 Alexander A. Wurm
Alexander A. Wurm Cristina Pina
Cristina Pina- 1Department of Medical Translational Oncology, National Center for Tumor Diseases (NCT) Dresden, Dresden, Germany
- 2Department of Genetics, University of Cambridge, Cambridge, United Kingdom
Acute myeloid leukemia is a hematopoietic neoplasm of dismal prognosis that results from the accumulation of immature myeloid blasts in the bone marrow and the peripheral blood. It is strongly dependent on epigenetic regulation for disease onset, maintenance and in response to treatment. Epigenetic regulation refers to the multiple chemical modifications of DNA or DNA-associated proteins that alter chromatin structure and DNA accessibility in a heritable manner, without changing DNA sequence. Unlike sequence-specific transcription factors, epigenetic regulators do not necessarily bind DNA at consensus sequences, but still achieve reproducible target binding in a manner that is cell and maturation-type specific. A growing body of evidence indicates that epigenetic regulators rely, amongst other factors, on their interaction with untranslated RNA molecules for guidance to particular targets on DNA. Non (protein)-coding RNAs are the most abundant transcriptional products of the coding genome, and comprise several different classes of molecules with unique lengths, conformations and targets. Amongst these, long non-coding RNAs (lncRNAs) are species of 200 bp to >100 K bp in length, that recognize, and bind unique and largely uncharacterized DNA conformations. Some have been shown to bind epigenetic regulators, and thus constitute attractive candidates to mediate epigenetic target specificity. Herein, we postulate that lncRNAs are central players in the unique epigenetic programming of AML and review recent evidence in support of this view. We discuss the value of lncRNAs as putative diagnostic, prognostic and therapeutic targets in myeloid leukemias and indicate novel directions in this exciting research field.
Foreword
The aim of this mini-review is to discuss emerging epigenetic roles of long non-coding RNAs (lncRNAs) in Acute Myeloid Leukemia (AML). AML has a characteristic dependency on chromatin and transcriptional regulators, which has been discussed in many reviews. To date, most of our understanding of epigenetic regulation and its participation in leukemogenesis is based on the enzymatic activities and protein-protein interactions of histone and DNA modifiers. However, a growing body of evidence suggests that recruitment, stability, and function of epigenetic factors can be mediated by non-protein-coding RNAs. In particular, there are incremental examples of the participation of the distinct class of long non-coding RNAs (lncRNAs) in AML epigenetic regulation, some of which have translational potential. In this context, we summarize common epigenetic alterations in AML that are relevant for understanding lncRNA potential contribution to the disease. We define lncRNAs and highlight their general roles of in normal development and in cancer. Finally, we review the developing body of literature on the participation of lncRNAs in epigenetic regulation of AML, and discuss their putative therapeutic relevance. We expect that the ideas advanced in this mini-review will be sedimented and extended in the coming years, thus positioning lncRNAs at the heart of epigenetic regulation and manipulation in AML.
Epigenetic Alterations are Common Features of Acute Myeloid Leukemia
AML is an aggressive malignancy of the non-lymphoid lineages of the blood. It is a heterogeneous disease, cellularly and molecularly, with an overarching theme of ectopic self-renewal and arrested differentiation potential at multiple levels of the hematopoietic tree (1). Akin to the normal hematopoietic system, leukemias are sustained by a small number of leukemia stem-like cells (LSC), which can be distinct from the normal hematopoietic stem cells (HSC) but also exhibit functional characteristics of self-renewal and (abnormal or hindered) differentiation, and are often quiescent (2–4). Emergence of LSC is dependent on individual or combined genetic mutations that broadly determine the cellular affiliation of the leukemia, and permit or impose ectopic self-renewal and a restricted differentiation potential into what constitutes the proliferative bulk of the leukemia (5, 6). Different genetic lesions confer distinct natural histories, and hence prognosis. From a molecular point of view, AML has several distinctive features. It has a low mutation burden, particularly in the more extensively analyzed coding genome (7). A significant number of these mutations target transcription factors (TFs) and/or epigenetic regulators, thus indicating a specific reliance on gene regulatory mechanisms (8). The strong dependence of AML on epigenetic regulation has recently been highlighted by the prognostic value of heterogeneous DNA methylation epi-alleles: these are established de novo after chemotherapy and independently of mutation, and may drive disease progression and relapse in a manner akin to genetic variegation (9).
Epigenetic regulators act by modulating the accessibility of DNA to the basal gene expression machinery without absolute sequence-specificity, but with at least some degree of heritability across cell divisions (10). Broadly speaking, they can: (1) activate or repress gene expression through specific enzymatic activities that add or remove active residues from DNA or histones (e.g., acetyl, methyl, phosphor residues); (2) recognize specific modifications and recruit additional enzymatic activities or regulatory machinery to extend or erase the modification; (3) directly interact with general TFs and the RNA polymerases to initiate, maintain or terminate transcription. They can be classified as writers—active residue depositors, erasers—active residue removers, or readers—active residue binders, all classes having been implicated in pathogenesis of cancer in general, and of AML in particular (11).
Epigenetic Alterations in AML Can Result From Mutations of Chromatin Regulators
Epigenetic regulators recurrently mutated in AML include: Histone acetyl-transferases (HATs)—namely EP300/CBP and members of the MYST/MOZ family, through point mutation or chromosomal translocation (12). Histone methyl-transferases—namely KMT2B/MLL1, a chromosomal translocation hotspot leading to a uniquely aggressive disease of bad prognosis (13). DNA methyl-transferases and demethylases—respectively DNMT3A (14, 15), and TET2 (16, 17) or the metabolic enzymes IDH1/2 (18, 19), that interfere with target-specific methylation, and can also reorganize DNA methylation patterns genome-wide (20, 21). Point mutations in DNMT3A have mutation-specific effects on DNA methylation (22). Point mutations in TET2 interfere with its capacity to demethylate DNA, with repressive consequences for gene expression (23). These are broadly mimicked by gain-of-function point mutations in the metabolic enzymes IDH1 and IDH2, which inhibit TET activity through accumulation of the methylation intermediate 5-hydroxy-methyl cytosine (5-hmC) (24). Mutations in the Cohesin complex perturb higher-order genome organization (25). Other commonly mutated genes affect gene expression regulators downstream of transcription, namely splicing and assembly of the translational machinery (8). In the latter group, the nucleus-to-cytoplasm pre-ribosomal transporter NPM1 is the most commonly targeted gene in AML (26).
Leukemogenic Programs Co-opt Non-mutated Epigenetic Regulators
Importantly, AML cells are also strictly dependent on a number of non-mutated epigenetic regulators that are co-opted by the oncogenic programs to maintain self-renewal and/or survival of the leukemic cells. Some of these epigenetic regulators are central to the leukemic program in specific mutation contexts. The H3K79me2/3 methyl-transferase DOT1L is recruited by several of the MLL fusion genes, e.g., MLLT1/ENL or MLLT3/AF9, as part of the RNA polymerase (Pol) II super-elongation complex to modify promoters and proximal gene bodies and activate transcription (27). Histone deacetylases (HDACs) are recruited by leukemic fusions involving the master regulator RUNX1 gene or its binding partner CBFB (in the so-called core-binding factor AMLs) to repress transcription at specific targets (28–30). Other epigenetic regulators are recruited more broadly across the AML mutational spectrum: the Bromodomain protein 4 (BRD4), a reader of lysine acetylation, was originally identified as a SEC participant in the context of MLL fusion AML (31), but has since been shown to exert a more general influence in maintaining AML gene expression programs, including in the frequent genetic contexts of NPM1 mutation (32), and of the oncogenic Fms-like Tyrosine Kinase 3 internal tandem duplication (FLT3-ITD) (33) that constitutively activates signaling from the hematopoietic growth factor receptor FLT3 (34). LSD1, or KDM1A, is a histone demethylase that removes mono and di-methylation from H3K4 and H3K9, with repressive or activating effects on gene expression (35, 36). Like BRD4, it was initially described as a requirement in MLL re-arranged AML (37), but has more recently been shown to inhibit AML cell differentiation more generally (38). We have added the histone acetyl-transferase KAT2A to the list of putative global requirements in AML. KAT2A is primarily required for acetylation of H3K9 in gene promoters (39), but it can also acetylate non-histone proteins, including transcription factors EGR2 (40) and C/EBPα (41), which are relevant for normal and malignant hematopoiesis. We identified KAT2A as a genetic vulnerability in a subset of AML cell lines in a genome-wide CRISPR drop-out screen, and showed a requirement for self-renewal and/or survival in patient samples across a spectrum of mutational events (42). It also a requirement in ES cells (43, 44) and induced pluripotent stem cells (45), and its interaction with the MYC oncogene may translate into AML. Recently, the methyl-transferase EZH2, a member of the Polycomb Repressive Complex 2 (PRC2) that deposits the repressive H3K27me3 modification, has been characterized as both a tumor suppressor and an oncogene in AML, depending on the stage of the disease (46). Specifically, loss of EZH2 promotes AML initiation, reflected in low-frequency loss-of-function mutations in myeloid neoplasms (47–49), and the observed acceleration of AML transformation from pre-malignant myeloproliferative neoplasms (MPN) (50, 51). On the other hand, established AML (52, 53), as well as chronic myeloid leukemia (CML) cells (54), depend on expression of EZH2 for disease maintenance. and there is potential for EZH2 inhibitors as a therapeutic strategy in established AML.
In summary, AML depends on multiple epigenetic regulators, in genetic abnormality, disease subtype, and disease stage-dependent manners, with and without mutation of the epigenetic regulators themselves. Unsurprisingly, this is an area of intensive research that holds promise in the development of novel anti-leukemia therapeutic strategies.
Long Non-coding RNAs are Largely Unexplored Transcriptional Species
It is known that only ~3% of the human transcriptome is translated into functional proteins (55). Thus, the vast majority of transcribed genes remain at the RNA level. The largest part of this group comprises ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs). The remaining fraction is referred to as “non-coding RNAs” (nc-RNAs) and can be stratified into distinct families, mainly according to the RNA length. There are small nc-RNAs, namely microRNAs, piwi-interacting RNAs or sno-RNAs, which range from below 40 to ~200–300 bp in length. At the other end of the spectrum, lncRNAs are a heterogeneous group that exhibit a wide size range from at least 200 bp and up to 100 kbp (56). There have been inconsistent attempts at lncRNAs classification, with tentative subdivisions on the basis of genomic localization or molecular function (57). As a potential confounder, several studies revealed strong structural, but not sequence, conservation of lncRNAs between different organisms (58), indicating that inter-species comparison such as used for protein-coding genes may not systematically predict the biological relevance of lncRNAs. LncRNAs have been associated with a plethora of regulatory functions, including regulation of gene expression and alternative splicing, DNA-RNA-protein interactions, and participation in histone-DNA complexes (59). A breakthrough observation in mouse embryonic development positioned lncRNAs as epigenetic regulators. Specifically, the lncRNA HOX transcript antisense intergenic RNA (HOTAIR), which is transcribed from the HOXC locus in a tissue-specific manner, was found to mediate transcriptional silencing of the HOXD gene locus through direct interaction with the PRC2. This provided the first evidence that non-coding RNAs could regulate epigenetic gene silencing. Subsequently, various other lncRNAs, e.g., Xist, RepA, Kcnq1ot1, Braveheart, or Malat-1, have been shown to interact with PRC2 in a multi-protein-RNA complex (60). Indeed, on the basis of global RNA immunoprecipitation (RIP) experiments, ~20% of human lncRNAs interact with PRC2 (61), highlighting the influence of the long non-coding RNAome on transcriptional activity.
LncRNAs Can Act as Epigenetic Drivers in Human Cancer
In contrast with other ncRNA species, e.g., microRNAs, which have been intensively studied in cancer, including leukemia (76, 77), lncRNAs have only recently been recognized as active players in initiation, maintenance and treatment response of human cancer (78).
In some instances, lncRNA contribution to tumor biology results from direct epigenetic regulation or participation in chromatin complexes. LncRNAs are actively transcribed by RNA-PolII (in some instances, RNA-PolIII), and their expression can be modulated by chromatin-modifying complexes. For example, the lncRNA Low Expression in Tumor (LET) is transcriptionally silenced in various solid cancers by Histone Deacetylase 3 (HDAC3)-mediated promoter inactivation (79). Also, the lncRNA Urothelial Cancer Associated 1 (UCA1) is regulated by ARID1A, a component of the SWI/SNF chromatin-remodeling complex (80), and itself interacts with multiple histone-modifying proteins to regulate target gene expression (81).
More importantly, lncRNAs can contribute to cancer by epigenetic activation or silencing of target loci in a cis or trans-regulatory manner. LncRNAs can modify gene expression in cis by altering chromatin structure or DNA methylation pattern of neighboring gene promoters. Approximately 40% of all human protein coding genes are co-expressed with a paired natural antisense transcript (NAT), a lncRNA subtype that is expressed from the opposite strand of the host gene (82). Host gene-NAT ratios are often tissue-dependent and have been shown to clearly define cancer subtypes (82). Antisense lncRNAs can protect paired host genes from epigenetic silencing by interacting with histone-modifying enzymes such as the PRC2 (60). In contrast, host gene-associated antisense RNAs can also silence paired protein-coding genes, as exemplified by the acetylcholinesterase (AChE) gene, which is silenced by AChE-AS lncRNA in hepatocellular carcinoma through triggering of histone methylation (83). Interactions between gene and antisense-RNA can impact disease progression, as in the increased chemotherapy resistance associated with transcriptional activation of GAS6-AS2 lncRNA in multiple cancers including AML (84). In addition to anti-sense pairing, lncRNAs can also exert cis-regulatory activity when transcribed in sense with a host gene. Exemplarily, the diRNA (DNMT1-interacting RNAs) lncRNA family modifies global DNA methylation patterns by protecting host gene promoters from DNMT1-driven de novo methylation after cell division (75).
There are several examples of trans-regulatory activities of lncRNAs that modify gene expression through chromatin remodeling. In gastric cancer, GClnc1 regulates a plethora of target genes by recruiting histone-modifying proteins KAT2A and WDR5 and activating gene expression (85). As discussed, KAT2A is a HAT with novel putative roles in AML maintenance. WDR5, itself an RNA binding protein, is an essential subunit of the MLL complex that drives H3K4me3 (86). WDR5 is also a component of the Ada-2-a-containing (ATAC) complex, one of the 2 complexes through which KAT2A exerts its HAT activity, suggesting an additional level of putative interaction between GClnc1 and the two histone modifiers in promoting gene expression. LncRNA-mediated trans-regulation of gene loci can equally lead to gene repression. Various lncRNAs including Xist, TUG1, and HOTAIR show the potential to interact with PRC2 and mediate target gene promoter inactivation in cancer (87–89). For instance, the p53 target lncRNA Taurine-Upregulated Gene 1 (TUG1) is dramatically downregulated in lung cancer. Mechanistically, it can silence the HOXB7 oncogene expression by recruiting EZH2 to the core promoter and repressing gene expression through deposition of H3K27me3 (89). In summary, lncRNAs are both mediators and targets of deregulated epigenetic cascades in human cancer, and can contribute to cancer initiation and progression at multiple levels.
Histone Modification in AML is Guided by LncRNAs
Knowledge about lncRNA function in AML onset, progression and response to treatment is in its infancy. LncRNAs have been shown to maintain or enhance LSC self-renewal, as exemplified by the dramatic reduction in the number of active LSC in an AML mouse model upon knockdown of lncRNA DANCR (90). They can promote leukemogenesis by maintaining active transcription, as described for LINC00152, which protects the elongating PolII subunit CDK9 from miR-193a induced silencing through a miRNA sponge-like effect (91). Globally, defined lncRNA expression patterns in AML bone marrow or peripheral blood samples exhibit strong correlation with clinical outcome and are suitable for AML subtype stratification (92–94). Important from a clinical perspective, individual lncRNA quantified in serum samples also have prognostic value and can be used as liquid biopsy-based biomarkers. For example, high serum levels of LINC00899 associate with reduced overall survival and can clearly discriminate between AML patients and healthy controls (95). Despite the clinical relevance of individual lncRNAs, to date no sequence-specific mutations in their genes (single nucleotide variants, SNV, insertions or deletions) have been described in AML. The same is true of another hematological malignancy, chronic lymphocytic leukemia (CLL), in which analysis of the non-coding genomic landscape of patients failed to identify recurrent mutations in lncRNA genes (96). This is in contrast with solid cancers, where various clinically and biologically relevant SNPs in lncRNA genes have been described (97). It is possible that lncRNA mutations are rare in leukemias and do not often contribute as leukemia drivers. Rather, lncRNAs may mediate malignant transformation events in hematopoietic cells through deregulated expression. Published examples are summarized in Table 1, and we will discuss them here in more detail. They illustrate similar regulatory mechanisms to those described in solid cancers, namely epigenetic deregulation of lncRNA expression and interaction with the chromatin-modifying machinery, as well as lncRNA-mediated cis and trans-regulation of individual loci.
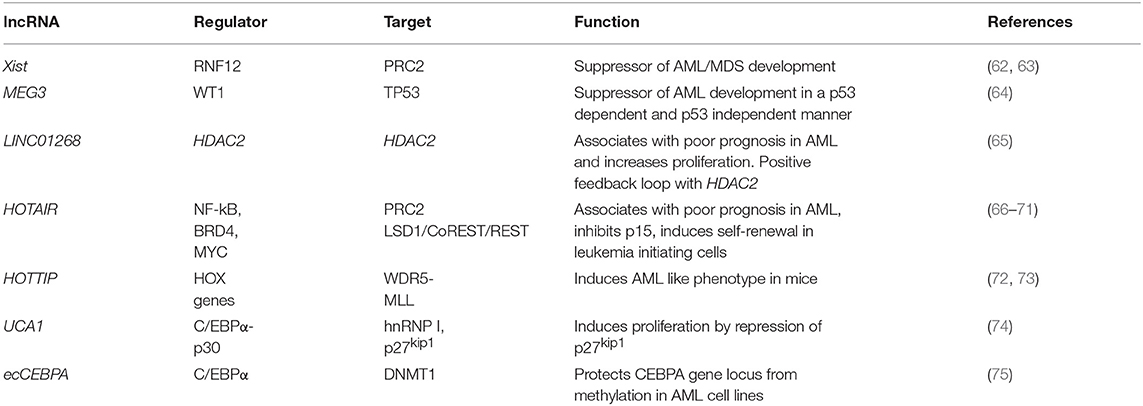
Table 1. Overview about lncRNA involved in epigenetic reprogramming in AML.
In the first category of epigenetic deregulation, it has been observed that AML-associated lncRNA genes have overall low DNA methylation and high H3K4me3 compared to healthy hematopoietic stem and progenitor cells (65). This results in higher lncRNA expression levels in AML and illustrates putative oncogene-type roles. Other lncRNAs can act as tumor suppressors, as shown for X-inactive specific transcript (Xist), the lncRNA central to X-chromosome inactivation (62): its depletion in HSCs and progenitors leads to the development of a myeloproliferative disorder through interaction with PRC2 (87). Also, epigenetic silencing or inactivating mutations of the Wilms' Tumor 1 (WT1) gene result in repression of Maternally Expressed Gene 3 (MEG3) lncRNA expression, a tumor suppressive lncRNA that inhibits tumor growth in a TP53 dependent and independent manner. Inactivation of MEG3 in turn promotes AML initiation (64). These examples illustrate the importance of epigenetic changes in the establishment of enhanced or aberrant lncRNA expression.
Locus regulation and downstream interaction with chromatin modifiers further illustrate the epigenetic role of lncRNAs in AML. For example, lncRNA LINC01268, located ~60 kb upstream of the HDAC2 gene, directly activates HDAC2 expression in a cis-regulatory manner. In turn HDAC2 triggers LINC01268 transcription to establish a positive feedback loop (65). One of the most prominent examples involves lncRNA HOTAIR, which associates with poor prognosis in AML (66). Mechanistically, HOTAIR binds simultaneously to PRC2 and to the LSD1/CoREST/REST complex and acts as a chromatin scaffold that couples PRC2-mediated repressive trimethylation of H3K27 with demethylation of H3K4 and the consequent inactivation of the specific loci (67). In AML, HOTAIR ensures the stem cell-like behavior of LSCs by mediating EZH2-driven H3K27me3 of the CDKN2B/P15-INK4b promoter. CDKN2B encodes for the P15 cyclin-dependent kinase inhibitor, and reducing its expression promotes proliferation and self-renewal (68). Maintenance of AML self-renewal is also achieved through induction of the lncRNA urothelial carcinoma associated 1 (UCA-1) in C/EBPα mutant AML. C/EBPα is a master transcription factor regulator of myeloid cell differentiation, which is recurrently mutated in AML. The mutant truncated p30-isoform of C/EBPα promotes UCA-1 expression, which in turns promotes leukemia proliferation through recruitment of the non-coding RNA-protein complex hnRNP I to repress the cyclin-dependent kinase inhibitor P27Kip1(74). HOXA distal transcript antisense RNA (HOTTIP) is a lncRNA located within the HOXA gene cluster (72). HOXA7-11 genes are central to AML self-renewal programs, namely in the context of MLL rearrangements and NPM1 mutation. Within the locus, HOTTIP activates expression of HOX genes in a candidate positive feedback loop: it guides the WDR5-MLL methyl-transferase complex to the respective promoters, resulting in deposition of the activating H3K4me3 modification (72). Accordingly, in a hematopoiesis-specific transgenic mouse model, increased HOTTIP expression leads to increased WBC and neutrophil counts, to a splenomegaly, and an altered self-renewal—differentiation balance of hematopoietic stem and progenitor cells (73). In summary, lncRNAs may contribute to AML initiation and maintenance through close regulatory interaction with the epigenetic machinery. Figure 1 illustrates these regulatory modes, which will continue to expand in coming years.
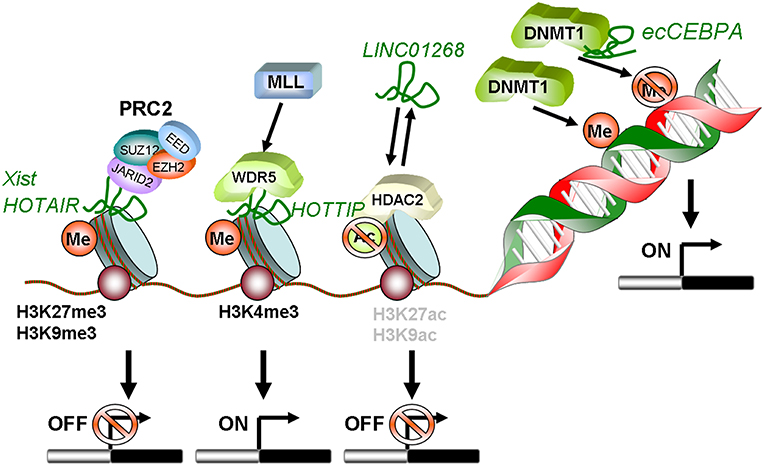
Figure 1. Schematic summary of lncRNAs involved in epigenetic regulation in AML. HOTAIR and Xist can recruit PRC2 to methylate histones on target gene promoters leading to transcriptional silencing. LINC01268 activates HDAC2, which in turn deacetylates H3 on K9 and K27. This also induces transcriptional silencing. In contrast, lncRNA HOTTIP guides WDR5 to H3K4 and recruits the MLL complex. This mediates tri-methylation and activation of transcription. The diRNA familiy member ecCEBPA directly interacts with DNMT1 and functions as a shield to protect the CEBPA promoter from DNA methylation to ensure constant expression.
Hypothesis, Conclusions, and Perspectives
The field of lncRNAs in cancer, including AML, is in its infancy. However, a growing body of data provides strong evidence for the role of lncRNAs as key mediators and effectors of epigenetic alterations in AML, which will add to more advanced research in the epigenetic field. Indeed, several epigenetic-modifying drugs have either been approved for AML treatment, or are in clinical trials with initial promising results (98). This notwithstanding, first-line therapy remains a combination of cytotoxic agents cytarabine and an anthracycline, which have an associated toxicity often not tolerated by the elderly patients more commonly affected by the disease. Newly-introduced chemicals offer therapeutic alternatives to frailer patients and have improved rates of remission, but have not yet improved survival. Detailed understanding of epigenetic and other regulatory mechanisms, and the design of individualized therapeutic regimens, are thus paramount in AML. LncRNAs emerge as promising new players in this task. This is illustrated in the case of DNA demethylating agents 5-azacytidine and decitabine, which have shown promising results in AML and related myelodysplastic syndromes (MDS) (99), but for which the molecular mechanisms are not fully uncovered and a significant proportion of patients fail to respond or acquire resistance (100). Although both agents show a non-random reproducible demethylation pattern in cell lines (101) they do not exclusively affect tumor-specific programs and might produce undesired side effects. Recently, diRuscio et al. unveiled a mechanism by which cells can use lncRNAs endogenously for target-specific demethylation (75), with direct potential therapeutic implications. In the example studied in AML cell lines, the ecCEBPA lncRNA directly binds to the DNMT1 protein and protects the CEBPA gene promoter from DNMT1-mediated de novo methylation after cell division. This mechanism tightly controls the transcriptional activity of the CEBPA host gene with promoter specificity; furthermore, it suggests a template for future targeted cancer therapies using lncRNAs as scaffolds to control demethylation of individual hypermethylated targets. In theory, this strategy can increase efficiency and decrease toxicity of untargeted DNA demethylating agents. In addition to demethylating therapies, HDAC inhibitors have also been developed as potential epigenetic therapeutic agents (102). However, their success remains limited, with challenges arising from poor drug selectivity, toxic side effects and lack of informative biomarkers to predict and guide selection of responding patients (103). LncRNAs have been proposed to regulate the balance between histone acetylation and deacetylation activities (104), and, similarly to specification of demethylation activities, they may potentially be used as molecular scaffolds to trigger target promoter/enhancer-specific chromatin acetylation, with fine and directed control of AML-relevant locus transcription.
In conclusion, preliminary data indicate strong potential for lncRNAs as biomarkers, prognostic factors, and therapeutic targets or mediators in AML, with the ability to individualize therapies, reduce toxicity and enhance treatment success, all of them urgent roles in this disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CP was a Leuka John Goldman Fellow for Future Science (JG2017) and the recipient of a Wellcome Trust (WT)/Institutional Strategic Support Fund (ISSF) award at the University of Cambridge.
References
1. Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. (2003) 3:89–101. doi: 10.1038/nrc989
2. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. (2019) 24:25–40. doi: 10.1016/j.stem.2018.11.017
3. Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. (2017) 129:1577–85. doi: 10.1182/blood-2016-10-696054
4. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
5. Grove CS, Vassiliou GS. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer? Dis Model Mech. (2014) 7:941–51. doi: 10.1242/dmm.015974
6. Martignoles JA, Delhommeau F, Hirsch P. Genetic hierarchy of acute myeloid leukemia: from clonal hematopoiesis to molecular residual disease. Int J Mol Sci. (2018) 19:E3850. doi: 10.3390/ijms19123850
7. Yi S, Lin S, Li Y, Zhao W, Mills GB, Sahni N. Functional variomics and network perturbation: connecting genotype to phenotype in cancer. Nat Rev Genet. (2017) 18:395–410. doi: 10.1038/nrg.2017.8
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
9. Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. (2016) 22:792–9. doi: 10.1038/nm.4125
10. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. (2009) 23:781–3. doi: 10.1101/gad.1787609
11. Fong CY, Morison J, Dawson MA. Epigenetics in the hematologic malignancies. Haematologica. (2014) 99:1772–83. doi: 10.3324/haematol.2013.092007
12. Gallipoli P, Giotopoulos G, Huntly BJ. Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Ther Adv Hematol. (2015) 6:103–19. doi: 10.1177/2040620715577614
13. Bach C, Slany RK. Molecular pathology of mixed-lineage leukemia. Future Oncol. (2009) 5:1271–81. doi: 10.2217/fon.09.96
14. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. (2010) 363:2424–33. doi: 10.1056/NEJMoa1005143
15. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. (2011) 43:309–15. doi: 10.1038/ng.788
16. Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. (2011) 29:1373–81. doi: 10.1200/JCO.2010.32.7742
17. Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. (2011) 118:3803–10. doi: 10.1182/blood-2011-02-339747
18. Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. (2010) 28:2348–55. doi: 10.1200/JCO.2009.27.3730
19. Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol. (2012) 5:5. doi: 10.1186/1756-8722-5-5
20. Gu T, Lin X, Cullen SM, Luo M, Jeong M, Estecio M, et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. (2018) 19:88. doi: 10.1186/s13059-018-1464-7
21. Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. (2014) 46:17–23. doi: 10.1038/ng.2836
22. Sandoval JE, Huang YH, Muise A, Goodell MA, Reich NO. Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners. J Biol Chem. (2019) 294:4898–910. doi: 10.1074/jbc.RA118.006795
23. Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. (2015) 29:910–22. doi: 10.1101/gad.260174.115
24. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
25. Fisher JB, McNulty M, Burke MJ, Crispino JD, Rao S. Cohesin mutations in myeloid malignancies. Trends Cancer. (2017) 3:282–93. doi: 10.1016/j.trecan.2017.02.006
26. Dovey OM, Cooper JL, Mupo A, Grove CS, Lynn C, Conte N, et al. Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood. (2017) 130:1911–22. doi: 10.1182/blood-2017-01-760595
27. Wang X, Chen CW, Armstrong SA. The role of DOT1L in the maintenance of leukemia gene expression. Curr Opin Genet Dev. (2016) 36:68–72. doi: 10.1016/j.gde.2016.03.015
28. Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood. (2014) 123:1341–52. doi: 10.1182/blood-2013-03-488114
29. Durst KL, Lutterbach B, Kummalue T, Friedman AD, Hiebert SW. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol. (2003) 23:607–19. doi: 10.1128/MCB.23.2.607-619.2003
30. Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, et al. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. (2001) 21:6470–83. doi: 10.1128/MCB.21.19.6470-6483.2001
31. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. (2011) 478:529–33. doi: 10.1038/nature10509
32. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. (2014) 28:311–20. doi: 10.1038/leu.2013.338
33. Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. (2014) 13:2315–27. doi: 10.1158/1535-7163.MCT-14-0258
34. Gilliland DG, Griffin JD. Role of FLT3 in leukemia. Curr Opin Hematol. (2002) 9:274–81. doi: 10.1097/00062752-200207000-00003
35. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. (2005) 437:436–9. doi: 10.1038/nature04020
36. Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep. (2014) 9:1618–27. doi: 10.1016/j.celrep.2014.11.008
37. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. (2012) 21:473–87. doi: 10.1016/j.ccr.2012.03.014
38. Magliulo D, Bernardi R, Messina S. Lysine-specific demethylase 1A as a promising target in acute myeloid Leukemia. Front Oncol. (2018) 8:255. doi: 10.3389/fonc.2018.00255
39. Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. (2011) 30:249–62. doi: 10.1038/emboj.2010.318
40. Wang Y, Yun C, Gao B, Xu Y, Zhang Y, Wang Y, et al. The lysine acetyltransferase GCN5 Is required for iNKT cell development through EGR2 acetylation. Cell Rep. (2017) 20:600–12. doi: 10.1016/j.celrep.2017.06.065
41. Bararia D, Kwok HS, Welner RS, Numata A, Sarosi MB, Yang H, et al. Acetylation of C/EBPalpha inhibits its granulopoietic function. Nat Commun. (2016) 7:10968. doi: 10.1038/ncomms10968
42. Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. (2016) 17:1193–205. doi: 10.1016/j.celrep.2016.09.079
43. Moris N, Edri S, Seyres D, Kulkarni R, Domingues AF, Balayo T, et al. Histone Acetyltransferase KAT2A stabilizes pluripotency with control of transcriptional heterogeneity. Stem Cells. (2018) 36:1828–38. doi: 10.1002/stem.2919
44. Wang L, Koutelou E, Hirsch C, McCarthy R, Schibler A, Lin K, et al. Dent: GCN5 regulates FGF signaling and activates selective MYC target genes during early embryoid body differentiation. Stem Cell Rep. (2018) 10:287–99. doi: 10.1016/j.stemcr.2017.11.009
45. Hirsch CL, Coban Akdemir Z, Wang L, Jayakumaran G, Trcka D, Weiss A, et al. Myc and SAGA rewire an alternative splicing network during early somatic cell reprogramming. Genes Dev. (2015) 29:803–16. doi: 10.1101/gad.255109.114
46. Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, Sasca D, et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med. (2019) 216:966–81. doi: 10.1084/jem.20181276
47. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. (2010) 42:722–6. doi: 10.1038/ng.621
48. Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. (2010) 24:1799–804. doi: 10.1038/leu.2010.167
49. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. (2010) 42:665–7. doi: 10.1038/ng.620
50. Shimizu T, Kubovcakova L, Nienhold R, Zmajkovic J, Meyer SC, Hao-Shen H, et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med. (2016) 213:1479–96. doi: 10.1084/jem.20151136
51. Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. (2011) 118:5227–34. doi: 10.1182/blood-2011-06-363424
52. Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. (2012) 120:1107–17. doi: 10.1182/blood-2011-11-394932
53. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci USA. (2012) 109:5028–33. doi: 10.1073/pnas.1202258109
54. Scott MT, Korfi K, Saffrey P, Hopcroft LE, Kinstrie R, Pellicano F, et al. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. (2016) 6:1248–57. doi: 10.1158/2159-8290.CD-16-0263
55. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. (2012) 489:101–8. doi: 10.1038/nature11233
56. Li CH, Chen Y. Targeting long non-coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol. (2013) 45:1895–910. doi: 10.1016/j.biocel.2013.05.030
57. Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol. (2017) 1008:1–46. doi: 10.1007/978-981-10-5203-3_1
58. Diederichs S. The four dimensions of noncoding RNA conservation. Trends Genet. (2014) 30:121–3. doi: 10.1016/j.tig.2014.01.004
59. Khorkova O, Hsiao J, Wahlestedt C. Basic biology and therapeutic implications of lncRNA. Adv Drug Deliv Rev. (2015) 87:15–24. doi: 10.1016/j.addr.2015.05.012
60. Davidovich C, Cech TR. The recruitment of chromatin modifiers by long noncoding RNAs, lessons from PRC2. RNA. (2015) 21:2007–22. doi: 10.1261/rna.053918.115
61. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. (2009) 106:11667–72. doi: 10.1073/pnas.0904715106
62. Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. (2013) 152:727–42. doi: 10.1016/j.cell.2013.01.034
63. Jonkers I, Barakat TS, Achame EM, Monkhorst K, Kenter A, Rentmeester E, et al. RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell. (2009) 139:999–1011. doi: 10.1016/j.cell.2009.10.034
64. Lyu Y, Lou J, Yang Y, Feng J, Hao Y, Huang S, et al. Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and -independent pathways. Leukemia. (2017) 31:2543–51. doi: 10.1038/leu.2017.116
65. Lei L, Xia S, Liu D, Li X, Feng J, Zhu Y, et al. Genome-wide characterization of lncRNAs in acute myeloid leukemia. Brief Bioinform. (2018) 19:627–35. doi: 10.1093/bib/bbx007
66. Wu S, Zheng C, Chen S, Cai X, Shi Y, Lin B, et al. Overexpression of long non-coding RNA HOTAIR predicts a poor prognosis in patients with acute myeloid leukemia. Oncol Lett. (2016) 10:2410–4. doi: 10.3892/ol.2015.3552
67. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. (2010) 329:689–93. doi: 10.1126/science.1192002
68. Gao S, Zhou B, Li H, Huang X, Wu Y, Xing C, et al. Long noncoding RNA HOTAIR promotes the self-renewal of leukemia stem cells through epigenetic silencing of p15. Exp Hematol. (2018) 67:32–40 e3. doi: 10.1016/j.exphem.2018.08.005
69. Ma MZ, Li CX, Zhang Y, Weng MZ, Zhang MD, Qin YY, et al. Long non-coding RNA HOTAIR, a c-Myc activated driver of malignancy, negatively regulates miRNA-130a in gallbladder cancer. Mol Cancer. (2014) 13:156. doi: 10.1186/1476-4598-13-156
70. Pastori C, Kapranov P, Penas C, Peschansky V, Volmar CH, Sarkaria JN, et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc Natl Acad Sci USA. (2015) 112:8326–31. doi: 10.1073/pnas.1424220112
71. Ozes AR, Miller DF, Ozes ON, Fang F, Liu Y, Matei D, et al. NF-kappaB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene. (2016) 35:5350–61. doi: 10.1038/onc.2016.75
72. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. (2011) 472:120–4. doi: 10.1038/nature09819
73. Luo L, Zhu G, Zha J, Yan B, Guo Y, Xu J, et al. Activation of hottip lncrna perturbs HSC function leading to AML like disease in mice. Blood. (2018) 132(Suppl. 1):3877. doi: 10.1182/blood-2018-99-110218
74. Hughes JM, Legnini I, Salvatori B, Masciarelli S, Marchioni M, Fazi F, et al. C/EBPalpha-p30 protein induces expression of the oncogenic long non-coding RNA UCA1 in acute myeloid leukemia. Oncotarget. (2015) 6:18534–44. doi: 10.18632/oncotarget.4069
75. Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. (2013) 503:371–6. doi: 10.1038/nature12598
76. Krakowsky RHE, Wurm AA, Gerloff D, Katzerke C, Brauer-Hartmann D, Hartmann JU, et al. miR-451a abrogates treatment resistance in FLT3-ITD-positive acute myeloid leukemia. Blood Cancer J. (2018) 8:36. doi: 10.1038/s41408-018-0070-y
77. Wurm AA, Zjablovskaja P, Kardosova M, Gerloff D, Brauer-Hartmann D, Katzerke C, et al. Disruption of the C/EBPalpha-miR-182 balance impairs granulocytic differentiation. Nat Commun. (2017) 8:46. doi: 10.1038/s41467-017-00032-6
78. Wang Z, Yang B, Zhang M, Guo W, Wu Z, Wang Y, et al. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that Interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell. (2018) 33:706–20 e9. doi: 10.1016/j.ccell.2018.03.006
79. Yang F, Huo XS, Yuan SX, Zhang L, Zhou WP, Wang F, et al. Repression of the long noncoding RNA-LET by histone deacetylase 3 contributes to hypoxia-mediated metastasis. Mol Cell. (2013) 49:1083–96. doi: 10.1016/j.molcel.2013.01.010
80. Guo X, Zhang Y, Mayakonda A, Madan V, Ding LW, Lin LH, et al. ARID1A and CEBPalpha cooperatively inhibit UCA1 transcription in breast cancer. Oncogene. (2018) 37:5939–51. doi: 10.1038/s41388-018-0371-4
81. Hu JJ, Song W, Zhang SD, Shen XH, Qiu XM, Wu HZ, et al. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci Rep. (2016) 6:23521. doi: 10.1038/srep23521
82. Balbin OA, Malik R, Dhanasekaran SM, Prensner JR, Cao X, Wu YM, et al. The landscape of antisense gene expression in human cancers. Genome Res. (2015) 25:1068–79. doi: 10.1101/gr.180596.114
83. Xi Q, Gao N, Zhang X, Zhang B, Ye W, Wu J, et al. A natural antisense transcript regulates acetylcholinesterase gene expression via epigenetic modification in Hepatocellular Carcinoma. Int J Biochem Cell Biol. (2014) 55:242–51. doi: 10.1016/j.biocel.2014.09.012
84. Bester AC, Lee JD, Chavez A, Lee YR, Nachmani D, Vora S, et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell. (2018) 173:649–64 e20. doi: 10.1016/j.cell.2018.03.052
85. Sun TT, He J, Liang Q, Ren LL, Yan TT, Yu TC, Tang JY, Bao YJ, Hu Y, Lin Y, Sun D, Chen YX, Hong J, Chen H, Zou W, Fang JY. LncRNA GClnc1 Promotes Gastric Carcinogenesis and May Act as a Modular Scaffold of WDR5 and KAT2A Complexes to Specify the Histone Modification Pattern. Cancer Discov. (2016) 6:784–801. doi: 10.1158/2159-8290.CD-15-0921
86. Yang YW, Flynn RA, Chen Y, Qu K, Wan B, Wang KC, et al. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. Elife. (2014) 3:e02046. doi: 10.7554/eLife.02046
87. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. (2008) 322:750–6. doi: 10.1126/science.1163045
88. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. (2007) 129:1311–23. doi: 10.1016/j.cell.2007.05.022
89. Zhang EB, Yin DD, Sun M, Kong R, Liu XH, You LH, et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. (2014) 5:e1243. doi: 10.1038/cddis.2014.201
90. Bill M, Papaioannou D, Karunasiri M, Kohlschmidt J, Pepe F, Walker CJ, et al. Expression and functional relevance of long non-coding RNAs in acute myeloid leukemia stem cells. Leukemia. (2019) doi: 10.1038/s41375-019-0429-5. [Epub ahead of print].
91. Zhang X, Tao W. Long noncoding RNA LINC00152 facilitates the leukemogenesis of acute myeloid leukemia by promoting CDK9 through miR-193a. DNA Cell Biol. (2019) 38:236–42. doi: 10.1089/dna.2018.4482
92. Mer AS, Lindberg J, Nilsson C, Klevebring D, Wang M, Gronberg H, et al. Expression levels of long non-coding RNAs are prognostic for AML outcome. J Hematol Oncol. (2018) 11:52. doi: 10.1186/s13045-018-0596-2
93. Garzon R, Volinia S, Papaioannou D, Nicolet D, Kohlschmidt J, Yan PS, et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc Natl Acad Sci USA. (2014) 111:18679–84. doi: 10.1073/pnas.1422050112
94. Schwarzer A, Emmrich S, Schmidt F, Beck D, Ng M, Reimer C, et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat Commun. (2017) 8:218. doi: 10.1038/s41467-017-00212-4
95. Wang Y, Li Y, Song HQ, Sun GW. Long non-coding RNA LINC00899 as a novel serum biomarker for diagnosis and prognosis prediction of acute myeloid leukemia. Eur Rev Med Pharmacol Sci. (2018) 22:7364–70. doi: 10.26355/eurrev_201811_16274
96. Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. (2015) 526:519–24. doi: 10.1038/nature14666
97. Minotti L, Agnoletto C, Baldassari F, Corra F, Volinia S. SNPs and somatic mutation on long non-coding RNA, new frontier in the cancer studies? High Throughput. (2018) 7:34. doi: 10.3390/ht7040034
98. Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. (2018) 58:187–207. doi: 10.1146/annurev-pharmtox-010716-105106
99. Bohl SR, Bullinger L, Rucker FG. Epigenetic therapy: azacytidine and decitabine in acute myeloid leukemia. Expert Rev Hematol. (2018) 11:361–71. doi: 10.1080/17474086.2018.1453802
100. Unnikrishnan A, Papaemmanuil E, Beck D, Deshpande NP, Verma A, Kumari A, et al. Integrative genomics identifies the molecular basis of resistance to azacitidine therapy in myelodysplastic syndromes. Cell Rep. (2017) 20:572–85. doi: 10.1016/j.celrep.2017.06.067
101. Hagemann S, Heil O, Lyko F, Brueckner B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS ONE. (2011) 6:e17388. doi: 10.1371/journal.pone.0017388
102. Min C, Moore N, Shearstone JR, Quayle SN, Huang P, van Duzer JH, et al. Selective inhibitors of histone deacetylases 1 and 2 synergize with azacitidine in acute myeloid leukemia. PLoS ONE. (2017) 12:e0169128. doi: 10.1371/journal.pone.0169128
103. McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res. (2018) 138:183–211. doi: 10.1016/bs.acr.2018.02.006
Keywords: acute myeloid leukemia, long non-coding RNA, chromatin regulation, epigenetic therapies, personalized medicine
Citation: Wurm AA and Pina C (2019) Long Non-coding RNAs as Functional and Structural Chromatin Modulators in Acute Myeloid Leukemia. Front. Oncol. 9:899. doi: 10.3389/fonc.2019.00899
Received: 17 June 2019; Accepted: 29 August 2019;
Published: 11 September 2019.
Edited by:
Sheng F. Cai, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Aniruddha Deshpande, Sanford Burnham Prebys Medical Discovery Institute, United StatesStanley Lee, Memorial Sloan Kettering Cancer Center, United States
Copyright © 2019 Wurm and Pina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Pina, Y3A1MzNAY2FtLmFjLnVr
†These authors have contributed equally to this work