 Anna Maria Militello1
Anna Maria Militello1 Teresa Zielli1
Teresa Zielli1 Daniela Boggiani1Maria Michiara1Nadia Naldi1Beatrice Bortesi1Paola Zanelli2Vera Uliana2Sara Giuliotti3
Daniela Boggiani1Maria Michiara1Nadia Naldi1Beatrice Bortesi1Paola Zanelli2Vera Uliana2Sara Giuliotti3 Antonino Musolino1,4*
Antonino Musolino1,4*- 1Breast Unit and Cancer Genetics Service, University Hospital of Parma, Parma, Italy
- 2Medical Genetics Unit, University Hospital of Parma, Parma, Italy
- 3Radiology Unit, University Hospital of Parma, Parma, Italy
- 4Gruppo Oncologico Italiano di Ricerca Clinica (GOIRC), Parma, Italy
Sensitivity to endocrine therapy of patients with estrogen receptor (ER)-positive metastatic breast cancer and germline BRCA1/2 mutations is not yet fully elucidated. Furthermore, the registration trials of CDK 4/6 inhibitors in combination with endocrine therapy lacked of a pre-specified subgroup analysis in BRCA1/2 mutation carriers. We report clinical history of two patients with BRCA-mutated, ER-positive metastatic breast cancer treated with letrozole plus the CDK 4/6 inhibitor palbociclib. Biological and clinical implications of the treatment outcome observed in the two cases are discussed with the knowledge of scientific evidence to date available. Overall, biological rationale, preclinical, and clinical data support the prominent role of CDK 4/6 inhibitors plus endocrine therapy, even in combination with PARP inhibitors, in the treatment of BRCA-mutated, ER-positive breast cancers. However, the interaction between Cyclin/CDK pathway, ER and BRCA is complex and evidences reported so far, albeit reliable, await confirmation in the context of future randomized clinical trials.
Background
Up to 10% of breast cancers (BCs) are attributed to pathogenic germline mutations in the BRCA1 and BRCA2 genes (1, 2). Several studies on over 10,000 cases with BRCA-associated BCs have reported estrogen receptor (ER)-positive rates of 20 and 77%, in BRCA1 and BRCA2 mutation carriers, respectively (3).
ER-positive BCs diagnosed in BRCA1/2 mutation carriers are frequently characterized by higher tumor grade and proliferation rate than ER-positive tumors occurring in non-carriers (4). Less is known about sensitivity to endocrine therapy (ET) of patients with ER-positive metastatic BC (MBC) and BRCA1/2 mutations (5, 6).
Three cyclin-dependent kinase (CDK) 4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) have recently been approved for the treatment of ER-positive MBC (7). However, the registration trials of CDK 4/6 inhibitors in combination with ET lacked of a pre-specified subgroup analysis in BRCA1/2 mutation carriers (8–10).
Here we report clinical history of two patients with BRCA-mutated, ER-positive MBC treated with letrozole plus palbociclib. Biological and clinical implications of the treatment outcome observed in the two cases are discussed with the knowledge of scientific evidence to date available.
Case Presentation 1
In 2012, a 59 year-old woman underwent left upper paracentral quadrantectomy for pT2N1M0, stage IIB, G3, BC [ER: 40%, progesterone receptor (PR): 0%, HER2-negative, ki67: 40%]. The patient received adjuvant chemotherapy with FEC (epirubicin, cyclophosphamide, and 5 FU) for 3 cycles and Docetaxel for 3 cycles. Then, in May 2013, she started adjuvant radiotherapy and ET with anastrozole. Due to the presence of strong family history (FH) of breast and ovarian cancer, BRCA genetic testing was performed with detection of the c.5332+1G>A germline, pathogenic BRCA1 mutation. The patient had prophylactic bilateral annessiectomy in October 2015.
In March 2016 the patient presented with sternal pain. Clinical examination revealed the presence of a chest-wall palpable mass. She had fine needle biopsy (FNB) of the sternal mass with cytological diagnosis of BC metastasis (ER: 60%, PR: 2%, HER2-negative). A total body computed tomography (CT) scan confirmed a unique metastatic bone lesion of the sternum. Anastrozole therapy was then stopped and on April 22, the patient started weekly chemotherapy with paclitaxel, which was interrupted after 2 months due to the CT evidence of an increase in size of the sternal lesion (longest diameter 67 vs. 57 mm). She was then enrolled in the experimental protocol “BRAVO” (11) and treated with the Poly(ADP-ribose) polymerase (PARP) inhibitor niraparib from August 2016 to February 2017, until the CT scan showed disease progression (best response: partial response [PR]). In view of the persistence of a single painful skeletal localization of disease, in March 2017 the patient underwent local radiotherapy and ET with fulvestrant was started concomitantly. After 2 months of therapy, the disease remained radiologically stable. In November 2017, the CT scan showed an increase of sternal lesion, and the CDK4/6 inhibitor, palbociclib, was added to ET. In June 2018, after 6 cycles of treatment, the CT scan showed stable disease (SD).
The patient is currently in good clinical condition (ECOG PS 0, no sternal pain) and continuing treatment. The latest CT scan (10 January 2019) showed a gradual reduction of the sternal lesion [best response by RECIST criteria (12): SD; Figures 1A,B]. At the last visit (April 2019), the duration of response (DOR) is 10 months.
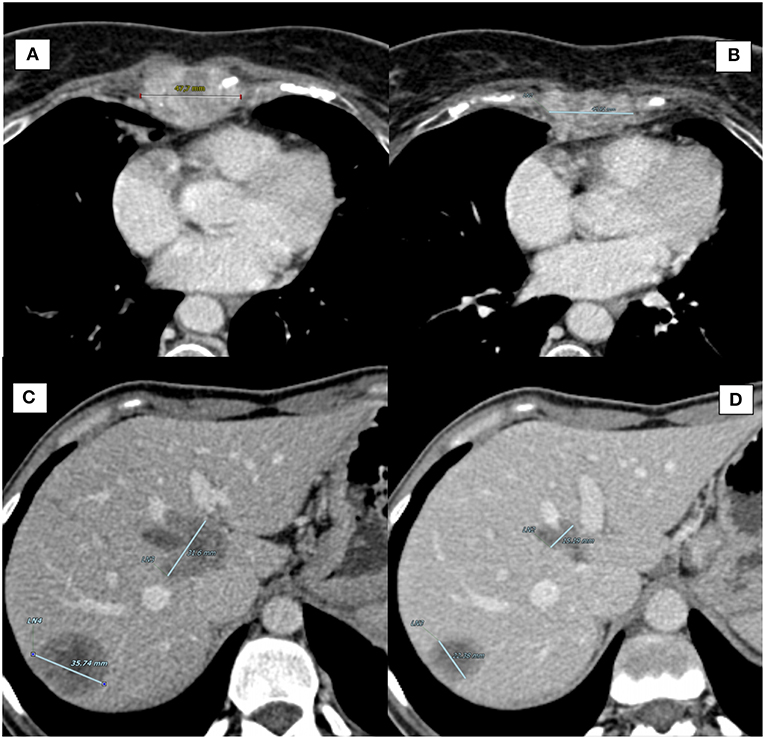
Figure 1. Computed Tomography (CT) scan of target lesions during therapy with CDK4/6 inhibitors. (A) Case report 1, sternal lesion at baseline (longest diameter: 47.7 mm). (B) Case report 1, sternal lesion at best response (longest diameter: 42.91 mm). (C) Case report 2, largest liver metastasis at baseline (longest diameter: 35.74 mm). (D) Case report 2, largest liver metastasis at best response (longest diameter: 22.38 mm).
Case Presentation 2
In August 2012, after self-detection of a left breast lump, a 32 year-old woman underwent fine biopsy of the breast nodule, which was positive for invasive ductal carcinoma. Due to the young age and the presence of a positive FH, BRCA genetic testing was performed with detection of the c.8878C>T germline pathogenic BRCA2 mutation. In October 2012 the patient underwent both left and prophylactic right mastectomy. The left breast pathology report revealed the presence of a pT1cN1M0, stage IIB, G2, BC (ER: 100%, PR: 40%, HER2 negative, ki67: 25%).
From December 2012 to March 2013, the patient received adjuvant chemotherapy with FEC for 3 cycles followed by docetaxel for 3 cycles. Then, she started adjuvant endocrine therapy with LHRH analog in combination with tamoxifen.
In June 2016, a blood exam showed an increase of CA 15-3 along with CT scan documenting suspicious chest-wall lymph-nodes, liver, and bone lesions. FNB and cytology of lymph-nodes confirmed the presence of BC cells (ER: 100%, PR: 35%, HER2-negative). In July 2016, the patient started first line chemotherapy with capecitabine, with good PR. However, in April 2017, the CT scan showed disease progression in lung, liver, and thoracic lymph-nodes. The patient started second-line chemotherapy with eribulin, which was interrupted after 3 cycles due to pulmonary and liver progression. From August 2017 to February 2018 the patient received 9 cycles of Carboplatin (best response: PR). The treatment was then interrupted for further disease progression (neoplastic lymphangitis with dyspnea). In April 2018, the patient began a new treatment with palbociclib plus letrozole and LHRH analog. After 5 months of treatment, she reported resolution of dyspnea with PS improvement, and a significant response was observed by CT scan [best response by RECIST critera (12): PR; Figures 1C,D]. At the last visit (April 2019), the patient is currently on treatment with no evidence of disease progression (DOR: 7 months).
Discussion
We reported clinical history of two patients with germline BRCA1/2 mutations and refractory ER-positive MBC who achieved durable response to the combination of palbociclib and ET.
There are multiple links between ER, BRCA, and CDK pathway (Figures 2, 3). Mote et al. suggested that altered expression of PR is a phenotype associated with mutations in BRCA1 and BRCA2 genes (13). Other in vitro studies suggested that the wild-type BRCA1 gene product interacts with and suppress the activity of ER-alpha (ESR1) through a direct physical and estrogen-independent interaction between the amino-terminal region of BRCA1 and the conserved carboxyl-terminal activation (AF-2) domain of ER-alpha (14). BRCA1 deleterious mutations abolish or reduce BRCA1 ability to inhibit ER-alpha activity (14, 15). It is therefore suggested that BRCA1 could function as a brake on ER-alpha driven proliferation and BRCA1 mutations can release that brake (16) (Figures 2, 3). Furthermore, Ma et al. showed that ER is also a target for the ubiquitin ligase activity of BRCA1 (17). Mutations of the ubiquitination sites abolish BRCA1-mediated inhibition of ER-alpha activity (17). BRCA1 is also able to inhibit P300-mediated ER acetylation, which is essential for the transactivation functions of ER (18). In addition to direct suppression of ER activity, wild-type BRCA1 also inhibits aromatase expression, thereby lowering estrogen levels and further reducing ER-alpha-mediated transcription (19). Overall, the higher activity of ER-alpha in BRCA mutation carriers could represent the rationale for a better sensitivity to ET in patients with ER-positive BC and BRCA1/2 germinal mutations (Figure 4).
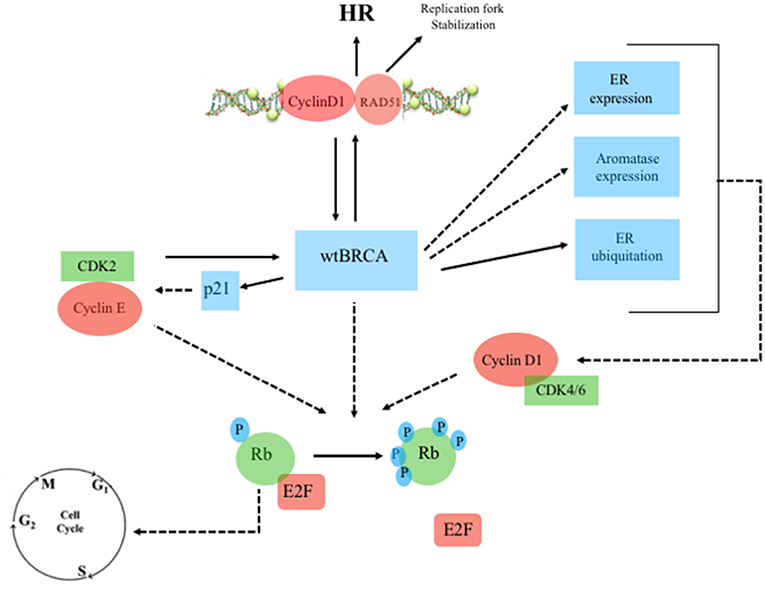
Figure 2. Wild-type BRCA, Estrogen Receptor and Cyclin/CDK pathway. Continuous line: activation, dashed line: inhibition. ERa, estrogen receptor-alpha; Rb, retinoblastoma; E2F, transcription factor; wtBRCA, wild-type BRCA; HR, homologous recombination; CDK, cyclin-dependent kinase; p21, endogenous CDK inhibitor; P, phosphorylation sites.
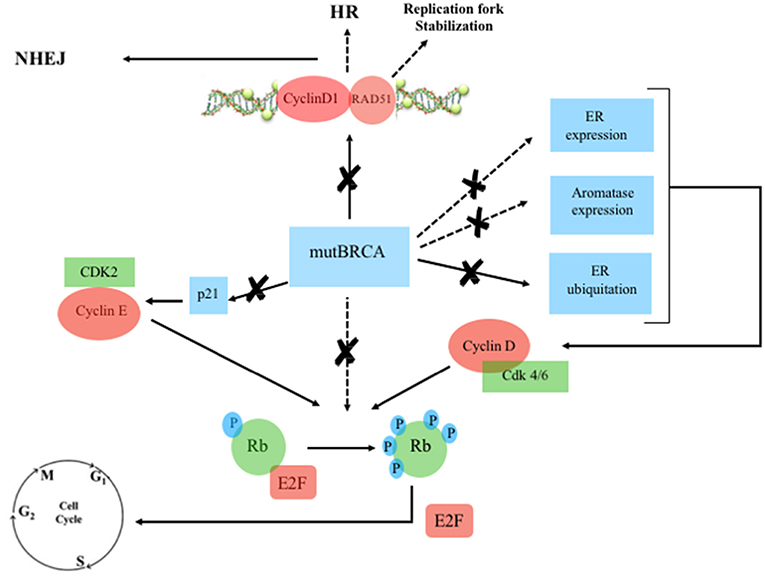
Figure 3. Mutated BRCA, Estrogen Receptor and Cyclin/CDK pathway. Continuous line: activation, dashed line: inhibition. ERa, estrogen receptor-alpha; Rb, retinoblastoma; E2F, transcription factor; mutBRCA, mutated BRCA; HR, homologous recombination; NHEJ, non-homologous end joining; CDK, cyclin-dependent kinase; p21, endogenous CDK inhibitor; P, phosphorylation sites.
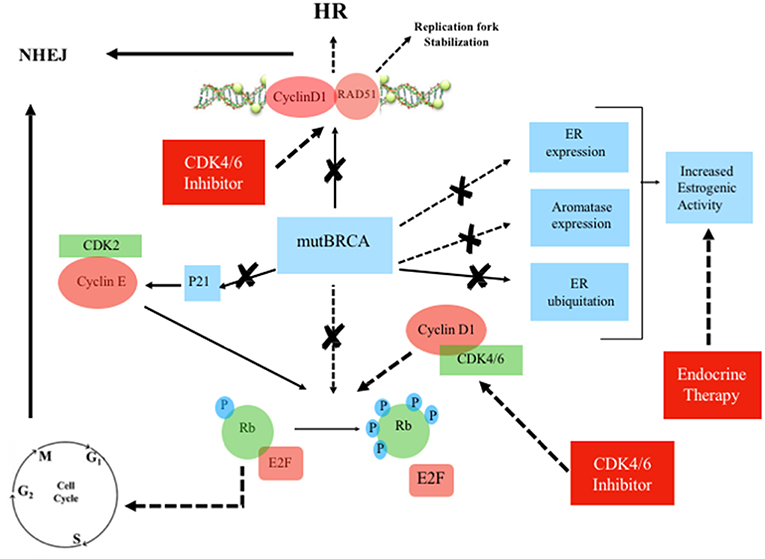
Figure 4. Mutated BRCA, Estrogen Receptor and Cyclin/CDK pathway in the presence of CDK4/6 inhibitor plus endocrine therapy. Continuous line: activation, dashed line: inhibition. ERa, estrogen receptor-alpha; Rb, retinoblastoma; E2F, transcription factor; mutBRCA, mutated BRCA; HR, homologous recombination; NHEJ, non-homologous end joining; CDK, cyclin-dependent kinase; p21, endogenous CDK inhibitor; P, phosphorylation sites.
The overexpression of cyclin D1 is found in ~50% of BCs (20), while amplification of its corresponding CCND1 gene has been observed in 9–30% of cases (21). Overexpression of cyclin D1 correlates with ER expression, PR expression, and luminal subtypes, with a favorable impact on overall survival (OS) in whole BC series containing all tumor phenotypes (21, 22). However, in the luminal A group, high expression of cyclin D1 has been associated with shorter disease-free survival (DFS), suggesting that the prognostic role of cyclin D1 depends on the molecular subtype (22). Patients with tumors with high amplification of CCND1, which are mostly of the luminal B subtype, have also been found been to have an increased risk of disease recurrence (21, 22). Cyclin D1 promotes the G 1/S-phase transition by binding and activating CDK4 and CDK6 (23), and CDK4/6 inhibitors effectively block the proliferation of sensitive cancer cells by inducing G1 cell cycle arrest (24). Cyclin D1 has also CDK-independent functions, such as ligand-dependent inhibition of the Androgen Receptor (AR) (25), and the promotion of transcription of the ER-alpha gene (26). Furthermore, Cyclin D1 stabilizes the interaction between ER and SRC-1 via direct binding to both proteins (27). ER also regulates the expression of the gene encoding cyclin D1 (28). There is therefore a positive feedback loop: activation of cyclin D1 leads to ER expression, which induces more cyclin D1 production (29). Wild-type BRCA is also involved in G1 cell cycle arrest. Aprelikova et al. showed that BRCA1 binds to hypophosphorylated RB, which interacts with the E2F transcription factor to block transcription and inhibit cell proliferation (30). In the presence of BRCA1 mutations, this antiproliferative control fails. However, in those cases, CDK4/6 inhibitors may restore the G1 arrest, preventing the cell from entering mitosis (31) (Figure 4). Somasundaram et al. also suggested that BRCA1 contributes to cell cycle arrest and tumor growth suppression through the induction of the CDK2 inhibitor p21 (32) (Figures 2, 3). CDK2 is a key bypass kinase of CDK4/6 inhibition and high mRNA expression of cyclin E1 (CCNE1), which activates CDK2, has been associated with resistance to palbociclib in the PALOMA-3 trial (33). It is interesting to note that, although the loss of BRCA1 may induce CDK2 activity through p21 inhibition, on the other hand, CCNE1 amplification and cyclin E1 protein overexpression have been reported to be mutually exclusive with BRCA1/2 mutations (34, 35).
Cyclin D1 also plays a cell-cycle-independent role in DNA repair. In response to DNA double-strand breaks (DSBs), it recruits proteins involved in the homologous recombination (HR) DSB repair pathway such as BRCA1 (36), RAD 51 (37), and BRCA2 (38). Interestingly, the recruitment itself of cyclin D1 to sites of DNA damage has been shown to be BRCA2-dependent (38) (Figures 2, 3). It is important to note that HR provides accurate recombination using a sister chromatid as a template, maintaining genomic stability. However, due to the need for a sister chromatid, HR is limited to the S-phase and G2-phase of cell cycle (39). By using CDK4/6 inhibitors, cells are arrested in G1 phase where they do not have an available sister chromatid in case of DNA damage and are dependent upon the compensatory, error-prone, non-homologous end joining (NHEJ) pathway, which is less accurate and may thus promote mutations and genomic instability (40). Furthermore, a decrease of HR activity during DNA damage may be induced by CDK4/6 inhibitors also in a cycle independent manner, due to the repression of critical factors for HR such as RAD51 (41). Therefore, in patients with HR deficiency (HRD), such as BRCA mutation carriers, who are treated with CDK 4/6 inhibitors, any DNA damage may cause cancer cell death since CDK4/6 inhibitors induce compensatory activation of the less accurate NHEJ pathway and impair recruitment of RAD51 (41) (Figure 4). These findings suggest that CDK inhibitors may exert synthetic lethal effect against BRCA-mutated, ER-positive BCs (37, 38).
PARP is constituted by several enzymes, which facilitate DNA repair in pathways involving single-strand breaks (SSBs) and base excision repair (BER) (42). In the presence of a PARP inhibitor, attempted DNA repair of SSB results in DSB formation. BRCA-proficient cells have the ability to repair the DSB, maintaining survival, but BRCA-deficient cells are unable to repair the accumulating DSBs which lead to cell synthetic lethality (42). The randomized, phase 3 OlympiAD and EMBRACA trials showed that, among patients with HER2-negative MBC and germline BRCA mutation, PARP inhibitors olaparib and talazoparib provided a significant progression-free survival (PFS) benefit over standard therapy (43, 44). Interestingly, subgroup analysis in the OlympiAD trial showed a greater efficacy of olaparib in patients with triple-negative breast cancer (TNBC) than in ER-positive ones, and also a better outcome in BRCA1 than in BRCA2 mutation carriers (43). However, in the final analysis of the study, OS, a secondary end point, was not significantly different between treatment arms both in the whole study population and among the predefined subgroups (45). At present, several ongoing trials are evaluating the combination of CDK inhibitors with PARP inhibitors (46). The combination of dinaciclib (a CDK 1, 2, 5, 9, and 12 inhibitor) with the PARP inhibitor veliparib has shown preliminary clinical benefit in patients with breast cancer, prostate cancer, ovarian cancer, and other gynecologic malignancies (47). By blocking CDK12, CDK1, or CDK2, BRCA wild-type cancer cells turn into HR-deficient cells through inhibition of BRCA1 expression and phosphorylation (48, 49) and, thus, become more susceptible to synthetic lethality induced by PARP inhibitors (46–49). A phase I/II trial of olaparib, palbociclib, and fulvestrant has been recently activated in BRCA-mutated patients with ER-positive, HER2-negative MBC1.
Conclusion
The Cyclin/CDK pathway has a crucial role in the regulation of cell cycle progression. In the last years, CDK4/6 inhibitors have changed the treatment landscape for ER-positive BC. The clinical reports here presented show a significant activity of CDK4/6 inhibitors in BRCA-mutated, ER-positive BC patients.
BRCA genes are involved in the regulation of DNA Repair Mechanism, but are also strategic for ER expression and function. In the presence of BRCA mutations, which abolish or reduce BRCA-dependent ER-alpha inhibition, the use of ET could restore the brake on ER-alpha driven proliferation. Moreover, due to BRCA mutations, cell cycle arrest fails, but CDK4/6 inhibitors could restore G1 arrest. The induction of G1 cell cycle arrest can be used to manipulate the activity of DNA repair pathways, especially in HR-deficient cells. In those cells, G1 cell cycle arrest may lead to a major activity of NHEJ, with consequent genomic instability and apoptosis.
Overall, biological rationale, preclinical, and clinical data support the prominent role of CDK 4/6 inhibitors plus endocrine therapy, even in combination with PARP inhibitors, in the treatment of BRCA-mutated, ER-positive breast cancers. However, the interaction between Cyclin/CDK pathway, ER and BRCA is complex and evidences reported so far, albeit reliable, await confirmation in the context of future randomized clinical trials.
Data Availability
All datasets generated for this study are included in the manuscript and/or the supplementary files.
Ethics Statement
The authors of this manuscript obtained patient consent for publication of clinical data and images. The patients' details were anonymized and signed consent forms were attached to the medical records. Due to the retrospective and non-interventional nature of the study, permission by the local ethics committee was not required.
Author Contributions
AMi and TZ contributed equally to the writing of the manuscript and designed the figures. AMu and DB were involved in planning and supervised the study. SG interpreted the radiological images. MM, NN, BB, PZ, and VU reviewed and approved the final version of this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnote
1. ^Olaparib, Palbociclib, and Fulvestrant in Patients With BRCA Mutation-associated, Hormone receptor-positive, HER2-negative metastatic Breast Cancer – NCT03685331. Available online at: https://clinicaltrials.gov/ct2/show/NCT03685331.
References
1. Malone KE, Daling JR, Doody DR, Hsu L, Bernstein L, Coates RJ, et al. Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast cancer in white and black American women ages 35 to 64 years. Cancer Res. (2006) 66:8297–308. doi: 10.1158/0008-5472.CAN-06-0503
2. Rennert G, Bisland-Naggan S, Barnett-Griness O, Bar-Joseph N, Zhang S, Rennert HS, et al. Clinical outcomes of breast cancer in carriers of BRCA1 and BRCA2 mutations. N Engl J Med. (2007) 357:115–23. doi: 10.1056/NEJMoa070608
3. Mavaddat N, Barrowdale D, Andrulis IL, Domchek SM, Eccles D, Nevanlinna H, et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol Biomarkers Prev. (2012) 21:134–47. doi: 10.1158/1055-9965.EPI-11-0775
4. Goodwin PJ, Phillips KA, West DW, Ennis M, Hopper JL, John EM, et al. Breast cancer prognosis in BRCA1 and BRCA2 mutation carriers: an International Prospective Breast Cancer Family Registry population-based cohort study. J Clin Oncol. (2012) 30:19–26. doi: 10.1200/JCO.2010.33.0068
5. Tung N, Wang Y, Collins LC, Kaplan J, Li H, Gelman R, et al. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: clinical risk factors and pathologic features. Breast Cancer Res. (2010) 12:R12. doi: 10.1186/bcr2478
6. Jonasson JG, Stefansson OA, Johannsson OT, Sigurdsson H, Agnarsson BA, Olafsdottir GH, et al. Oestrogen receptor status, treatment and breast cancer prognosis in Icelandic BRCA2 mutation carriers. Br J Cancer. (2016) 115:76. doi: 10.1038/bjc.2016.249
7. O'leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. (2016) 13:417. doi: 10.1038/nrclinonc.2016.26
8. Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. (2017) 35:3638–46. doi: 10.1200/JCO.2017.75.6155
9. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. (2016) 375:1738–48. doi: 10.1056/NEJMoa1609709
10. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. (2016) 17:425–39. doi: 10.1016/S1470-2045(15)00613-0
11. Tryfonidis K, Bogaerts J, Martell RE, Sledge GW, Balmaña J, Audeh MW, et al. A phase III randomized trial of niraparib versus physician's choice in previously treated, HER2-negative, germline-BRCA mutated breast cancer patients: intergroup study EORTC-1307-BCG and BIG5-13. J Clin Oncol. (2017) 32(15_Suppl):TPS659. doi: 10.1200/jco.2014.32.15_suppl.tps659
12. Schwartz LH, Litière S, de Vries E, Ford R, Gwyther S, Mandrekar S, et al. RECIST 1.1-Update and clarification: from the RECIST committee. Eur J Cancer. (2016) 62:132–7. doi: 10.1016/j.ejca.2016.03.081
13. Mote PA, Leary JA, Avery KA, Sandelin K, Chenevix-Trench G, Kirk JA, et al. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer. (2004) 39:236–48. doi: 10.1002/gcc.10321
14. Fan S, Wang JA, Yuan R, Ma Y, Meng Q, Erdos MR, et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. (1999) 284:1354–56. doi: 10.1126/science.284.5418.1354
15. Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. (2001) 20:77. doi: 10.1038/sj.onc.1204073
16. Gorski JJ, Kennedy RD, Hosey AM, Harkin DP. The complex relationship between BRCA1 and ERα in hereditary breast cancer. Clin Cancer Res. (2009) 15:1514–8. doi: 10.1158/1078-0432.CCR-08-0640
17. Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-α. Mol Endocrinol. (2010) 24:76–90. doi: 10.1210/me.2009-0218
18. Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, et al. p300 Modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. (2002) 62:141–51.
19. Hu Y, Ghosh S, Amleh A, Yue W, Lu Y, Katz A, et al. Modulation of aromatase expression by BRCA1: a possible link to tissue-specific tumor suppression. Oncogene. (2005) 24:343. doi: 10.1038/sj.onc.1208985
20. Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, Nicholson RI, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. (1999) 5:2069–76.
21. Lundberg A, Lindström LS, Li J, Harrell JC, Darai-Ramqvist E, Sifakis EG, et al. The long-term prognostic and predictive capacity of cyclin D1 gene amplification in 2305 breast tumours. Breast Cancer Res. (2019) 21:34. doi: 10.1186/s13058-019-1121-4
22. Ortiz AB, Garcia D, Vicente Y, Palka M, Bellas C, Martin P. Prognostic significance of cyclin D1 protein expression and gene amplification in invasive breast carcinoma. PLoS ONE. (2017) 12:e0188068. doi: 10.1371/journal.pone.0188068
23. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. (2004) 145:5439–47. doi: 10.1210/en.2004-0959
24. Ribnikar D, Volovat SR, Cardoso F. Targeting CDK4/6 pathways and beyond in breast cancer. Breast. (2019) 43:8–17. doi: 10.1016/j.breast.2018.10.001
25. Reutens AT, Fu M, Wang C, Albanese C, McPhaul MJ, Sun Z, et al. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol Endocrinol. (2001) 15:797–811. doi: 10.1210/mend.15.5.0641
26. Casimiro MC, Wang C, Li Z, Di Sante G, Willmart NE, Addya S, et al. Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Mol Endocrinol. (2013) 27:1415–28. doi: 10.1210/me.2013-1065
27. Zwijsen RM, Buckle RS, Hijmans EM, Loomans CJ, Bernards R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev. (1998) 12:3488–98. doi: 10.1101/gad.12.22.3488
28. Griekspoor A, Margarido TC, Zwart W, Michalides R. Review of: BRCA1 and cyclin D1: gate keepers in hormone responsive tissues? Breast Cancer Online. (2006) 9:1–3. doi: 10.1017/S1470903106005098
29. Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y, et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor α activity. Cancer Res. (2005) 65:6557–67. doi: 10.1158/0008-5472.CAN-05-0486
30. Aprelikova ON, Fang BS, Meissner EG, Cotter S, Campbell M, Kuthiala A, et al. BRCA1-associated growth arrest is RB-dependent. Proc Natl Acad Sci USA. (1999) 96:11866–71. doi: 10.1073/pnas.96.21.11866
31. Murphy CG, Dickler MN. The role of CDK4/6 inhibition in breast cancer. Oncologist. (2015) 20:483–90. doi: 10.1634/theoncologist.2014-0443
32. Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Zhang H, et al. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21 WAF1/CiPl. Nature. (1997) 389:187. doi: 10.1038/38291
33. Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J Clin Oncol. (2019) 37:1169–78. doi: 10.1200/JCO.18.00925
34. Etemadmoghadam D, Weir BA, Au-Yeung G, Alsop K, Mitchell G, George J, et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc Natl Acad Sci USA. (2013) 110:19489–94. doi: 10.1073/pnas.1314302110
35. Zhao ZM, Yost SE, Hutchinson KE, Li SM, Yuan YC, Noorbakhsh J, et al. CCNE1 amplification is associated with poor prognosis in patients with triple negative breast cancer. BMC Cancer. (2019) 19:96. doi: 10.1186/s12885-019-5290-4
36. Di Sante G, Di Rocco A, Pupo C, Casimiro MC, Pestell RG. Hormone-induced DNA damage response and repair mediated by cyclin D1 in breast and prostate cancer. Oncotarget. (2017) 8:81803. doi: 10.18632/oncotarget.19413
37. Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, et al. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res. (2010) 70:8802–11. doi: 10.1158/0008-5472.CAN-10-0312
38. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. (2011) 474:230. doi: 10.1038/nature10155
39. Toss A, Cortesi L. Molecular mechanisms of PARP inhibitors in BRCA-related ovarian cancer. J Cancer Sci Ther. (2013) 5:409–16. doi: 10.4172/1948-5956.1000234
40. Weil MK, Chen A. PARP inhibitor treatment in ovarian and breast cancer. Curr Probl Cancer. (2011) 35:7. doi: 10.1016/j.currproblcancer.2010.12.002
41. Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. (2012) 287:29075–87. doi: 10.1074/jbc.M112.365494
42. Zimmer AS, Gillard M, Lipkowitz S, Lee JM. Update on PARP inhibitors in breast cancer. Curr Treat Options Oncol. (2018) 19:2. doi: 10.1007/s11864-018-0540-2
43. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. (2017) 377:523–33. doi: 10.1056/NEJMoa1706450
44. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
45. Robson ME, Tung N, Conte P, Im SA, Senkus E, Xu B, et al. OlympiAD final overall survival and tolerability results: olaparib versus chemotherapy treatment of physician's choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. (2019) 30:558–66. doi: 10.1093/annonc/mdz012
46. Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: clinical utility and possibilities of overcoming resistance. Gynecol Oncol. (2017) 147:695–704. doi: 10.1016/j.ygyno.2017.10.003
47. Shapiro GI, Do KT, Tolaney SM, Hilton JF, Cleary JM, Wolanski A, et al. Abstract CT047: Phase 1 dose-escalation study of the CDK inhibitor dinaciclib in combination with the PARP inhibitor veliparib in patients with advanced solid tumors. Cancer Res. (2017) 77(13Suppl):CT047. doi: 10.1158/1538-7445.AM2017-CT047
48. Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. (2011) 17:875. doi: 10.1038/nm.2377
Keywords: BRCA, CDK4/6 inhibitor, breast cancer, estrogen receptor, cyclin D1, endocrine therapy, homologous recombination
Citation: Militello AM, Zielli T, Boggiani D, Michiara M, Naldi N, Bortesi B, Zanelli P, Uliana V, Giuliotti S and Musolino A (2019) Mechanism of Action and Clinical Efficacy of CDK4/6 Inhibitors in BRCA-Mutated, Estrogen Receptor-Positive Breast Cancers: Case Report and Literature Review. Front. Oncol. 9:759. doi: 10.3389/fonc.2019.00759
Received: 27 April 2019; Accepted: 29 July 2019;
Published: 13 August 2019.
Edited by:
Massimo Broggini, Istituto Di Ricerche Farmacologiche Mario Negri, ItalyReviewed by:
Luigi Formisano, University of Naples Federico II, ItalyFilippo Montemurro, Fondazione del Piemonte per l'Oncologia, Istituto di Candiolo (IRCCS), Italy
Copyright © 2019 Militello, Zielli, Boggiani, Michiara, Naldi, Bortesi, Zanelli, Uliana, Giuliotti and Musolino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonino Musolino, YW50b25pbm9tdXNvbGlub0Bob3RtYWlsLmNvbQ==