: 1.5 μM, MK2 inhibitor IV 4.75 μM + bortezomib 46.2 nM, MK2 inhibitor IV 4.75 μM + dexamethasone 6.6 μM, MK2 inhibitor IV 4.75 μM + doxorubicin 0.3 μM. *p < 0.05, **p < 0.01. Data are expressed as mean ± SD.
Downregulation of MK2 Improves the Survival Time of 5TMM Mice
To further evaluate the therapeutic potential of the MK2 inhibitor in animal models, survival analyses were performed on 5TMM mice based on the MK2 inhibitor IV (20 mg/kg) treatment. As shown in Figure 5, Kaplan-Meier survival curves showed that mice with high MK2 expression had inferior outcomes than those with low MK2 expression though using MK2 inhibitor IV (p < 0.05). Taken together, it further indicates that MK2 may serve as a prognostic marker in MM.
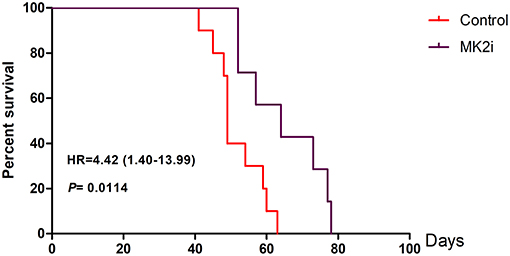
Figure 5. MK2 inhibitor IV exerts a great function on improving survival time of 5TMM mice. MK2 inhibitor IV was intraperitoneally injected at the dose of 20 mg/kg three times a week lasting for 78 days until all the mice were dead (Tuesday, Thursday, and Saturday). Data are expressed as mean ± SD.
Discussion
Increasing evidence has shown that activation of p38 MAPK signaling displays a critical role in promoting hematopoietic growth leading to hematologic malignancies in some hematologic systems (18). It has been reported that p38 MAPK regulates Hsp27 through MK2, which confers resistance to bortezomib and dexamethasone in MM cells (19). Amplification of MK2 is closely associated with tumorigenesis and the development of various cancers, like prostate cancer (18), gastrointestinal stromal tumors (20) and bladder cancer (5). However, the role of MK2 in MM is still poorly understood. In the current research, GEP database revealed the impressive increment of MK2 expression levels in MM cells, which is in accordance with the previous study on lung cancer (21). More importantly, patients with high MK2 expression suffered from worse survival outcomes than MK2 low-expressing patients. These studies suggest that MK2 is a novel therapeutic option for MM treatment.
Our work indicated that decreased MK2 expression induced cell growth inhibition, while increased MK2 expression promoted cell proliferation and ABC transporter drug pump activation, which implied a positive function in tumorigenesis and development (Figures 2, 3). It is served as a different angle about the function of MK2 exerted on tumor cells, for the majority reports focus on cell invasion via modulation of matrix metalloproteinase and actin cytoskeleton (5, 7, 22). Inspiringly, inferred from the data associated with MK2 inhibitor IV, we found the inhibitor manifesting remarkable reduction of MM cellular growth, colony formation and ABC transporter drug pump (Figure 3). Furthermore, this process could be mediated by pAKT level (Figure 3A), which was confirmed by our team and other groups (15, 23, 24). However, whether MK2 directly or indirectly phosphorylated AKT in MM cells needs further investigation. In addition, blockade of MK2 activates cleaved PARP and Caspase-3 resulting in triggering cell apoptosis, thus MK2 inhibitor may be a promising agent for MM treatment in clinics.
It has been known that MK2 inhibitors are in preclinical development (25), in which both MK2 inhibitor III and PF-3644022 declined cell survival when combined with mitomycin-c treatment, unlike their pro-survival effects upon application with gemcitabine collectively (26). Meanwhile, cisplatin as well as the topoisomerase II inhibitor doxorubicin combined with MK2 inhibitors were reported as exhibiting increased cytotoxicity (11, 27). In this paper, we traced the influence of MK2 inhibitor IV on increasing the efficacy of bortezomib, doxorubicin, and dexamethasone on MM cell proliferation. The results showed that using MK2 inhibitor IV alone was more effective than bortezomib or doxorubicin. Our study first claimed that combining MK2 inhibitor IV with the anticancer agents (dexamethasone/doxorubicin/bortezomib) had better curative efficacy than single agents. Interestingly, researchers have corroborated that some complex factors like tumor genotype and DNA repair machineries are considered as the manipulators in determining the effect of combination therapy regimens (28, 29). In view of the difficulties in accurate predictions, any cytotoxic drug should be carefully monitored about its collaboration or antagonism with MK2 inhibitors, before applying such drug combinations to the clinics.
In conclusion, our findings illustrate the oncogenetic functions of MK2 in MM, suggesting it is a novel molecular marker. Combining the MK2 inhibitor with bortezomib, doxorubicin or dexamethasone has significant inhibition effect on MM cells. Targeting MK2 may afford a new therapeutic approach to MM. Future studies is undergoing to get better understanding in the interaction of MK2 inhibitor and clinical drugs in vivo and elaborate its mechanism at the subcellular level.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2658.
Ethics Statement
This study was carried out in accordance with the recommendations of National Research Council guidance's for the care and use of laboratory animals, and approved by Animal Ethical and Experimental Committee of Nanjing University of Chinese Medicine (ACU170501). The protocol was approved by Animal Ethical and Experimental Committee of Nanjing University of Chinese Medicine.
Author Contributions
CG and MG conceived the manuscript and provided critical input. MG wrote the manuscript. YYu, YuZ, RW, JH, MS, and YaZ performed the experiments. DS and ZF provided technical counseling on experiments. CG, YYa, JQ, and FL reviewed the data and edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (81770220, 81600177, 81670200) to CG and YYa, The 2016 outstanding youth fund of Jiangsu Province (BK20160048) to YYa, Natural Science Foundation of Jiangsu Province (BK20161041) to CG, National key research and development program-precision medicine sub-program (2016YFC0905900) to YYa, The Priority Academic Program Development of Jiangsu Higher Education Institutions for Chinese Medicine, Innovation Team of Six Talent Peaks Project in Jiangsu Province (TD-SWYY-015).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
MAPK, mitogen-activated protein kinase; MM, multiple myeloma; GEP, gene expression profiling; aCGH, array-based comparative genomichybridization.
References
1. Kyle RA, Rajkumar SV. Multiple myeloma. Blood. (2008) 111:2962–72. doi: 10.1182/blood-2007–10-078022
2. Stein R, Smith MR, Chen S, Zalath M, Goldenberg DM. Combining milatuzumabwith bortezomib, doxorubicin, or dexamethasone improves responses in multiple myeloma cell lines. Clin Cancer Res. (2009) 15:2808–17. doi: 10.1158/1078–0432.CCR-08–1953
3. Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. (2014) 28:1122–8. doi: 10.1038/leu.2013.313
4. Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. (2012) 26:149–57. doi: 10.1038/leu.2011.196
5. Kumar B, Koul S, Petersen J, Khandrika L, Hwa JS, Meacham RB, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. (2010) 70:832–41. doi: 10.1158/0008–5472.CAN-09–2918
6. Lin SP, Lee YT, Wang JY, Miller SA, Chiou SH, Hung MC, et al. Survival of cancer stem cells under hypoxia and serum depletion via decrease in PP2A activity and activation of p38-MAPKAPK2-Hsp27. PLoS ONE. (2012) 7:e49605. doi: 10.1371/journal.pone.0049605
7. Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez JB, Pitman D, et al. Distinct cellular functions of MK2. Mol Cell Biol. (2002) 22:4827–35. doi: 10.1128/MCB.22.13.4827–4835.2002
8. Huang X, Zhu X, Chen X, Zhou W, Xiao D, Degrado S, et al. A three-step protocol for lead optimization: quick identification of key conformational features and functional groups in the SAR studies of non-ATP competitive MK2 (MAPKAPK2) inhibitors. Bioorg Med Chem Lett. (2012) 22:65–70. doi: 10.1016/j.bmcl.2011.11.074
9. Huang X, Shipps GW Jr, Cheng CC, Spacciapoli P, Zhang X, McCoy MA, et al. Discovery and hit-to-lead optimization of non-ATP competitive MK2 (MAPKAPK2) inhibitors. ACS Med Chem Lett. (2011) 2:632–7. doi: 10.1021/ml200113y
10. Felix RS, Colleoni GW, Caballero OL, Yamamoto M, Almeida MS, Andrade VC, et al. SAGE analysis highlights the importance of p53csv, ddx5, mapkapk2 and ranbp2 to multiple myeloma tumorigenesis. Cancer Lett. (2009) 278:41–8. doi: 10.1016/j.canlet.2008.12.022
11. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. (2007) 11:175–89. doi: 10.1016/j.ccr.2006.11.024
12. Hopker K, Hagmann H, Khurshid S, Chen S, Hasskamp P, Seeger-Nukpezah T, et al. AATF/Che-1 acts as a phosphorylation-dependent molecular modulator to repress p53-driven apoptosis. EMBO J. (2012) 31:3961–75. doi: 10.1038/emboj.2012.236
13. Yu YL, Chen DQ, Li YN, Yang WQ, Tu JS, Shen Y. Improving the topical ocular pharmacokinetics of lyophilized cyclosporine A-loaded micelles: formulation, in vitro and in vivo studies. Drug Deliv. (2018) 25:888–99. doi: 10.1080/10717544.2018.1458923
14. Lv G, Sun D, Zhang J, Xie X, Wu X, Fang W, et al. Lx2–32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B. (2017) 7:52–8. doi: 10.1016/j.apsb.2016.06.005
15. Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. (2013) 4:342–59. doi: 10.1177/1947601913507951
16. Alici E, Konstantinidis KV, Aints A, Dilber MS, Abedi-Valugerdi M. Visualization of 5T33 myeloma cells in the C57BL/KaLwRij mouse: establishment of a new syngeneic murine model of multiple myeloma. Exp Hematol. (2004) 32:1064–72. doi: 10.1016/j.exphem.2004.07.019
17. Gu C, Cheng H, Yang H, Bian Y, Wang Y, Zhang Y, et al. MK2 is a therapeutic target for high-risk multiple myeloma. Haematologica. (2018). doi: 10.3324/haematol.2017.182121. [Epub ahead of print].
18. Feng Y, Wen J, Chang CC. p38 Mitogen-activated protein kinase and hematologic malignancies. Arch Pathol Lab Med. (2009) 133:1850–6. doi: 10.1043/1543–2165-133.11.1850
19. Yasui H, Hideshima T, Ikeda H, Jin J, Ocio EM, Kiziltepe T, et al. BIRB 796 enhances cytotoxicity triggered by bortezomib, heat shock protein (Hsp) 90 inhibitor, and dexamethasone via inhibition of p38 mitogen-activated protein kinase/Hsp27 pathway in multiple myeloma cell lines and inhibits paracrine tumour growth. Br J Haematol. (2007)136:414–23. doi: 10.1111/j.1365–2141.2006.06443.x
20. Birner P, Beer A, Vinatzer U, Stary S, Hoftberger R, Nirtl N, et al. MAPKAP kinase 2 overexpression influences prognosis in gastrointestinal stromal tumors and associates with copy number variations on chromosome 1 and expression of p38 MAP kinase and ETV1. Clin Cancer Res. (2012) 18:1879–87. doi: 10.1158/1078–0432.CCR-11–2364
21. Liu B, Yang L, Huang B, Cheng M, Wang H, Li Y, et al. A functional copy-number variation in MAPKAPK2 predicts risk and prognosis of lung cancer. Am J Hum Genet. (2012) 91:384–90. doi: 10.1016/j.ajhg.2012.07.003
22. Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. (2006) 25:2987–98. doi: 10.1038/sj.onc.1209337
23. McGuire VA, Gray A, Monk CE, Santos SG, Lee K, Aubareda A, et al. Cross talk between the Akt and p38alpha pathways in macrophages downstream of Toll-like receptor signaling. Mol Cell Biol. (2013) 33:4152–65. doi: 10.1128/MCB.01691–12
24. Fiore M, Forli S, Manetti F. Targeting mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2, MK2) medicinal chemistry efforts to lead small molecule inhibitors to clinical trials. J Med Chem. (2016) 59:3609–34. doi: 10.1021/acs.jmedchem.5b01457
25. Li Y, Kopper F, Dobbelstein M. Inhibition of MAPKAPK2/MK2 facilitates DNA replication upon cancer cell treatment with gemcitabine but not cisplatin. Cancer Lett. (2018) 428:45–54. doi: 10.1016/j.canlet.2018.04.030
26. Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. (2010) 40:34–49. doi: 10.1016/j.molcel.2010.09.018
27. Kopper F, Bierwirth C, Schon M, Kunze M, Elvers I, Kranz D, et al. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc Natl Acad Sci USA. (2013) 110:16856–61. doi: 10.1073/pnas.1304355110
28. Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem. (2007) 282:21598–608. doi: 10.1074/jbc.M611316200
Keywords: multiple myeloma, MK2, inhibitor, proliferation, combination
Citation: Guo M, Sun D, Fan Z, Yuan Y, Shao M, Hou J, Zhu Y, Wei R, Zhu Y, Qian J, Li F, Yang Y and Gu C (2019) Targeting MK2 Is a Novel Approach to Interfere in Multiple Myeloma. Front. Oncol. 9:722. doi: 10.3389/fonc.2019.00722
Received: 19 February 2019; Accepted: 19 July 2019;
Published: 08 August 2019.
Edited by:
Ritu Gupta, All India Institute of Medical Sciences, IndiaReviewed by:
Jerome Moreaux, UMR9002 Institut de Génétique Humaine (IGH), FranceMaria Chaudhry, The Ohio State University, United States
Copyright © 2019 Guo, Sun, Fan, Yuan, Shao, Hou, Zhu, Wei, Zhu, Qian, Li, Yang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Li, eXgwMjEwMjFAc2luYS5jb20=; Ye Yang, eWFuZ3llODc2QHNpbmEuY29t; Chunyan Gu, Z3VjaHVueWFuQG5qdWNtLmVkdS5jbg==
†These authors have contributed equally to this work