 Hayley R. Walsh
Hayley R. Walsh Brianne M. Cruickshank
Brianne M. Cruickshank Justin M. Brown
Justin M. Brown Paola Marcato
Paola Marcato- 1Department of Pathology, Dalhousie University, Halifax, NS, Canada
- 2Department of Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
Within heterogeneous tumors, cancer stem cell (CSC) populations exhibit the greatest tumor initiation potential, promote metastasis, and contribute to therapy resistance. For breast cancer specifically, CSCs are identified by CD44highCD24low cell surface marker expression and increased aldehyde dehydrogenase activity. In general, bulk breast tumor cells possess altered energetics characterized by aerobic glycolysis. In contrast, breast CSCs appear to have adaptive metabolic plasticity that allows these tumor-initiating cells to switch between glycolysis and oxidative phosphorylation, depending on factors present in the tumor microenvironment (e.g., hypoxia, reactive oxygen species, availability of glucose). In this article, we review the regulatory molecules that may facilitate the metabolic plasticity of breast CSCs. These regulatory factors include epigenetic chromatin modifiers, non-coding RNAs, transcriptional repressors, transcription factors, energy and stress sensors, and metabolic enzymes. Furthermore, breast cancer cells acquire CSC-like characteristics and altered energetics by undergoing epithelial-mesenchymal transition (EMT). This energy costly process is paired with reprogrammed glucose metabolism by epigenetic modifiers that regulate expression of both EMT and other metabolism-regulating genes. The survival advantage imparted to breast CSCs by metabolic plasticity suggests that targeting the factors that mediate the energetic switch should hinder tumorigenesis and lead to improved patient outcomes.
Introduction
Relative to non-transformed cells, transformed cells possess unique metabolic requirements (1). Specifically, highly proliferative cells require more energy and biomolecules to support rapid division (1). To accomplish this, transformed cells must increase the production of biomolecules needed and extract elevated quantities of nutrients from the surrounding environment (2). Most cancer cells reprogram metabolic pathways to facilitate the use of aerobic glycolysis as their primary energy source in lieu of oxidative phosphorylation (3). This change is accompanied by an increase in glucose uptake to support the high-energy needs of highly proliferative cancer cells. Although aerobic glycolysis remains inefficient for ATP production, these cells will preferentially initiate aerobic glycolysis even when oxygen is present (Warburg effect) (4). Explanations for this are still unclear; however, some have suggested that the transformed cell switches or reprograms its metabolic machinery to increase biomass instead of maximizing ATP production (5). Others suggest a more integrated approach where cancer cells upregulate glycolysis in response to intermittent hypoxia, producing a large amount of lactate, and increasing the acidity of the microenvironment. In response to this, cancer cells develop acid resistance, which conveys a growth advantage over other cells (3).
For breast tumors specifically, many recent studies are linking glycolytic metabolism to increased tumor progression. For example, plasminogen activator inhibitor 1 (PAI1) promotes migration by stimulating cytoskeletal rearrangement, mitochondrial fragmentation, and glycolytic metabolism in triple-negative breast cancer (TNBC) cells (6). Glycolic metabolite methylglyoxal promotes extracellular matrix remodeling and activation of the pro-migratory signaling pathway, MEK/ERK/SMAD1 in estrogen receptor-positive (ER+) and TNBC cell lines (7). Inversely, treatment of breast cancer cells and xenografts with natural compound cantharidin inhibited migration, invasion, angiogenesis, and metastasis by increasing oxygen consumption and suppressing aerobic glycolysis (8). Furthermore, increased oxygen consumption and dependence on oxidative metabolism through the expression of good prognostic marker B-cell lymphoma two associated death promoter (BAD) resulted in large but non-aggressive breast tumors (9).
Conversely, there is also evidence to support the hypothesis that impeding oxidative phosphorylation with resveratrol is detrimental to breast cancer cells (10). Similar findings regarding the requirement for oxidative phosphorylation for cancer progression have also been reported (11–13). A possible explanation for the contradicting evidence is that cancer cells can express a hybrid phenotype where both glycolytic and oxidative states co-exist, allowing these cells to adapt to the dynamic tumor microenvironment (14). Undoubtedly, distinct subpopulations of cancer cells within tumors (e.g., cancer stem cells, CSCs, vs. non-CSCs), that have a varying capacity for tumorigenicity, metastasis, and proliferation, would also differ in their energetic and metabolic needs. The shifting energy demands inherent of the evolving tumor microenvironment is best met with metabolic plasticity, which increasing evidence suggests, is often associated with CSC populations.
Breast Cancer Stem CELLS: Inherent Metabolic Plasticity
In a heterogeneous tumor consisting of cancer cells and non-cancer cells (e.g., resident fibroblasts, cancer-associated fibroblasts (CAFs), immune cells), CSCs are the most tumorigenic cancer cell population, illustrated by their capacity to initiate new tumors with high efficiency (15). Tumor cell populations enriched for CSCs can be defined by cancer-specific cell surface markers (e.g., CD44highCD24low for breast cancer). Alternatively, increased aldehyde dehydrogenase (ALDH) activity detected by the Aldefluor assay is commonly used to identify CSCs across tumor types. ALDH1A3 and ALDH1A1 are the primary contributors of the Aldefluorhigh activity associated with breast CSCs (16). Of the breast cancer subtypes, TNBC/basal-like breast cancers have higher percentages of CSCs, which may contribute to their aggressiveness and the worse patient outcomes associated with these breast cancers (17–23). In terms of mitigating the risk of recurrence, the resistance of CSCs to chemotherapies, radiotherapy, and possibly immunotherapies is an increasing concern (24–29). Therapies that target CSCs may reduce the risk of relapse. Signaling pathways [Notch, Wnt, and Hedgehog (30)] and enzymes [e.g., ALDHs (31)] associated with CSCs, are also mediators of tumorigenicity, metastasis, and therapy resistance and are under investigation as potential avenues for therapeutic intervention (32). In recent years targeting CSCs through their metabolic vulnerabilities has been proposed (33).
CSCs can be distinguished from bulk tumor cells based on their dependence on specific metabolic pathways (1). Cancer cells that regulate their glucose metabolism in response to adverse tumor microenvironments are described as metabolically plastic and typically have CSC-associated characteristics such as metastasis and chemo/radio-resistance (34). There is conflicting evidence regarding the dependence of the CSCs of breast (and other tumor types) on glycolysis vs. oxidative phosphorylation. Reports have demonstrated that chemically inhibiting glycolysis reduces populations of breast cancer cells with CSC characteristics (35, 36). Furthermore, during epithelial-mesenchymal transition (EMT), breast cancer cells undergo glycolytic metabolic reprogramming where they acquire CSC-like characteristics and exhibit increased tumorigenicity (37). This EMT/epigenetic and metabolic reprogramming of CSC-like cells are linked by key EMT-initiating factor Snail. A transcriptional repressor, Snail, recruits methyltransferase G9a (increases repressive H3K9me2 and decreases activating H3K9ac histone marks), and DNA methyltransferases (DNMT) to promoters, resulting in hypermethylated silenced genes. These epigenetically silenced genes include epithelial-associated cell adhesion molecule, E-cadherin, and glycolytic antagonist, fructose-1,6-biphosphatase (FBP1), which contribute to EMT development and the switch to glycolytic metabolism in breast cancer cells (37). Inversely, FBP1 expression prevents Snail-mediated EMT (37). Importantly, downregulation of oxidative phosphorylation is intimately linked to the expression of EMT gene signatures (across 20 different cancers, including breast cancer), and metastatic cancers associated with the worse clinical results (38). Although not specifically studied by Gaude and Frezza (38), the well-established connection between EMT and CSCs (39), may indirectly implicate breast CSCs with decreased oxidative phosphorylation. The most commonly downregulated gene in their comprehensive study was mitochondrial respiratory chain enzyme, succinate dehydrogenase complex subunit D (SDHD) (38). In breast tumors downregulation of SDH complex subunit C (SDHC) is associated with EMT, and expression of pro-EMT transcription factors Twist-related protein 1 (TWIST1) and Snail resulted in lower mitochondrial mass and respiration (40). Furthermore, glycolytic phosphoglucose isomerase (PGI)/autocrine motility factor (AMF) is associated with EMT in breast cancer cells and the expression of mesenchymal markers zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2 (41). Hence, there are plenty of studies suggesting that there is an intimate link between the shift toward glycolytic metabolism and EMT/CSC-like characteristics.
In contrast to the studies described above, there is also ample evidence of breast CSCs utilizing oxidative phosphorylation as their primary metabolic program. Vlashi et al. reported that breast CSCs rely on oxidative phosphorylation, while their more differentiated progeny exhibit a glycolytic phenotype (42). Similarly, Banerjee et al. demonstrated that bulk tumor cells depend chiefly on glycolysis, while tumors enriched in breast CSCs rely mainly on oxidative phosphorylation (43). They restricted glycolysis in the breast cancer cells by adapting the cells to fructose, which increased CSCs (i.e., cells with high ALDH activity and the CD44high/CD24low phenotype), invasiveness, and chemoresistance. Similarly, a different study showed that breast CSCs are dependent on oxidative phosphorylation (44), while another study suggests breast CSCs rely on alternative metabolic processes such as fatty acid oxidation (45). The contradicting findings concerning breast CSCs displaying predominant glycolytic vs. oxidative metabolism may be a result of the differing microenvironments that the studies were completed in, which in turn evokes the metabolic plasticity phenotype of CSCs (Figure 1). In agreement with this, breast CSC populations demonstrated increased metabolic plasticity by having enhanced ability to boost oxidative phosphorylation upon glycolysis inhibition (46). In the initial culturing of patient-derived lines, CSCs seem to preferentially utilize oxidative phosphorylation rather than aerobic glycolysis (47). Hence it is possible that CSC metabolic reprogramming in a culturing environment that is glucose-enriched, favors glycolysis, while glucose depleted conditions shift the metabolic balance toward oxidative phosphorylation. Determining the cellular factors responsible for metabolic plasticity will allow for more effective targeting of CSC populations within heterogeneous breast tumors.
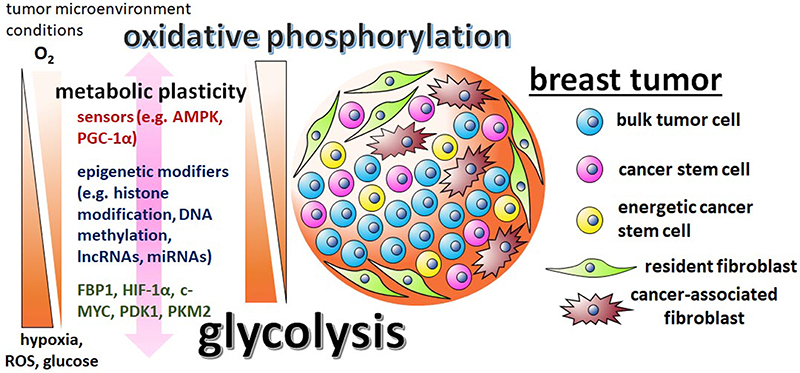
Figure 1. The metabolic plasticity of breast CSCs is dictated by the varying conditions of the tumor microenvironment and mediated by sensors, epigenetic modifiers, and metabolic enzymes. There are contradictory reports regarding breast CSC metabolism, with some studies suggesting the cells are predominately glycolytic, while other studies suggest that breast CSCs utilize primarily oxidative phosphorylation pathways. These discrepancies are possibly reflective of the differing tumor microenvironments in which the cells were assayed in and the metabolic/epigenetic plasticity inherent of CSCs. Influential factors in the tumor microenvironment include oxygen, ROS, and glucose levels. The initiation of the varying metabolic phenotypes associated with breast CSCs depend on regulatory factors, like stress sensors AMPK and PGC-1α, epigenetic modifiers G9a, DNMTs, lncRNA H19, miRNAs let-7 and miR-21, master regulator HIF-1α, glycolytic enzymes PDK1 and PKM2, and glycolytic antagonist FBP1.
Regulators of the Metabolic Phenotypes Associated With Breast Cancer Stem Cells
Essential to metabolic plasticity is the regulation of mitochondria biogenesis. Mitochondria are the hubs that harbor the enzymes that drive the tricarboxylic acid cycle, oxidative phosphorylation, fatty acid oxidation, biosynthesis of nucleotides, amino acids and lipids, and calcium homeostasis (48). Mitochondrial biogenesis is mediated by a diverse signaling network. Key factors include AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1α), which both function as energy and stress sensors (49). PGC-1α is a co-transcriptional regulator that induces mitochondrial biogenesis by activating transcription factors such as peroxisome proliferator-activated alpha (PPARα), estrogen-related receptor alpha (ERRα), nuclear respiratory factor 1 (NRF1), and NRF2 in breast cancer (50). Increased PGC-1α leads to increased mitochondrial biogenesis, oxidative phosphorylation, and generation of ATP. The requirement of PGC-1α for the oxidative phosphorylation energetics associated with CSCs was first shown in pancreatic cancer (51). More recently, increased PGC-1α was demonstrated in the proteomics analysis of a newly characterized breast CSC sub-population termed energetic cancer stem cells [e-CSCs, Figure 1, Table 1; (59)]. These breast CSCs are characterized by increased glycolysis, oxidative phosphorylation, and mitochondria size.
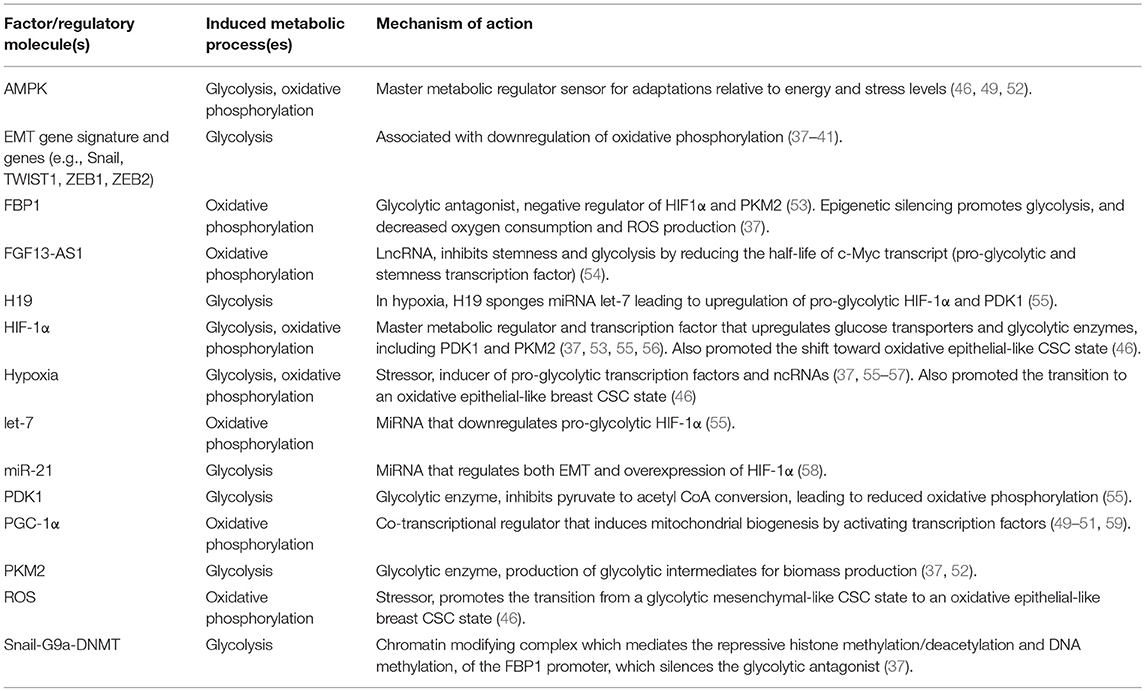
Table 1. Factors that promote or regulate the metabolic reprograming of CSCs or cancer cells with stemness features in breast tumors.
The metabolic/oxidative cellular stressors hypoxia and reactive oxygen species (ROS) present in the tumor microenvironment drive the need for metabolic plasticity (Table 1, Figure 1). These stressors can promote the transition of ROSlow mesenchymal-like quiescent breast CSC state to a ROShigh epithelial-like proliferative breast CSC state. Inhibition of glycolysis by 2-deoxy-D-glucose shifted the proportion of CSC populations toward the more oxidative epithelial-like state and decreased the glycolytic mesenchymal-like state (46). This metabolic transition is mediated by activation of the master metabolism regulator axis, AMPK-hypoxia-inducible factor 1 alpha (S-1α) and reversed by the addition of ROS inhibitor N-acetylcysteine (Table 1).
The hypoxia-induced transcription factor HIF-1α is linked to the Warburg effect through its upregulation of glucose transporters, elevated expression of enzymes involved in glycolysis, and inhibition of oxidative phosphorylation, resulting in HIF-1α-mediated glycolysis in cancer cells (60). In breast cancer, negative regulation of HIF-1α by FBP1 results in decreased growth, migration, glucose consumption, and lactate production (53). Epigenetic silencing of FBP1 promoted glycolysis, increased glucose uptake, macromolecule biosynthesis, sustained ATP production under hypoxic conditions, and inhibited oxygen use and ROS production by suppressing mitochondrial complex I activity (Table 1) (37). Importantly, the loss of FBP1 also promoted the CSC-like phenotype in the breast cancer cells by increasing the interaction of Wnt signaling molecules β-catenin and transcription factor t-cell factor (TCF). HIF-1α promotes glycolytic reprogramming at least in part through its regulation of key glycolytic enzymes in glucose metabolism, pyruvate dehydrogenase kinase 1 (PDK1), and pyruvate kinase isoform M2 (PKM2) (56). For example, when PKM2 activity is inhibited by FBP1, glycolytic intermediates for biomass synthesis are not produced (37). Treatment of breast tumor xenografts with diallyl disulfide suppressed breast CSCs and glucose metabolism in a mechanism implicating PKM2 and AMPK (52). In the hypoxic regions of breast tumor xenografts, PDK1 is increased in breast CSC populations, where it activates glycolysis and promotes CSC characteristic in breast tumor xenografts (55). The study on PDK1 also highlighted the essential role that epigenetic modifiers, non-coding RNAs, play in the metabolic plasticity of breast CSCs.
Non-Coding RNAs As Mediators of Breast Cancer Stem Metabolic Plasticity
Non-coding RNAs are key mediators of metabolic reprogramming, with early evidence illustrating the essential role of microRNAs (miRNAs). For example, hypoxia-induced miR-210 reduces mitochondrial respiration in cancer cells, including breast cancer (57). Similarly, inflammation-induced miR-155 promotes glycolysis by regulating expression of glycolytic enzyme hexokinase two in breast cancer cells (61).
The dysregulation of non-coding RNAs, miRNAs and long non-coding RNAs (lncRNAs), result in gene expression changes that are critical in not only metabolic reprogramming of breast cancer, but also EMT and progression to metastasis, CSC maintenance, and response to therapy (58, 62–64). LncRNAs are non-protein coding transcripts >200 nucleotides in length. Notably, the number of lncRNAs expressed in humans is at least equivalent to the number of protein-coding genes (65, 66). It was once thought that these molecules had little functional relevance; however, characterization of some lncRNAs has revealed they typically function in gene regulation by acting as activators or decoys for transcription factors, guides, recruiters of chromatin-modifying complexes, and miRNA sponges (67). Due to their tissue-specific expression patterns and selective expression in certain cancers, lncRNAs represent attractive therapeutic targets (68).
In terms of evidence for the role of lncRNAs in the metabolic reprogramming of breast cancer, there are several recent examples. LincRNA-p21 plays a critical role in hypoxia-induced glycolysis in breast cancer. The hypoxia-responsive lncRNA binds HIF-1α, preventing its ubiquitination and degradation, leading to the accumulation of the key hypoxic mediator of glycolysis (69). Similarly, lactate-induced HIF-1α-stabilizing long non-coding RNA (HISLA) blocks the interaction of prolyl hydroxylase domain-containing protein 2 (PHD2) and HIF-1α, which inhibits the hydroxylation and degradation of HIF-1α (70). In TNBC, long intergenic non-coding RNA for kinase activation (LINK-A) promotes glycolysis by mediating phosphorylation and stabilization of HIF-1α (71).
For breast CSCs specifically, the evidence that miRNAs and lncRNAs play essential roles in mediating the metabolic plasticity of the cells is starting to accumulate. This is well-exemplified by how they mediate the effects of PDK1 and HIF-1α in breast CSC-specific glycolysis (55). LncRNA H19 sponges miRNA let-7 and HIF-1α is a target of the miRNA. The sponging of let-7 by H19 resulted in upregulation of HIF-1α, which then increased the expression of glycolytic enzyme PDK1. Therefore, the dysregulation of the lncRNA H19 is the initiating event that results in that metabolic reprogramming of CSCs under hypoxic conditions. Similarly, HIF-1α is regulated by miR-21 in the CSC-like cells of breast cancer cells, with antagonism of the miRNA suppressing EMT, CSC-like qualities and HIF-1α (58). FGF13-AS1 may play a critical role in the glycolytic reprogramming of breast CSCs. This lncRNA is downregulated in breast tumors and inhibits glycolysis and stemness by reducing the half-life of c-Myc mRNA (54). Stemness-associated c-Myc is a transcription factor and target of HIF-1α and induces the expression of glycolytic genes, leading to altered glucose consumption (72). It is very likely that as the functions of more lncRNAs are revealed, further connections between these epigenetic regulatory molecules, stemness, and altered energetics in breast cancer will continue to be made.
Conclusions
Most evidence supports the hypothesis that within a heterogeneous tumor, breast CSCs likely occupy a unique metabolic state relative to other cancer cells, mediated by the increased epigenetic and metabolic plasticity of the cells. Like other solid tumors, breast cancers generally possess genetic modifications that facilitate increased glycolysis in the presence of ample glucose, and under normoxic conditions, CSCs can utilize oxidative phosphorylation. Hence, due to the metabolic adaptability of CSCs, targeting a single metabolic pathway is likely insufficient for tumor eradication. For example, glycolytic inhibitors such as dichloroacetate and phloretin are proposed novel anti-cancer drugs that rely upon the metabolic reprogramming of cancer cells (73–75). However, the results of a recent phase I/II clinical trial reported modest responses when patients with tumors with altered metabolic activity were treated with targeted pentose phosphate pathway, glycolysis, and amino acid depleting inhibitors (76). It is unclear how breast CSC populations would be affected by such treatments; however, based on current knowledge of the metabolic plasticity of the cells, it seems unlikely that CSCs would be depleted by such strategies. This suggests that targeting breast CSCs based on the metabolic reprogramming of tumors requires identification of the factors that impart CSCs with metabolic plasticity (Table 1). For example, to target key metabolic plasticity reprogrammer HIF-1α, various drugs have been proposed; aminoflavone, 2-methoxyestradiol, and synthetic oligonucleotides inhibit its expression, acriflavine inhibits HIF dimerization, echinomycin inhibits its DNA binding activity and chetomin blocks its transcriptional activity (77). Alternatively, one could use hypoxia prodrugs (e.g., evofosfamide, tirapazamine) which are activated in hypoxic environments. Novel therapeutic strategies that target HIF-1 α and other key regulators will limit the capacity for the metabolic rewiring of breast CSCs and render these tumor-initiating cells more vulnerable to the varying conditions of the tumor microenvironment.
Author Contributions
HW, BC, and JB drafted the manuscript, figure, and table. PM edited and conceived the manuscript figure and table.
Funding
Support was provided by grant funding to PM from the Canadian Institutes of Health Research (CIHR, PJT 162313). BC was supported by Killam Laureate and Nova Scotia Research and Innovation Graduate Scholarships. JB was supported by a scholarship from the Beatrice Hunter Cancer Research Institute and the Terry Fox Research Institute.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. (2016) 114:1305–12. doi: 10.1038/bjc.2016.152
2. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
3. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. (2004) 4:891–9. doi: 10.1038/nrc1478
4. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. (2011) 11:325–37. doi: 10.1038/nrc3038
5. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
6. Humphries BA, Buschhaus JM, Chen YC, Haley HR, Qyli T, Chiang B, et al. Plasminogen activator inhibitor 1 (PAI1) promotes actin cytoskeleton reorganization and glycolytic metabolism in triple negative breast cancer. Mol Cancer Res. 17:1142–54. (2019). doi: 10.1158/1541-7786.MCR-18-0836
7. Nokin MJ, Bellier J, Durieux F, Peulen O, Rademaker G, Gabriel M, et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. (2019) 21:11. doi: 10.1186/s13058-018-1095-7
8. Pan Y, Zheng Q, Ni W, Wei Z, Yu S, Jia Q, et al. Breaking glucose transporter 1/pyruvate kinase M2 glycolytic loop is required for cantharidin inhibition of metastasis in highly metastatic breast cancer. Front Pharmacol. (2019) 10:590. doi: 10.3389/fphar.2019.00590
9. Mann J, Githaka JM, Buckland TW, Yang N, Montpetit R, Patel N, et al. Non-canonical BAD activity regulates breast cancer cell and tumor growth via 14-3-3 binding and mitochondrial metabolism. Oncogene. (2019) 38:3325–39. doi: 10.1038/s41388-018-0673-6
10. Rodríguez-Enríquez S, Pacheco-Velázquez SC, Marín-Hernández Á, Gallardo-Pérez JC, Robledo-Cadena DX, Hernández-Reséndiz I, et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol Appl Pharmacol. (2019) 370:65–77. doi: 10.1016/j.taap.2019.03.008
11. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. (2014) 16:992–1003. doi: 10.1038/ncb3039
12. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, et al. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene. (2015) 34:3908–16. doi: 10.1038/onc.2014.321
13. Viale A, Corti D, Draetta GF. Tumors and mitochondrial respiration: a neglected connection. Cancer Res. (2015) 75:3687–91. doi: 10.1158/0008-5472.CAN-15-0491
14. Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, et al. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. (2017) 77:1564–74. doi: 10.1158/0008-5472.CAN-16-2074
15. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. (2006) 58:267–84. doi: 10.1146/annurev.med.58.062105.204854
16. Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. (2011) 29:32–45. doi: 10.1002/stem.563
17. Li H, Ma F, Wang H, Lin C, Fan Y, Zhang X, et al. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int J Biol Mark. (2013) 28:357–64. doi: 10.5301/JBM.5000048
18. Giatromanolaki A, Sivridis E, Fiska A, Koukourakis MI. The CD44+/CD24- phenotype relates to ‘triple-negative’ state and unfavorable prognosis in breast cancer patients. Med Oncol. (2011) 28:745–52. doi: 10.1007/s12032-010-9530-3
19. Wu Y, Sarkissyan M, Elshimali Y, Vadgama JV. Triple negative breast tumors in african-american and hispanic/latina women are high in CD44+, low in CD24+, and have loss of PTEN. PLoS ONE. (2013) 8:e78259. doi: 10.1371/journal.pone.0078259
20. Tsang JY, Huang YH, Luo MH, Ni YB, Chan SK, Lui PC, et al. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res Treat. (2012) 136:407–17. doi: 10.1007/s10549-012-2271-6
21. Perrone G, Gaeta LM, Zagami M, Nasorri F, Coppola R, Borzomati D, et al. In situ identification of CD44+/CD24- cancer cells in primary human breast carcinomas. PLoS ONE. (2012) 7:e43110. doi: 10.1371/journal.pone.0043110
22. Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. (2011) 64:937–44. doi: 10.1136/jcp.2011.090456
23. Idowu MO, Kmieciak M, Dumur C, Burton RS, Grimes MM, Powers CN, et al. CD44+/CD24-/lowcancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum Pathol. (2012) 43:364–73. doi: 10.1016/j.humpath.2011.05.005
24. Thomas M, Coyle KM, Sultan M, Vaghar-Kashani A, Marcato P. Chemoresistance in cancer stem cells and strategies to overcome resistance. Chemother Open Access. (2014) 3:1. doi: 10.4172/2167-7700.1000125
25. Sultan M, Vidovic D, Paine AS, Huynh TT, Coyle KM, Thomas ML, et al. Epigenetic silencing of TAP1 in aldefluor + breast cancer stem cells contributes to their enhanced immune evasion. Stem Cells. (2018) 36:641–54. doi: 10.1002/stem.2780
26. Sultan M, Coyle KM, Vidovic D, Thomas ML, Gujar S, Marcato P. Hide-and-seek: the interplay between cancer stem cells and the immune system. Carcinogenesis. (2017) 38:107–18. doi: 10.1093/carcin/bgw115
27. Mathews LA, Cabarcas SM, Farrar WL. DNA repair: the culprit for tumor-initiating cell survival? Cancer Metastasis Rev. (2011) 30:185–97. doi: 10.1007/s10555-011-9277-0
28. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
29. Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. (2008) 26:2839–45. doi: 10.1200/JCO.2007.15.1829
30. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. (2011) 8:97–106. doi: 10.1038/nrclinonc.2010.196
31. Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. (2011) 10:1378–84. doi: 10.4161/cc.10.9.15486
32. Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta. (2013) 1830:2481–95. doi: 10.1016/j.bbagen.2012.11.008
33. Jagust P, de Luxán-Delgado B, Parejo-Alonso B, Sancho P. Metabolism-based therapeutic strategies targeting cancer stem cells. Front Pharmacol. (2019) 10:203. doi: 10.3389/fphar.2019.00203
34. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. (2016) 18:203. doi: 10.1186/s13058-016-0712-6
35. O'Neill S, Porter RK, McNamee N, Martinez VG, O'Driscoll L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci Rep. (2019) 9:3788. doi: 10.1038/s41598-019-39789-9
36. Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. (2014) 21:124–35. doi: 10.1038/cdd.2013.131
37. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. (2013) 23:316–31. doi: 10.1016/j.ccr.2013.01.022
38. Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun. (2016) 7:13041. doi: 10.1038/ncomms13041
39. Wang SS, Jiang J, Liang XH, Tang YL. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. (2015) 8:2973–80. doi: 10.2147/OTT.S91863
40. Røsland GV, Dyrstad SE, Tusubira D, Helwa R, Tan TZ, Lotsberg ML, et al. Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC. Cancer Metab. (2019) 7:6. doi: 10.1186/s40170-019-0197-8
41. Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, et al. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. (2011) 71:3400–9. doi: 10.1158/0008-5472.CAN-10-0965
42. Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, Chan M, et al. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res Treat. (2014) 146:525–34. doi: 10.1007/s10549-014-3051-2
43. Banerjee A, Arvinrad P, Darley M, Laversin SA, Parker R, Rose-Zerilli MJJ, et al. The effects of restricted glycolysis on stem-cell like characteristics of breast cancer cells. Oncotarget. (2018) 9:23274–88. doi: 10.18632/oncotarget.25299
44. Jain S, Wolf DA. Abstract LB-397: SMIP004-7: a novel oxidative phosphorylation inhibitor selectively kills breast cancer stem-like cells with metabolic vulnerabilities. in Molecular and Cellular Biology / Genetics 78, LB-397-LB-397 (American Association for Cancer Research, 2018). doi: 10.1158/1538-7445.AM2018-LB-397
45. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. (2017) 27:136–150.e5. doi: 10.1016/j.cmet.2017.11.001
46. Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. (2018) 28:69–86.e6. doi: 10.1016/j.cmet.2018.06.006
47. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA. (2011) 108:16062–67. doi: 10.1073/pnas.1106704108
48. Cannino G, Ciscato F, Masgras I, Sánchez-Martín C, Rasola A. Metabolic plasticity of tumor cell mitochondria. Front Oncol. (2018) 8:333. doi: 10.3389/fonc.2018.00333
49. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. (2010) 47:69–84. doi: 10.1042/bse0470069
50. Bost F, Kaminski L. The metabolic modulator PGC-1α in cancer. Am J Cancer Res. (2019) 9:198–211.
51. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
52. Xie X, Huang X, Tang H, Ye F, Yang L, Guo X, et al. Diallyl disulfide inhibits breast cancer stem cell progression and glucose metabolism by targeting CD44/PKM2/AMPK signaling. Curr Cancer Drug Targets. (2018) 18:592–9. doi: 10.2174/1568009617666171024165657
53. Shi L, He C, Li Z, Wang Z, Zhang Q. FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1α. Neoplasma. (2017) 64:535–42. doi: 10.4149/neo_2017_407
54. Ma F, Liu X, Zhou S, Li W, Liu C, Chadwick M, et al. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. (2019) 450:63–75. doi: 10.1016/j.canlet.2019.02.008
55. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene. (2018) 37:1062–74. doi: 10.1038/onc.2017.368
56. Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells. (2014) 32:364–76. doi: 10.1002/stem.1552
57. Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. (2009) 10:273–84. doi: 10.1016/j.cmet.2009.08.015
58. Han M, Wang Y, Liu M, Bi X, Bao J, Zeng N, et al. MiR-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1α expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci. (2012) 103:1058–64. doi: 10.1111/j.1349-7006.2012.02281.x
59. Fiorillo M, Sotgia F, Lisanti MP. “Energetic” cancer stem cells (e-CSCs): a new hyper-metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front Oncol. (2018) 8:677. doi: 10.3389/fonc.2018.00677
60. Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y, et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer. (2017) 16:130. doi: 10.1186/s12943-017-0699-3
61. Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. (2012) 31:1985–98. doi: 10.1038/emboj.2012.45
62. Vincent A, Van Seuningen I. On the epigenetic origin of cancer stem cells. Biochim Biophys Acta. (2012) 1826:83–8. doi: 10.1016/j.bbcan.2012.03.009
63. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. (2010) 31:27–36. doi: 10.1093/carcin/bgp220
64. Vidovic D, Huynh TT, Konda P, Dean C, Cruickshank BM, Sultan M, et al. ALDH1A3-regulated long non-coding RNA NRAD1 is a potential novel target for triple-negative breast tumors and cancer stem cells. Cell Death Differ. (2019) 1. doi: 10.1038/s41418-019-0362-1. [Epub ahead of print].
65. Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K, et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. (2013) 41:D246–51. doi: 10.1093/nar/gks915
66. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. (2013) 193:651–69. doi: 10.1534/genetics.112.146704
67. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. (2012) 81:145–66. doi: 10.1146/annurev-biochem-051410-092902
68. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. (2011) 1:391–407. doi: 10.1158/2159-8290.CD-11-0209
69. Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol Cell. (2014) 53:88–100. doi: 10.1016/j.molcel.2013.11.004
70. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. (2019) 21:498–510. doi: 10.1038/s41556-019-0299-0
71. Lin A, Li C, Xing Z, Hu Q, Liang K, Han L, et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat Cell Biol. (2016) 18:213–24. doi: 10.1038/ncb3295
72. Peixoto J, Lima J. Metabolic traits of cancer stem cells. Dis Model Mech. (2018) 11:dmm033464. doi: 10.1242/dmm.033464
73. Pathak RK, Marrache S, Harn DA, Dhar S. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem Biol. (2014) 9:1178–87. doi: 10.1021/cb400944y
74. El Sayed SM, Baghdadi H, Ahmed NS, Almaramhy HH, Mahmoud AA, El-Sawy SA, et al. Dichloroacetate is an antimetabolite that antagonizes acetate and deprives cancer cells from its benefits: a novel evidence-based medical hypothesis. Med Hypotheses. (2019) 122:206–9. doi: 10.1016/j.mehy.2018.11.012
75. Kim MS, Kwon JY, Kang NJ, Lee KW, Lee HJ. Phloretin induces apoptosis in H-Ras MCF10A human breast tumor cells through the activation of p53 via JNK and p38 mitogen-activated protein kinase signaling. Ann N Y Acad Sci. (2009) 1171:479–83. doi: 10.1111/j.1749-6632.2009.04692.x
76. van der Mijn JC, Panka DJ, Geissler AK, Verheul HM, Mier JW. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. (2016) 4:14. doi: 10.1186/s40170-016-0154-8
Keywords: breast cancer, metabolic plasticity, CSCs, incRNAs, miRNAs
Citation: Walsh HR, Cruickshank BM, Brown JM and Marcato P (2019) The Flick of a Switch: Conferring Survival Advantage to Breast Cancer Stem Cells Through Metabolic Plasticity. Front. Oncol. 9:753. doi: 10.3389/fonc.2019.00753
Received: 09 June 2019; Accepted: 26 July 2019;
Published: 20 August 2019.
Edited by:
Dayanidhi Raman, College of Medicine and Life Sciences, University of Toledo, United StatesReviewed by:
Ada Pesapane, Department of Clinical Medicine and Surgery, University of Naples Federico II, ItalyMasahiko Tanabe, The University of Tokyo, Japan
Copyright © 2019 Walsh, Cruickshank, Brown and Marcato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Marcato, cGFvbGEubWFyY2F0b0BkYWwuY2E=
†These authors have contributed equally to this work