 Jiaojiao Zhou
Jiaojiao Zhou Qishan Chen3
Qishan Chen3 Huihui Chen
Huihui Chen- 1Department of Surgical Oncology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2The Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, Hangzhou, China
- 3Department of Cardiology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 4Department of Clinical Medicine, Hangzhou Medical College, Hangzhou, China
Breast cancer stem cells have been known to contribute immensely to the carcinogenesis of the breast and therapeutic resistance in the clinic. Current studies show that the population of breast cancer stem cells is heterogeneous, involving various cellular markers and regulatory signaling pathways. In addition, different subtypes of breast cancer exhibit distinct subtypes and frequencies of breast cancer stem cells. In this review, we provide an overview of the characteristics of breast cancer stem cells, including their various molecular markers, prominent regulatory signaling, and complex microenvironment. The cellular origins of breast cancer are discussed to understand the heterogeneity and diverse differentiations of stem cells. Importantly, we also outline the recent advances and controversies in the therapeutic implications of breast cancer stem cells in different subtypes of breast cancer.
Introduction
Breast cancer is the most commonly reported type of cancer in women worldwide, and a leading cause of morbidity and mortality (1). With the morbidity and mortality associated with breast cancer, more newer therapeutic approaches are warranted. The complexity and heterogeneity of breast cancer render its treatment challenging. Gene expression profiling comprehensively demonstrated molecular portraits of breast cancer (2, 3), defining tumors as luminal-like, HER2-positive, or basal-like. Clinically, the heterogeneous breast cancer is categorized into four distinct molecular subgroups (2, 4): Luminal A and luminal B breast cancers are broadly defined as those with estrogen receptor (ERs) positive expression, which response to the antiestrogen therapy. The HER2 positive breast cancer is the subtype with high amplification of HER2 gene. The triple negative breast cancer (TNBCs, usually basal-like), lacking the expression of ER, progesterone receptor (PR), and HER2 (5), always has an increased incidence of germline BRCA1/2 mutations (6, 7). Compared to the other subtypes, TNBC is highly heterogenous, and usually have higher incidence of hematogenous metastasis. Based on a large cohort of 465 primary TNBC tumors, TNBC are now classified into transcriptome-based subtypes: luminal androgen receptor, immunomodulatory, basal-like immune-suppressed, and mesenchymal-like (8).
Two models have emerged to explain the heterogeneity of breast tumors (9). One is the clonal evolution model (10), which postulated that random mutation and clonal selection give rise to cellular heterogeneity within breast tumors. Another is the cancer stem cell model (11), which posited that the cellular diversity and tumor hierarchy are generated by the breast cancer stem cells (BCSCs). In both of the two models, tumor microenvironment plays an important role in equipping the evolution of breast cancer cells.
BCSCs (12) are defined as a small fraction of cells capable of self-renewal and propagation of the heterogeneous populations of breast cancer cells. The concept of BCSCs revealed the cellular origin, tumor maintenance, and progression of breast cancer. Clinically, BCSCs are considered to be responsible for the development of resistance to treatment and cancer relapse, through their virtue of relative resistance to radiation, cytotoxic chemotherapy and molecular targeted therapy (13).
In the present review, we provide an overview of the current advances in stem cells and the cellular origins of breast cancer. We summarized information regarding the various molecular markers of BCSCs, BCSC portraits among different subtypes of breast cancer, and associations between BCSCs, and the tumor microenvironment. Moreover, considering the contribution of BCSCs to the development of resistance to treatment and tumor recurrence, we also focused on regulatory pathways and the related therapeutic implications of BCSCs.
Molecular Markers and Tumorigenic Features of BCSCs
BCSCs are defined as a limited group of breast cancer-initiating cells, possessing properties of self-renewal, and differentiation potential (14). In cancer research, BCSCs are usually characterized as cells that are able to form the transplantable tumors and re-establish tumor heterogeneity (15). A panel of molecular markers was used to identify the BCSCs (Table 1). Among these, CD44+/CD24−/low (16, 17) and ALDH1+ (aldehyde dehydrogenase1) (18) are the most commonly used markers. Indeed, the CD44+/CD24−/low and ALDH1+ phenotype cells are two distinct subpopulations of BCSCs (18). Different gene expression profiles (34) have shown that CD44+/CD24−/low marked a mesenchymal and quiescent type of BCSCs (EMT-BCSCs, EMT: epithelial-mesenchymal transition) (35), while ALDH1+ labeled an epithelial and proliferative type of BCSCs (MET-BCSCs, MET: mesenchymal-epithelial transition). According to the fostered concept of transient EMT-MET switches in metastatic tumor cells (36), these two subgroups of BCSCs can be accepted as two dynamic states of BCSCs. It has been recently demonstrated that the transition between these two states of BCSCs (EMT and MET-BCSCs) can be reversely regulated by cytokine signaling such as Id1 (inhibitor of DNA binding 1) (37).
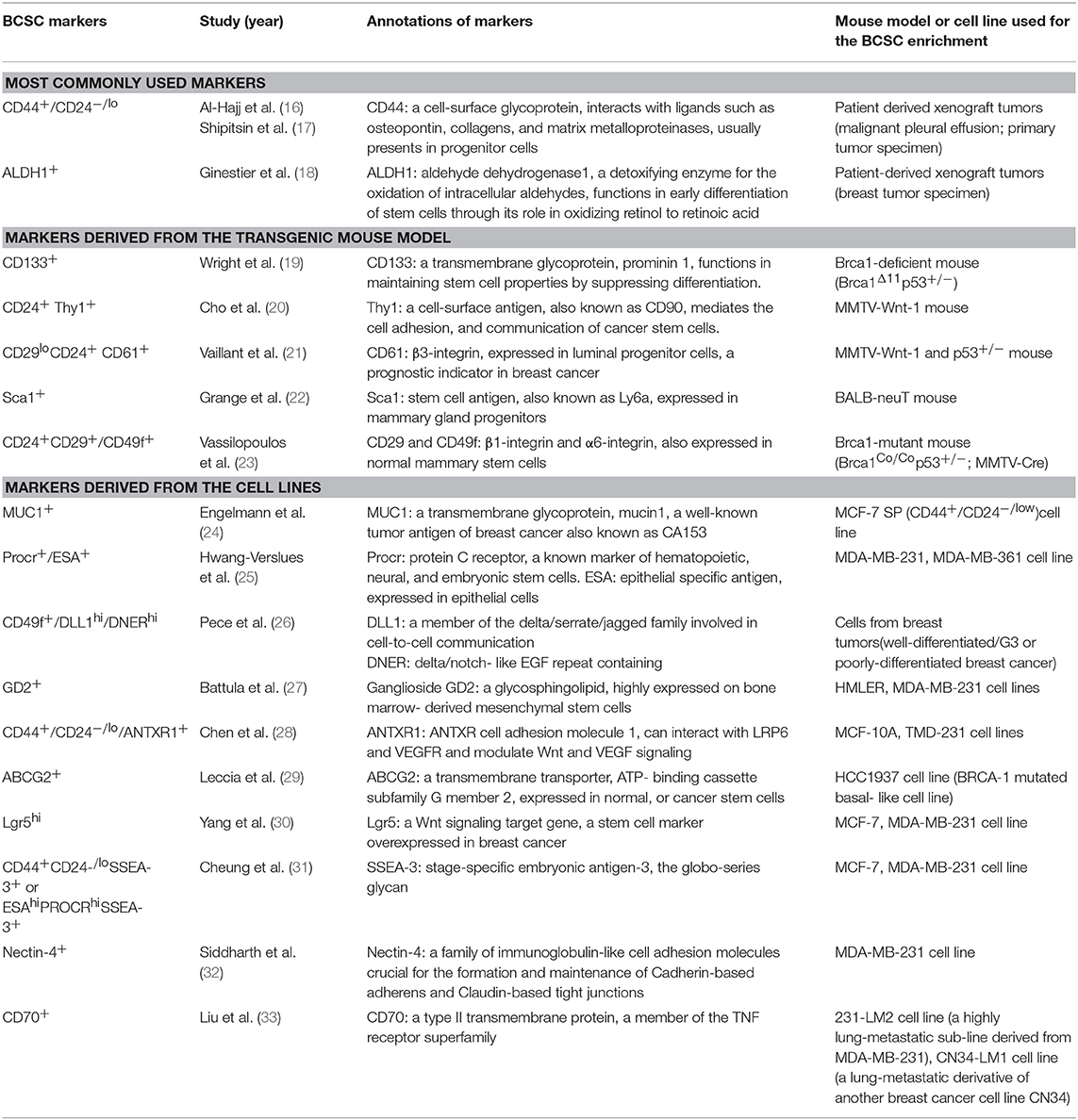
Table 1. Markers used to identify breast cancer stem cells, derived from breast cancer cell lines, transgenic mouse models, and patient-derived tumors.
Numerous other BCSCs markers were identified in different mouse models and breast cancer cell lines (Table 1). Wright et al. (19) reported that BRCA1-deficient mouse mammary tumors—which mimic the BRCA1-associated breast cancer—harbor heterogeneous BCSCs subpopulations including CD133+ (prominin1) and CD44+/CD24− stem cells. Vassilopoulos et al. (23) found that CD24+CD29+/CD49f+ enriched BCSC population in BRCA1-mutant mice displayed enhanced metastatic potential. BCSCs in breast tumors of MMTV-Wnt-1 and p53+/−mice were identified by the marker of Thy1 (cell surface antigen also known as CD90) (20) and CD61 (21). In addition, Sca-1+ (stem cell antigen 1) marked a BCSC subpopulation in the BALB-neuT mouse model (22).
Various BCSC markers were also identified from breast cancer cell lines such as MUC1 (also known as CA153) (24), Procr+/ESA+ (epithelial specific antigen) (25), DLL1+/DNER+ (delta-like canonical Notch ligand1/delta/notch-like EGF repeat containing) (26), GD2 (27), ANTXR1 (ANTXR cell adhesion molecule 1) (28), ABCG2 (ATP-binding cassette subfamily G member 2) (29), Lgr5 (leucine rich repeat containing G protein-coupled receptor 5) (30), SSEA-3 (stage-specific embryonic antigen-3) (31), Nectin-4 (nectin cell adhesion molecule 4) (32), and CD70 (33) (Table 1).
Considering the heterogeneity of breast cancer, the variety of BCSCs markers observed in different studies may be attributed to different levels of breast cancer hierarchy. Moreover, it may be explained as the results of dynamic states of BCSCs, regulated by the microenvironment. Further in vivo and patient-derived xenograft studies are required for the definitive identification of BCSCs.
Signaling Pathways Regulating BCSCs
Given their self-renewal and tumor-initiating properties, BCSCs have emerged as the “ringleader” for the development of therapeutic resistance in breast cancer (38). Therefore, BCSC-related therapeutic options, such as targeting the main regulatory signaling pathways in BCSCs, have recently been developed for the treatment of breast cancer (Table 2), especially in case with therapeutic resistance.
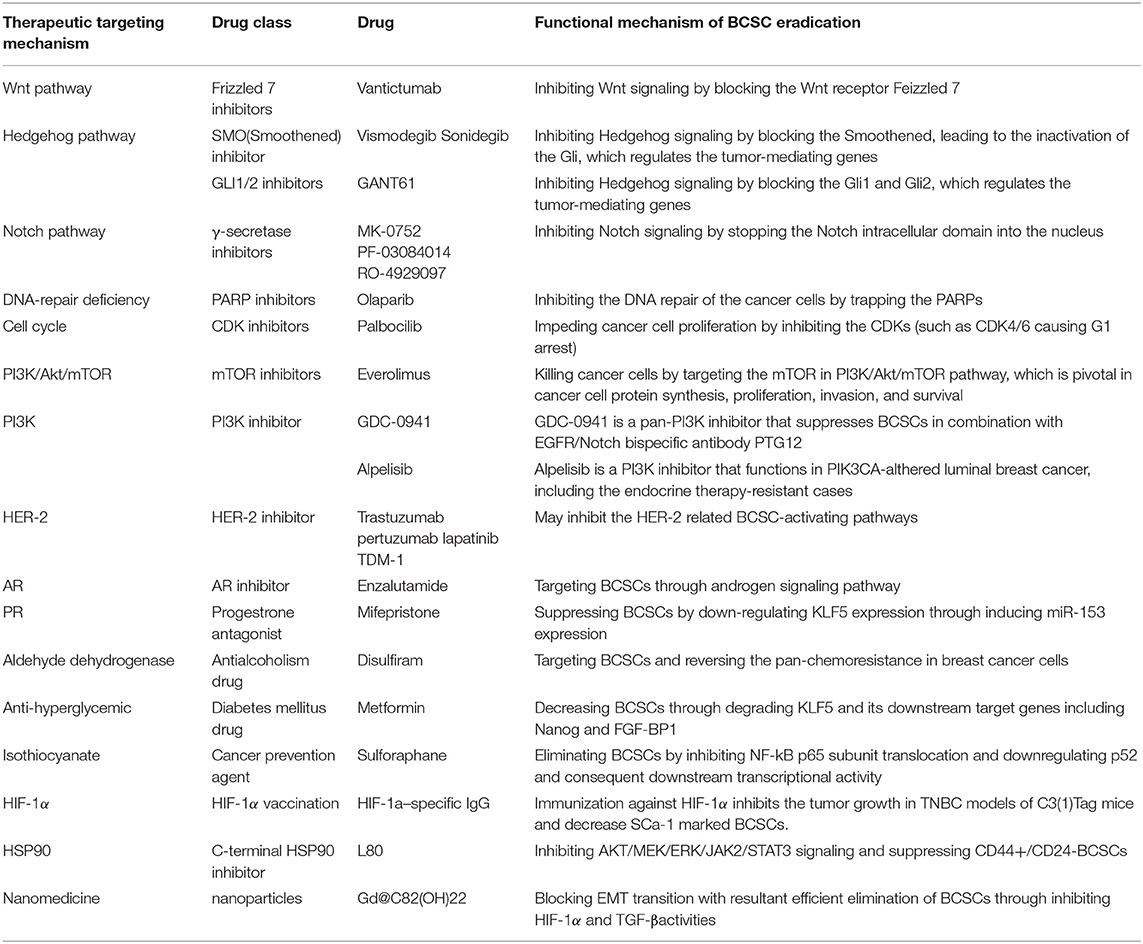
Table 2. Breast cancer stem cells-targeted therapies in the treatment of breast cancer and their potential mechanism of breast cancer stem cell eradication.
The major signaling pathways (39) regulating BCSCs include Wnt (40), Notch (41), and Hedgehog (42) (Figure 1). Inhibitors blocking these signaling were developed as the BCSC-targeting therapies (Figure 1). (1) Increased activation of the Wnt pathway is usually found in BCSCs, leading to the nuclear translocation of cytosolic β-catenin to activate the Wnt-targeted genes, by binding the TCF/LEF (T cell factor/lymphoid enhancing factor) family. Recently, the Wnt heterodimer receptor (i.e., FZD7 and LRP6: frizzled-7 and lipoprotein receptor related protein-6) was found to be up-regulated in the TNBCs. Knockdown of this receptor suppressed tumor growth (43). Targeted medicines for the inhibition of the FZD7 receptor (i.e., Vantictumab) have been developed (44, 45). (2) The Hedgehog pathway is also crucial in BCSCs. Binding of Hedgehog to the Patched alleviates its inhibition of Smoothened. The activated Smoothened subsequently releases Gli to regulate Hedgehog target genes (46). Higher expression of Smoothened was found in a BCSC subpopulation (CD44+/CD24− cells) (47) and the inhibitor of Smoothened (i.e., Vismodegib, Sonidegib) has already been investigated in clinical trials (48, 49). Besides, the anti-cancer stem cell activities of Gli1/2 inhibitor (GANT61) were also proved in TNBCs (50). (3) The Notch pathway serves as a key signaling cascade involved in the maintenance of BCSC phenotype. Notch ligands (e.g., Delta-like 1, 3, 4, and Jagged 1, 2) binding to Notch receptors result in the release of Notch intracellular domain (NCID) (41). With the help of γ-secretase, NCID translocates into the nucleus to activate several downstream effectors (41). McGowan et al. (51) demonstrated that CD44hi/CD24loBCSCs contributed to the brain metastases of breast cancer, partially arisen from increased Notch activity. Clinically, drugs (e.g., MK-0752, RO-4929097, and PF-03084010) (52) targeting the γ-secretase—through the mechanism of curbing Notch signaling by stopping the NCID into the nucleus—are currently underway for the treatment of breast cancer.
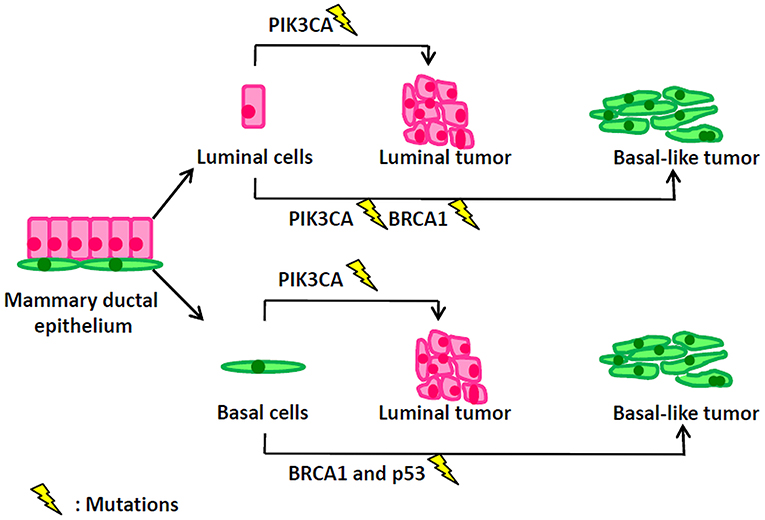
Figure 1. Mutation events in different types of mammary cells lead to distinct types of breast tumors.
MicroRNAs Regulating BCSCs
Epigenetic regulation of microRNAs is important in BCSCs (53). MiR-200 family was proved to be a critical regulator for BCSCs growth and function. MiR-200c can strongly suppress the clonogenicity of BCSCs by targeting BMI1 (BMI1 proto-oncogene, polycomb ring finger) (54). MiR-200b directly acts on Suz12 (SUZ12 polycomb repressive complex 2 subunit), and the miR-200b-Suz12-cadherin pathway functions on BCSC growth (55). MicroRNA-200c/141 was found to regulate the heterogeneity of the BCSCs and promote the EMT-like BCSC generation, by targeting HIPK1 (homeodomain-interacting protein kinase 1)/β-catenin axis (56). Let-7 was found to regulate BCSCs by silencing H-RAS (HRas proto-oncogene, GTPase) and HMGA2 (high mobility group AT-hook 2), resulting in reduction of self-renewal and enhancement of differentiation of BCSCs (57). Similar to let-7, reduction of microRNA-30 in BCSCs contributes to the maintenance of self-renewal capacity in BCSCs, by targeting UBC9 (ubiquitin-conjugating enzyme 9) and ITGB3 (integrin b3) (58). In addition, in BCSCs, miR-600 acts as a bimodal switch that regulates WNT Signaling through SCD1 (stearoyl desaturase 1), balancing the self-renewal, and differentiation of BCSCs (59). More microRNAs and their clusters need to be investigated in BCSCs, especially for their roles in stem cell maintenance of self-renewal, differentiation, and EMT transition. Clarifying the microRNA regulation of BCSCs can further advance or understanding of the roles of BCSCs in breast cancer progression.
BCSCs and the Tumor Microenvironment
BCSCs are located in the tumor microenvironment, which is also called as BCSC “niche.” The BCSC niche plays a vital role in sustaining the function of BCSCs. It is a complex network, containing stroma cells [such as mesenchymal stem cells (MSCs), cancer associated fibroblasts (CAFs), adipocytes, endothelial cells, and immune cells], extracellular matrix (ECM) components, cytokines, growth factors, and physical factors (such as hypoxia).
MSCs are the multipotent mesenchymal stomal cells which can be recruited from the bone marrow or normal breast stroma. It has been shown that bone marrow-derived MSCs can expand the BCSC population through cytokine loops involving IL6 (interleukin 6) and CXCL7 (pro-platelet basic protein) (60). It was also found that MSCs can propagate traits of BCSCs by promoting the contact-dependent upregulation of microRNA-199a and subsequent repression of FOXP2 (forkhead-box P2) (61). CAFs are activated fibroblasts in the tumor-hosting niche, which can promote cancer progression, especially that of breast cancer. It was reported that CAFs can regulate BCSCs through factors such as CCL2 (monocyte chemotactic protein-1) (62), IL-6 and IL-8 (63). Moreover, autophagic CAFs can promote stemness of luminal breast cancer cells by releasing HMGB1 (high-mobility group box 1) (64). Conversely, BCSCs can also regulate CAFs via signaling such as Hedgehog, in which CAFs subsequently promote the expansion and self-renewal of BCSCs (49, 65). Immune cells, especially the tumor-associated macrophages (TAMs) –are closely associated with tumor propagation. Tumor cells produce M-CSF (macrophage colony-stimulating factor) to expand TAMs, while TAMs produce TNFα and TGFβ to facilitate CSCs (66). In breast cancer, TAMs can promote BCSCs through a paracrine EGFR/Stat3/Sox-2 signaling pathway (67), while upregulation of HAS2 (hyaluronan synthase 2) in CD44+/CD24−/ESA+ BCSCs can enhance the interaction between BCSCs and TAMs, resulting in the BCSC growth (68). Other immune cells, such as tumor-infiltrating lymphocytes (TILs, including CD4+, CD8+, and FOXP3+ TILs) are also closely correlated with BCSC phenotypes, therapeutic response, and prognosis in breast cancer (69). Recent research by machine learning identified that immune microenvironment content and PD-L1 levels associated with the stemness of breast cancer (70). Adipocytes constitute a major component of the breast stroma which provide pro-tumorigenic signals in breast cancer (71). In the Goto et al. study (72)—using a PDX (Patient derived xenograft) model—it was found that adipose tissue secreted adipsin to enhance BCSC properties in breast cancer. Endothelial cells are necessary in tumor angiogenesis, which is important for nutrient and oxygen supply in the tumor microenvironment. Independently of their vascular functions, Ghiabi et al. (73) found that endothelial cells can enrich the CD44+/CD24− stem population in breast cancer.
The ECM is a three-dimensional network of extracellular macromolecules that confines tumor cells in the microenvironment, thereby sustaining tissue homeostasis. It was illustrated that stiffness of the ECM influenced breast cancer cells through the YAP/TAZ (Yes-associated protein/Transcriptional coactivator with PDZ-binding motif) (74). Hpoxia is very important in sustaining the quiescent state of stem cells by activating the hypoxia-inducible factor (HIF). In breast cancer, hypoxia induces the BCSC phenotype through HIF-dependent and ALKBH5-mediated m(6)-demethylation of NANOG mRNA (75).
Cellular Origins of Breast Cancer
The concept of “cellular origins” is closely associated to—but quite distinct—from the notion of “cancer stem cells” (76). The BCSC concept highlights a tiny population of the breast tumor-initiating cells that can maintain tumorigenesis and seed metastases, while the notion of cellular origins emphasizes the original normal cell types in the breast which generate a full-blown tumor. The diversity of the phenotypes displayed by breast cancer cells stimulates interests in investigating the cellular origins of this disease.
Historically, in breast cancer, the name “luminal” of luminal-A/B subtypes and the name “basal-like” of basal-like subtype are derived from similarities in transcriptomes between breast tumors and the corresponding normal mammary luminal or basal epithelium. However, the real cellular origins of luminal and basal-like breast cancer are greatly different from their naming rules. Oncogenic events in different types of mammary cells lead to distinct types of breast tumors (Figure 1). PIK3CA (α-catalytic subunit of PI3K) mutations occur in 30% of breast cancers, including both luminal and basal-like tumors. However, Meyer et al. (77) reported that the mutant PIK3CA in mammary luminal progenitors generated heterogeneous tumors of both luminal and basal differentiation. Van Keymeulen et al. (78) also found that expression of the PI3KCA mutant in luminal cells—marked by CK8—induced the luminal or basal-like breast tumors, while its expression in basal cells—marked by CK5—gave rise to the luminal tumors. BRCA1 basal-like breast cancers may originate from basal stem cells. However, interestingly, Molyneux et al. (79) demonstrated that deletion of BRCA1 in mammary luminal epithelial cells—targeted by Blg—can generate basal-like breast tumors, phenocopying the human BRCA1-associated breast cancers, while the deficiency of BRCA1 in basal cells– targeted by CK14– can only generate the malignant adenomyoepitheliomas which are rare in human BRCA1-associated breast cancer. Furthermore, Tao et al. (80) depicted that the CK8+ luminal cells carrying the Etv6-NTRK3 fusion oncogene can induce the heterogeneous tumors with the expression of luminal and basal markers. Compelling evidence showed that luminal progenitors can serve as the cellular origins of both luminal- and basal-like human breast cancers, while the distinct genetic mutations—occurring in the transformation of luminal progenitors—are probably determinant of the eventual luminal-like or basal-like tumor phenotypes (81). Genetic sequencing results have illustrated different mutation profiles between luminal-like and basal-like tumors. The luminal-like tumors present distinct mutations, such as PIK3CA (82, 83), GATA3 (84, 85) and FOXA1 (84), while the basal-like tumors exhibit high rates of p53 and BRCA1 mutations. Using conditional mouse models, Liu et al. showed that somatic loss of both BRCA1 and p53 did result in the development of basal-like breast cancer (86).
The cellular origins of the rare type of breast cancers, such as metaplastic carcinoma should also be mentioned. Molecularly, this metaplastic subtype is similar to claudin-low breast cancer. Keller et al. (87) found that the transformation of CD10+ basal cells gave rise to rare metaplastic tumors. McCarthy et al. (88) demonstrated that these metaplastic tumors frequently harbored the p53 mutation and aberrant BRCA1 expression.
BCSCs Among Different Subtypes of Breast Cancer and Their Therapeutic Implications
Different subtypes of breast cancer exhibit different abundances of BCSCs, as well as varying proportions of epithelial or mesenchymal BCSC subtypes. It is commonly recognized that BCSCs are much more enriched in the TNBCs and HER2 subtypes vs. luminal breast cancer. In the model illustrated by Brooks et al. (9), claudin-low TNBCs are characterized by a high proportion of mesenchymal BCSCs with CD44+/CD24−/lo expression, while basal-like TNBCs contain a subcomponent of mesenchymal BCSCs and a higher proportion of epithelial ALDH1+ BCSCs. Liu et al. (89) compared the transcriptional profiles of epithelial or mesenchymal BCSC subtypes in TNBCs and found that the bi-BCSC subgroup (i.e., ALDH1+ and CD44+/CD24−/lo) was highly purified, with expression of prognostic genes such as P4HA2, PTGR1 and RAB40B.
TNBCs harbor the highest proportion of BCSCs compared with other subtypes, contributing to the poor prognosis associated with this subtype (90). Currently, the only established treatment against TNBCs is the cytotoxic chemotherapy; however a considerable number of patients develop resistance (5, 91). Recently, a PARP (poly-ADP-polymerase) inhibitor—through the underlying mechanism of inhibiting DNA repair, has demonstrated good efficacy against BRCA1-associated breast cancer, which usually refers to TNBCs. In breast cancer cell lines, PARP inhibitor (Olaparib) can significantly decrease the proportion of BCSCs with CD44+/CD24−/low/ESA+ cell surface marker, indicating the potential activity of PARP inhibitor in anticancer stem cells (92) (Figure 2, Table 2). However, Liu et al. (93) recently reported that there were some BCSCs in BRCA1-mutant TNBCs which were relatively resistant to PARP inhibitor (Olaparib), and reduction of RAD51 can sensitize these BCSCs to Olaparib treatment. Clinical benefits from the EGFR inhibitor (94) and a pan-PI3K inhibitor (95) have been reported in TNBC patients, while Fu et al. (96) further reported that EGFR/Notch bispecific antibody PTG12 in combination with pan-PI3K inhibitor GDC-0941 exerted a stronger antitumor effect in TNBC tumors by inhibiting the stem cell–like subpopulation and reducing tumor-initiating cell frequency (Figure 2, Table 2). Up to half of all TNBCs express androgen receptor (AR) (97). AR-targeted therapies (Enzalutamide) can decrease a BCSC-like population in TNBC cell lines (98), indicating Enzalutamide may enhance the efficacy of chemotherapy by targeting a BCSC like cell population (Figure 2, Table 2). In TNBCs, hypoxia-driven BCSCs abated the effectiveness of paclitaxel-based chemotherapy and antiangiogenic agents (e.g., VEGF inhibitors bevacizumab), through HIF-1α (hypoxia-inducible factors 1α). In pre-clinical models, the co-administration of HIF-1α inhibitors was able to overcome BCSC-related resistance (99, 100). HIF-1α vaccination can also inhibit tumor growth in TNBC models of C3(1)Tag mice and decrease SCa-1 marked BCSCs (101) (Table 2). Recently, Cho et al. (102) reported that L80, which is the C-ring truncated deguelin derivative as a C-terminal HSP90 inhibitor, can effectively target BCSC-like trait in TNBCs, together with obvious reduction in CD44+/CD24− cancer cell population, ALDH1 activity and mammosphere forming-ability (Table 2).
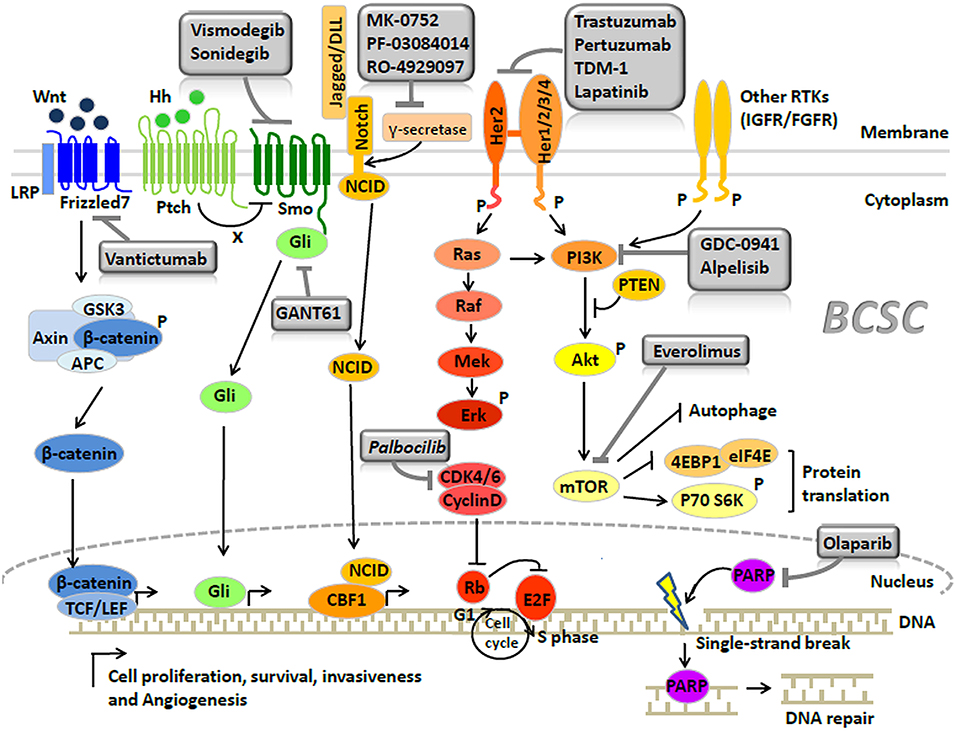
Figure 2. Signaling pathways and breast cancer stem cell-targeted agents in breast cancer (Copyright: Springer Nature 2014 and 2016).
Some drugs were repurposed as the BCSC inhibitors in TNBCs (Table 2). Disulfiram, an antialcoholism drug, was found to target BCSCs and reverse the acquired pan-chemoresistance in TNBC cell lines (103) (Table 2). Mifepristone, a progesterone antagonist for abortion, was reported to suppress BCSCs in TNBC tumors by down-regulating Krüppel-like factor 5 (KLF5) expression, which is a stem cell transcription factor over-expressed in basal type TNBC (104) (Table 2). Metformin, a first-line drug for type 2 diabetes mellitus, was found by the same group that can decrease the BCSC population in TNBC, also by targeting KLF5 for degradation (105) (Table 2). Besides, sulforaphane, a cancer prevention agent, was found to preferentially eliminate BCSCs by inhibiting NF-kB p65 subunit translocation and downregulating p52 (106) (Table 2). And Sulforaphane can also reverse taxane-induced ALDH+ BCSC enrichment (106). Interestingly, novel medicines such as nanomedicines, Gd@C82(OH)22 nanoparticles, were found possess intrinsic inhibitory activity against BCSCs in claudin-low TNBC cell lines (107).
In Brook's model, HER2 breast cancer is characterized by a high proportion of epithelial ALDH1+ BCSCs (9). HER2 is a crucial regulator in BCSCs (108). Recent studies indicated that the regulation of BCSCs by HER2 was not observed only in HER2 breast cancer but extended to all the subtypes of breast cancer (109). To some extent, the remarkable clinical efficacy of HER-2 inhibitors (e.g., trastuzumab, pertuzumab, lapatinib, and TDM-1) was attributed to target BCSCs (109) (Figure 2, Table 2). Nevertheless, a proportion of HER-2 amplified breast cancers continue to eventually develop drug resistance, probably due to the PTEN (phosphatase and tensin homolog) loss or activation of PIK3CA mutation (110). A recent study performed by Sun et al. (111) showed that MEOX1 (mesenchyme homeobox 1) may be a novel target in BCSCs of PTEN-deficient trastuzumab-resistant breast cancers. However, the precise role of BCSCs in the development of resistance to HER-2 inhibitors remains elusive. Thus, further investigation is required to elucidate this process.
Luminal A breast cancer had the lowest proportion of BCSCs among all the breast cancer subtypes, presenting the best prognosis (9). Luminal B breast cancer displayed a certain proportion of BCSCs, which was lower than those observed in TNBC or HER2 breast cancer. A proportion of luminal-B patients have poor prognosis, supposed to be the result of containing a proportion of the BCSCs (112). The presence of the BCSCs is also regarded as the main cause of resistance to hormonal therapy in luminal breast cancer (113, 114), which can be regulated by the CyclinD-CDK4/6 (cyclin-dependent kinase 4/6) complex (115, 116) and mTOR signaling (117) (Figure 2, Table 2). Use of CDK4/6 (i.e., palbocilib) (118) or mTOR inhibitor (i.e., everolimus) (119) significantly improves the survival of patients who develop endocrine resistance. Recently, the PI3K inhibitor (i.e., alpelisib) was also proved to have great clinical activity in PIK3CA-altered luminal breast cancers, including the endocrine therapy-resistant cases (120).
As described above, BCSCs are of great therapeutic relevance, particularly in overcoming resistance to treatment. Although pre-clinical models and clinical trials yielded promising results in targeting BCSCs, the efficacy and safety of BCSC-targeted therapy requires further evaluation. BCSCs share the similar markers and signaling pathways with mammary stem cells. Currently, it is unclear whether BCSC-targeting agents may also target normal mammary stem cells, resulting in severe treatment-related side effects. Besides, the clinical trial data have shown that addition of BCSCs- targeting agents to chemotherapy or hormonal therapy improved treatment efficacy. However, the combination of BCSC-targeting agents and different conventional therapies requires further investigation.
Conclusions and Perspectives
Impressive advances have been witnessed in understanding the carcinogenesis of breast cancer, in which BCSCs hypothesis provided very important models. BCSC-targeted therapy may eradicate the cancer stem cells, which are regarded as the “the seed of tumor.” Therefore, indubitably, the clinical relevance of BCSCs is a primary concern. However, the BCSC population is heterogeneous, involving various cellular markers and regulatory signaling pathways. In addition, the BCSC microenvironment is complex. These facts render BCSC-targeted therapy difficult. Also, different subtypes of breast cancer exhibit distinct BCSC subtypes and abundances. This evidence calls for appropriate BCSC- targeted regimens against different subtypes of breast cancer. Moreover, considering that BCSCs only constitute a small fraction of the tumor cells, the classic clinical endpoints of tumor shrinkage may be inappropriate to assess the efficacy of BCSC-targeted therapy. Recent studies have indicated that the detection of BCSCs markers in tumor biopsies or even in circulating tumor cells may be a favorable option (121, 122).
In conclusion, despite challenges ahead, the comprehensive understandings of stem cells and the cellular origins of breast cancer have assisted and will continue to assist treating physicians in ultimately overcoming the stubborn aspects of breast cancer.
Author Contributions
JZ, YZ, and QC mainly drafted the manuscript. JZ and YC supervised and edited the manuscript. YZ, HC, and LQ participated in drawing the pictures. QC edited the language of the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the funding of National Natural Science Foundation of China (Grant Nos. 81702866 and 81700240), Foundation of the Education Department of Zhejiang Province (Grant No. Y201636451), the Key Program of the Natural Science Foundation of Zhejiang Province (Grant No. LZ16H160002), and the Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All the people contributed to this manuscript were listed in the authorship. No other acknowledgments.
Abbreviations
BCSCs, Breast cancer stem cells; CD44, CD44 molecule; CD24, heat-stable antigen; ALDH1, Aldehyde dehydrogenase1; EMT, epithelial-mesenchymal transition; MET, mesenchymal-epithelial transition; Id1, inhibitor of DNA binding 1; CD133, prominin1; CD29, β1-integrin; CD49f:, α6-integrin; Thy1, Thy-1 cell surface antigen, CD90; CD61, 1, Integrin subunit beta 3, ITGB3; Sca-1, Stem cell antigen 1; MUC1, also known as CA153; ESA, Epithelial specific antigen; DLL1, Delta like canonical Notch ligand1; DNER, Delta/notch like EGF repeat containing; GD2, Ganglioside GD2; ANTXR1, ANTXR cell adhesion molecule 1; ABCG2, ATP binding cassette subfamily G member 2; Lgr5, leucine rich repeat containing G protein-coupled receptor 5; SSEA-3, stage-specific embryonic antigen-3; Nectin-4, nectin cell adhesion molecule 4; CD70, CD70 molecule; TCF/LEF, T cell factor/lymphoid enhancing factor; FZD7, Frizzled-7; LRP6, Lipoprotein receptor related protein-6; MaSCs, Mammary stem cells; NCID, Notch intracellular domain; BMI1, BMI1 proto-oncogene, polycomb ring finger; Suz12, SUZ12 polycomb repressive complex 2 subunit; HIPK1, Homeodomain-interacting protein kinase 1; H-RAS, HRas proto-oncogene, GTPase; HMGA2, High mobility group AT-hook 2; UBC9, Ubiquitin-conjugating enzyme 9; ITGB3, Integrin b3; SCD1, Stearoyl desaturase 1; MSCs, Mesenchymal stem cells; CAFs, Cancer associated fibroblasts; ECM, Extracellular matrix; IL6, interleukin 6; CXCL7, pro-platelet basic protein; FOXP2, forkhead-box P2; CCL2, Monocyte chemotactic protein-1; HMGB1, High-mobility group box 1; TAMs, Tumor associated macrophages; M-CSF, Macrophage colony-stimulating factor; TNFα, Tumor necrosis factor alpha; TGFβ, Transforming growth factor beta; CSCs, Cancer stem cells; EGFR/Stat3/Sox-2, Epidermal growth factor receptor/ signal transducer and activator of transcription 3/ SRY-box 2; HAS2, Hyaluronan synthase 2; TILs, Tumor-infiltrating lymphocytes; CD4, CD4 molecule; CD8, CD8 molecule; FOXP3, forkhead box P3; PDX, Patient derived xenograft; YAP/TAZ, Yes-associated protein/Transcriptional coactivator with PDZ-binding motif; HIF, hypoxia inducible factor; ALKBH5, AlkB homolog 5; NANOG, Nanog homeobox; PIK3CA, α-catalytic subunit of PI3K; CK8, Keratin 8; CK5, Keratin5; CK14, Keratin14; BRCA1, DNA repair associated; GATA3, GATA binding protein 3; FOXA1, Forkhead box A1; CD10, 0, Membrane metalloendopeptidase, MME; ER, Estrogen receptor; TNBC, Triple negative breast cancer; P4HA2, Prolyl 4-hydroxylase subunit alpha 2; PTGR1, Prostaglandin reductase 1; RAB40B, Member RAS oncogene family; PARP, poly-ADP-polymerase; HIF-1α, Hypoxia-inducible factors 1α; AR, androgen receptor; KLF5, Krüppel-like factor 5; CDK4/6, Cyclin-dependent kinase 4/6; PTEN, Phosphatase and tensin homolog; MEOX1, Mesenchyme homeobox 1.
References
2. Perou C, Sørlie T, Eisen M, van de Rijn M, Jeffrey S, Rees C, et al. Molecular portraits of human breast tumours. Nature. (2000) 406:747–52. doi: 10.1038/35021093
3. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. (2001) 98:10869–74. doi: 10.1073/pnas.191367098
4. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
5. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. (2016) 13:674–90. doi: 10.1038/nrclinonc.2016.66
6. Foulkes WD. Germline BRCA1 Mutations and a basal epithelial phenotype in breast cancer. CancerSpectr Knowl Environ. (2003) 95:1482–5. doi: 10.1093/jnci/djg050
7. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. (2016) 16:110–20. doi: 10.1038/nrc.2015.21
8. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. (2019) 35:428–,40 e5. doi: 10.1016/j.ccell.2019.02.001
9. Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. (2015) 17:260–71. doi: 10.1016/j.stem.2015.08.014
10. Greaves M, Maley CC. Clonal evolution in cancer. Nature. (2012) 481:306–13. doi: 10.1038/nature10762
11. Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells–old concepts, new insights. Cell Death Differ. (2008) 15:947–58. doi: 10.1038/cdd.2008.20
12. Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. (2012) 47:570–84. doi: 10.1016/j.molcel.2012.06.014
13. Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. (2010) 28:4006–12. doi: 10.1200/JCO.2009.27.5388
14. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. (2013) 13:727–38. doi: 10.1038/nrc3597
15. Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. (2003) 9:702–12. doi: 10.1038/nm0603-702
16. Al-Hajj M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
17. Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. (2007) 11:259–73. doi: 10.1016/j.ccr.2007.01.013
18. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
19. Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. (2008) 10:R10. doi: 10.1186/bcr1855
20. Cho RW, Wang X, Diehn M, Shedden K, Chen GY, Sherlock G, et al. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells. (2008) 26:364–71. doi: 10.1634/stemcells.2007-0440
21. Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. (2008) 68:7711–7. doi: 10.1158/0008-5472.CAN-08-1949
22. Grange C, Lanzardo S, Cavallo F, Camussi G, Bussolati B. SCA-1 identifies the tumor-initiating cells in mammary tumors of BALB-neuT transgenic mice. Neoplasia. (2008) 10:1433–43. doi: 10.1593/neo.08902
23. Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. (2014) 33:5477–82. doi: 10.1038/onc.2013.516
24. Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. (2008) 68:2419–26. doi: 10.1158/0008-5472.CAN-07-2249
25. Hwang-Verslues W, Kuo W, Chang P, Pan C, Wang H, Tsai S, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS ONE. (2009) 4:e8377. doi: 10.1371/journal.pone.0008377
26. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. (2010) 140:62–73. doi: 10.1016/j.cell.2009.12.007
27. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. (2012) 122:2066–78. doi: 10.1172/JCI59735
28. Chen D, Bhat-Nakshatri P, Goswami C, Badve S, Nakshatri H. ANTXR1, a stem cell-enriched functional biomarker, connects collagen signaling to cancer stem-like cells and metastasis in breast cancer. Cancer Res. (2013) 73:5821–33. doi: 10.1158/0008-5472.CAN-13-1080
29. Leccia F, Del Vecchio L, Mariotti E, Di Noto R, Morel A, Puisieux A, et al. ABCG2, a novel antigen to sort luminal progenitors of BRCA1- breast cancer cells. Mol Cancer. (2014) 13:213. doi: 10.1186/1476-4598-13-213
30. Yang L, Tang H, Kong Y, Xie X, Chen J, Song C, et al. LGR5 promotes breast cancer progression and maintains stem-like cells through activation of Wnt/beta-catenin signaling. Stem Cells. (2015) 33:2913–24. doi: 10.1002/stem.2083
31. Cheung S, Chuang P, Huang H, Hwang-Verslues W, Cho C, Yang W, et al. Stage-specific embryonic antigen-3 (SSEA-3) and β3GalT5 are cancer specific and significant markers for breast cancer stem cells. Proc Natl Acad Sci USA. (2016) 113:960–5. doi: 10.1073/pnas.1522602113
32. Siddharth S, Goutam K, Das S, Nayak A, Nayak D, Sethy C, et al. Nectin-4 is a breast cancer stem cell marker that induces WNT/beta-catenin signaling via Pi3k/Akt axis. Int J Biochem Cell Biol. (2017) 89:85–94. doi: 10.1016/j.biocel.2017.06.007
33. Liu L, Yin B, Yi Z, Liu X, Hu Z, Gao W, et al. Breast cancer stem cells characterized by CD70 expression preferentially metastasize to the lungs. Breast Cancer. (2018) 25:706–16. doi: 10.1007/s12282-018-0880-6
34. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. (2014) 2:78–91. doi: 10.1016/j.stemcr.2013.11.009
35. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
36. Brabletz T. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell. (2012) 22:699–701. doi: 10.1016/j.ccr.2012.11.009
37. Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, et al. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. (2013) 5:1228–42. doi: 10.1016/j.celrep.2013.11.014
38. Al-Ejeh F, Smart CE, Morrison BJ, Chenevix-Trench G, Lopez JA, Lakhani SR, et al. Breast cancer stem cells: treatment resistance and therapeutic opportunities. Carcinogenesis. (2011) 32:650–8. doi: 10.1093/carcin/bgr028
39. Yang F, Xu J, Tang L, Guan X. Breast cancer stem cell: the roles and therapeutic implications. Cell Mol Life Sci. (2016) 74:951. doi: 10.1007/s00018-016-2334-7
40. Monteiro J, Gaspar C, Richer W, Franken PF, Sacchetti A, Joosten R, et al. Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis. (2014) 35:2–13. doi: 10.1093/carcin/bgt279
41. Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. (2011) 10:9–15. doi: 10.1158/1535-7163.MCT-10-0677
42. Habib JG, O'Shaughnessy JA. The hedgehog pathway in triple-negative breast cancer. Cancer Med. (2016) 5:2989–3006. doi: 10.1002/cam4.833
43. King T, Suto M, Li Y. The Wnt/β-catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. (2012) 113:13–18. doi: 10.1002/jcb.23350
44. Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene. (2011) 30:4437–46. doi: 10.1038/onc.2011.145
45. Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M, Sinha N. Therapeutic targets of triple-negative breast cancer: a review. Br J Pharmacol. (2015) 172:4228–37. doi: 10.1111/bph.13211
46. Chen Y, Jiang J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. (2013) 23:186–200. doi: 10.1038/cr.2013.10
47. Wang L, Duan W, Kang L, Mao J, Yu X, Fan S, et al. Smoothened activates breast cancer stem-like cell and promotes tumorigenesis and metastasis of breast cancer. Biomed Pharmacother. (2014) 68:1099–104. doi: 10.1016/j.biopha.2014.09.012
48. Merchant AA, Matsui W. Targeting Hedgehog–a cancer stem cell pathway. Clin Cancer Res. (2010) 16:3130–40. doi: 10.1158/1078-0432.CCR-09-2846
49. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. (2018) 9:2897. doi: 10.1038/s41467-018-05220-6
50. Koike Y, Ohta Y, Saitoh W, Yamashita T, Kanomata N, Moriya T, et al. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer. (2017) 24:683–93. doi: 10.1007/s12282-017-0757-0
51. McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS, et al. Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res. (2011) 9:834–44. doi: 10.1158/1541-7786.MCR-10-0457
52. Olsauskas-Kuprys R, Zlobin A, Osipo C. Gamma secretase inhibitors of Notch signaling. Onco Targets Ther. (2013) 6:943–55. doi: 10.2147/OTT.S33766
53. Liu S, Clouthier SG, Wicha MS. Role of microRNAs in the regulation of breast cancer stem cells. J Mammary Gland Biol Neoplasia. (2012) 17:15–21. doi: 10.1007/s10911-012-9242-8
54. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. (2009) 138:592–603. doi: 10.1016/j.cell.2009.07.011
55. Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. (2010) 39:761–72. doi: 10.1016/j.molcel.2010.08.013
56. Liu B, Du R, Zhou L, Xu J, Chen S, Chen J, et al. miR-200c/141 Regulates Breast Cancer Stem Cell Heterogeneity via Targeting HIPK1/beta-Catenin Axis. Theranostics. (2018) 8:5801–13. doi: 10.7150/thno.29380
57. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. Let-7 regulates self-renewal and tumorigenicity of breast cancer cells. Cell. (2007) 131:1109–23. doi: 10.1016/j.cell.2007.10.054
58. Yu F, Deng H, Yao H, Liu Q, Su F, Song E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. (2010) 29:4194–204. doi: 10.1038/onc.2010.167
59. El Helou R, Pinna G, Cabaud O, Wicinski J, Bhajun R, Guyon L, et al. miR-600 acts as a bimodal switch that regulates breast cancer stem cell fate through WNT signaling. Cell Rep. (2017) 18:2256–68. doi: 10.1016/j.celrep.2017.02.016
60. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. (2011) 71:614–24. doi: 10.1158/0008-5472.CAN-10-0538
61. Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC, et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell. (2014) 15:762–74. doi: 10.1016/j.stem.2014.10.001
62. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. (2012) 72:2768–79. doi: 10.1158/0008-5472.CAN-11-3567
63. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. (2011) 121:3804–9. doi: 10.1172/JCI57099
64. Zhao X, Lin Y, Jiang J, Tang Z, Yang S, Lu L, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. (2017) 243:376–89. doi: 10.1002/path.4958
65. Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, et al. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. (2017) 77:2134–47. doi: 10.1158/0008-5472.CAN-15-3490
66. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. (2015) 16:225–38. doi: 10.1016/j.stem.2015.02.015
67. Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, et al. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells. (2013) 31:248–58. doi: 10.1002/stem.1281
68. Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. (2012) 72:537–47. doi: 10.1158/0008-5472.CAN-11-1678
69. Seo AN, Lee HJ, Kim EJ, Kim HJ, Jang MH, Lee HE, et al. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br J Cancer. (2013) 109:2705–13. doi: 10.1038/bjc.2013.634
70. Malta T, Sokolov A, Gentles A, Burzykowski T, Poisson L, Weinstein J, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. (2018) 173:338–54. doi: 10.1016/j.cell.2018.03.034
71. Wolfson B. Adipocyte activation of cancer stem cell signaling in breast cancer. World J Biol Chem. (2015) 6:39. doi: 10.4331/wjbc.v6.i2.39
72. Goto H, Shimono Y, Funakoshi Y, Imamura Y, Toyoda M, Kiyota N, et al. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene. (2018) 38:1. doi: 10.1038/s41388-018-0477-8
73. Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, Rafii S, et al. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE. (2014) 9:e112424. doi: 10.1371/journal.pone.0112424
74. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. (2011) 474:179–83. doi: 10.1038/nature10137
75. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. (2016) 113:E2047–56. doi: 10.1073/pnas.1602883113
76. Skibinski A, Kuperwasser C. The origin of breast tumor heterogeneity. Oncogene. (2015) 34:5309–16. doi: 10.1038/onc.2014.475
77. Meyer DS, Brinkhaus H, Muller U, Muller M, Cardiff RD, Bentires-Alj M. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. (2011) 71:4344–51. doi: 10.1158/0008-5472.CAN-10-3827
78. Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, Giraddi RR, et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature. (2015) 525:119–23. doi: 10.1038/nature14665
79. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. (2010) 7:403–17. doi: 10.1016/j.stem.2010.07.010
80. Tao L, van Bragt MP, Laudadio E, Li Z. Lineage tracing of mammary epithelial cells using cell-type-specific cre-expressing adenoviruses. Stem Cell Rep. (2014) 2:770–9. doi: 10.1016/j.stemcr.2014.04.004
81. Zhang M, Lee AV, Rosen JM. The cellular origin and evolution of breast cancer. Cold Spring Harb Perspect Med. (2017) 7:a027128. doi: 10.1101/cshperspect.a027128
82. Miller TW. Initiating breast cancer by PIK3CA mutation. Breast Cancer Res. (2012) 14:301. doi: 10.1186/bcr3103
83. Sabine VS, Crozier C, Brookes CL, Drake C, Piper T, van de Velde CJ, et al. Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol. (2014) 32:2951–8. doi: 10.1200/JCO.2013.53.8272
84. Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. (2006) 127:1041–55. doi: 10.1016/j.cell.2006.09.048
85. Usary J, Llaca V, Karaca G, Presswala S, Karaca M, He X, et al. Mutation of GATA3 in human breast tumors. Oncogene. (2004) 23:7669–78. doi: 10.1038/sj.onc.1207966
86. Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci USA. (2007) 104:12111–6. doi: 10.1073/pnas.0702969104
87. Keller P, Arendt L, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci USA. (2012) 109:2772–7. doi: 10.1073/pnas.1017626108
88. McCarthy A, Savage K, Gabriel A, Naceur C, Reis-Filho JS, Ashworth A. A mouse model of basal-like breast carcinoma with metaplastic elements. J Pathol. (2007) 211:389–98. doi: 10.1002/path.2124
89. Liu M, Liu Y, Deng L, Wang D, He X, Zhou L, et al. Transcriptional profiles of different states of cancer stem cells in triple-negative breast cancer. Mol Cancer. (2018) 17:65. doi: 10.1186/s12943-018-0809-x
90. Yu KD., Zhu R, Zhan M, Rodriguez AA, Yang W, Wong S, et al. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin Cancer Res. (2013) 19:2723–33. doi: 10.1158/1078-0432.CCR-12-2986
91. Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. (2008) 68:3243–50. doi: 10.1158/0008-5472.CAN-07-5480
92. Shimo T, Kurebayashi J, Kanomata N, Yamashita T, Kozuka Y, Moriya T, et al. Antitumor and anticancer stem cell activity of a poly ADP-ribose polymerase inhibitor olaparib in breast cancer cells. Breast Cancer. (2014) 21:75–85. doi: 10.1007/s12282-012-0356-z
93. Liu Y, Burness M, Martin-Trevino R, Guy J, Bai S, Harouaka R, et al. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin Cancer Res. (2017) 23:514–22. doi: 10.1158/1078-0432.CCR-15-1348
94. Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. (2012) 30:2615–23. doi: 10.1200/JCO.2010.34.5579
95. Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. (2012) 30:282–90. doi: 10.1200/JCO.2011.36.1360
96. Fu W., Lei C., Yu Y., Liu S., Li T., Lin F., et al. (2019) EGFR/Notch antagonists enhance the response to inhibitors of the PI3K-Akt pathway by decreasing tumor-initiating cell frequency. Clin. Cancer Res. 25, 2835–47. doi: 10.1158/1078-0432.CCR-18-2732
97. Barton VN, D'Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. (2015) 14:769–78. doi: 10.1158/1535-7163.MCT-14-0926
98. Barton VN, Christenson JL, Gordon MA, Greene LI, Rogers TJ, Butterfield K, et al. Androgen receptor supports an anchorage-independent, cancer stem cell-like population in triple-negative breast cancer. Cancer Res. (2017) 77:3455–3466. doi: 10.1158/0008-5472.CAN-16-3240
99. Conley S, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath A, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. (2012) 109:2784–9. doi: 10.1073/pnas.1018866109
100. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. (2014) 111:E5429–38. doi: 10.1073/pnas.1421438111
101. Cecil D, Slota M, O'Meara M, Curtis B, Gad E, Dang Y, et al. Immunization against HIF-1α inhibits the growth of basal mammary tumors and targets mammary stem cells in vivo. Clin Cancer Res. (2017) 23:3396–404. doi: 10.1158/1078-0432.CCR-16-1678
102. Cho TM, Kim JY, Kim YJ, Sung D, Oh E, Jang S, et al. C-terminal HSP90 inhibitor L80 elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Cancer Lett. (2019) 447:141–53. doi: 10.1016/j.canlet.2019.01.029
103. Liu P, Kumar IS, Brown S, Kannappan V, Tawari PE, Tang JZ, et al. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br J Cancer. (2013) 109:1876–85. doi: 10.1038/bjc.2013.534
104. Liu R, Shi P, Nie Z, Liang H, Zhou Z, Chen W, et al. Mifepristone suppresses basal triple-negative breast cancer stem cells by down-regulating KLF5 expression. Theranostics. (2016) 6:533–44. doi: 10.7150/thno.14315
105. Shi P, Liu WT, Wang H, Li F, Zhang H, Wu Y, et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. (2017) 3:17010. doi: 10.1038/celldisc.2017.10
106. Burnett JP, Lim G, Li Y, Shah RB, Lim R, Paholak HJ, et al. Sulforaphane enhances the anticancer activity of taxanes against triple negative breast cancer by killing cancer stem cells. Cancer Lett. (2017) 394:52–64. doi: 10.1016/j.canlet.2017.02.023
107. Liu Y, Chen C, Qian P, Lu X, Sun B, Zhang X, et al. Gd-metallofullerenol nanomaterial as non-toxic breast cancer stem cell-specific inhibitor. Nat Commun. (2015) 6:5988. doi: 10.1038/ncomms6988
108. Dittrich A, Gautrey H, Browell D, Tyson-Capper A. The HER2 signaling network in breast cancer–like a spider in its web. J Mammary Gland Biol Neoplasia. (2014) 19:253–70. doi: 10.1007/s10911-014-9329-5
109. Korkaya H, Wicha MS. HER2 and breast cancer stem cells: more than meets the eye. Cancer Res. (2013) 73:3489–93. doi: 10.1158/0008-5472.CAN-13-0260
110. Dave B, Migliaccio I, Gutierrez MC, Wu MF, Chamness GC, Wong H, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol. (2011) 29:166–73. doi: 10.1200/JCO.2009.27.7814
111. Sun L, Burnett J, Gasparyan M, Xu F, Jiang H, Lin C, et al. Novel cancer stem cell targets during epithelial to mesenchymal transition in PTEN-deficient trastuzumab-resistant breast cancer. Oncotarget. (2016) 7:51408–22. doi: 10.18632/oncotarget.9839
112. Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA. (2008) 105:5774–9. doi: 10.1073/pnas.0706216105
113. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. (2009) 106:13820–5. doi: 10.1073/pnas.0905718106
114. Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. (2014) 6:66–79. doi: 10.1002/emmm.201303411
115. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. (2009) 11:R77. doi: 10.1186/bcr2419
116. Lamb R, Lehn S, Rogerson L, Clarke RB, Landberg G. Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor-dependent divergent functions in breast cancer migration and stem cell-like activity. Cell Cycle. (2013) 12:2384–94. doi: 10.4161/cc.25403
117. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci USA.. (2007) 104:16158–63. doi: 10.1073/pnas.0702596104
118. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. (2015) 16:25–35. doi: 10.1016/S1470-2045(14)71159-3
119. Baselga J, Campone M, Piccart M, Burris Hr, Rugo H, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med.. (2012) 366:520–9. doi: 10.1056/NEJMoa1109653
120. Juric D, Janku F, Rodon J, Burris HA, Mayer IA, Schuler M, et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. (2018) 5:e184475. doi: 10.1001/jamaoncol.2018.4475
121. Gkountela S, Aceto N. Stem-like features of cancer cells on their way to metastasis. Biol Direct. (2016) 11:33. doi: 10.1186/s13062-016-0135-4
Keywords: breast cancer, stem cell, cellular origins, therapeutic implications, heterogeneity
Citation: Zhou J, Chen Q, Zou Y, Chen H, Qi L and Chen Y (2019) Stem Cells and Cellular Origins of Breast Cancer: Updates in the Rationale, Controversies, and Therapeutic Implications. Front. Oncol. 9:820. doi: 10.3389/fonc.2019.00820
Received: 28 January 2019; Accepted: 09 August 2019;
Published: 28 August 2019.
Edited by:
Amit K. Tiwari, University of Toledo, United StatesReviewed by:
Vishwa Mohini Khare, University of Pennsylvania, United StatesMd Kamal Hossain, University of Toledo, United States
Dayanidhi Raman, University of Toledo, United States
Copyright © 2019 Zhou, Chen, Zou, Chen, Qi and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaojiao Zhou, emhvdWpqQHpqdS5lZHUuY24=; Yiding Chen, eWRjaGVuQHpqdS5lZHUuY24=