 Weina Yu
Weina Yu Li Yang
Li Yang Ting Li
Ting Li Yi Zhang
Yi Zhang- 1Biotherapy Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Cancer Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 3Henan Key Laboratory for Tumor Immunology and Biotherapy, Zhengzhou, China
- 4School of Life Sciences, Zhengzhou University, Zhengzhou, China
Cadherin family includes lists of transmembrane glycoproteins which mediate calcium-dependent cell-cell adhesion. Cadherin-mediated adhesion regulates cell growth and differentiation throughout life. Through the establishment of the cadherin-catenin complex, cadherins provide normal cell-cell adhesion and maintain homeostatic tissue architecture. In the process of cell recognition and adhesion, cadherins act as vital participators. As results, the disruption of cadherin signaling has significant implications on tumor formation and progression. Altered cadherin expression plays a vital role in tumorigenesis, tumor progression, angiogenesis, and tumor immune response. Based on ongoing research into the role of cadherin signaling in malignant tumors, cadherins are now being considered as potential targets for cancer therapies. This review will demonstrate the mechanisms of cadherin involvement in tumor progression, and consider the clinical significance of cadherins as therapeutic targets.
Introduction
Cadherin was first described by Hyafil and Peyrieras in the 1980s as “Uvomorulin” in mouse and L-CAM in chicken (1, 2) and the mouse gene was later cloned (3). Subsequently, several other cadherins were found and analyzed, including N-cadherin (CDH1) (4), which is mainly expressed in neural tissue, and P-cadherin (CDH3), which was first characterized from placental tissue (5). Many subtypes have since been identified in other species and there are now more than 100 known members in this superfamily (6).
Functionally, cadherins act as modulators during organism growth. First, tissue cohesion in organisms depends largely on cadherins. Radice et al. (7) found that mice lacking N-cadherin (CDH2) developed heart defects and died in utero. Moreover, cadherins affect cell polarization, by differentiating between cell populations during development (8); this function might has a relationship with intrinsic characteristics of the ectodomains of cadherins (9).
The research to date has focused on the relationship between cadherins and malignant tumors, and drugs targeting cadherins have been developed and tested in clinical trials. In this review, we intend to summarize the mechanisms whereby cadherins mediate tumor formation and progression, and to discuss their potential use in clinical treatment of cancer patients.
Types of Cadherin
Cadherins can be classified into several subtypes: type I classical cadherins such as E-cadherin, N-cadherin, and P-cadherin; type II classical cadherins such as VE-cadherin (CDH5) and OB-cadherin (CDH11) (9, 10); the desmosomal cadherins (11, 12); the seven-pass transmembrane cadherins (13, 14); FAT and dachsous (DCHS) group cadherins (15–17); and protocadherins (PCDHs) (18–21) (Table 1). Nearly all cadherins are transmembrane proteins with three components: (1) an extracellular cadherin domain (EC) responsible for homotypic cadherin-cadherin interaction; (2) a single-pass transmembrane domain (absent in seven-pass transmembrane cadherins); and (3) a cytoplasmic domain acts as a connector between cell surface and cytokeleton (21, 29) (Figure 1).
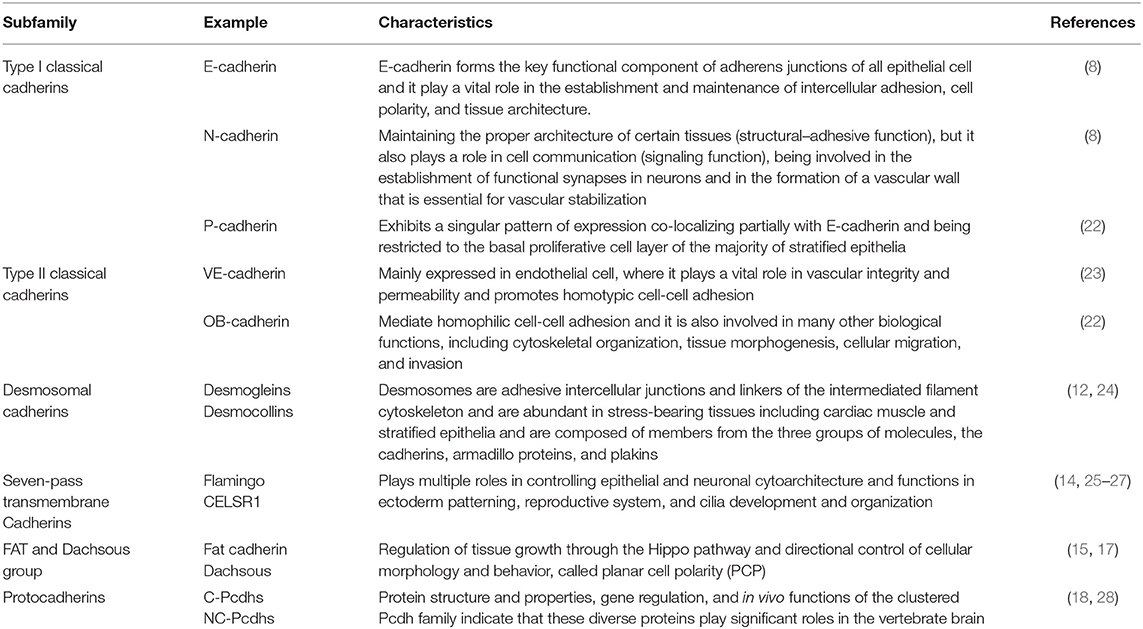
Table 1. The cadherin superfamily.
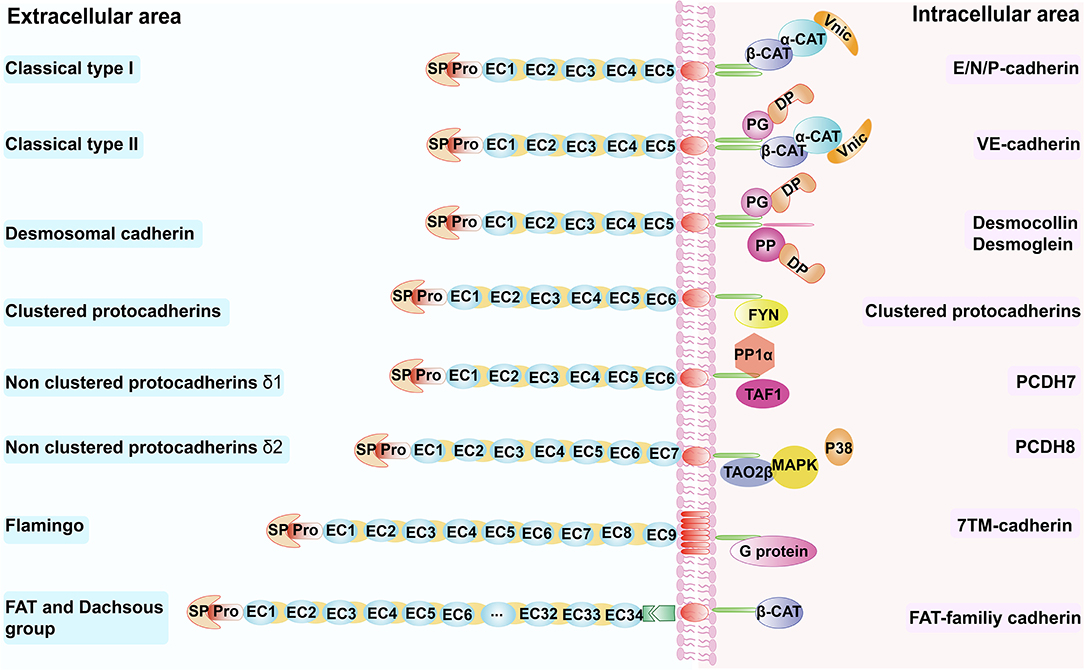
Figure 1. Schematic overview of the cadherin superfamily. Schematic overview of the cadherin superfamily depicting representative molecules for the respective subfamilies. β-CAT, β-catenin, α-CAT, α-catenin; Vinc, vinculin; DP, desmoplakin; PG, plakoglobin; PP, plakophilins; TAF1, template-activating factor 1; TAO2β, thousand and one amino acid protein kinase 2β; EC, extracellular cadherin repeats; SP, signal peptide; Pro, pro-peptide; FYN, Src-family kinase; PP1α, protein phosphatase-1α; MAPK, mitogen-activated protein kinase.
Classical cadherins like E-cadherin, P-cadherin and N-cadherin, which translate the intercellular contact signals involved in cellular organization, are mediated mainly by the catenins, namely α-catenin, β-catenin, and γ-catenin, which are membrane undercoat proteins that link the tail of cadherins in the cytoplasm (30, 31) (Figure 1). VE-cadherin binds to other proteins such as plakoglobin, which binds and recruits a desmosomal plaque protein named desmoplakin to the cell surface (22); vinculin also participates in this process. In tissues that undergo greater mechanical strain, such as the epidermis and myocardium, desmosomal cadherins function through plakoglobin, desmoplakin, and the armadillo family member plakophilins, to mediate the linkage between the cytoskeleton and these proteins (21). Protocadherin can be divided into two subfamilies—clustered and non-clustered protocadherins (Table 1). Clustered protocadherins have a unique binding site for non-receptor tyrosine kinase Fyn, one of the Src kinases families in the cytoplasm. Non-clustered protocadherins include three types: δ1 (e.g., PCDH7), δ2 (e.g., PCDH8), and ε (e.g., PCDH15). PCDH7 has three motifs, all of which can interact with the histone-regulating protein template-activating factor 1 and protein phosphatase-1α, and is inactivated when it binds to the CM3 motif. PCDH8 can interact with thousand and one amino acid protein kinase 2β through its intracellular domain, thereby activating the p38 MAPK pathway, which may enhance endocytosis of N-cadherin (32) (Figure 1). The flamingo cadherin differs from the other cadherins—it has a transmembrane domain that spans the plasma membrane seven times, somewhat like that of G proteins, and participates in many cellular signaling pathways (33) (Figure 1). Fat and dachsous cadherins are very large cell-adhesion molecules (Table 1); a recent study showed the involvement of Fat cadherin in the Wnt-signaling pathway with the help of β-catenin (34) (Figure 1).
Cadherins and Tumorigenesis
In the developmental process of multicellular organisms, the maintaining of cellular and tissue morphogenesis mainly depends on cell-cell adhesion (35), which initiated and maintained by adheren junction. The signaling mediated by adhesion between cell-cell and cell-extracellular matrix is associated with gene regulation in normal tissue homeostasis (36, 37). Disturbance of cell-cell and cell-extracellular matrix adhesion and the subsequent changes in adhesion-mediated signaling pathways would induce malignant phenotypes in normal cells (38).
As a major contributor to cell–cell adhesion in epithelial tissues, abnormal expression of cadherins is closely related to tumorigenesis. β- and γ-catenin (39, 40) are bridges that mediate linkage of classical cadherins to the actin cytoskeleton through their cytoplasmic domains (41). It has been shown that cell–cell adhesion is markedly reduced when a well-differentiated benign adenoma with apico-basal polarization, a feature of normal epithelial cells, becomes an invasive carcinoma by losing its normal membrane polarization (42, 43). Many signaling pathways are involved in this process (44). Epithelial–mesenchymal transition (EMT), a phenotype transition, is a driving force in tumorigenesis (43, 45, 46).
E-cadherin and N-cadherin are founding members of the cadherin superfamily and act as crucial regulators in the process of tumor development. E-cadherin is essential in maintaining epithelial tissue integrity and providing strength to maintain polarization of the epithelial cell layers (6). N-cadherin is highly expressed in mesenchymal cells and neural tissue (7). N-cadherin promotes increased cell motility and migration, by interacting with epidermal growth factor receptor (EGFR) 1 via the 88-amino acid region of the EC4 domain (47). During malignancy, cell–cell adhesion mediated by E-cadherin is lost (48), and this process can be reversed when E-cadherin is re-established (48, 49). These studies propose the novel view that EMT can participate in the development of malignant tumors.
Many factors can lead to E-cadherin loss. Some of cancer associated germline mutations had been reported to interfere the expression and function of cadherin (50–52). Additionally, in many epithelial malignancies like esophagus, melanoma, and hepatocellular carcinoma, the expression of E-cadherin is decreased by the hypermethylation of promoter (53). Besides, the frequent loss of p120 expression would cause the degradation of E-cadherin in lung cancer (54, 55). As a result, the expression of β-catenin would upregulate along with the reduction of E-cadherin and downstream signaling pathways associated with tumorigenesis in nuclear would be activated by the elevated β-catenin. However, E-cadherin loss by itself is insufficient to stimulate the following signaling pathway due to the decrease mechanism of β-catenin which mediated by Wnt signaling pathway (Figure 2A). In this degradation process, glycogen synthase kinase (GSK) phosphorylates β-catenin and induces its ubiquitination and degradation in the 26 S proteasome to keep the low level of β-catenin in cytoplasm, as well as cell nucleus (56). After Wnt proteins bind to Frizzled, the signaling pathway is activated (Figure 2A). The disheveled protein then binds, resulting in failure of degradation of the complex and block the degradation effects casein kinase 1 α and GSK3 on β-catenin. Thus, β-catenin accumulates in the cytoplasm and is translocated to the nucleus, where it competitively inhibits a suppresser of T-cell factor/lymphoid enhancer factor (TCF/LEF) and could active Wnt-responsive genes. Additionally, some co-activators in transcription, such as p300, CREB-binding protein and B-cell lymphoma 9, could be stimulated at the same time. N-cadherin has a physical association with and could stimulate the MAPK/ERK signaling pathway, which participates in tumorigenesis (Figure 2A).
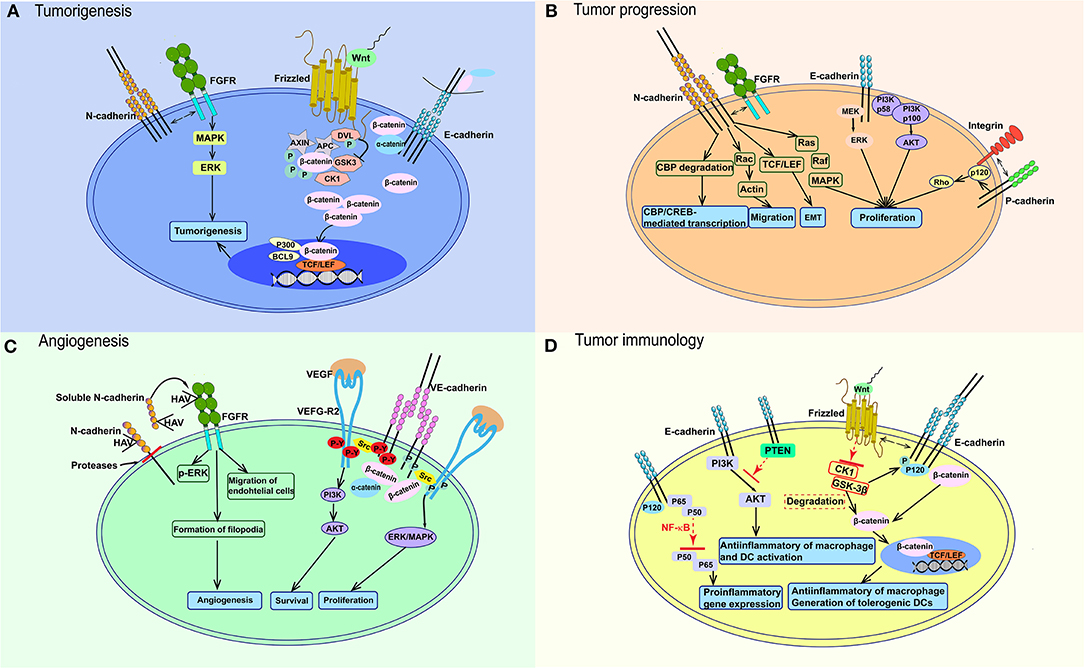
Figure 2. Signaling pathways participating in the function of cadherins in cancer. (A) E-cadherin loss lead to the upregulation of β-catenin in cytoplasm. After Wnt binds to Frizzled, it blocks the effects of CK1α and GSK3 on β-catenin and cause the accumulation of β-catenin in cytoplasm and nucleus, which activates TCF/LEF and co-activators. N-cadherin could affect tumorigenesis through interacting with FGFR and stimulating MAPK/ERK pathway. (B) Wnt-β-catenin pathway also works in tumor progression like in (A). E-cadherin could also activate PI3K-AKT and MEK-ERK pathways. N-cadherin activates Ras-MAPK pathway, TCF/LEF transcription factor, etc. P-cadherin could increase tumor migration through interacting with integrin. (C) In endothelial cells, VEGF-VEGFR2 activates VE-cadherin and Src, then leads to VE-cadherin phosphorylation on tyrosines, which would promote endothelial cell proliferation through PI3K/AKT signaling. ERK/MAPK signaling pathway would be stimulated without tyrosines phosphorylation. sN-cadherin induces by proteases could bind to FGFR and phosphorylates ERK to stimulate angiogenesis. (D) The Wnt-β-catenin signaling pathway also works in immune cells, which stimulates anti-inflammatory macrophages and tolerogenic DCs. E-cadherin mediates anti-inflammatory activation of macrophages and DCs through PI3K/AKT and NF-κB. FGFR, fibroblast growth factor receptor; DVL, disheveled protein; P, phosphorylation; APC, adenomatosis polyposis coli; MAPK, mitogen-activated protein-kinase; ERK, extracellular signal-regulated kinase; GSK3, glycogen synthase kinase; BCL9, B-cell lymphoma 9; TCF/LEF, T-cell factor/lymphoid enhancer factor; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; CBP, CREB-binding protein; HAV, His–Ala–Val; VEGF, vascular endothelial growth factor; VEGF-R2, vascular endothelial growth factor receptor 2; P-Y, phosphorylation on tyrosin; PTEN, gene of phosphate and tension homology deleted on chromsome ten; NF-κB, nuclear factor κB; CK1α, casein kinase 1 α; TGF-β, Transforming growth factor beta.
One possible explanation for the role of EMT in this transition is that ectopically expressed N-cadherin may promote the migration of epithelial cells with a similar cadherin expression profile, and thereafter act in an entirely different way (57). On the other hand, altered cadherin expression may affect intracellular signaling, causing depolarization of cells and enhanced migratory properties (58). Many factors participate in this process. For example, The E-cadherin/β-catenin complex functions as an intercellular junction and is regulated by phosphorylation. Phosphorylation by Src or epithelial growth factor receptor (59) destroys the binding between β-catenin and cadherin, and subsequently enhances the expression of cytoplasmic β-catenin, which activates Wnt/β-catenin signaling. It has been reported that abnormal activation of this pathway may induce tumor formation, by binding with TCF/LEF-type transcription factors and stimulating cancer-related genes such as c-MYC and CCND1, which may affect the cell cycle and promote tumorigenesis (Figure 2) (60). N-cadherin has also been reported to be physically associated with various members of the fibroblast growth factor receptor (FGFR) family, and to participate in increasing the cell-surface receptor level and sustaining MAPK/ERK signaling, thus increasing cellular motility and invasion (61, 62).
Other types of cadherins have similar effects in malignant tumor formation. For example, the role of P-cadherin in human cancer has been debated for years (63, 64). Moreover, loss of P-cadherin has a significant correlation with histological tumor staging (65). In a P-cadherin-null mouse model, mammary glands developed hyperplasia, dysplasia, and abnormal lymphoid infiltration (66), demonstrating that P-cadherin expression is important in maintaining normal tissue. Similarly, Hardy et al. (67) found that in colonic lesions, regulation of P-cadherin differs from that of E-cadherin during colonocyte transformation; P-cadherin was expressed in low-malignancy potential lesions, indicating that P-cadherin is unlikely to participate directly in tumorigenesis. Similarly, H-cadherin participate in the carcinogenesis of colorectal cancer (68).
Therefore, cadherins function not only as the molecules of cell-cell adhesion but also as factors which induce tumorigenesis predominantly through Wnt/β-catenin or other signaling pathways.
Cadherins and Tumor Progression
Once cell-cell adhesion was disrupted, adhesion-mediated signaling pathways would be changed subsequently. Some characteristics in invasive metastatic cancer, such as cell mobility, would be affected as well (35). Cadherins have been implicated in tumorigenesis, and research into their roles in tumor progression should not be neglected. It is widely accepted that EMT of epithelial cells results in the feature of strong cell-cell adhesion and more motile and invasive characteristics (69, 70). A feature of EMT is the downregulation of E-cadherin and parallel upregulation of other cadherins, like N-cadherin, which plays a key role during early invasion and metastasis (71, 72).
E-cadherin and Tumor Progression
Many researches have proved that E-cadherin expression is often lost in advanced tumors; this may be associated, at least in some instances, with increased metastasis and recurrence (73). It had been demonstrated in vitro that downregulation of E-cadherin is related to the loss of epithelial phenotype (74, 75) and the acquisition of the mesenchymal phenotype, which is associated with invasive behavior; this has been validated using 3D gels and heart explants (76). Further, partial or complete reversal of this process occurs when E-cadherin is constitutively produced (49, 74, 77–79).
The E-cadherin–catenin complex was the first signaling pathway to be studied. Many factors affecting this complex participate in reduced cell adhesion, which is of great importance during tumor metastasis (80); these factors include reduction, loss, or redistribution of E-cadherin, and mutation or reduced transcription of the related genes (81). Similarly, many factors lead to downregulation of E-cadherin-mediated cell adhesion; these include hepatocyte growth factor, epithelial growth factor, and TGF-β, which are thought to initiate EMT and upregulate EMT-related transcriptional factors such as Snail, Slug, and T-wist (82–86) (Figure 2). This downregulation reduces cell–cell adhesion, and an oncogenic stimulus via β-catenin, further stimulating the canonical Wnt signaling pathway, and finally stimulating TCF-regulated genes. TCF-regulated genes, which are key factors in cell proliferation and invasion, including CCND1, MMP-7, MMP14, FN1, c-MYC, LGR5, PLAUR etc. (87–90). The non-canonical Wnt pathway may also influence cancer metastasis, and overexpression of Wnt5a is responsible for enhanced cell migration and resistance to treatment (91, 92) (Figure 2). Further, crosstalk involved in cell–cell communication and cell-matrix adhesion can be regulated by several signaling pathways that affect the migratory and invasive ability of cells by disrupting adherens junctions when integrins are activated (93). For example, a downstream factor of integrin focal adhesion kinase–Src regulates TGF-β-induced EMT (94), and is involved in coupling integrin activation with TGF-β-induced EMT (95–97). E-cadherin also plays a role in cancer progression itself, partially through the PI3K-AKT and MEK-ERK pathways. Integrin functions as a cell–cell adhesion molecular and its interaction with P-cadherin increases migration (Figure 2B).
However, Kowalski et al. (98) hold the view that abnormal E-cadherin expression is more common in invasive ductal carcinomas with the potential to develop distant metastases. E-cadherin expression in more consistent and often more frequent in distant metastases than in the primary cancer. Therefore, it is possible that E-cadherin protein is re-expressed once cancer cells reach distant sites. In the study from Bukholm et al. (99), 95% metastatic lymph nodes were highly expressed E-cadherin. The expression or re-expression of E-cadherin in distant metastases of invasive cancer suggests that it may contribute to the establishment of metastatic foci (100).
N-cadherin and Tumor Progression
The other key factor involved in EMT is N-cadherin. A major consequence of EMT is the loss of stable adhesive junctions of epithelial cells, the reversion of normal polarity among cells, and the destruction of epithelial tissue structure, therefore accelerating the migration of malignant tumor cells toward adjacent tissues (101, 102). Unlike E-cadherin, N-cadherin upregulation enhances the migratory and invasive capacities of tumor cells (103). N-cadherin is as important as E-cadherin in development and adulthood. Besides, during tumor progression, N-cadherin can affect cell survival, facilitate the process of EMT and migration/invasion by recruiting signaling molecules. Crosstalk of N-cadherin with other membrane protein such as fibroblast growth factor receptor (FGFR) also activates signaling cascades that affect cell proliferation, invasion, and cell–cell adhesion (Figure 2B). In normal epithelial cells, N-cadherin is absent or expressed at low levels, and aberrant expression is associated with malignancies and tumor progression (71, 104–106). Using an orthotopic mouse model, Shintani et al. (107) showed that, compared to N-cadherin cancer cell lines, N-cadherin-knockdown cancer cells formed significantly smaller primary tumors and micrometastases in lungs. A similar result was found by Bouvet et al. (108). In E-cadherin–positive cancer cell lines, overexpression of N-cadherin made cells more invasive both in vitro and in vivo, but did not cause parallel downregulation of E-cadherin (109, 110). These results suggest that N-cadherin may promote cancer metastasis even without the presence of E-cadherin (103).
Many signaling pathways participate in the process of malignancy. Hulit et al. (111) found that mammary epithelium tumors had greater metastatic potential in PyVmT transgenic mice with N-cadherin expression than in those without it. Further, PyVmT transgenic mice with N-cadherin expression showed greater ERK activation, and migration and invasion, and increased matrix metalloprotein-9 expression, than those without N-cadherin expression (111). Further, when FGFR was blocked in the PyVmT transgenic mice with N-cadherin expression, motility and invasion were reduced, as was ERK phosphorylation. Similar results have been found in several other cell lines that co-express N-cadherin and FGFR-1 (112), suggesting that the FGFR-N-cadherin signaling pathways might be general routes involved in metastasis. These results reveal a signaling cascade, N-cadherin/FGFR/MAPK/ERK/ matrix metalloprotein-9, in which N-cadherin mediates metastasis (103).
Other Cadherins and Tumor Progression
Aside from the two classical cadherins, other types of cadherins can also participate in metastasis. Despite its role in the maintenance of tissue architecture (113, 114), studies are increasingly revealing that P-cadherin dysfunction is strongly associated with tumorigenesis and confers the malignant phenotype in cancer cells (115–118). Mandeville et al. (119) revealed that P-cadherin can modulate the migratory ability of bladder carcinoma cells in vitro. Similarly, Taniuchi et al. (120) demonstrated that, in the vast majority of pancreatic ductal adenocarcinoma cells examined, overexpression of P-cadherin altered the subcellular localization of the p120 catenin, and increased the steady-state activity of Rho GTPases. Those changes were associated with increased motility of pancreatic ductal adenocarcinoma cells. In the liver metastatic site of colon cancer, P-cadherin expression was much higher compared to primary site; P-cadherin knockdown affected wound healing, proliferation, and colony formation, and inhibited liver metastatic foci formation in vivo (121).
OB-cadherin belongs to the type II cadherin subfamily, and is related to a poor prognosis (104, 122). OB-cadherin interacts with α- and β-catenin in vivo (122, 123) and may play a role in connecting to the cell-matrix, especially in promoting cell extension. Therefore, OB-cadherin may participate in promoting invasion and metastasis of malignant tumor via the interaction with fibroblasts or osteoblasts in the stroma and bone (122).
Bartolomé et al. pointed out that the Arg-Gly-Asp motifs of VE-cadherin activate the α2β1 integrin, and then promote cell adhesion, cancer invasion, proliferation, and lung metastasis (124). It has been revealed that higher expression of VE-cadherin leads to poor prognosis in breast cancer and melanoma patients. These results suggest the novel concept that therapies that target cadherin-specific Arg-Gly-Asp motifs may control distant metastasis in multiple cancers.
The role of PCDHs in cancer progression is complicated. Several studies have mentioned that PCDHs could induce apoptosis of tumor cells and prevent malignant tumor from proliferating and metastasis (125). This point of view has been approved in colon cancer (126). Similarly, based on in vitro testing, restoration of PCDH9 expression has effects on cell cycle and could strength apoptosis (127). So do PCDH20 and PCDH17 (128–130). When PCDHs are lost, tumor cells gain a more aggressive phenotype. For example, loss of PCDH10 significantly enhances the proliferation of myeloma cells (131) and may act as a prognostic marker for the reduced survival of colorectal cancer patients (132). More specifically, when PCDH17 was lost, it could stimulate EGFR/MEK/ERK signaling pathway and accelerate the progression of liver cancer (130). Recent research has elucidated the oncogenic activity of PCDHs. It has been reported that PCDH7 is significantly increased in breast cancer and has a positive correlation with brain metastases (133). Further, Chen et al. and Terry et al. found PCDH-PC, a cytoplasmically retained variant of PCDH11Y, was upregulated in advanced prostate cancer and metastasis (134, 135). The specific mechanisms whereby PCDHa affect cancer remain to be studied.
In brief, cadherins, especially E-cadherin and N-cadherin, participate in EMT and thus regulate tumor invasion and metastasis.
Cadherins and Angiogenesis
Malignant tumors consume much more energy and oxygen than normal tissues and need fast-growing vascular networks to support the proliferation of tumor (136). In this process, various types of pro-angiogenic factors are secreted by tumor cells and stromal cells and help building abnormal vascular networks which are disorganized, immature and permeable, leading to inadequately perfused malignant tumors (137). In the development of malignant tumor, cadherins do more than just providing adhesion. They interact with many cytoskeletal and signaling molecules by transferring signals into cells. Endothelial cells present different cadherins which may transfer specific signals and have distinct functional roles.
VE-cadherin, originally called cadherin-5 (138), is mainly located in vascular endothelial cells and is indispensable for vascular building during embryonic development (139, 140). In addition to being a transmembrane component of the endothelial cadheren junction, it maintains the integrity and permeability of the endothelium and vascular system (141, 142). Thus, a burst of VE-cadherin expression may be required in promoting and sustaining vascular growth.
VE-cadherin promotes tumor progression mainly by facilitating tumor angiogenesis (143). VE-cadherin could interact with vascular endothelial growth factor receptor (VEGFR)-2 (Figure 2C) and stimulate TGF-β signaling pathway to enhance cell proliferation (144). Many growth factors such as VEGF could induce the tyrosine phosphorylation of VE-cadherin, β-catenin and p120, which would increase vascular permeability in vitro system (145). VE-cadherin phosphorylation through the VEGF/VEGF R2 pathway needs Src activity (146) (Figure 2C). In angiogenic tissues, VE-cadherin-associated Src phosphorylation is increased (143) and inhibition of Src inhibits VEGF-stimulated vascular permeability (147). With the help of other phosphatases, VEGF 2R and Src lead to the phosphorylation of VE-cadherin on tyrosines (143) and this association would induce endothelial cell survival through PI3K/AKT pathway (Figure 2C). However, in the absence of protein tyrosine phosphatase activity, ERK/MAPK pathway is activated to stimulate cell proliferation (Figure 2C) (148). Similarly, VE-cadherin can interact with the TGF-receptor complex (149), platelet-derived growth factor β-receptor (150), or FGFR1 (151), among others, and may affect the proliferation of endothelial cells and stability of vascular structures.
N-cadherin functions mainly as a promoter of malignant tumors. However, the role it plays in tumor angiogenesis should not be underestimated. N-cadherin affects vascular tissue mainly by interacting with pericytes and vascular smooth muscle cells in the endothelium (152, 153). Unlike VE-cadherin, which predominates in vascular epithelial cells, N-cadherin predominates in pericytes. In chickens, once N-cadherin is neutralized or lost, defective pericyte adhesion occurs causing aberrant vascular development (154). The amount of VE-cadherin expressed depends on N-cadherin, and N-cadherin affects vascular morphogenesis by regulating VE-cadherin (155), suggesting that N-cadherin is a crucial regulator of angiogenesis.
Nalla et al. (156) found that N-cadherin knockdown suppressed the expression of several angiogenic molecules and reduced the angiogenic ability of PC3 cells (156), which indicates that endothelial cell is not the only target of N-cadherin, angiogenesis could also be promoted by N-cadherin through regulating chemokines and angiogenic molecules. Doherty et al. (157, 158) have found that N-cadherin has an His–Ala–Val (HAV) sequence in the extracellular domain directly linked to the HVA sequence present on FGFR (Figure 2C) and propose that trans-interactions between N-cadherin molecules on cell membranes trigger cis-interactions between N-cadherin complexes and FGFR monomers, subsequently causing FGFR dimerization and activation. Thus, N-cadherin-mediated endothelial cell adhesion may facilitate angiogenesis. Similarly, a soluble 90 kDa fragment of N-cadherin promoted angiogenesis in an FGFR-dependent manner (159). Proteases cleave the extracellular part of N-cadherin and produce soluble N-cadherin. Soluble N-cadherin can phosphorylate ERK and stimulate angiogenesis (Figure 2C).
In conclusion, cadherins play a vital role in tumor angiogenesis mainly through binding to VEGFR or platelet-derived growth factor receptor in order to affect vascular structures.
Cadherins and Tumor Immune Response
A tumor is not a one step result; the role of host immunity in the formation and progression of maglinant tumor s is complex and should not be negelected. Solid tumors are commonly infiltrated by a large number of immune cells which funtion seems pretty paradoxical (81). Once the microcolonies of malignant cells were detected by immune system, innate and adaptive immune cells would be able to eradicate them. However, genome plasticity renders some tumor cells the ability to proliferate continuously and thus cause an equilibrium between the generating and eradicating of tumor cells. The continuing proliferation of tumor cells would finally cause a broken equilibrium along with detectable tumor followed by metastasis (160). Concomitantly, surviving tumor clones control immune responses by specifically suppressing anti-tumor effector mechanisms and accumlating immunosuppressive immune cell subsets, inculding DCs, tumor-associated macrophages and Tregs at the tumor site (81).
Viollet et al. (161) hold the view that E-cadherin participates in the maintaining of epithelial barrier and prevents potentially harmful agents from inner tissue. This is the immunologic function of E-cadherin. Furthermore, E-cadherin might take part in the regulation of epithelial innate immune function through multiple chemokines and cytokines and modulating transepithelial transfer of immunes cells (162). Recent studies have shown that cadherins are highly expressed in several leukocytes, such as dendritic cells (DCs), macrophages and Langerhans cells (161).
DCs are important participators in adaptive immunity. The roles of cadherins in DCs have been investigated. It is widely recognized that E-cadherin is expressed in Langerhans cells, a type of immature DC (163). As DCs mature, their expression of E-cadherin is reduced to facilitate the migration of DCs to the lymphoid organs (164). Infiltration of DCs occurs in solid tumors, and E-cadherin expression in DCs may be upregulated by tumor cells; this upregulation is thought to inhibit the function of DCs in presenting tumor antigens (165). Jiang et al. (166) showed that bone marrow-derived DC (BMDC) express E-cadherin, and BMDC activation would be induced when E-cadherin-depended DC-DC interaction was disrupted. This would upregulate the expression of costimulatory molecules and CCR7, downregulate the macropinocytosis, and lead to the migration of MHC II molecules from lysosomes to cell surface. At the same time, loss of E-cadherin results in the accumulation of cytoplasmic β-catenin and stimulates the following signaling pathways like TCF. Manicassamy et al. (167) showed that β-catenin deletion in DC would accumulates Th1 and Th17 cell infiltation in intestine of mice, and decrease the number of regulatory T cells. This indicates that E-cadherin plays a significant role in DCs and may affect their functions in the tumor microenvironment.
Macrophages are involved in the whole process of life via phagocytic ability and producing immunoregulatory mediators and trophic. They could also recognize, engulf and destruct pathogens to facilitate tissue development and recognition and maintain homeostasis (168). According to the research of Rehli et al. (169), IL-4 could induce the expression of E-cadherin, which was considered as a marker of M2 macrophages, in mouse bone marrow-derived macrophages (in vitro) and several mice models (in vivo) (170).
In specimens of sarcoidosis and foreign body granulomas from humans, E-cadherin is expressed in skin granuloma macrophages (171). In an in vitro experiment, IL-4/IL-13 induced macrophages to express E-cadherin (172, 173). In Th2-driven diseases, IL-4 and IL-3 are crucial for inducing E-cadherin in both mouse and human macrophages (172). However, in the tumor microenvironment, E-cadherin expression is higher and independent of IL-4/IL-13 in M1-like macrophages, in contrast to the situation in M2-like macrophages (174). One possible reason is that M2-like macrophages are preindominantly accumulated in the hypoxic areas of tumor tissue, which reported to reduce E-cadherin expression (175). Further, E-cadherin may contribute to the process of fusion and engulfment in macrophages: β-catenin is barely detectable in naïve macrophages due to the absence of E-cadherin, but is found to be stable in the E-cadherin/β-catenin complex after stimulated with IL-4, which could enhance the fusion and engulfment of pathogens in macrophages in a STAT5-dependent way (172, 176). Thus, E-cadherin might act as a promoter in helping macrophages to kill tumor cells.
The classical cadherin pathways, including Wnt, PI3K, Rho-family GTPases, and NF-κB, operate in macrophages and DCs. Besides, Wnt/β-catenin has been reported to contribute to the anti-inflammatory phenotype of macrophages and tolerogenic state of DCs (177). When Wnt binds to Frizzled, the phosphorylation of β-catenin by casein kinase 1 and GSK-3β is inhibited, leading to an upregulation of nuclear β-catenin and activation of TCF/LEF-dependent gene expression, which stimulates anti-inflammatory macrophages and tolerogenic DCs (Figure 2D). And at the same time, other pathways can also influence macrophage and DC functioning. It has been shown that E-cadherin could suppress the activity of macrophages and DCs via PI3K/AKT signaling pathway. In addition, NF-κB, a vital transcription factor, could also be regulated by E-cadherin-catenin in macrophages and DCs, using p120 catenin as a docking site and working as a suppresser (Figure 2D).
On the surface of other immune cells, including CD8+ T, NK, and regulatory T cells, there are various ligands that can bind to the E-cadherin expressed on DCs and macrophages. For example, killer cell lectin-like receptor G1 (KLRG1) binds to the N-terminal homodimeric interface of E-cadherin EC1 (178). KLRG1 is expressed mainly on the surface of mature NK cells, and its activation inhibits NK cytotoxicity (179, 180). Furthermore, KLRG1 has been found on CD4+ T cells and CD8+ T cells (181, 182) and regulates T cell responses to suboptimal conditions. The integrin αE chain, also known as CD103, is a ligand expressed mainly on intraepithelial αβ and γδ T cells, and mediates the adhesion between E-cadherin+ epithelial cells and T cells in vitro (183) (Figure 2).
In shortly, besides the effect on tumor cells, cadherins also participate in regulating various immune cells in the tumor microenvironment. However, further study is needed to illuminate the role of cadherins in tumor-associated immune cells.
Therapeutic Targets in Cancer
It has been reported that many signaling pathways or proteins are abnormally activated in malignant tumor cells (184–186), so molecular-targeted therapy has been developed and achieved pretty good response in cancer patients recent years (187, 188). Those molecular-targeted drugs are able to kill cancer cell selectively with decreased toxicity on normal cell, which is the significant improvement comparing with traditional anticancer drugs (189). Targeted drugs inhibit specific signaling pathways that contribute to the malignant phenotype of cancer cells and limit the progression of malignant tumors. Cadherins, vital molecules in tumorigenesis and cancer progression, could be also worked as therapeutic targets in cancer treatment.
Inhibition of Angiogenesis
Generation of new vascular network tissue is of great importance for tumors because of their high proliferative rate. As a result, targeting tumor blood vessels is a feasible strategy in cancer therapy. Specific antibodies of VE-cadherin have effects on vascular permeability, which suggests that targeting VE-cadherin is feasible in controlling vascular barrier function (190). Further, blocking of VE-cadherin-mediated endothelial-cell adhesion affected microvessel stability (191, 192).
Liao et al. (193) were the first to reveal that BV13, a monoclonal antibody of VE-cadherin that binds to EC4, inhibits angiogenesis, tumor growth, and metastasis. However, administration of the monoclonal antibody (mAb) BV13 in vivo markedly increased permeability in the lungs and heart (194). To resolve this problem, another mAb, BV14, was developed; this did not significantly increase the permeability of the lungs, heart, and other organs, even after repeated high doses, and even in animals carrying well-developed tumors. BV14 and BV13 were equally effective in inhibiting angiogenesis in the mouse cornea and downregulating the growth of hemangioma and C6 glioma (Table 2). Corada et al. (202) tested five different mAbs directed at different extracellular domains of VE-cadherin, to investigate their functions and side effects. They found that mAb Cad5, which binds to the EC1 domain of VE-cadherin, was the most effective in increasing permeability. EC1 may be important in mediating cell–cell interaction, and thus the binding of an antibody to it would disrupt the integrity of vessels. Another mAb, E4G10, binds to the N-terminal sequences of VE-cadherin; unlike BV13, which binds to all vasculature, E4G10 binds only to the tumor neovasculature (191, 195). Therefore, future research should focus on finding ways to disrupt endothelial junctions in growing vessels, without affecting stabilized junctions in normal vessels (192, 195). These findings demonstrate that VE-cadherin plays a crucial role in postnatal angiogenesis, and further validate VE-cadherin as a potential target for antiangiogenic treatment.
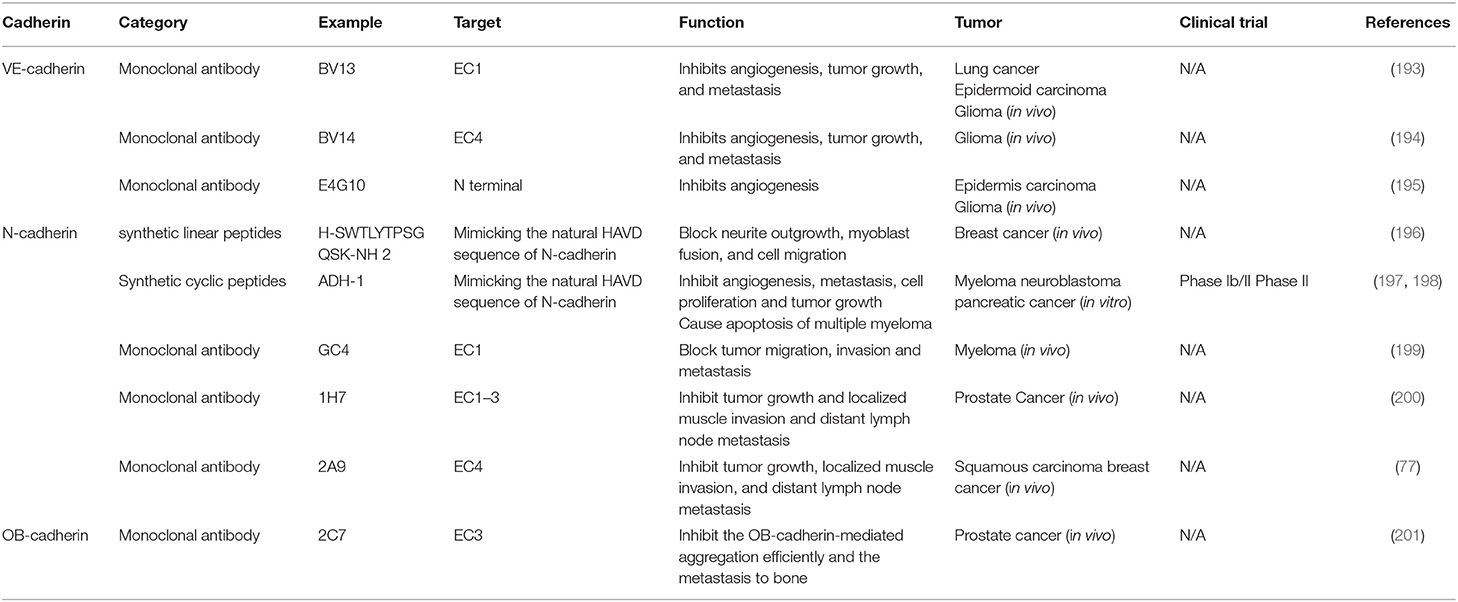
Table 2. An overview of cadherin as a therapeutic target for cancer.
As with VE-cadherin, it has long been known that N-cadherin occurs on endothelial cells, but its role in vascular structure is only now becoming a focus of research. Inhibition of N-cadherin function destabilizes microvessels structure (154, 203). For example, direct blockage of N-cadherin impedes the adhesive complex between endothelial cells and pericytes, and thus induces hemorrhage (154). Several types of antagonists targeting N-cadherin have been discovered. Three types of antagonists are based on the cell adhesion recognition sequence HAV: synthetic linear peptides, synthetic cyclic peptides, and non-peptidyl peptidomimetics (204). ADH-1, a synthetic cyclic peptide harboring the HAV motif, has been shown to inhibit angiogenesis in various model systems (197), including the chick chorioallantoic membrane assay (205). When ADH-1 is used in a tumor-bearing mouse model, the hemorrhage that it causes is limited to tumor blood vessels, without affecting normal vessels. Therefore, it is possible that the antiangiogenic properties of ADH-1 induced the massive apoptosis we observed in the tumors that we studied (107). Another N-cadherin antagonist, H-SWTLYTPSGQSK-NH 2 (196), has recently been discovered; it may inhibit endothelial cell tube formation in vitro, indicating that it has antiangiogenic properties. In 2010, two monoclonal antibodies of N-cadherin were developed that inhibit the invasiveness and proliferation of N-cadherin-expressing cancer cells in vitro and suppress tumor growth and lymph-node metastasis in vivo (200).
Although VE-cadherin and N-cadherin are promising targets in tumor therapy, they have some shortcomings. Toxicity is one of the questions needed to be solved in the future, as they both widely expressed in normal tissues like vascular, heart, peripheral nerve and liver. Loss of VE-cadherin might cause hemorrhage and loss of N-cadherin can disrupt the structure of organs, leading to severe side effects. More precise targeting is high on the agenda, so do further preclinical and clinical testing to confirm the safety.
Blockade of Metastasis
As N-cadherin involves in cancer metastasis, N-cadherin antagonists are showing promise as therapeutic drugs for inhibiting cell adhesion and modulating cell signaling, thus preventing tumor metastasis. As mentioned above, ADH-1 is a stable cyclic peptide harboring an HAV motif, which similarly inhibited N-cadherin-dependent functioning (197). It has been reported that ADH-1 can cause apoptosis in several cancer cells (107, 206, 207) and inhibit tumor cell migration at sub-cytotoxic concentrations (207–209). In a pre-clinical mouse model, ADH-1 not only inhibited primary tumor growth, but also prevented localized tumor invasion and metastasis (210). Further, ADH-1 has entered clinical testing, and there are two completed Phase I studies (211). Combination of ADH-1 and various cytotoxic agents has shown great promise: positive results in animal models, the absence of severe side effects, and promising antitumor effects in Phase I studies. There are also several monoclonal antibodies, such as GC4, directed against N-cadherin, that can block N-cadherin-dependent tumor migration and invasion in vitro and metastasis in vivo (199, 212, 213). Besides, GC-4-mediated blocking could increase the sensitivity to imatinib in CML, which means a potential therapeutic strategy in tyrosine kinase inhibitor resistance (214). 1H7 (targeting N-cadherin EC1–3) and 2A9 (targeting N-cadherin EC4) are newfound antibodies targeting N-cadherin and have been proved effectiveness in treating prostate cancer in mouse model. Both slowed down tumor growth and reduced local invasion and distant metastasis in lymph nodes (200, 215).
OB-cadherin might also be effective as a therapeutic target. Kaur et al. (216) found that OB-cadherin knockdown markedly reduced the survival ability of cancer cells in vitro and colony-forming abilities when there were no exogenous growth factors. Therefore, OB-cadherin is a potential target to reduce the aggressiveness and motility of cancer, and develop specific drugs to target it is an urgent priority. To explore this possibility, Lee et al. (201) generated a panel of monoclonal antibodies (mAbs) against the OB-cadherin extracellular domain; one of these, mAb 2C7, inhibited the osseous metastasis of PC3-MM 2 cells (Table 2).
Taken together, cadherins targeted therapy could effectively limit the progression and angiogenesis of cancer, and further studies should be focused on elevating the specificity and effectiveness, as well as the combination therapies.
Conclusions
In summary, cadherins are key factors in maintaining normal tissue development. However, abnormal expression is found in tumorigenesis, development and metastasis. Therefore, a better understanding of cadherins is critical for cancer clinical applications, especially as therapeutic targets. In this review, we have discussed the various types of cadherins, their effects on tumor growth, and the clinical application of cadherins. Targeting cadherins is a promising strategy for cancer treatment and further studies and clinical experiments are needed to make it come true.
Author Contributions
WY and LY contributed equally to preparing and writing the manuscript. WY and TL prepared the figures and tables. LY and YZ designed, reviewed, and edited the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China, grant numbers U1804281, 81771781, and 81602024, and was further supported by State's Key Project of Research and Development Plan, grant number 2016YFC1303500.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Peyrieras N, Hyafil F, Louvard D, Ploegh HL, Jacob F. Uvomorulin: a nonintegral membrane protein of early mouse embryo. Proc Natl Acad Sci USA. (1983) 80:6274–7. doi: 10.1073/pnas.80.20.6274
2. Hyafil F, Babinet C, Jacob F. Cell-cell interactions in early embryogenesis: a molecular approach to the role of calcium. Cell. (1981) 26:447–54. doi: 10.1016/0092-8674(81)90214-2
3. Schuh R, Vestweber D, Riede I, Ringwald M, Rosenberg UB, Jackle H, et al. Molecular cloning of the mouse cell adhesion molecule uvomorulin: cDNA contains a B1-related sequence. Proc Natl Acad Sci USA. (1986) 83:1364–8. doi: 10.1073/pnas.83.5.1364
4. Nose A, Nagafuchi A, Takeichi M. Isolation of placental cadherin cDNA: identification of a novel gene family of cell-cell adhesion molecules. EMBO J. (1987) 6:3655–61. doi: 10.1002/j.1460-2075.1987.tb02698.x
5. Vieira AF, Paredes J. P-cadherin and the journey to cancer metastasis. Mol Cancer. (2015) 14:178. doi: 10.1186/s12943-015-0448-4
6. Stemmler MP. Cadherins in development and cancer. Mol Biosyst. (2008) 4:835–50. doi: 10.1039/b719215k
7. Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol. (1997) 181:64–78. doi: 10.1006/dbio.1996.8443
8. Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. (1991) 251:1451–5. doi: 10.1126/science.2006419
9. Goodwin M, Yap AS. Classical cadherin adhesion molecules: coordinating cell adhesion, signaling and the cytoskeleton. J Mol Histol. (2004) 35:839–44. doi: 10.1007/s10735-004-1833-2
10. Kemler R. Classical cadherins. Semin Cell Biol. (1992) 3:149–55. doi: 10.1016/S1043-4682(10)80011-X
11. Kottke MD, Delva E, Kowalczyk AP. The desmosome: cell science lessons from human diseases. J Cell Sci. (2006) 119:797–806. doi: 10.1242/jcs.02888
12. Yin T, Green KJ. Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol. (2004) 15:665–77. doi: 10.1016/S1084-9521(04)00091-6
13. Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. (2007) 8:11–20. doi: 10.1038/nrn2043
14. Goffinet AM, Tissir F. Seven pass Cadherins CELSR1–3. Semin Cell Dev Biol. (2017) 69:102–10. doi: 10.1016/j.semcdb.2017.07.014
15. Tanoue T, Takeichi M. New insights into Fat cadherins. J Cell Sci. (2005) 118:2347–53. doi: 10.1242/jcs.02398
16. Redies C, Vanhalst K, Roy F. delta-Protocadherins: unique structures and functions. Cell Mol Life Sci. (2005) 62:2840–52. doi: 10.1007/s00018-005-5320-z
17. Ahmed AF, de Bock CE, Sontag E, Hondermarck H, Lincz LF, Thorne RF. FAT1 cadherin controls neuritogenesis during NTera2 cell differentiation. Biochem Biophys Res Commun. (2019) 514:625–31. doi: 10.1016/j.bbrc.2019.04.197
18. Frank M, Kemler R. Protocadherins. Curr Opin Cell Biol. (2002) 14:557–62. doi: 10.1016/S0955-0674(02)00365-4
19. Junghans D, Haas IG, Kemler R. Mammalian cadherins and protocadherins: about cell death, synapses and processing. Curr Opin Cell Biol. (2005) 17:446–52. doi: 10.1016/j.ceb.2005.08.008
20. Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. (1999) 97:779–90. doi: 10.1016/S0092-8674(00)80789-8
21. Sannigrahi MK, Srinivas S, Rakshit S. The prospects of cadherin-23 as a mediator of homophilic cell-cell adhesion. Adv Exp Med Biol. (2018) 1112:99–105. doi: 10.1007/978-981-13-3065-0_8
22. Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. (2001) 114:629–41.
23. Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. J Clin Invest. (1996) 98:1949–53. doi: 10.1172/JCI118997
24. Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Muller EJ, et al. Mechanisms of plakoglobin-dependent adhesion: desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem. (2005) 280:40355–63. doi: 10.1074/jbc.M506692200
25. Devenport D, Fuchs E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol. (2008) 10:1257–68. doi: 10.1038/ncb1784
26. Ravni A, Qu Y, Goffinet AM, Tissir F. Planar cell polarity cadherin Celsr1 regulates skin hair patterning in the mouse. J Invest Dermatol. (2009) 129:2507–9. doi: 10.1038/jid.2009.84
27. Boutin C, Labedan P, Dimidschstein J, Richard F, Cremer H, Andre P, et al. A dual role for planar cell polarity genes in ciliated cells. Proc Natl Acad Sci USA. (2014) 111:E3129–38. doi: 10.1073/pnas.1404988111
28. Hirayama T, Yagi T. Regulation of clustered protocadherin genes in individual neurons. Semin Cell Dev Biol. (2017) 69:122–30. doi: 10.1016/j.semcdb.2017.05.026
29. Wheelock MJ, Soler AP, Knudsen KA. Cadherin junctions in mammary tumors. J Mammary Gland Biol Neoplasia. (2001) 6:275–85. doi: 10.1023/A:1011319507155
30. Gumbiner BM, McCrea PD. Catenins as mediators of the cytoplasmic functions of cadherins. J Cell Sci Suppl. (1993) 17:155–8. doi: 10.1242/jcs.1993.Supplement_17.22
31. Nathaniel Clarke D, Lowe CJ, James Nelson W. The cadherin-catenin complex is necessary for cell adhesion and embryogenesis in Nematostella vectensis. Dev Biol. (2019) 447:170–81. doi: 10.1016/j.ydbio.2019.01.007
32. Yasuda S, Tanaka H, Sugiura H, Okamura K, Sakaguchi T, Tran U, et al. Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron. (2007) 56:456–71. doi: 10.1016/j.neuron.2007.08.020
33. Formstone CJ. 7TM-Cadherins: developmental roles and future challenges. Adv Exp Med Biol. (2010) 706:14–36. doi: 10.1007/978-1-4419-7913-1_2
34. Nie D, Wang Z, Zhang Y, Pang D, Ouyang H, Li LI. Fat-1 gene inhibits human oral squamous carcinoma cell proliferation through downregulation of β-catenin signaling pathways. Exp Ther Med. (2016) 11:191–6. doi: 10.3892/etm.2015.2847
35. Wickstrom SA, Niessen CM. Cell adhesion and mechanics as drivers of tissue organization and differentiation: local cues for large scale organization. Curr Opin Cell Biol. (2018) 54:89–97. doi: 10.1016/j.ceb.2018.05.003
36. Conacci-Sorrell M, Zhurinsky J, Ben-Ze'ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. (2002) 109:987–91. doi: 10.1172/JCI0215429
37. Clement R, Dehapiot B, Collinet C, Lecuit T, Lenne PF. Viscoelastic dissipation stabilizes cell shape changes during tissue morphogenesis. Curr Biol. (2017) 27:3132–42.e3134. doi: 10.1016/j.cub.2017.09.005
38. Basu S, Cheriyamundath S, Ben-Ze'ev A. Cell-cell adhesion: linking Wnt/β-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Res. (2018) 7:F1000. doi: 10.12688/f1000research.15782.1
39. Ozawa M, Ringwald M, Kemler R. Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. Proc Natl Acad Sci USA. (1990) 87:4246–50. doi: 10.1073/pnas.87.11.4246
40. Jou TS, Stewart DB, Stappert J, Nelson WJ, Marrs JA. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc Natl Acad Sci USA. (1995) 92:5067–71. doi: 10.1073/pnas.92.11.5067
41. Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. (1995) 92:8813–7. doi: 10.1073/pnas.92.19.8813
42. Cavallaro U, Christofori G. Multitasking in tumor progression: signaling functions of cell adhesion molecules. Ann N Y Acad Sci. (2004) 1014:58–66. doi: 10.1196/annals.1294.006
43. Christofori G. Changing neighbours, changing behaviour: cell adhesion molecule-mediated signalling during tumour progression. EMBO J. (2003) 22:2318–23. doi: 10.1093/emboj/cdg228
44. Molitoris BA, Nelson WJ. Alterations in the establishment and maintenance of epithelial cell polarity as a basis for disease processes. J Clin Invest. (1990) 85:3–9. doi: 10.1172/JCI114427
45. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. (2006) 127:679–95. doi: 10.1016/j.cell.2006.11.001
46. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. (2006) 7:131–42. doi: 10.1038/nrm1835
47. Williams EJ, Williams G, Howell FV, Skaper SD, Walsh FS, Doherty P. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J Biol Chem. (2001) 276:43879–86. doi: 10.1074/jbc.M105876200
48. Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. (1994) 1198:11–26. doi: 10.1016/0304-419X(94)90003-5
49. Vleminckx K, Vakaet L Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. (1991) 66:107–19. doi: 10.1016/0092-8674(91)90143-M
50. Pinheiro H, Bordeira-Carrico R, Seixas S, Carvalho J, Senz J, Oliveira P, et al. Allele-specific CDH1 downregulation and hereditary diffuse gastric cancer. Hum Mol Genet. (2010) 19:943–52. doi: 10.1093/hmg/ddp537
51. Petrova YI, Spano MM, Gumbiner BM. Conformational epitopes at cadherin calcium-binding sites and p120-catenin phosphorylation regulate cell adhesion. Mol Biol Cell. (2012) 23:2092–108. doi: 10.1091/mbc.e11-12-1060
52. Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell. (2016) 27:3233–44. doi: 10.1091/mbc.E16-01-0058
53. Winter JM, Ting AH, Vilardell F, Gallmeier E, Baylin SB, Hruban RH, et al. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin Cancer Res. (2008) 14:412–8. doi: 10.1158/1078-0432.CCR-07-0487
54. Wang M, Liu Y, Liu S, Wang E. [Abnormal expression of p120-catenin and E-cadherin is significantly correlated with malignant phenotype of human lung cancer.]. Zhongguo Fei Ai Za Zhi. (2009) 12:306–11. doi: 10.3779/j.issn.1009-3419.2009.04.09
55. Mortazavi F, An J, Dubinett S, Rettig M. p120-catenin is transcriptionally downregulated by FOXC2 in non-small cell lung cancer cells. Mol Cancer Res. (2010) 8:762–74. doi: 10.1158/1541-7786.MCR-10-0004
57. Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. (2004) 4:118–32. doi: 10.1038/nrc1276
58. Cavallaro U, Schaffhauser B, Christofori G. Cadherins and the tumour progression: is it all in a switch? Cancer Lett. (2002) 176:123–8. doi: 10.1016/S0304-3835(01)00759-5
59. Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. (1999) 274:36734–40. doi: 10.1074/jbc.274.51.36734
60. Fan G, Ye D, Zhu S, Xi J, Guo X, Qiao J, et al. RTL1 promotes melanoma proliferation by regulating Wnt/β-catenin signalling. Oncotarget. (2017) 8:106026–37. doi: 10.18632/oncotarget.22523
61. Cavallaro U, Christofori G. Cell adhesion in tumor invasion and metastasis: loss of the glue is not enough. Biochim Biophys Acta. (2001) 1552:39–45. doi: 10.1016/S0304-419X(01)00038-5
62. Nguyen T, Mege RM. N-Cadherin and Fibroblast Growth Factor Receptors crosstalk in the control of developmental and cancer cell migrations. Eur J Cell Biol. (2016) 95:415–26. doi: 10.1016/j.ejcb.2016.05.002
63. Van Marck V, Stove C, Jacobs K, Van den Eynden G, Bracke M. P-cadherin in adhesion and invasion: opposite roles in colon and bladder carcinoma. Int J Cancer. (2011) 128:1031–44. doi: 10.1002/ijc.25427
64. Jacobs K, Feys L, Vanhoecke B, Van Marck V, Bracke M. P-cadherin expression reduces melanoma growth, invasion, and responsiveness to growth factors in nude mice. Eur J Cancer Prev. (2011) 20:207–16. doi: 10.1097/CEJ.0b013e3283429e8b
65. Bauer R, Valletta D, Bauer K, Thasler WE, Hartmann A, Muller M, et al. Downregulation of P-cadherin expression in hepatocellular carcinoma induces tumorigenicity. Int J Clin Exp Pathol. (2014) 7:6125–32. doi: 10.1055/s-0032-1332048
66. Radice GL, Ferreira-Cornwell MC, Robinson SD, Rayburn H, Chodosh LA, Takeichi M, et al. Precocious mammary gland development in P-cadherin-deficient mice. J Cell Biol. (1997) 139:1025–32. doi: 10.1083/jcb.139.4.1025
67. Hardy RG, Tselepis C, Hoyland J, Wallis Y, Pretlow TP, Talbot I, et al. Aberrant P-cadherin expression is an early event in hyperplastic and dysplastic transformation in the colon. Gut. (2002) 50:513–9. doi: 10.1136/gut.50.4.513
68. Ye M, Huang T, Li J, Zhou C, Yang P, Ni C, et al. Role of CDH13 promoter methylation in the carcinogenesis, progression, and prognosis of colorectal cancer: a systematic meta-analysis under PRISMA guidelines. Medicine. (2017) 96:e5956. doi: 10.1097/MD.0000000000005956
69. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. (2003) 15:740–6. doi: 10.1016/j.ceb.2003.10.006
70. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
71. Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res. (2004) 10:4125–33. doi: 10.1158/1078-0432.CCR-0578-03
72. Jolly MK, Ware KE, Xu S, Gilja S, Shetler S, Yang Y, et al. E-cadherin represses anchorage-independent growth in sarcomas through both signaling and mechanical mechanisms. Mol Cancer Res. (2019) 17:1391–402. doi: 10.1158/1541-7786.MCR-18-0763
73. Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Persp Biol. (2009) 1:a003129. doi: 10.1101/cshperspect.a003129
74. Behrens J, Mareel MM, Van Roy FM, Birchmeier W. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol. (1989) 108:2435–47. doi: 10.1083/jcb.108.6.2435
75. Li J, Di Russo J, Hua X, Chu Z, Spatz JP, Wei Q. Surface immobilized E-cadherin mimetic peptide regulates the adhesion and clustering of epithelial cells. Adv Healthc Mater. (2019) 8:e1801384. doi: 10.1002/adhm.201801384
76. Chen WC, Obrink B. Cell-cell contacts mediated by E-cadherin (uvomorulin) restrict invasive behavior of L-cells. J Cell Biol. (1991) 114:319–27. doi: 10.1083/jcb.114.2.319
77. Kim JB, Islam S, Kim YJ, Prudoff RS, Sass KM, Wheelock MJ, et al. N-Cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J Cell Biol. (2000) 151:1193–206. doi: 10.1083/jcb.151.6.1193
78. Navarro P, Gomez M, Pizarro A, Gamallo C, Quintanilla M, Cano A. A role for the E-cadherin cell-cell adhesion molecule during tumor progression of mouse epidermal carcinogenesis. J Cell Biol. (1991) 115:517–33. doi: 10.1083/jcb.115.2.517
79. Dang DK, Makena MR, Llongueras JP, Prasad H, Ko M, Bandral M, et al. A Ca2+-ATPase regulates E-cadherin biogenesis and epithelial-mesenchymal transition in breast cancer cells. Mol Cancer Res. (2019) 17:1735–47. doi: 10.1158/1541-7786.MCR-19-0070
80. Beavon IR. The E-cadherin-catenin complex in tumour metastasis: structure, function and regulation. Eur J Cancer. (2000) 36:1607–20. doi: 10.1016/S0959-8049(00)00158-1
81. Cali B, Molon B, Viola A. Tuning cancer fate: the unremitting role of host immunity. Open Biol. (2017) 7:170006. doi: 10.1098/rsob.170006
82. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. (2000) 2:76–83. doi: 10.1038/35000025
83. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. (2008) 19:4875–87. doi: 10.1091/mbc.e08-05-0506
84. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. (2007) 7:415–28. doi: 10.1038/nrc2131
85. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. (2002) 2:442–54. doi: 10.1038/nrc822
86. Pal I, Rajesh Y, Banik P, Dey G, Dey KK, Bharti R, et al. Prevention of epithelial to mesenchymal transition in colorectal carcinoma by regulation of the E-cadherin-β-catenin-vinculin axis. Cancer Lett. (2019) 452:254–63. doi: 10.1016/j.canlet.2019.03.008
87. Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis–a look outside the nucleus. Science. (2000) 287:1606–9. doi: 10.1126/science.287.5458.1606
88. Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, et al. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. (1999) 18:2883–91. doi: 10.1038/sj.onc.1202627
89. Gradl D, Kuhl M, Wedlich D. The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol Cell Biol. (1999) 19:5576–87. doi: 10.1128/MCB.19.8.5576
90. Alsaleem M, Toss MS, Joseph C, Aleskandarany M, Kurozumi S, Alshankyty I, et al. The molecular mechanisms underlying reduced E-cadherin expression in invasive ductal carcinoma of the breast: high throughput analysis of large cohorts. Mod Pathol. (2019) 32:967–76. doi: 10.1038/s41379-019-0209-9
91. Pukrop T, Klemm F, Hagemann T, Gradl D, Schulz M, Siemes S, et al. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc Natl Acad Sci USA. (2006) 103:5454–9. doi: 10.1073/pnas.0509703103
92. Dissanayake SK, Wade M, Johnson CE, O'Connell MP, Leotlela PD, French AD, et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem. (2007) 282:17259–71. doi: 10.1074/jbc.M700075200
93. Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. (2013) 126:393–401. doi: 10.1242/jcs.100115
94. Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. (2008) 314:143–52. doi: 10.1016/j.yexcr.2007.09.005
95. Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem. (2001) 276:46707–13. doi: 10.1074/jbc.M106176200
96. Galliher AJ, Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. (2006) 8:R42. doi: 10.1186/bcr1524
97. Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. (2009) 11:R68. doi: 10.1186/bcr2360
98. Rakha EA, Abd El Rehim D, Pinder SE, Lewis SA, Ellis IO. E-cadherin expression in invasive non-lobular carcinoma of the breast and its prognostic significance. Histopathology. (2005) 46:685–93. doi: 10.1111/j.1365-2559.2005.02156.x
99. Bukholm IK, Nesland JM, Borresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients [seecomments]. J Pathol. (2000) 190:15–9. doi: 10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L
100. Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. (2003) 5:R217–22. doi: 10.1186/bcr651
101. Kourtidis A, Lu R, Pence LJ, Anastasiadis PZ. A central role for cadherin signaling in cancer. Exp Cell Res. (2017) 358:78–85. doi: 10.1016/j.yexcr.2017.04.006
102. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. (1998) 392:190–3. doi: 10.1038/32433
103. Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann N Y Acad Sci. (2004) 1014:155–63. doi: 10.1196/annals.1294.016
104. Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, et al. Cadherin switching in human prostate cancer progression. Cancer Res. (2000) 60:3650–4. doi: 10.5980/jpnjurol.91.92
105. Choi Y, Lee HJ, Jang MH, Gwak JM, Lee KS, Kim EJ, et al. Epithelial-mesenchymal transition increases during the progression of in situ to invasive basal-like breast cancer. Hum Pathol. (2013) 44:2581–9. doi: 10.1016/j.humpath.2013.07.003
106. Lascombe I, Clairotte A, Fauconnet S, Bernardini S, Wallerand H, Kantelip B, et al. N-cadherin as a novel prognostic marker of progression in superficial urothelial tumors. Clin Cancer Res. (2006) 12:2780–7. doi: 10.1158/1078-0432.CCR-05-2387
107. Shintani Y, Fukumoto Y, Chaika N, Grandgenett PM, Hollingsworth MA, Wheelock MJ, et al. ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int J Cancer. (2008) 122:71–7. doi: 10.1002/ijc.23027
108. Bouvet M, Yang M, Nardin S, Wang X, Jiang P, Baranov E, et al. Chronologically-specific metastatic targeting of human pancreatic tumors in orthotopic models. Clin Exp Metastasis. (2000) 18:213–8. doi: 10.1023/A:1006767405609
109. Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. (1999) 147:631–44. doi: 10.1083/jcb.147.3.631
110. Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. (2000) 148:779–90. doi: 10.1083/jcb.148.4.779
111. Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. (2007) 67:3106–16. doi: 10.1158/0008-5472.CAN-06-3401
112. Takeichi M. Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol. (1993) 5:806–11. doi: 10.1016/0955-0674(93)90029-P
113. Pizarro A, Gamallo C, Benito N, Palacios J, Quintanilla M, Cano A, et al. Differential patterns of placental and epithelial cadherin expression in basal cell carcinoma and in the epidermis overlying tumours. Br J Cancer. (1995) 72:327–32. doi: 10.1038/bjc.1995.333
114. Fujita M, Furukawa F, Fujii K, Horiguchi Y, Takeichi M, Imamura S. Expression of cadherin cell adhesion molecules during human skin development: morphogenesis of epidermis, hair follicles and eccrine sweat ducts. Arch Dermatol Res. (1992) 284:159–66. doi: 10.1007/BF00372710
115. Hibi K, Goto T, Mizukami H, Kitamura YH, Sakuraba K, Sakata M, Saito M, et al. Demethylation of the CDH3 gene is frequently detected in advanced colorectal cancer. Anticancer Res. (2009) 29:2215–7.
116. Hibi K, Kitamura YH, Mizukami H, Goto T, Sakuraba K, Sakata M, et al. Frequent CDH3 demethylation in advanced gastric carcinoma. Anticancer Res. (2009) 29:3945–7.
117. Paredes J, Albergaria A, Oliveira JT, Jeronimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res. (2005) 11:5869–77. doi: 10.1158/1078-0432.CCR-05-0059
118. Xi Y, Zhang X, Yang Z, Zhang X, Guo Q, Zhang Z, et al. Prognositic significance of P-cadherin expression in breast cancer: protocol for a meta-analysis. Medicine (Baltimore). (2019) 98:e14924. doi: 10.1097/MD.0000000000014924
119. Mandeville JA, Silva Neto B, Vanni AJ, Smith GL, Rieger-Christ KM, Zeheb R, et al. P-cadherin as a prognostic indicator and a modulator of migratory behaviour in bladder carcinoma cells. BJU Int. (2008) 102:1707–14. doi: 10.1111/j.1464-410X.2008.08115.x
120. Taniuchi K, Nakagawa H, Hosokawa M, Nakamura T, Eguchi H, Ohigashi H, et al. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer Res. (2005) 65:3092–9. doi: 10.1158/0008.5472.CAN-04-3646
121. Sun L, Hu H, Peng L, Zhou Z, Zhao X, Pan J, et al. P-cadherin promotes liver metastasis and is associated with poor prognosis in colon cancer. Am J Pathol. (2011) 179:380–90. doi: 10.1016/j.ajpath.2011.03.046
122. Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA, Byers SW. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. (1999) 59:947–52.
123. Satriyo PB, Bamodu OA, Chen JH, Aryandono T, Haryana SM, Yeh CT, et al. Cadherin 11 inhibition downregulates β-catenin, deactivates the canonical WNT signalling pathway and suppresses the cancer stem cell-like phenotype of triple negative breast cancer. J Clin Med. (2019) 8:E148. doi: 10.3390/jcm8020148
124. Bartolome RA, Torres S, Isern de Val S, Escudero-Paniagua B, Calvino E, Teixido J, et al. VE-cadherin RGD motifs promote metastasis and constitute a potential therapeutic target in melanoma and breast cancers. Oncotarget. (2017) 8:215–27. doi: 10.18632/oncotarget.13832
125. Shan M, Su Y, Kang W, Gao R, Li X, Zhang G. Aberrant expression and functions of protocadherins in human malignant tumors. Tumour Biol. (2016) 37:12969–81. doi: 10.1007/s13277-016-5169-9
126. Asad M, Wong MK, Tan TZ, Choolani M, Low J, Mori S, et al. FZD7 drives in vitro aggressiveness in Stem-A subtype of ovarian cancer via regulation of non-canonical Wnt/PCP pathway. Cell Death Dis. (2014) 5:e1346. doi: 10.1038/cddis.2014.302
127. Wang C, Tao B, Li S, Li B, Wang X, Hu G, et al. Characterizing the role of PCDH9 in the regulation of glioma cell apoptosis and invasion. J Mol Neurosci. (2014) 52:250–60. doi: 10.1007/s12031-013-0133-2
128. Chen T, Long B, Ren G, Xiang T, Li L, Wang Z, et al. Protocadherin20 acts as a tumor suppressor gene: epigenetic inactivation in nasopharyngeal carcinoma. J Cell Biochem. (2015) 116:1766–75. doi: 10.1002/jcb.25135
129. Wu JC, Wang FZ, Tsai ML, Lo CY, Badmaev V, Ho CT, et al. Se-Allylselenocysteine induces autophagy by modulating the AMPK/mTOR signaling pathway and epigenetic regulation of PCDH17 in human colorectal adenocarcinoma cells. Mol Nutr Food Res. (2015) 59:2511–22. doi: 10.1002/mnfr.201500373
130. Dang Z, Shangguan J, Zhang C, Hu P, Ren Y, Lv Z, et al. Loss of protocadherin-17 (PCDH-17) promotes metastasis and invasion through hyperactivation of EGFR/MEK/ERK signaling pathway in hepatocellular carcinoma. Tumour Biol. (2016) 37:2527–35. doi: 10.1007/s13277-015-3970-5
131. Xu Y, Yang Z, Yuan H, Li Z, Li Y, Liu Q, et al. PCDH10 inhibits cell proliferation of multiple myeloma via the negative regulation of the Wnt/β-catenin/BCL-9 signaling pathway. Oncol Rep. (2015) 34:747–54. doi: 10.3892/or.2015.4056
132. Jao TM, Tsai MH, Lio HY, Weng WT, Chen CC, Tzeng ST, et al. Protocadherin 10 suppresses tumorigenesis and metastasis in colorectal cancer and its genetic loss predicts adverse prognosis. Int J Cancer. (2014) 135:2593–603. doi: 10.1002/ijc.28899
133. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. (2009) 459:1005–9. doi: 10.1038/nature08021
134. Chen MW, Vacherot F, De La Taille A, Gil-Diez-De-Medina S, Shen R, Friedman RA, et al. The emergence of protocadherin-PC expression during the acquisition of apoptosis-resistance by prostate cancer cells. Oncogene. (2002) 21:7861–71. doi: 10.1038/sj.onc.1205991
135. Terry S, Maille P, Baaddi H, Kheuang L, Soyeux P, Nicolaiew N, et al. Cross modulation between the androgen receptor axis and protocadherin-PC in mediating neuroendocrine transdifferentiation and therapeutic resistance of prostate cancer. Neoplasia. (2013) 15:761–72. doi: 10.1593/neo.122070
136. Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. (2017) 20:409–26. doi: 10.1007/s10456-017-9562-9
137. Wu JB, Tang YL, Liang XH. Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy. Onco Targets Ther. (2018) 11:6901–9. doi: 10.2147/OTT.S172042
138. Suzuki S, Sano K, Tanihara H. Diversity of the cadherin family: evidence for eight new cadherins in nervous tissue. Cell Regul. (1991) 2:261–70. doi: 10.1091/mbc.2.4.261
139. Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, et al. Role of vascular endothelial-cadherin in vascular morphogenesis. Development. (1999) 126:2093–102.
140. Kouklis P, Konstantoulaki M, Malik AB. VE-cadherin-induced Cdc42 signaling regulates formation of membrane protrusions in endothelial cells. J Biol Chem. (2003) 278:16230–6. doi: 10.1074/jbc.M212591200
141. Breviario F, Caveda L, Corada M, Martin-Padura I, Navarro P, Golay J, et al. Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler Thromb Vasc Biol. (1995) 15:1229–39. doi: 10.1161/01.ATV.15.8.1229
142. Zhang R, Li R, Tang Y. Soluble vascular endothelial cadherin: a promising marker of critical illness? Crit Care. (2019) 23:57. doi: 10.1186/s13054-019-2343-7
143. Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. Trends Cardiovasc Med. (2006) 16:55–9. doi: 10.1016/j.tcm.2005.11.008
144. Labelle M, Schnittler HJ, Aust DE, Friedrich K, Baretton G, Vestweber D, et al. Vascular endothelial cadherin promotes breast cancer progression via transforming growth factor beta signaling. Cancer Res. (2008) 68:1388–97. doi: 10.1158/0008-5472.CAN-07-2706
145. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. (2008) 121:2115–22. doi: 10.1242/jcs.017897
146. Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. (2004) 113:885–94. doi: 10.1172/JCI200420702
147. Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. (1999) 4:915–24. doi: 10.1016/S1097-2765(00)80221-X
148. Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol. (2003) 161:793–804. doi: 10.1083/jcb.200209019
149. Rudini N, Felici A, Giampietro C, Lampugnani M, Corada M, Swirsding K, et al. VE-cadherin is a critical endothelial regulator of TGF-beta signalling. EMBO J. (2008) 27:993–1004. doi: 10.1038/emboj.2008.46
150. Hou WH, Liu IH, Tsai CC, Johnson FE, Huang SS, Huang JS. CRSBP-1/LYVE-1 ligands disrupt lymphatic intercellular adhesion by inducing tyrosine phosphorylation and internalization of VE-cadherin. J Cell Sci. (2011) 124:1231–44. doi: 10.1242/jcs.078154
151. Giampietro C, Taddei A, Corada M, Sarra-Ferraris GM, Alcalay M, Cavallaro U, et al. Overlapping and divergent signaling pathways of N-cadherin and VE-cadherin in endothelial cells. Blood. (2012) 119:2159–70. doi: 10.1182/blood-2011-09-381012
152. Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. (2003) 314:15–23. doi: 10.1007/s00441-003-0745-x
153. Zhuo H, Zhao Y, Cheng X, Xu M, Wang L, Lin L, et al. Tumor endothelial cell-derived cadherin-2 promotes angiogenesis and has prognostic significance for lung adenocarcinoma. Mol Cancer. (2019) 18:34. doi: 10.1186/s12943-019-0987-1
154. Gerhardt H, Wolburg H, Redies C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev Dyn. (2000) 218:472–9. doi: 10.1002/1097-0177(200007)218:3<472::AID-DVDY1008>3.0.CO;2-#
155. Luo Y, Radice GL. N-cadherin acts upstream of VE-cadherin in controlling vascular morphogenesis. J Cell Biol. (2005) 169:29–34. doi: 10.1083/jcb.200411127
156. Nalla AK, Estes N, Patel J, Rao JS. N-cadherin mediates angiogenesis by regulating monocyte chemoattractant protein-1 expression via PI3K/Akt signaling in prostate cancer cells. Exp Cell Res. (2011) 317:2512–21. doi: 10.1016/j.yexcr.2011.07.024
157. Viollet C, Doherty P. CAMs and the FGF receptor: an interacting role in axonal growth. Cell Tissue Res. (1997) 290:451–5. doi: 10.1007/s004410050952
158. Saffell JL, Williams EJ, Mason IJ, Walsh FS, Doherty P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron. (1997) 18:231–42. doi: 10.1016/S0896-6273(00)80264-0
159. Derycke L, Morbidelli L, Ziche M, De Wever O, Bracke M, Van Aken E. Soluble N-cadherin fragment promotes angiogenesis. Clin Exp Metastasis. (2006) 23:187–201. doi: 10.1007/s10585-006-9029-7
160. Fridman WH, Dieu-Nosjean MC, Pages F, Cremer I, Damotte D, Sautes-Fridman C, et al. The immune microenvironment of human tumors: general significance and clinical impact. Cancer Microenviron. (2013) 6:117–22. doi: 10.1007/s12307-012-0124-9
161. Van den Bossche J, Malissen B, Mantovani A, De Baetselier P, Van Ginderachter JA. Regulation and function of the E-cadherin/catenin complex in cells of the monocyte-macrophage lineage and DCs. Blood. (2012) 119:1623–33. doi: 10.1182/blood-2011-10-384289
162. Nawijn MC, Hackett TL, Postma DS, van Oosterhout AJ, Heijink IH. E-cadherin: gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. (2011) 32:248–55. doi: 10.1016/j.it.2011.03.004
163. Jiang Y, Wan T, Chen G, Xiu F, Xia D, Zhang W, et al. DC-CLM, a cadherin-like molecule cloned from human dendritic cells, inhibits growth of breast cancer cells. J Cancer Res Clin Oncol. (2003) 129:57–64. doi: 10.1007/s00432-002-0404-8
164. Riedl E, Stockl J, Majdic O, Scheinecker C, Knapp W, Strobl H. Ligation of E-cadherin on in vitro-generated immature Langerhans-type dendritic cells inhibits their maturation. Blood. (2000) 96:4276–84.
165. Padovan E, Terracciano L, Certa U, Jacobs B, Reschner A, Bolli M, et al. Interferon stimulated gene 15 constitutively produced by melanoma cells induces E-cadherin expression on human dendritic cells. Cancer Res. (2002) 62:3453–8.
166. Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. (2007) 27:610–24. doi: 10.1016/j.immuni.2007.08.015
167. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. (2010) 329:849–53. doi: 10.1126/science.1188510
168. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. (2009) 9:259–70. doi: 10.1038/nri2528
169. Rehli M, Sulzbacher S, Pape S, Ravasi T, Wells CA, Heinz S, et al. Transcription factor Tfec contributes to the IL-4-inducible expression of a small group of genes in mouse macrophages including the granulocyte colony-stimulating factor receptor. J Immunol. (2005) 174:7111–22. doi: 10.4049/jimmunol.174.11.7111
170. Ghassabeh GH, De Baetselier P, Brys L, Noel W, Van Ginderachter JA, Meerschaut S, et al. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood. (2006) 108:575–83. doi: 10.1182/blood-2005-04-1485
171. Wanat KA, Rosenbach M, Zoiber AF, Zhang PJ, Schaffer A. E-cadherin is expressed by mono- and multinucleated histiocytes in cutaneous sarcoidal and foreign body granulomas. Am J Dermatopathol. (2014) 36:651–4. doi: 10.1097/DAD.0b013e31828de7e0
172. Van den Bossche J, Bogaert P, van Hengel J, Guerin CJ, Berx G, Movahedi K, et al. Alternatively activated macrophages engage in homotypic and heterotypic interactions through IL-4 and polyamine-induced E-cadherin/catenin complexes. Blood. (2009) 114:4664–74. doi: 10.1182/blood-2009-05-221598
173. Van den Bossche J, Laoui D, Naessens T, Smits HH, Hokke CH, Stijlemans B, et al. E-cadherin expression in macrophages dampens their inflammatory responsiveness in vitro, but does not modulate M2-regulated pathologies in vivo. Sci Rep. (2015) 5:12599. doi: 10.1038/srep12599
174. Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
175. Schietke R, Warnecke C, Wacker I, Schodel J, Mole DR, Campean V, et al. The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF-1. J Biol Chem. (2010) 285:6658–69. doi: 10.1074/jbc.M109.042424
176. Moreno JL, Mikhailenko I, Tondravi MM, Keegan AD. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: contribution of E-cadherin. J Leukoc Biol. (2007) 82:1542–53. doi: 10.1189/jlb.0107058
177. Schaale K, Neumann J, Schneider D, Ehlers S, Reiling N. Wnt signaling in macrophages: augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur J Cell Biol. (2011) 90:553–9. doi: 10.1016/j.ejcb.2010.11.004
178. Li Y, Hofmann M, Wang Q, Teng L, Chlewicki LK, Pircher H, et al. Structure of natural killer cell receptor KLRG1 bound to E-cadherin reveals basis for MHC-independent missing self recognition. Immunity. (2009) 31:35–46. doi: 10.1016/j.immuni.2009.04.019
179. Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. (2006) 203:289–95. doi: 10.1084/jem.20051986
180. Robbins SH, Nguyen KB, Takahashi N, Mikayama T, Biron CA, Brossay L. Cutting edge: inhibitory functions of the killer cell lectin-like receptor G1 molecule during the activation of mouse NK cells. J Immunol. (2002) 168:2585–9. doi: 10.4049/jimmunol.168.6.2585
181. Robbins SH, Terrizzi SC, Sydora BC, Mikayama T, Brossay L. Differential regulation of killer cell lectin-like receptor G1 expression on T cells. J Immunol. (2003) 170:5876–85. doi: 10.4049/jimmunol.170.12.5876
182. Beyersdorf N, Ding X, Tietze JK, Hanke T. Characterization of mouse CD4 T cell subsets defined by expression of KLRG1. Eur J Immunol. (2007) 37:3445–54. doi: 10.1002/eji.200737126
183. Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. (1994) 372:190–3. doi: 10.1038/372190a0
184. Yu Y, Feng XH. TGF-beta signaling in cell fate control and cancer. Curr Opin Cell Biol. (2019) 61:56–63. doi: 10.1016/j.ceb.2019.07.007
185. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. (2019) 18:26. doi: 10.1186/s12943-019-0954-x
186. Galluzzi L, Spranger S, Fuchs E, Lopez-Soto A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. (2019) 29:44–65. doi: 10.1016/j.tcb.2018.08.005
187. Olgen S. Overview on anticancer drug design and development. Curr Med Chem. (2018) 25:1704–19. doi: 10.2174/0929867325666171129215610
188. Kumar B, Singh S, Skvortsova I, Kumar V. Promising targets in anti-cancer drug development: recent updates. Curr Med Chem. (2017) 24:4729–52. doi: 10.2174/0929867324666170331123648
189. Zajac M, Muszalska I, Jelinska A. New molecular targets of anticancer therapy - current status and perspectives. Curr Med Chem. (2016) 23:4176–220. doi: 10.2174/0929867323666160814002150
190. Wilson CW, Ye W. Regulation of vascular endothelial junction stability and remodeling through Rap1-Rasip1 signaling. Cell Adhes Migr. (2014) 8:76–83. doi: 10.4161/cam.28115
191. May C, Doody JF, Abdullah R, Balderes P, Xu X, Chen CP, et al. Identification of a transiently exposed VE-cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood. (2005) 105:4337–44. doi: 10.1182/blood-2005-01-0010
192. Corada M, Zanetta L, Orsenigo F, Breviario F, Lampugnani MG, Bernasconi S, et al. A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood. (2002) 100:905–11. doi: 10.1182/blood.V100.3.905
193. Liao F, Li Y, O'Connor W, Zanetta L, Bassi R, Santiago A, et al. Monoclonal antibody to vascular endothelial-cadherin is a potent inhibitor of angiogenesis, tumor growth, and metastasis. Cancer Res. (2000) 60:6805–10.
194. Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci USA. (1999) 96:9815–20. doi: 10.1073/pnas.96.17.9815
195. Liao F, Doody JF, Overholser J, Finnerty B, Bassi R, Wu Y, et al. Selective targeting of angiogenic tumor vasculature by vascular endothelial-cadherin antibody inhibits tumor growth without affecting vascular permeability. Cancer Res. (2002) 62:2567–75.
196. Devemy E, Blaschuk OW. Identification of a novel N-cadherin antagonist. Peptides. (2008) 29:1853–61. doi: 10.1016/j.peptides.2008.06.025
197. Williams E, Williams G, Gour BJ, Blaschuk OW, Doherty P. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. (2000) 275:4007–12. doi: 10.1074/jbc.275.6.4007
198. Blaschuk OW. N-cadherin antagonists as oncology therapeutics. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140039. doi: 10.1098/rstb.2014.0039
199. Harrison OJ, Corps EM, Berge T, Kilshaw PJ. The mechanism of cell adhesion by classical cadherins: the role of domain 1. J Cell Sci. (2005) 118:711–21. doi: 10.1242/jcs.01665
200. Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. (2010) 16:1414–20. doi: 10.1038/nm.2236
201. Lee YC, Bilen MA, Yu G, Lin SC, Huang CF, Ortiz A, et al. Inhibition of cell adhesion by a cadherin-11 antibody thwarts bone metastasis. Mol Cancer Res. (2013) 11:1401–11. doi: 10.1158/1541-7786.MCR-13-0108
202. Corada M, Liao F, Lindgren M, Lampugnani MG, Breviario F, Frank R, et al. Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood. (2001) 97:1679–84. doi: 10.1182/blood.V97.6.1679
203. Burg N, Swendeman S, Worgall S, Hla T, Salmon JE. Sphingosine 1-phosphate receptor 1 signaling maintains endothelial cell barrier function and protects against immune complex-induced vascular injury. Arthritis Rheumatol. (2018) 70:1879–89. doi: 10.1002/art.40558
204. Blaschuk OW. Discovery and development of N-cadherin antagonists. Cell Tissue Res. (2012) 348:309–13. doi: 10.1007/s00441-011-1320-5
205. Blaschuk OW, Sullivan R, David S, Pouliot Y. Identification of a cadherin cell adhesion recognition sequence. Dev Biol. (1990) 139:227–9. doi: 10.1016/0012-1606(90)90290-Y
206. Sadler NM, Harris BR, Metzger BA, Kirshner J. N-cadherin impedes proliferation of the multiple myeloma cancer stem cells. Am J Blood Res. (2013) 3:271–85.
207. Lammens T, Swerts K, Derycke L, De Craemer A, De Brouwer S, De Preter K, et al. N-cadherin in neuroblastoma disease: expression and clinical significance. PLoS ONE. (2012) 7:e31206. doi: 10.1371/journal.pone.0031206
208. Kelland L. Drug evaluation: ADH-1, an N-cadherin antagonist targeting cancer vascularization. Curr Opin Mol Ther. (2007) 9:86–91.
209. Erez N, Zamir E, Gour BJ, Blaschuk OW, Geiger B. Induction of apoptosis in cultured endothelial cells by a cadherin antagonist peptide: involvement of fibroblast growth factor receptor-mediated signalling. Exp Cell Res. (2004) 294:366–78. doi: 10.1016/j.yexcr.2003.11.033
210. Mrozik KM, Cheong CM, Hewett D, Chow AW, Blaschuk OW, Zannettino AC, et al. Therapeutic targeting of N-cadherin is an effective treatment for multiple myeloma. Br J Haematol. (2015) 171:387–99. doi: 10.1111/bjh.13596
211. Mariotti A, Perotti A, Sessa C, Ruegg C. N-cadherin as a therapeutic target in cancer. Expert Opin Investig Drugs. (2007) 16:451–65. doi: 10.1517/13543784.16.4.451
212. Groen RW, de Rooij MF, Kocemba KA, Reijmers RM, de Haan-Kramer A, Overdijk MB, et al. N-cadherin-mediated interaction with multiple myeloma cells inhibits osteoblast differentiation. Haematologica. (2011) 96:1653–61. doi: 10.3324/haematol.2010.038133
213. Volk T, Volberg T, Sabanay I, Geiger B. Cleavage of A-CAM by endogenous proteinases in cultured lens cells and in developing chick embryos. Dev Biol. (1990) 139:314–26. doi: 10.1016/0012-1606(90)90301-X
214. Eiring AM, Khorashad JS, Anderson DJ, Yu F, Redwine HM, Mason CC, et al. β-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. (2015) 29:2328–37. doi: 10.1038/leu.2015.196
215. Gao Y, Qu Y, Zhou Q, Ma Y. SIRT6 inhibits proliferation and invasion in osteosarcoma cells by targeting N-cadherin. Oncol Lett. (2019) 17:1237–44. doi: 10.3892/ol.2018.9676
Keywords: cadherin signaling, tumorigenesis, tumor progression, angiogenesis, tumor immune response, therapeutic target
Citation: Yu W, Yang L, Li T and Zhang Y (2019) Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front. Oncol. 9:989. doi: 10.3389/fonc.2019.00989
Received: 14 May 2019; Accepted: 16 September 2019;
Published: 04 October 2019.
Edited by:
Paolo E. Porporato, University of Turin, ItalyReviewed by:
Kenji Mandai, Kitasato University, JapanXiang Yu, University of Pennsylvania, United States
Copyright © 2019 Yu, Yang, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhang, eWl6aGFuZ0B6enUuZWR1LmNu
†These authors have contributed equally to this work