 Sangita Sridharan1†
Sangita Sridharan1† Cory M. Howard1†
Cory M. Howard1† Augustus M. C. Tilley1†
Augustus M. C. Tilley1† Boopathi Subramaniyan1†
Boopathi Subramaniyan1† Amit K. Tiwari2
Amit K. Tiwari2 Randall J. Ruch1
Randall J. Ruch1 Dayanidhi Raman1*
Dayanidhi Raman1*- 1Department of Cancer Biology, University of Toledo, Toledo, OH, United States
- 2Department of Pharmacology and Experimental Therapeutics, University of Toledo, Toledo, OH, United States
Breast cancer stem cells (BCSCs) play a vital role in tumor progression and metastasis. They are heterogeneous and inherently radio- and chemoresistant. They have the ability to self-renew and differentiate into non-BCSCs. These determinants of BCSCs including the plasticity between the mesenchymal and epithelial phenotypes often leads to minimal residual disease (MRD), tumor relapse, and therapy failure. By studying the resistance mechanisms in BCSCs, a combinatorial therapy can be formulated to co-target BCSCs and bulk tumor cells. This review addresses breast cancer stemness and molecular underpinnings of how the cancer stemness can lead to pharmacological resistance. This might occur through rewiring of signaling pathways and modulated expression of various targets that support survival and self-renewal, clonogenicity, and multi-lineage differentiation into heterogeneous bulk tumor cells following chemotherapy. We explore emerging novel and alternative molecular targets against BC stemness and chemoresistance involving survival, drug efflux, metabolism, proliferation, cell migration, invasion, and metastasis. Strategic targeting of such vulnerabilities in BCSCs may overcome the chemoresistance and increase the longevity of the metastatic breast cancer patients.
Introduction
Breast cancer (BC) is the second leading cause of death in women among the cancer mortalities. With the new estimates in 2019, 3 out of 10 women (30%) will develop BC in her lifetime and 1 in 7 (15%) will succumb to BC (1). Mortality in BC patients is mainly due to metastasis to the lungs, bone and the brain. Breast cancer is a heterogeneous disease with differential expression of several molecular markers. Luminal BC expresses estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). The triple-negative breast cancer (TNBC) subtype lacks the expression of all of the above three markers. Based on the gene expression profile, TNBC is classified into basal-like1 (BL1) and basal-like2 (BL2), mesenchymal (M), mesenchymal stem-like (MSL), immunomodulatory (IM), and luminal androgen receptor (LAR) types (2, 3). The pathological complete response (pCR) rate with BL1, BL2, LAR, and MSL tumors are 52, 0, 10, and 23%, respectively (4). Moreover, intra- and inter-tumor heterogeneity (clonal diversity), and plasticity observed in TNBC (5–11) leads to chemoresistance, tumor relapse, and poor patient outcome. Both luminal and TNBC are reported to contain a small subpopulation of cells amidst the bulk tumor cells called breast cancer stem cells (BCSCs) or tumor initiating cells. BCSCs are capable of self-renewal, tumor initiation and differentiation into bulk tumor cells. BCSCs are intrinsically chemoresistant and can repopulate the tumor following chemotherapy and ionizing radiation. This ultimately leads to therapy failure, distant metastasis, or metastasis of the metastases, tumor relapse and mortality. This is especially true in luminal HER2+ and TNBC tumor types. TNBC is highly lethal (5-year mortality >75%), characterized by aggressive growth, therapy failure, and lack of successful targeted therapies (12, 13). TNBC patients often exhibit initial sensitivity to neoadjuvant chemotherapy, but eventually become refractory to such therapy presumably due to BCSCs. BCSCs are thus clinically important and there is an unmet need to co-target BCSCs along with bulk tumor cells. The co-targeting approach may overcome chemoresistance, molecular and metabolic plasticity, and most importantly reduce mortality and improve longevity in metastatic BC patients (14–18). In order to design effective and rational therapies against BCSCs, it is imperative to find novel and actionable molecular targets to combine effectively with current available therapies. This review focuses on emerging molecular targets that could become potential BCSC-directed precision therapies to overcome clinical chemoresistance.
Heterogeneity in BC and Its Influence on Clinical Outcome
The heterogeneity in BC cells can arise by stochastic genetic or epigenetic (clonal evolution) changes. The paracrine interactions of the tumor cells with its microenvironment (TME) can also confer phenotypic and functional heterogeneity based on the spatiotemporal dynamics (location of the tumor and continual changes in the cellular and acellular TME) of the constantly evolving tumors in the same patient (19–22). Metabolic heterogeneity has been reported to exist in BC organoids by employing optical metabolic imaging (OMI) similar to in vivo situation (23). OMI has been suggested to predict any potentially unresponsive subpopulation of cells within the tumor.
Heterogeneity exists among BCSCs as well (24). By isolating BCSCs based on high flavin content, energetic BCSCs (e-BCSCs) were identified with a higher glycolytic activity and a larger mitochondrial mass (25). On the contrary, quiescent BCSCs (qBCSCs) have been reported based on the epigenetic activities (26). Mesenchymal and epithelial phenotypes of heterogeneous BCSCs have been described contributing to differential chemoresistance (27). Notch-Jagged signaling has been proposed to contribute to heterogeneity in BCSCs with more mesenchymal BCSCs at the invasive edge and the hybrid epithelial/mesenchymal (E/M) BCSCs in the center of the tumor (24). Interestingly, ITGB4+-enriched BCSCs have been reported to reside in an intermediate E/M phenotypic state (28). Mathematical modeling coupled with data on single-cell sequencing of BCSCs has been suggested to dissect the heterogeneity. This will also help our understanding of the replication and invasive dynamics of BC cells during cancer progression and importantly in response to therapy (29).
Single cell sequencing (sc-seq) technology (single-cell genomics and transcriptomics) has pioneered our understanding of intra-tumoral genetic heterogeneity, the cancer genome evolution and also phenotypic diversity (30–32). Understanding molecular and genetic variations at the single cell level and as an ensemble in the tumor will provide mechanisms of chemoresistance. Chemoresistance and relapse can also occur in patients undergoing combination chemotherapy. In such cases, tapping the circulating tumor cells (CTCs) by liquid biopsy would enable assessment of the tumor cells for any molecular or genetic changes following chemotherapy. Many of the CTCs are BCSCs and one can examine for ratios of BCSCs to tumor cells (CD44 vs. CD24 and ALDH staining) before, during and after therapy. The isolated CTCs/BCSCs can be subjected to sc-seq for genomic, epigenomic, and transcriptomic analysis. Using this approach, continuously activated T-cells were identified in the cellular TME. Additionally, it revealed a co-existence of M1 and M2 macrophage polarization genes in the same cell indicating that macrophages fall along a spectrum between the two states (33). Also, aldehyde dehydrogenase (ALDH+) positive BCSCs at the single cell level analysis, exhibited hybrid epithelial/mesenchymal phenotype with a gene expression associated with aggressive TNBC (34). Identification of biomarkers predictive of therapy response and emergence of resistance following therapy based on sc-seq would prove valuable (17).
tRNA as Predictive Biomarkers in BCSCs
Transfer RNA (tRNA)-derived small non-coding RNAs (tDRs) are novel small non-coding RNAs (sncRNA) that have been demonstrated in some human diseases and biological processes. BCSCs isolated by the expression of CD44+/CD24−/low surface markers were tested for tDR expression profiles in TNBC and non-TNBC types by RNA sequencing (RNA-Seq). Among a total of 1,327 differentially expressed tDRs, 18 were upregulated and 54 were downregulated in the TNBC group. The expression level of tDR-000620 was consistently lower in BCSCs derived from TNBC cell lines and patient serum samples. Interestingly, tDR-000620 expression (p = 0.002) and the node status (p = 0.001) groups were statistically significant with recurrence-free survival (35).
tRNA-derived fragments (tRF) also serve as predictive biomarkers (36). tRF-30-JZOYJE22RR33 and tRF-27-ZDXPHO53KSN were correlated with trastuzumab resistance (37). The tDRs such as tDR-0009 [derived from transfer RNA (tRNA)Gly−GCC−1−1] and tDR-7336 (derived from tRNA Gly−GCC−1−2) were significantly upregulated when the SUM-1315 cell line was subjected to hypoxic conditions. The protein-protein interaction network from the STRING database identified that tDR-0009 may be involved in imparting chemoresistance to TNBC cells through the regulation of STAT3 activation. Specific tDRs act as regulatory factors in hypoxia-induced chemoresistance in TNBC, and they could serve as predictive biomarkers (38). In HER2-overexpressing breast cancer, there is an ongoing clinical trial evaluating molecular biomarkers to predict the efficacy of the Trastuzumab therapy and recurrence (NCT03521245).
Breast Cancer Stem Cells
BCSCs through their self-renewal capacity can initiate tumorigenesis, contribute to primary tumor progression, local invasion, and distant metastases (39). Historically, CSCs have been described as a “side population” (SP) by flow cytometric analyses based on the exclusion of the Hoechst dye by the drug transporters in CSCs. This reflects their capability to exclude xenobiotics including anti-cancer drugs to outside of the cell. There is spatial and temporal variability in the expression of stemness markers by BCSCs such as CD44 (Hyaluronan receptor) (39), CD133 (40, 41), CD49f+ (Integrin-α6) (42), epithelial cell adhesion molecule (EpCAM), chemokine receptor CXCR4, transcription factors [SRY (sex determining region Y) box 2—SOX2, homeobox protein Nanog, and octamer-binding transcription factor 4 (OCT4)] and aldehyde dehydrogenase (ALDH) activity (39). A small fraction of BCSCs express both CD44 and ALDH markers and are considered highly metastatic (39, 43). Interestingly, there are 2 isoforms of CD44 with opposite functions. The standard isoform of CD44 (CD44s) promotes BCSC stemness whereas the CD44 variant form (CD44v) opposes it (44). SOX2 works in conjunction with cyclin-dependent kinases 4/6 to transactivate the Cyclin D1 promoter, which facilitates proliferation and clonogenicity (45, 46). In TNBC, SOX2 promotes proliferation, and metastasis (47). SOX2 also promotes tamoxifen resistance (48) and a SOX2-SOX9 signaling axis was reported to maintain BCSCs (49). Resistance to tamoxifen by ER+-BCSCs was attributed to SOX9-FXYD Domain Containing Ion Transport Regulator 3 (FXYD3)-Src axis (50). Basically, significantly upregulated expression of FXYD3 is crucial for mediating tamoxifen resistance in ER+-BCSCs. FXYD3 is critical for the nuclear localization of SOX9 which in turn directly promotes the expression of FXYD3 forming a positive feedback loop. The trimeric complex consisting of FXYD3, ER-α and c-Src which transduces non-genomic estrogen signaling which facilitates the activity of ER+-BCSCs. Nanog is also involved in the maintenance of pluripotency and self-renewal of BCSCs. An increased expression of Nanog serves as a prognostic indicator and was suggested to be co-expressed with the CD133 marker (22, 51–53). OCT4 expression has been suggested to be a worse prognostic marker for surgical TNBC patients (54). Expression of SOX2, Nanog and OCT4 transcription factors correlated with poor differentiation, advanced BC stage and worst survival in BC patients with HER2 positivity (55). The expression of cell surface and subcellular markers of BCSCs is not a static property as they change in response to their microenvironment. Mesenchymal and epithelial phenotypes of BCSCs have been described with distinct gene expression profiles and contribute to heterogeneity and differential chemoresistance (27). The differential characteristics between these cells are described in Table 1. A hybrid version of BCSCs has been suggested to exist with both epithelial and mesenchymal stem cells markers in the center of the tumor (24, 28). Generally, mesenchymal BCSCs are more resistant to chemotherapy than the epithelial type (69). Interconversion between them occurs at a slow rate which we call “stem cell buffering” (SCB) (27, 70). The innate plasticity of BCSCs, thus contributes to tumor heterogeneity and chemoresistance. The BCSCs can dynamically oscillate between a bulk tumor cell type and stemness state based on temporal and spatial context in the microenvironment around the BCSC (22). For example, chemotherapy may first induce a BCSC phenotype conversion from bulk tumor cells. Following cessation of therapy, cells may revert to bulk tumor cells. Additionally, there is heterogeneity in BCSC pools in which the subsets of BCSCs have differing abilities ranging from quiescence, chemoresistance, interconversion between epithelial to mesenchymal types, proliferation, local invasion and metastasis. Thus, there is remarkable genetic and/or epigenetic heterogeneity and cellular plasticity in BCSCs and bulk tumor cells presenting clinical challenges. Thus, it is imperative to develop targeted therapies against the “mosaic nature” of BCSCs along with the co-targeting of bulk tumor cells.
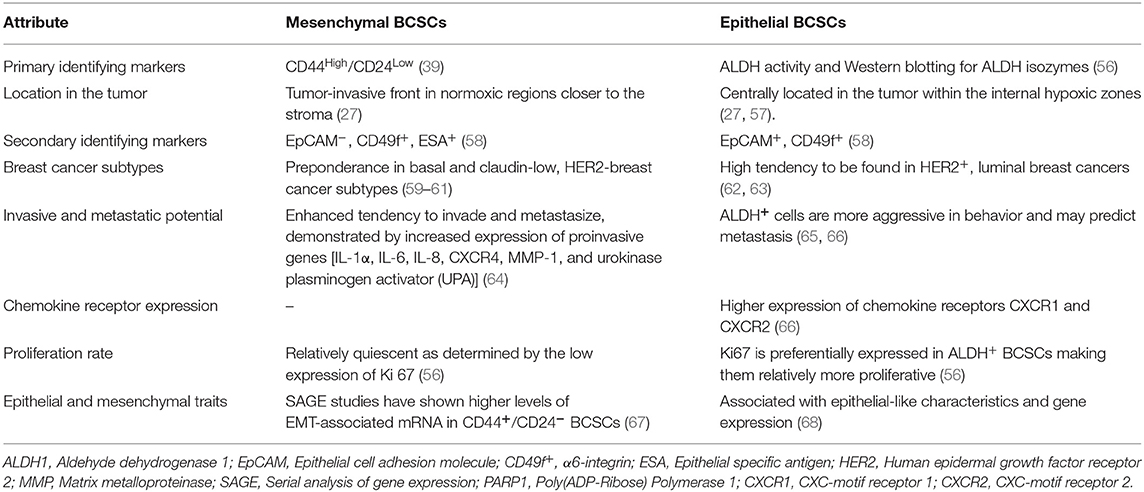
Table 1. Differential characteristics of Mesenchymal vs. Epithelial BCSCs.
The Link Between Epithelial-Mesenchymal Transition (EMT) and BCSCs
The mechanistic evidence suggests that EMT and the acquisition of BC stemness are correlated (69). Following the experimental activation of the EMT program (HMLER cells), induction of the autocrine signaling loops that were known to associate with cancer stemness were observed. Importantly, blocking the autocrine pathways was sufficient to abolish the CSC properties. This brings out the causal link between EMT and induction of BC stemness. The EMT program can also contribute to cancer stemness through its effects on intracellular signaling pathways. For instance, EMT-transcription factor (EMT-TF) Snail1 has been reported to diminish the expression of p53 in tumor cells through the formation of a ternary complex consisting of a Snail1, histone deacetylase 1 (HDAC1) and p53. This ternary association leads to deacetylation of p53 and its degradation (71). TGF-β signaling pathway has also been demonstrated to induce the expression of EMT-TFs such as Twist, Snail1, and Slug (72). In early breast cancer patients, a spectrum of EMT phenotypes in circulating tumor cells (CTCs) has been reported (73).
The BC Stemness State Imparts Therapy Resistance in the Clinics
BCSCs possess the intrinsic ability to survive cytotoxic therapy through a variety of mechanisms. They include upregulation of anti-apoptotic proteins, activation of alternate survival pathways, drug efflux or ATP-binding cassette (ABC) transporters, detoxification/reduction of reactive oxygen species (ROS) (18, 74), and an enhanced capacity for DNA repair (75, 76). Myeloid cell leukemia 1 (MCL1) is one of the key proteins involved in the survival of BCSCs (77, 78). Both MYC and the anti-apoptotic protein MCL1 co-operate in BCSCs to promote chemoresistance through mitochondrial oxidative phosphorylation (79). Treatment of TNBC with “mammalian target of rapamycin complex 1/2” (mTORC1/2) inhibitors led to sustained drug-resistance in Notch1-dependent BCSCs (80). Interestingly, the Notch-mediated tumor-stroma-inflammatory network promoted tumor invasiveness and secretion of the chemokine CXCL8. CXCL8 promotes BC stemness through its action on the chemokine receptors CXCR1 and CXCR2 on BCSCs (81). The survival and resistance through upregulation and rewiring of alternate pathways in breast cancer is provided in Table 2.
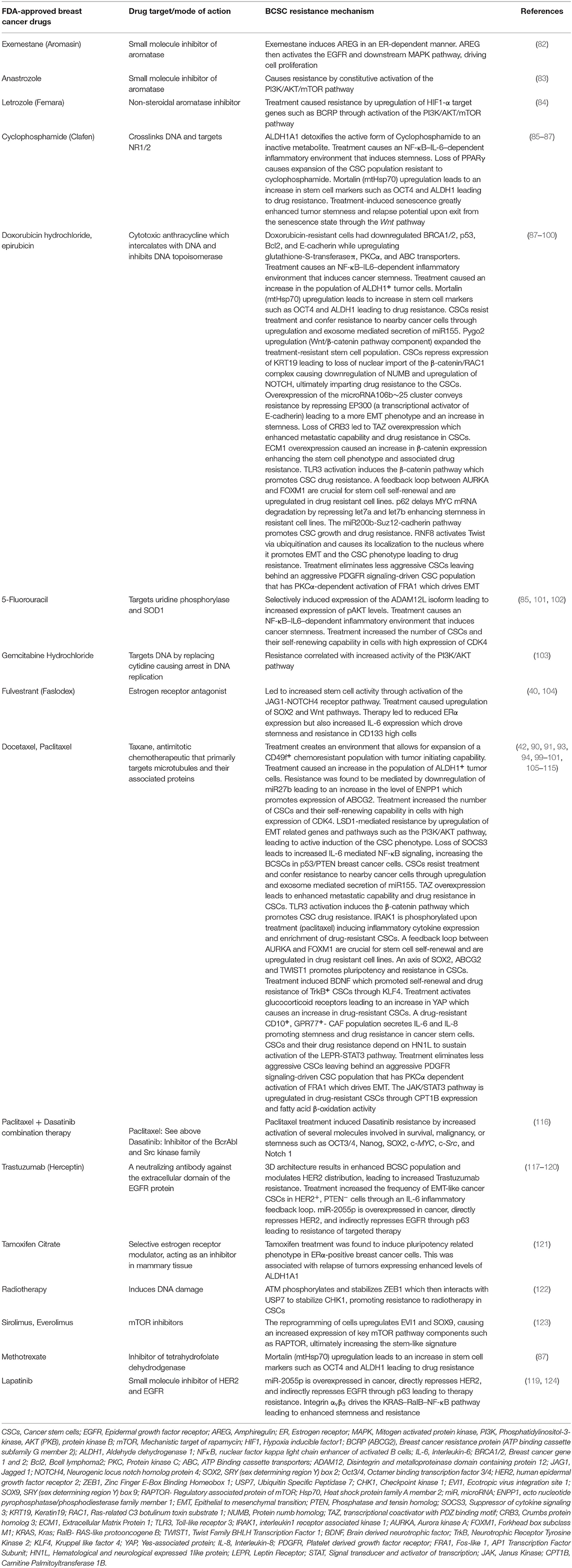
Table 2. Resistance mechanisms encountered in BCSCs during or after therapy.
Resistance Arising From BCSCs and the Tumor Microenvironment
Currently, there are no clearly defined, targeted inhibitors for BCSCs established for successful BCSC-directed therapy. One of the key considerations for such targeted therapy is that the selected targets should be enriched in BCSCs. If the target is not enriched, at least the relative susceptibility of BCSCs should be present. Alternatively, dual targeting of BCSCs and bulk tumor cells with synergistic inhibitors may prevent activation of alternative survival pathways and subsequent chemoresistance. Generally, one should avoid targeting molecular nodes that are common to both BCSCs and normal mammary stem cells (MaSCs) or other stem cells in the body. At the least, the developed inhibitors should be minimally toxic to MaSCs. Alternatively, one has to specifically deliver drugs that have efficacy against BCSCs by employing a targeted delivery approach such as nanotechnology (tumor-homing nanoparticles or nanospheres) (125). In addition to paracrine input from bulk tumor cells, BCSCs depend on the surrounding tumor microenvironment (TME) called the “BCSC niche.” The “BCSC niche” is currently a high value therapeutic target. BCSCs interact constantly with the cellular component of the niche including neutrophils, macrophages, endothelial, and endothelial progenitor cells, mesenchymal stem cells and carcinoma associated fibroblasts (CAFs) (126–128). The signaling cues from the acellular TME such as cytokines, chemokines, growth factors, and some hormones activate many signaling pathways in BCSCs and form attractive targets in BCSCs (Figures 1, 2). For example, the chemokine CXCL8 or interleukin-8 (IL-8) and the hormone erythropoietin activated survival signaling pathways protect BCSCs following chemotherapy (129–131). Inflammatory components from TME also feed into BCSCs. Inhibition of cyclooxygenase-2 (COX-2) led to blockade of TGFβ-induced enrichment of two morphologically distinct BCSC populations; CD44hi/CD24lo and ALDH+ (43, 132). The role of hedgehog (Hh), Notch, and Wnt signaling pathways in CSCs has been reviewed previously (53). Additionally, efflux transporters are also implicated in clonogenicity, pluripotency, and survival of BCSCs against cytotoxic chemotherapy (133, 134).
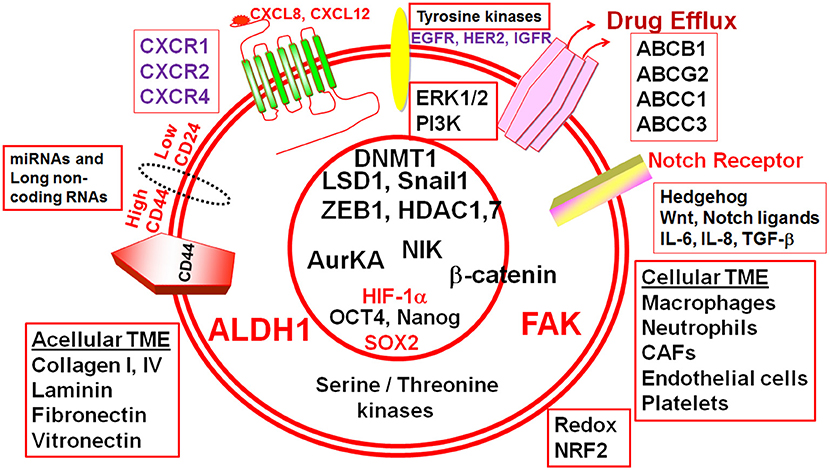
Figure 1. Cellular and acellular tumor microenvironment (TME) shape the response of breast cancer stem cells (BCSCs). The schematic diagram depicts different molecular players that execute the functionality of BCSCs and form potential actionable molecular targets in BCSCs. ALDH1, Aldehyde dehydrogenase 1; CD44, Cluster differentiation antigen 44; CD24, Cluster differentiation antigen 24; EGFR, Epidermal growth factor receptor; HER2, Human epidermal growth factor receptor 2; IGFR, Insulin-like growth factor receptor; CXCR1, CXC-motif receptor 1; CXCR2, CXC-motif receptor 2; CXCR4, CXC-motif receptor 4; ERK1/2, Extracellular signal regulated kinase1/2; PI3K, Phosphatidylinositol-3-kinase, FAK, Focal adhesion kinase; HIF-1α, Hypoxia inducible factor-1α; ABC, ATP Binding cassette transporters; BCRP (ABCG2), Breast cancer resistance protein (ATP binding cassette subfamily G member 2); ABCB1, ATP binding cassette subfamily B member 1; ABCC, ATP binding cassette subfamily C member 1; ABCC3, ATP binding cassette subfamily C member 3; IL-6, Interleukin-6; IL-8, Interleukin-8; SOX2, SRY (sex determining region Y) box 2; OCT4, Octamer binding transcription factor 4; ZEB1, Zinc Finger E-Box Binding Homeobox 1; miR, microRNA; AURKA, Aurora kinase A; NIK, NF-kB inducible kinase; AurKA, Aurora Kinase A; LSD1, Lysine-specific demethylase1; HDAC1, Histone deacetylase1; HDAC7, Histone deacetylase7; DNMT1, DNA methyltransferase1; TGF-β, Transforming growth factor-β; NRF2, NF-E2-related factor 2.
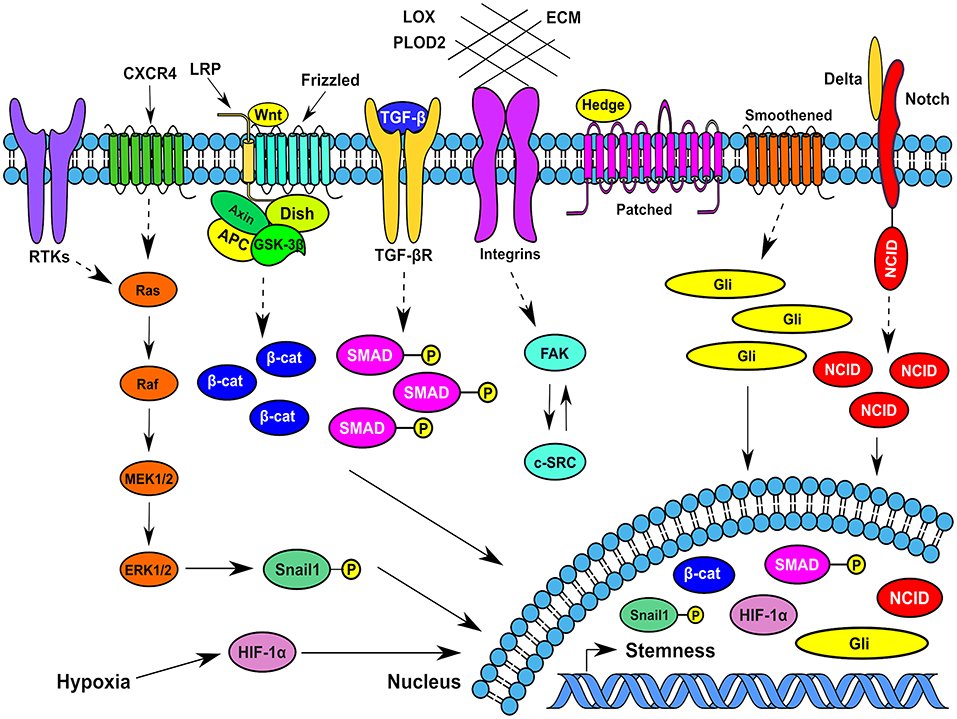
Figure 2. Typical signaling pathways operating in breast cancer stem cells at different spatiotemporal contexts in the tumor microenvionment. Several signaling pathways that function in BCSCs impinge on transcription factors such as Snail1, β-catenin, Gli1, HIF-1α, phospho-SMADs, and Notch intracellular domain that traverses to the nucleus and increase or maintain the breast cancer stemness. CXCR4, CXC-motif receptor 4; RTKs, Receptor tyrosine kinases; MEK1/2, Mitogen activated protein kinase kinase 1/2; ERK1/2, Extracellular signal regulated kinase1/2; HIF-1α, Hypoxia inducible factor-1α; β-cat, β-catenin; ECM, Extracellular Matrix; FAK, Focal adhesion kinase; Gli- a transcription factor; SMAD, Homologous to Caenorhabditis elegans SMA (“small” worm phenotype) and Drosophila MAD (“Mothers Against Decapentaplegic”); Dish, Disheveled; Hedge, Hedgehog; APC-Adenomatous Polyposis Coli; c-SRC, cellular protooncogene similar to viral sarcoma; GSK-3β, Glycogen synthase kinase-3β; LPR, Lipoprotein receptor related protein; TGF-β, Transforming growth factor-β; NCID, Notch intracellular domain; LOX, Lysyl oxidase; PLOD2, Procollagen-lysine,2-oxoglutarate 5-dioxygenase.
Emerging Targets for Breast Cancer Stem Cells
Efflux Transporters
The intrinsic multidrug resistance (MDR) in BCSCs determines the efficacy of chemotherapy. One of the key characteristics that differentiate BCSCs from normal cells is an increased expression of ATP-binding cassette (ABC) efflux transporters. Upregulated expression of ABC transporters may contribute tremendously to chemoresistance. It is imperative to target these transporters in BCSCs without running into toxicity problems. Of the 49 ABC transporters (ABCT) known, ABCB1 [P-glycoprotein (Pgp) or multidrug resistance protein 1 (MDR1)], ABCC1 [(multidrug resistance-associated protein 1 (MRP1)], ABCC3, and ABCG2 [(breast cancer resistance protein (BCRP)] protect BCSCs from drugs by exporting them out of the cells. Among these, ABCG2 and ABCB1 serve as functional cell surface markers for BCSCs. Preclinical studies indicated that genetic deletion of ABCG2 significantly reduced the number of normal SP cells but nearly ablated them in mammary glands of Abcb1a/1b−/−; Abcg2−/− mice. Also, knockdown of ABCC3 led to a reduction in stemness. Additionally, there was a reduction in the number of BCSCs bearing CD44 on their cell surface. Importantly, the knockdown of ABCC3 demonstrated reduced formation of primary tumors (tumor initiating ability) and more susceptibility to doxorubicin in a xenograft mouse model (135). Hypoxic regions observed in a rapidly growing tumor can induce the expression of the transcription factor hypoxia inducible factor1-α (HIF1-α). HIF1-α can in turn induce the expression of ABCB1 which results in an expansion of the BCSCs via paracrine activation by interleukin-6 (IL-6) and CXCL8 (136). It is reported that ABC transporters may increase chemoresistance through expansion of BCSCs (75, 120). So, targeting of MDR is highly important because even if the BCSC targets or signaling nodes are highly sensitive, the effective drugs may simply be exported out. So achieving effective intracellular therapeutic concentration in BCSCs is crucial in eliminating them.
Several approaches have been attempted to overcome MDR including (i) ABC gene silencing by anti-sense oligonucleotide (ASO)inhibitors (ii) Inhibition of the functionality of ABCT through competitive and allosteric modulators (iii) miRNA-mediated downregulation (iv) Targeted inhibition of receptor tyrosine kinases (v) Nanoparticle-mediated delivery of inhibitors (vi) Transcriptional and post-translational regulation of ABCTs and (vii) Signaling pathways affecting them (76, 137). A few examples of the aforementioned approaches that were successful are provided here. Specifically, for instance, “third generation” ABCB1 modulators, such as elacridar (GF120918), laniquidar (R101933), zosuquidar (LY335979), and tariquidar (XR9576) with only nanomolar concentrations of the inhibitors required to inhibit ABCB1-mediated pumping of drugs out of tumor cells effectively. Elacridar was also found to inhibit ABCG2 transporter (138, 139). Therefore, this compound may be useful in treating MDR tumors that express multiple ABC transporters, specifically targeting CSCs (139). Tyrosine-kinase inhibitors (TKIs) such as erlotinib, lapatinib, imatinib, and nilotinib at clinically achievable concentrations, modulate the ATPase activity of ABC transporters, inhibiting the active drug export (140). Specifically, suppression of ABCB1 and/or ABCG2 by TKIs has been demonstrated in several studies, though the detailed mechanism remains unclear (141–144). The caveat is that overcoming MDR mediated by ABC transporters may prove difficult as selective targeting of the BCSCs is vital to avoid any detrimental side effects on normal breast, hematopoietic stem cell populations and the central nervous system. One potential approach would be to define a “therapeutic window” that selectively eliminates BCSCs without affecting normal stem cells. Another challenge is the observed compensatory mechanisms between ABC transporters. For example, several ABCTs have overlapping substrates, i.e., redundant substrate recognition which can lead to cross-resistance to specific drugs/xenobiotics. Such compensatory changes by ABCTs are problematic and will accentuate the tumor growth and chemoresistance. Understanding how functional redundancy among ABCTs contribute to self-renewal of BCSCs is immensely important (145). Finally, drugs targeting ABCTs (e.g., Lapatinib) may be delivered to BCSCs through nanocarriers and using antibodies like Trastuzumab directed against HER2. Alternatively, a combinatorial therapy involving low dose inhibition of ABCTs and another key target such as DNA repair capacity, ALDH/CD44 activity, reactive oxygen species (ROS), anti-apoptotic signaling nodes, key proteins that modulate autophagy/senescence, epigenetic modulators, modulators of EMT, and metastasis or radiotherapy may prove beneficial.
G-Protein Coupled Receptors (GPCRs)
CXCR1 and CXCR2
Chemokine receptors CXCR1 and CXCR2 are Gαi-coupled receptors that generally play a role in chemotaxis of neutrophils, macrophages, and endothelial cells in a physiological microenvironment. In the breast cancer setting, they play a vital role in survival of BCSCs before and after chemotherapy (129–131). CXCL8 can activate both CXCR1 and CXCR2. These receptors not only feed into their own cell survival pathways, but also transactivate epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) in BCSCs (146). Selective targeting of BCSCs with CXCR1 inhibitors would also facilitate reduction in pro-tumor stromal cells that express CXCR1 (147, 148). Antagonizing CXCR1 either by CXCR1-neutralizing antibody or by the small molecule inhibitor Repertaxin selectively depleted BCSCs than bulk tumor cells in vitro. This was followed by massive apoptosis of bulk tumor cells through FASL/FAS signaling through FAK/AKT/FOXO3A pathway. Furthermore, in a xenograft model, Repertaxin reduced the primary tumor burden and metastasis. It was proposed that CXCR1 blockade may selectively eliminate BCSCs (147). The chemotherapeutic drug Reparixin is an allosteric inhibitor of CXCR1 and CXCR2. A phase Ib trial (NCT02001974) has been conducted to evaluate the efficacy of reparixin in inhibiting CXCR1 and CXCR2 and any attendant toxicity (149). In this trial, co-administration of paclitaxel and reparixin appeared to be safe and tolerable in metastatic breast cancer. Additionally, the trial had demonstrated responses in the enrolled population. Based on the favorable outcome, this was taken for further study in a randomized Phase II trial and the results are awaited (NCT02370238).
CXCR4
The chemokine receptor CXCR4 is also a Gαi-coupled receptor. Physiologically, this chemokine receptor generally plays a role in embryogenesis. CXCR7, another Gαi-coupled receptor, shapes the CXCL12 (ligand) gradient for embryonic cells expressing CXCR4. As a result, these cells migrate and form different regions of the embryo. This system is recapitulated by the tumor cells. The chemokine receptor CXCR4 is expressed in BCSCs and is a key chemokine receptor involved in metastasis of breast cancer (150, 151) and forms a target in restraining or removal of BCSCs. Activation of this receptor is thought to facilitate the metastasis of mesenchymal BCSCs. A system-wide analysis of phosphorylation events identified a novel signaling pathway emanating from CXCR4 that activates protein kinase A (PKA) probably through atypical A kinase anchoring protein (AKAP). Active PKA feeds into MAP kinase-activated protein kinase 2-like (MAPKAP2) pathway which eventually stimulates the extracellular signal-regulated kinase (ERK) pathway in BCSCs (150, 152). Activation of ERK2 is critical as ERK2 is known to directly phosphorylate the TF Snail1 and induce its nuclear translocation (153). Nuclear-localized Snail1 is stable and functions as a TF which can generate and maintain BCSCs. Knockdown of CXCR4 abrogated tumor growth in mouse xenograft model (154). Moreover, in mouse mammary carcinoma model, CXCR4 was found to regulate both primary and metastatic breast cancer (155). Recently, the anti-neoplastic agent Balixafortide (a potent and selective CXCR4 antagonist) (Polyphor) in combination with Eribulin (non-taxane, anti-microtubule drug) has been successfully employed in stage IV breast cancer in a phase Ib/proof of concept clinical trial (NCT01837095). During the dose-escalation phase of the trial, the drug combination was tolerated well and no dose-limiting toxicities were observed. The objective response was observed in 30% (16 out of 54) and stable disease in an additional 46% (25 out of 54) of the stage IV patients. Based on this, balixafortide has been fast-track designated by food and drug administration (FDA) for its use in advanced, metastatic breast cancer (MBC) (156). The trial outcome suggests that balixafortide-eribulin combination chemotherapy has promising potential in heavily pretreated patients with MBC and warrants further investigation through randomized trials.
Enantiomeric RNA (L-RNA) aptamers mimicking ligands of receptors can be employed to inhibit activation of key signaling pathways. The RNA aptamer mimicking the ligand CXCL12, NOX-A12 (Olaptesed pegol), seems to control the activation of CXCR4 (157). NOX-A12 binds to two key sites in CXCL12 in order to disrupt its activity and target them for degradation. This has entered into clinical trial in patients (Noxxon Pharma-AG) (158). These will be less toxic compared to the use of small molecule inhibitors or even immunotherapy. Interestingly, combined employment of NOX-A12 and PD-1 blockade enhanced T cell and NK cell infiltration (159) and there are some ongoing clinical trials for the combination therapy involving NOX-A12 and PD-1 inhibitors in different types of cancer.
Tyrosine Kinases
Human Epidermal Growth Factor Receptor 2 (HER2)
HER2 may play a role in the expansion of BCSCs in luminal cell lines and HER2+ breast tumors by upregulating drug transporters and the chemokine receptor CXCR4. HER2 amplification is linked to an early onset of metastasis through increases in the efficiency of mammosphere formation and expansion of the ALDH+ cell population (146, 148). The inhibition of HER2 decreases the invasive and tumorigenic potential of breast cancer cells (160), but HER2 modulation in BCSC could produce resistance to HER2 inhibitors such as Trastuzumab (120). In this situation, the employment of Pertuzumab which inhibits HER2 dimerization with other HER receptors may overcome the resistance. Pharmacological inhibition of HER2 using Lapatinib reversed the MDR mediated by ABCB1 and ABCG2 by directly inhibiting their transport function (142). This result suggests a possible link between ABC transporters and HER2 signaling. The mammosphere formation efficiency (MFE) was reduced regardless of HER2 status and more pronounced in BCSCs with HER2 expression by decreasing their proliferation but not self-renewal (161).
There are many ongoing clinical trials that target HER2 in combination with other approaches. HER2-sensitized dendritic cell (DC) vaccine will be employed to improve the response to breast cancer therapy and in particular preventing recurrence (NCT03630809). A phase II randomized study has started to evaluate the efficacy of anti-PD1 therapy (Pembrolizumab) with concurrent alphavirus-like replication particles containing self-amplifying replicon RNA for HER2 (VRP-HER2) vaccine in increasing the tumor infiltrating and peripheral blood immune response upon administration of the VRP-HER2 vaccine. This is for patients with advanced HER2-overexpressing breast cancer (NCT03632941). There are also numerous ongoing peptide- or domain-based anti-HER2 vaccine clinical trials (NCT02276300, NCT03793829, NCT01632332, and NCT01526473).
Focal Adhesion Kinase (FAK) and Rho GTPases
Bidirectional signaling operates between Rho GTPases and the focal adhesion kinase (FAK). Rho GTPases are reported to govern a variety of cellular processes including a prominent role in the regulation of cell migration. The typical Rho family members such as RhoA, Rac1, and Cdc42 function by cycling between an active GTP-bound and inactive GDP-bound conformations. They are regulated by guanine nucleotide exchange factors (GEFs), GTPase-accelerating proteins (GAPs), and GDP-dissociation inhibitors (GDIs). Among the Rho family members, Ras homolog gene family member C (RhoC) has been reported to impart tumor cell plasticity and is essential for metastasis (162–165). Functionally, RhoC co-ordinates cell motility and actomyosin contractility. RhoA and RhoC have been demonstrated to display a reciprocal relationship in TNBC cells. RhoA impedes tumor cell invasion while RhoC promotes it (166). With regard to breast cancer stemness, the expression of RhoC segregates with ALDH positivity and it impacts the frequency of CSCs found in a previous tissue microarray where 136 breast cancer tissues were analyzed (164). When RhoC was knocked down (RhoC-KD) in ALDH+ cells, tumor initiation was severely impaired (i.e., no induction in the RhoC-KD group vs. 5/9 tumors formed in non-silencing control when 50 CSCs are injected in each group) (164). RhoC has been suggested to work through α5-integrin and activate Src-FAK signaling cascade in regulating metastasis (167).
FAK plays a critical role in BCSCs and forms and attractive target. Inhibition of FAK signaling seems to selectively target BCSCs (168). Mammary epithelial-specific ablation of FAK suppresses tumorigenesis by targeting BCSCs (169). Interestingly, FAK forms a ternary complex with the cytosolic connexin26 and the transcription factor Nanog. This ternary complex is involved in the self-renewal of BCSCs of TNBC origin (170) thus forming an attractive target in BCSCs. ST8SIA1 regulates ganglioside GD2 expression in BCSCs. Interestingly, ST8SIA1 is highly expressed in primary TNBC. Genetic ablation of ST8SIA1 inhibited mammosphere formation in BCSCs. Importantly, T8SIA1-KO TNBC cells were inhibited in its tumorigenic capacity in a mouse xenograft model. Mechanistically, this process involved activation of the FAK-AKT-mTOR signaling pathway in GD2+-BCSCs (171). In another study, inhibition of FAK activity by VS-4718 or VS-6063 preferentially targeted BCSCs in cell lines as well as ex vivo cultured human primary breast cancer specimens. In a mouse xenograft TNBC model, administration of VS-4718 or VS-6063 reduced the BCSCs in the tumor significantly. The tumor-initiating ability is also reduced in the limiting dilution assay in vivo (172). Anti-FAK inhibitor Defactinib along with anti-PD1 therapy is in a clinical trial against solid tumors (NCT02758587).
Serine-Threonine Kinases
Cyclin-Dependent Kinase 4/6
SOX2 can elevate the level of Cyclin D1 through up regulation of its transcripts through transactivation. Cyclin D1 would bind to cyclin-dependent kinase 4/6 (CDK4/6) and form the Cyclin D1-CDK4/6 complex that activates BCSC proliferation and clonogenicity. Inhibiting CDK4/6 with Palbociclib would prevent CDK4/6 activation and would thereby nullify SOX2-directed Cyclin D1. CDK4/6 inhibitors are also promising in chemoresistant cases of HER2+-breast cancer. Importantly, blocking the activity of CDK4/6 synergized with immune checkpoint blockade enhanced the cancer cell immunogenicity and subsequent clearance by cytotoxic T-cells (173–175). Blocking CDK4 activity reduced the stemness and efficiently eliminated chemoresistant cells (101). So CDK4/6 is an attractive target if Cyclin D1 expression is high in the tumor biopsy. A clinical trial is in place targeting CDK4/6 (SHR6390) and HER2 (Pyrotinib) in advanced breast cancer (NCT03993964). There is an ongoing clinical targeting CDK4/6 only (NCT03310879).
Aurora Kinase A
Aurora kinases are a family of mitotic serine/threonine protein kinases comprised of Aurora A (AURKA), Aurora B (AURKB) and Aurora C (AURKC) kinases. AURKA is involved in duplication of centrosomes and AURKB orchestrates mitotic events (176–178). The transcription factor Forkhead box subclass M1 (FOXM1) recruits nuclear AURKA to transactivate FOXM1 target genes in a kinase-independent manner in BCSCs (99). The positive feedback loop with co-operation between AURKA and FOXM1 sustains a high level of expression of both proteins. Both AURKA and FOXM1 promote maintenance and self-renewal of BCSCs (99). Additionally, the nuclear AURKA interacts with heterogeneous nuclear ribonucleoprotein K (hnRNPK) and activates the MYC promoter leading to expression of MYC. As a result, the stemness of BCSCs is enhanced (179). Aberrant AURKA activity can induce phosphorylation of SMAD5 (homolog 5 of the Drosophila protein, mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein Sma) that subsequently promotes the expression of CD44 leading to gain of chemoresistance (180).
There is an ongoing phase Ib trial examining Aurora A Inhibitor (Alisertib; MLN8237) in combination with a dual TORC1/2 inhibitor (MLN0128) in patients with advanced solid tumors with an expansion cohort in metastatic TNBC (NCT02719691).
NF-κB Inducing Kinase (NIK)
The “nuclear factor of kappa light polypeptide gene enhancer in activated B cells” (NF-κB) pathway has been implicated in transcriptional regulation of genes related to survival, proliferation, angiogenesis, metastasis, and immune responses (181). NF-κB inducing kinase (NIK) or Mitogen-activated protein kinase kinase kinase 14 (MAP3K14) is reported to enhance stem cell markers, and growth in BCSCs in vitro and in vivo (182). NIK can activate both canonical and non-canonical pathways by inducing phosphorylation and degradation of inhibitor of κB (IκB). The canonical pathway is mediated by the transcriptional activity of the p50:p65 dimer, whereas the non-canonical pathway is transcriptionally controlled by the p52:RelB dimer (183, 184). Physiologically, NIK plays an important role in the maintenance of the embryonic pluripotent stem cell state and mammary gland development. This may suggest a potential role for NIK in maintenance of BCSCs (185–188). NIK-IKKα was shown to regulate ErbB2-induced mammary tumorigenesis in a preclinical model through the nuclear export of p27/kip1 which supports the proliferation and expansion of BCSCs (189). Recently, NIK was shown to regulate the expression of genes linked to stemness through activation of ERK1/2 and the NF-κB pathways along with the correlative expression between ALDH1 and NIK in breast cancer patients tissue samples and the knockdown of NIK impaired tumorigenic potential (182).
Epigenetic Targets
Epigenetic modifications play a key role in self-renewal, heterogeneity (190) and plasticity of BCSCs (191). Adaptive chromatin remodeling (methylation/demethylation of gene promoters and different lysine residues in histones) may result in differential regulation of proteins leading to chemoresistance and plasticity. An upregulation of drug transporters would lead to chemoresistance and increased viability following therapy. Modulation of transcription factor (TF) networks have been observed in BCSCs. Poised chromatin at “Zinc Finger E-Box Binding Homeobox 1” (ZEB1) sites was reported to play a role in generating CSCs in response to ligands in the TME (192). Snail1 up regulates expansion and activity of BCSCs through repression of p53 (71). The pluripotency factor SOX2 has been implicated in up regulating the activity of the multidrug transporter ABCG2 and the TF Twist1 (112). The level of SOX2 also correlated with the tumor size and expression of epidermal growth factor receptor (EGFR) and cyclin-dependent kinase 5/6 (CDK5/6). Histone deacetylases (HDAC), HDAC1 and HDAC7, are selectively amplified in BCSCs and so these can be targeted (193) either individually or by combination therapy (DNMT and HDAC inhibitors). Histone lysine-specific demethylase 1 (LSD1 or KDM1) is involved in stemness and can serve as a potential target in BCSCs (114). The key clinical advantage is that the epigenetic states are reversible and this vulnerability should be clinically targeted.
The Anti-HDAC6 inhibitor (KA2507) is being examined clinically in patients with PD-L1 expressing solid tumors which have relapsed or are refractory to prior treatment (NCT03008018). A phase I trial is in place targeting LSD1 with the inhibitor (Seclidemstat) in patients with advanced solid tumors (NCT03895684).
Quiescent BCSCs (qBCSCs)
Quiescent CSCs play important roles in tumor dormancy, relapse and resistance to therapy. SET domain-containing protein 4 (SETD4) was demonstrated to be important for the maintenance of qBCSCs. SETD4 trimethylates the side chain of 2nd lysine residue of histone H4. This creates the formation of H4K20me3 (heterochromatin) on the promoter regions leading to silencing of genes that regulate qBCSCs. SETD4-generated qBCSCs were resistant to therapy and promoted tumor relapse in a mouse model and correlated with malignancy and chemoresistance in patients. Importantly, qBCSC underwent asymmetric division into a small quiescent BCSC and a bigger and active daughter cell that proliferates and generates normal tumor cells. Single-cell sequence analysis indicated that SETD4+-qBCSCs cluster together among the heterogeneous BCSCs (26).
Non-coding RNAs
MicroRNAs and long non-coding RNAs
Micro RNAs (miRs) and long non-coding RNAs (lncRNAs) play a key role in the sustenance and also the heterogeneity of BCSCs in TNBC (194). miR-600 acts as a bimodal switch and pushes BCSCs into differentiation and vice versa when miR level was regulated (195). miR-519d overcomes cisplatin-resistance in BCSCs by downregulating the expression of MCL1 making them less viable. miR-199a directly repressed nuclear receptor corepressor (NCOR) and this protected BCSCs from interferon-based induction of senescence and differentiation (196). miR-100 inhibits self-renewal of BCSCs and tumorigenesis (197). LncRNA H19 is responsible for glycolysis and maintenance of BCSCs (198, 199). LncRNA HOTAIR is upregulated in BCSCs derived from MCF7 and MDA-MB-231 cells. HOTAIR transcriptionally downregulates miR-34a level which spares degradation of SOX2 mRNA and in turn increased SOX2 protein levels contributing BC stemness (200). Similar to HOTAIR, the lnc RNA “metastasis-associated lung adenocarcinoma transcript-1” (MALAT-1) plays a critical role in maintaining the BC stemness. First, the level of MALAT-1 was higher in BCSCs than the parental MCF7 cells. Silencing of MALAT-1 led to reduction in the number of BCSCs and the mammosphere formation efficiency. Furthermore, there was reduced proliferation, colony formation, migration and invasion of BCSCs in vitro (201). Targeting of lnc RNA NRAD1 produced cells with less BCSC characteristics (202). Interestingly, mesenchymal stem/stromal cells trigger a lncRNA LINC01133 pathway in neighboring TNBC cells which upregulates pluripotency factor “Kruppel-Like Factor 4” (KLF4). This pushes tumor cells into cancer stemness (203). Also, lnc RNA FEZF1-AS1 has been shown to promote BC stemness and tumorigenesis via targeting miR-30a/Nanog axis (204). Long non-coding RNA in the aldehyde dehydrogenase 1 A pathway (NRAD1) has been suggested to be a potential target in TNBC and BCSCs. Targeting of NRAD1 using the ASO approach resulted in reduced cell survival, tumor growth, and the number of cells with CSC characteristics (202).
“Metabostemness”
CSCs employ either glycolysis or mitochondrial oxidative phosphorylation (OXPHOS) depending on the temporality and the microenvironment or the niche in which they are placed. In the quiescent mode, BCSCs utilize glycolytic pathway for their energy needs. In the proliferative state, BCSCs employ OXPHOS mode of energy derivatization (18, 74). So targeting the metabolic flexibility of BCSCs between OXPHOS and glycolysis may force them into a unilateral OXPHOS or glycolytic mode and may sensitize them to anti-CSC inhibitors. A two “metabolic hit” strategy has been proposed for the eradication of CSCs. Doxycycline has been shown to impair the mitochondrial respiration and a second hit targeting the glycolysis will be effective in elimination of CSCs (205).
“Energetic” Breast Cancer Stem Cells (e-BCSCs)
Based on the energetic profile, a new subset of hyper-metabolic, proliferative BCSCs (called e-BCSCs) driven by mitochondrial energy has been identified. This reflects the presence of metabolic heterogeneity in BCSCs. These eBCSCs are more glycolytic with elevated oxidative metabolism and increased mitochondrial mass. These were ALDH+ with enhanced anchorage-independent growth and NRF2-mediated anti-oxidant response signature. The e-BCSCs can be effectively targeted by OXPHOS and CDK4/6 inhibitors. Therefore, mitochondrial inhibitors to target this subset of highly active BCSCs should be developed (25, 206).
A small molecule inhibitor against mitochondrial electron transport chain complex I (IACS-010759) has been demonstrated to inhibit cell growth in 13 of the 16 TNBC cell lines employed. An ongoing clinical trial (NCT03291938) is in place with IACS-010759 in advanced breast cancer patients (207). Another similar drug (ME-344) against mitochondrial complex I is in phase I clinical trial in breast cancer patients (NCT02806817).
Redox Pathways
NF-E2-Related Factor 2 (NRF2)
Newer data has identified NF-E2-related factor 2 (NRF2) transcription factor as a novel biomarker for BCSCs. One study has shown that NRF2 expression increases in drug resistant BCSCs (208). NRF2 is a master regulator of cell redox homeostasis. It performs its regulatory function by up regulating genes that have an antioxidant response element (ARE). The work of Wu et al. (208) revealed that NRF2 conferred resistance to multiple drugs in BCSCs by keeping ROS level reduced during the drug treatment. In a recent discovery, CD44+-BCSCs showed co-localization of NRF2 with CD44, and found that NRF2 expression was dictated by CD44-p62 signaling (209). Importantly, co-inhibition of NRF2 or downstream thioredoxin and glutathione antioxidant pathways and glycolysis has been shown tom induce terminal differentiation of both mesenchymal and epithelial BCSCs and induction of apoptosis (74). Furthermore, this inhibition suppressed tumor growth, tumor-initiating potential and importantly metastasis by eliminating both mesenchymal and epithelial BCSCs (74). Additionally, NRF2 has been implicated in other CSC types including ovarian (210) and acute myeloid leukemia (AML) (211).
Miscellaneous
Sirtuin1 (SIRT1)
Sirtuin1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase involved in both cellular stress and longevity. The hallmark function of SIRT1 is to enhance cell survival through the deacetylation and inactivation of p53 (212). An increase in SIRT1 expression levels in drug resistant cancer cell lines induce deacetylation and activation of FOXO1 which upregulates drug transporters MDR1 (213) and MRP2 (214). SIRT1 is a key facilitator in stem cell biology as well. For instance, mouse embryonic stem cells which lack SIRT1 have a delayed capacity to differentiate through the ability of SIRT1 to repress the expression of Dnmt3l (215). In addition, SIRT1 inhibits p53-mediated suppression of Nanog, a pluripotency transcription factor involved in the maintenance of BCSCs (216). SIRT1 is upregulated in CD44+/CD24− mesenchymal BCSCs (217). In another report, SIRT1 is downregulated in ALDH1+ epithelial BCSCs and is reported to stabilize the EMT inducer PRRX1 and indirectly inhibits the stemness factor KLF4 (218). Future studies will need to elucidate the role of SIRT1 in mesenchymal and epithelial BCSCs and clear out the controversies involved. Alternatively, relative abundance of other SIRT isoforms may contribute to differential outcome observed. A mechanistic understanding of the role of SIRT1 in each type of BCSCs will justify future therapeutic intervention as both small molecule activators and inhibitors are commercially available for SIRT1 (219). Lastly, it is also interesting to note that c-MYC has been reported to activate SIRT1 which in turn promotes c-MYC function (220).
Targeting Other Signaling and Survival Pathways in BCSCs
The MCL1 inhibitor S63845 has been reported to have success against BCSCs arising out of HER2+ and TNBC (221). Targeting the Wnt/β-catenin signaling pathway showed promising results in reducing the metastatic potential by altering BCSC activity in a preclinical mouse model (222). Moreover, loss of the tumor suppressor Liver-kinase B1 (LKB1) led to an increase in the number of BCSCs (223) and elevated expression of OCT4, Nanog, and SOX2. Interestingly, a plant bioactive molecule called “Honokiol” effectively upregulated LKB1 protein levels that abrogated the stem phenotype (224). In addition, co-targeting of Notch ligand production and IL-6 receptor in human breast cancer cell lines and PDX xenografts was beneficial in reducing the number of BCSCs (225). A caveat would be targeting the Notch pathway may be detrimental to the immune system (226). Finally, Insulin-like growth factor-2 (IGF-2) induced NF-κB activity and blockade of the IGF-2 signaling reduced tumorigenesis in a PDX model enriched with BCSCs (227).
Tinkering With Lysosomes
Drug screening identified salinomycin as a selective agent against BCSCs by sequestering iron in lysosomes that led to ferroptosis of CSCs (228, 229). In particular, C20-O-acylated analogs of salinomycin performed better in terms of efficacy (230). Ferroptotic agents have been shown to selectively kill BCSCs (231). The anti-malarial drug chloroquine (CQ) was reported as a sensitizing agent to paclitaxel through inhibition of autophagy in TNBC cells. This reduced the number of CD44hi/CD24−/low-BCSCs in both preclinical and clinical settings (232). Mechanistically, CQ worked to inhibit the Janus kinase 2 (Jak2)-signal transducer and activator of transcription 3 (JAK2-STAT3) signaling pathway. (233). The downside of CQ is that it favors the accumulation of CD3+/CD4+/FOXP3+ regulatory T cells (Tregs) (234).
Concluding Remarks
In this review, we have explored the molecular origin of BC stemness and chemoresistance and have identified several emerging molecular targets that are vital for BCSCs. These targets could be employed to overcome chemoresistance mediated by BCSCs. By simultaneous targeting both BCSCs and bulk tumor cell populations, the problem arising out of interconversion between bulk tumor and stemness state could be contained. This will prevent tumor relapse and increase patient longevity. Additionally, metabolic vulnerabilities should be combined with novel pharmacological targets. Overall, combinatorial therapy involving emergent vulnerable nodes in receptor and redox signaling pathways, survival, self-renewal, drug efflux transporters, and metabolism would pave the way for effective modalities of therapy and attain favorable prognosis in the metastatic breast cancer.
Author Contributions
SS: contributed to introduction, Table 1, NIK section, and oversaw several sections. CH: contributed a section on SIRT1 and drew Figure 2. AMCT: composed Table 2 and a section on NRF2. BS: contributed a section on AurK. AKT: contributed a section on efflux transporters and edited the whole manuscript. RR: edited the manuscript and contributed intellectual concepts. DR: contributed to all sections, wrote all other sections not mentioned above, edited and intellectually co-ordinated the complete manuscript and drew Figure 1.
Funding
This manuscript has been supported in part by National Institute of Health (NIH)/National Cancer Institute (NCI) grant R21CA202176 (to DR), Ohio Cancer Research foundation (OCR) (to DR), University of Toledo startup grants (F110796 to DR and F110760 to AKT), a grant from Susan G. Komen Breast Cancer Foundation (CCR18548498 to AKT) and College of Graduate Studies (COGS) Fellowship, University of Toledo (to CH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Max Wicha for his insightful comments on the manuscript.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
2. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. (2011) 121:2750–67. doi: 10.1172/JCI45014
3. Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. (2014) 232:142–50. doi: 10.1002/path.4280
4. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. (2013) 19:5533–40. doi: 10.1158/1078-0432.CCR-13-0799
5. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. (2012) 486:395–9. doi: 10.1038/nature10933
6. Valent P, Bonnet D, Wohrer S, Andreeff M, Copland M, Chomienne C, et al. Heterogeneity of neoplastic stem cells: theoretical, functional, and clinical implications. Cancer Res. (2013) 73:1037–45. doi: 10.1158/0008-5472.CAN-12-3678
7. Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. (2015) 17:260–71. doi: 10.1016/j.stem.2015.08.014
8. Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. (2015) 21:751–9. doi: 10.1038/nm.3886
9. Yang F, Wang Y, Li Q, Cao L, Sun Z, Jin J, et al. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis. (2017) 38:900–9. doi: 10.1093/carcin/bgx071
10. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. (2017) 32:169–84 e167. doi: 10.1016/j.ccell.2017.07.005
11. Echeverria GV, Powell E, Seth S, Ge Z, Carugo A, Bristow C, et al. High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat Commun. (2018) 9:5079. doi: 10.1038/s41467-018-07406-4
12. Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. (2017) 389:2430–42. doi: 10.1016/S0140-6736(16)32454-0
13. Geyer FC, Pareja F, Weigelt B, Rakha E, Ellis IO, Schnitt SJ, et al. The spectrum of triple-negative breast disease: high- and low-grade lesions. Am J Pathol. (2017) 187:2139–51. doi: 10.1016/j.ajpath.2017.03.016
14. Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. (2004) 14:43–7. doi: 10.1016/j.gde.2003.11.007
15. Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. Cancer Lett. (2013) 341:56–62. doi: 10.1016/j.canlet.2013.06.003
16. Luo M, Wicha MS. Metabolic plasticity of cancer stem cells. Oncotarget. (2015) 6:35141–2. doi: 10.18632/oncotarget.6177
17. Banerjee A, Birts CN, Darley M, Parker R, Mirnezami AH, West J, et al. Stem cell-like breast cancer cells with acquired resistance to metformin are sensitive to inhibitors of NADH-dependent CtBP dimerization. Carcinogenesis. (2019) 40:871–82. doi: 10.1093/carcin/bgy174
18. Luo M, Wicha MS. Targeting cancer stem cell redox metabolism to enhance therapy responses. Semin Radiat Oncol. (2019) 29:42–54. doi: 10.1016/j.semradonc.2018.10.003
19. Galdiero MR, Marone G, Mantovani A. Cancer inflammation and cytokines. Cold Spring Harb Perspect Biol. (2018) 10:883–99. doi: 10.1101/cshperspect.a028662
20. Galdiero MR, Varricchi G, Loffredo S, Mantovani A, Marone G. Roles of neutrophils in cancer growth and progression. J Leukoc Biol. (2018) 103:457–64. doi: 10.1002/JLB.3MR0717-292R
21. Eiro N, Gonzalez LO, Fraile M, Cid S, Schneider J, Vizoso FJ. Breast cancer tumor stroma: cellular components, phenotypic heterogeneity, intercellular communication, prognostic implications and therapeutic opportunities. Cancers. (2019) 11:E664. doi: 10.3390/cancers11050664
22. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. (2019) 24:25–40. doi: 10.1016/j.stem.2018.11.017
23. Sharick JT, Jeffery JJ, Karim MR, Walsh CM, Esbona K, Cook RS, et al. Cellular metabolic heterogeneity in vivo is recapitulated in tumor organoids. Neoplasia. (2019) 21:615–26. doi: 10.1016/j.neo.2019.04.004
24. Bocci F, Gearhart-Serna L, Boareto M, Ribeiro M, Ben-Jacob E, Devi GR, et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc Natl Acad Sci USA. (2019) 116:148–57. doi: 10.1073/pnas.1815345116
25. Fiorillo M, Sotgia F, Lisanti MP. “Energetic” Cancer Stem Cells (e-CSCs): a new hyper-metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front Oncol. (2018) 8:677. doi: 10.3389/fonc.2018.00677
26. Ye S, Ding YF, Jia WH, Liu XL, Feng JY, Zhu Q, et al. SET domain-containing protein 4 epigenetically controls breast cancer stem cell quiescence. Cancer Res. (2019) 79:4729–43. doi: 10.1158/0008-5472.CAN-19-1084
27. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. (2014) 2:78–91. doi: 10.1016/j.stemcr.2013.11.009
28. Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci USA. (2017) 114:E2337–46. doi: 10.1073/pnas.1618298114
29. Sehl ME, Wicha MS. Modeling of interactions between cancer stem cells and their microenvironment: predicting clinical response. Methods Mol Biol. (2018) 1711:333–49. doi: 10.1007/978-1-4939-7493-1_16
30. Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. (2017) 17:557–69. doi: 10.1038/nrc.2017.58
31. Gao R, Kim C, Sei E, Foukakis T, Crosetto N, Chan LK, et al. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat Commun. (2017) 8:228. doi: 10.1038/s41467-017-00244-w
32. Yang Z, Li C, Fan Z, Liu H, Zhang X, Cai Z, et al. Single-cell sequencing reveals variants in ARID1A, GPRC5A and MLL2 driving self-renewal of human bladder cancer stem cells. Eur Urol. (2017) 71:8–12. doi: 10.1016/j.eururo.2016.06.025
33. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. (2018) 174:1293–308 e1236. doi: 10.1016/j.cell.2018.05.060
34. Colacino JA, Azizi E, Brooks MD, Harouaka R, Fouladdel S, McDermott SP, et al. Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling. Stem Cell Rep. (2018) 10:1596–609. doi: 10.1016/j.stemcr.2018.03.001
35. Feng W, Li Y, Chu J, Li J, Zhang Y, Ding X, et al. Identification of tRNA-derived small noncoding RNAs as potential biomarkers for prediction of recurrence in triple-negative breast cancer. Cancer Med. (2018) 7:5130–44. doi: 10.1002/cam4.1761
36. Sun C, Fu Z, Wang S, Li J, Li Y, Zhang Y, et al. Roles of tRNA-derived fragments in human cancers. Cancer Lett. (2018) 414:16–25. doi: 10.1016/j.canlet.2017.10.031
37. Sun C, Yang F, Zhang Y, Chu J, Wang J, Wang Y, et al. tRNA-derived fragments as novel predictive biomarkers for trastuzumab-resistant breast cancer. Cell Physiol Biochem. (2018) 49:419–31. doi: 10.1159/000492977
38. Cui Y, Huang Y, Wu X, Zheng M, Xia Y, Fu Z, et al. Hypoxia-induced tRNA-derived fragments, novel regulatory factor for doxorubicin resistance in triple-negative breast cancer. J Cell Physiol. (2019) 234:8740–51. doi: 10.1002/jcp.27533
39. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
40. Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun. (2016) 7:10442. doi: 10.1038/ncomms10442
41. Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. (2017) 77:1927–41. doi: 10.1158/0008-5472.CAN-16-2129
42. Gomez-Miragaya J, Palafox M, Pare L, Yoldi G, Ferrer I, Vila S, et al. Resistance to taxanes in triple-negative breast cancer associates with the dynamics of a CD49f+ tumor-initiating population. Stem Cell Rep. (2017) 8:1392–407. doi: 10.1016/j.stemcr.2017.03.026
43. Isman FK, Kucukgergin C, Dasdemir S, Cakmakoglu B, Sanli O, Seckin S. Association between SDF1-3'A or CXCR4 gene polymorphisms with predisposition to and clinicopathological characteristics of prostate cancer with or without metastases. Mol Biol Rep. (2012) 39:11073–9. doi: 10.1007/s11033-012-2010-4
44. Zhang H, Brown RL, Wei Y, Zhao P, Liu S, Liu X, et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. (2019) 33:166–79. doi: 10.1101/gad.319889.118
45. Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. (2008) 283:17969–78. doi: 10.1074/jbc.M802917200
46. Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Transl Med. (2014) 3:19. doi: 10.1186/2001-1326-3-19
47. Liu P, Tang H, Song C, Wang J, Chen B, Huang X, et al. SOX2 promotes cell proliferation and metastasis in triple negative breast cancer. Front Pharmacol. (2018) 9:942. doi: 10.3389/fphar.2018.00942
48. Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. (2014) 6:66–79. doi: 10.1002/emmm.201303411
49. Domenici G, Aurrekoetxea-Rodriguez I, Simoes BM, Rabano M, Lee SY, Millan JS, et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene. (2019) 38:3151–69. doi: 10.1038/s41388-018-0656-7
50. Xue Y, Lai L, Lian W, Tu X, Zhou J, Dong P, et al. SOX9/FXYD3/Src axis is critical for ER(+) breast cancer stem cell function. Mol Cancer Res. (2019) 17:238–49. doi: 10.1158/1541-7786.MCR-18-0610
51. Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. (2005) 104:2255–65. doi: 10.1002/cncr.21432
52. Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. (2013) 6:1207–20. doi: 10.2147/OTT.S38114
53. Ramos EK, Hoffmann AD, Gerson SL, Liu H. New opportunities and challenges to defeat cancer stem cells. Trends Cancer. (2017) 3:780–96. doi: 10.1016/j.trecan.2017.08.007
54. Zhang JM, Wei K, Jiang M. OCT4 but not SOX2 expression correlates with worse prognosis in surgical patients with triple-negative breast cancer. Breast Cancer. (2018) 25:447–55. doi: 10.1007/s12282-018-0844-x
55. Yang F, Zhang J, Yang H. OCT4, SOX2, and NANOG positive expression correlates with poor differentiation, advanced disease stages, and worse overall survival in HER2(+) breast cancer patients. Onco Targets Ther. (2018) 11:7873–81. doi: 10.2147/OTT.S173522
56. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
57. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. (2012) 109:2784–9. doi: 10.1073/pnas.1018866109
58. Liu S, Clouthier SG, Wicha MS. Role of microRNAs in the regulation of breast cancer stem cells. J Mammary Gland Biol Neoplasia. (2012) 17:15–21. doi: 10.1007/s10911-012-9242-8
59. Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. (2008) 10:R53. doi: 10.1186/bcr2108
60. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. (2009) 106:13820–5. doi: 10.1073/pnas.0905718106
61. Sun H, Jia J, Wang X, Ma B, Di L, Song G, et al. CD44+/CD24- breast cancer cells isolated from MCF-7 cultures exhibit enhanced angiogenic properties. Clin Transl Oncol. (2013) 15:46–54. doi: 10.1007/s12094-012-0891-2
62. Morimoto K, Kim SJ, Tanei T, Shimazu K, Tanji Y, Taguchi T, et al. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. (2009) 100:1062–8. doi: 10.1111/j.1349-7006.2009.01151.x
63. Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer Res. (2013) 73:1635–46. doi: 10.1158/0008-5472.CAN-12-3349
64. Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. (2006) 8:R59. doi: 10.1186/bcr1610
65. Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. (2009) 69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741
66. Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. (2009) 13:2236–52. doi: 10.1111/j.1582-4934.2008.00455.x
67. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
68. Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. (2015) 21:1301–10. doi: 10.2174/1381612821666141211120604
69. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
70. Zhu Y, Luo M, Brooks M, Clouthier SG, Wicha MS. Biological and clinical significance of cancer stem cell plasticity. Clin Transl Med. (2014) 3:32. doi: 10.1186/s40169-014-0032-3
71. Ni T, Li XY, Lu N, An T, Liu ZP, Fu R, et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nat Cell Biol. (2016) 18:1221–32. doi: 10.1038/ncb3425
72. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
73. Markiewicz A, Topa J, Nagel A, Skokowski J, Seroczynska B, Stokowy T, et al. Spectrum of epithelial-mesenchymal transition phenotypes in circulating tumour cells from early breast cancer patients. Cancers. (2019) 11:E59. doi: 10.3390/cancers11010059
74. Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. (2018) 28:69–86.e66. doi: 10.1016/j.cmet.2018.06.006
75. Berghuis D, Schilham MW, Santos SJ, Savola S, Knowles HJ, Dirksen U, et al. The CXCR4-CXCL12 axis in Ewing sarcoma: promotion of tumor growth rather than metastatic disease. Clin Sarcoma Res. (2012) 2:24. doi: 10.1186/2045-3329-2-24
76. McIntosh K, Balch C, Tiwari AK. Tackling multidrug resistance mediated by efflux transporters in tumor-initiating cells. Expert Opin Drug Metab Toxicol. (2016) 12:633–44. doi: 10.1080/17425255.2016.1179280
77. Lang JY, Hsu JL, Meric-Bernstam F, Chang CJ, Wang Q, Bao Y, et al. BikDD eliminates breast cancer initiating cells and synergizes with lapatinib for breast cancer treatment. Cancer Cell. (2011) 20:341–56. doi: 10.1016/j.ccr.2011.07.017
78. De Blasio A, Pratelli G, Drago-Ferrante R, Saliba C, Baldacchino S, Grech G, et al. Loss of MCL1 function sensitizes the MDA-MB-231 breast cancer cells to rh-TRAIL by increasing DR4 levels. J Cell Physiol. (2019) 234:18432–47. doi: 10.1002/jcp.28479
79. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. (2017) 26:633–47.e637. doi: 10.1016/j.cmet.2017.09.009
80. Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, et al. Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and notch-dependent cancer stem cell population. Cancer Res. (2016) 76:440–52. doi: 10.1158/0008-5472.CAN-15-1640-T
81. Liubomirski Y, Lerrer S, Meshel T, Morein D, Rubinstein-Achiasaf L, Sprinzak D, et al. Notch-mediated tumor-stroma-inflammation networks promote invasive properties and CXCL8 expression in triple-negative breast cancer. Front Immunol. (2019) 10:804. doi: 10.3389/fimmu.2019.00804
82. Wang X, Masri S, Phung S, Chen S. The role of amphiregulin in exemestane-resistant breast cancer cells: evidence of an autocrine loop. Cancer Res. (2008) 68:2259–65. doi: 10.1158/0008-5472.CAN-07-5544
83. Vilquin P, Villedieu M, Grisard E, Larbi SB, Ghayad SE, Heudel P-E, et al. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int. J. Cancer. (2013) 133:1589–602. doi: 10.1002/ijc.28182
84. Kazi AA, Gilani RA, Schech AJ, Chumsri S, Sabnis G, Shah P, et al. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. (2014) 16:R15. doi: 10.1186/bcr3609
85. Saha S, Mukherjee S, Khan P, Kajal K, Mazumdar M, Manna A, et al. Aspirin suppresses the acquisition of chemoresistance in breast cancer by disrupting an NFκB–IL6 signaling axis responsible for the generation of cancer stem cells. Cancer Res. (2016) 76:2000–12. doi: 10.1158/0008-5472.CAN-15-1360
86. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2017) 553:96. doi: 10.1038/nature25167
87. Yun C-O, Bhargava P, Na Y, Lee J-S, Ryu J, Kaul SC, et al. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci Rep. (2017) 7:42016. doi: 10.1038/srep42016
88. Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell. (2010) 39:761–72. doi: 10.1016/j.molcel.2010.08.013
89. Lee H-J, Li C-F, Ruan D, Powers S, Thompson PA, Frohman MA, et al. The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through twist activation. Mol. Cell. (2016) 63:1021–33. doi: 10.1016/j.molcel.2016.08.009
90. Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein Kinase C and#x3b1; is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell. (2013) 24:347–64. doi: 10.1016/j.ccr.2013.08.005
91. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, et al. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. (2009) 15:4234–41. doi: 10.1158/1078-0432.CCR-08-1479
92. Zhou Y, Hu Y, Yang M, Jat P, Li K, Lombardo Y, et al. The miR-106b~25 cluster promotes bypass of doxorubicin-induced senescence and increase in motility and invasion by targeting the E-cadherin transcriptional activator EP300. Cell Death Differ. (2013) 21:462. doi: 10.1038/cdd.2013.167
93. Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G, et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene. (2014) 34:681. doi: 10.1038/onc.2014.5
94. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, et al. β-Catenin and NF-κB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. (2014) 22:298. doi: 10.1038/cdd.2014.145
95. Lee KM, Nam K, Oh S, Lim J, Kim RK, Shim D, et al. ECM1 regulates tumor metastasis and CSC-like property through stabilization of β-catenin. Oncogene. (2015) 34:6055. doi: 10.1038/onc.2015.54
96. Tsou S-H, Chen T-M, Hsiao H-T, Chen Y-H. A critical dose of doxorubicin is required to alter the gene expression profiles in MCF-7 cells acquiring multidrug resistance. PLoS ONE. (2015) 10:e0116747. doi: 10.1371/journal.pone.0116747
97. Xu LZ, Li SS, Zhou W, Kang ZJ, Zhang QX, Kamran M, et al. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene. (2016) 36:304. doi: 10.1038/onc.2016.202
98. Zhang ZM, Wu JF, Luo QC, Liu QF, Wu QW, Ye GD, et al. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/β-catenin pathway. Oncogene. (2016) 35:4787. doi: 10.1038/onc.2016.10
99. Yang N, Wang C, Wang Z, Zona S, Lin SX, Wang X, et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene. (2017) 36:3428–40. doi: 10.1038/onc.2016.490
100. Santos JC, Lima NDS, Sarian LO, Matheu A, Ribeiro ML, Derchain SFM. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci Rep. (2018) 8:829. doi: 10.1038/s41598-018-19339-5
101. Dai M, Zhang C, Ali A, Hong X, Tian J, Lo C, et al. CDK4 regulates cancer stemness and is a novel therapeutic target for triple-negative breast cancer. Sci Rep. (2016) 6:35383. doi: 10.1038/srep35383
102. Wang X, Wang Y, Gu J, Zhou D, He Z, Wang X, et al. ADAM12-L confers acquired 5-fluorouracil resistance in breast cancer cells. Sci Rep. (2017) 7:9687. doi: 10.1038/s41598-017-10468-x
103. Yang XL, Lin FJ, Guo YJ, Shao ZM, Ou ZL. Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways. OncoTargets Ther. (2014) 7:1033–42. doi: 10.2147/OTT.S63145
104. Simões BM, O'Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. (2015) 12:1968–77. doi: 10.1016/j.celrep.2015.08.050
105. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. (2011) 147:759–72. doi: 10.1016/j.cell.2011.09.048
106. Liu Y, Choi DS, Sheng J, Ensor JE, Liang DH, Rodriguez-Aguayo C, et al. HN1L promotes triple-negative breast cancer stem cells through LEPR-STAT3 pathway. Stem Cell Rep. (2018) 10:212–27. doi: 10.1016/j.stemcr.2017.11.010
107. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemnessCell. (2018) 172:841–56.e16. doi: 10.1016/j.cell.2018.01.009
108. Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene. (2014) 34:671–80. doi: 10.1038/onc.2014.4
109. Yin B, Ma ZY, Zhou ZW, Gao WC, Du ZG, Zhao ZH, et al. The TrkB+ cancer stem cells contribute to post-chemotherapy recurrence of triple-negative breast cancers in an orthotopic mouse model. Oncogene. (2014) 34:761–70. doi: 10.1038/onc.2014.8
110. Takahashi R-U, Miyazaki H, Takeshita F, Yamamoto Y, Minoura K, Ono M, et al. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat Commun. (2015) 6:7318. doi: 10.1038/ncomms8318
111. Wee ZN, Yatim SMJM, Kohlbauer VK, Feng M, Goh JY, Bao Y, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. (2015) 6:8746. doi: 10.1038/ncomms9746
112. Mukherjee P, Gupta A, Chattopadhyay D, Chatterji U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Sci Rep. (2017) 7:9170. doi: 10.1038/s41598-017-08971-2
113. Sorrentino G, Ruggeri N, Zannini A, Ingallina E, Bertolio R, Marotta C, et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat Commun. (2017) 8:14073. doi: 10.1038/ncomms14073
114. Boulding T, McCuaig RD, Tan A, Hardy K, Wu F, Dunn J, et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci Rep. (2018) 8:73. doi: 10.1038/s41598-017-17913-x
115. Wang T., Fahrmann J.F., Lee H., Li Y.J., Tripathi S.C., Yue C., et al. (2018). JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27, 136–50 e135. doi: 10.1016/j.cmet.2017.11.001
116. Jeong Y-J, Kang JS, Lee SI, So DM, Yun J, Baek JY, et al. Breast cancer cells evade paclitaxel-induced cell death by developing resistance to dasatinib. Oncol Lett. (2016) 12:2153–8. doi: 10.3892/ol.2016.4852
117. Korkaya H, Kim G-I, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 Inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. (2012) 47:570–84. doi: 10.1016/j.molcel.2012.06.014
118. Burnett JP, Korkaya H, Ouzounova MD, Jiang H, Conley SJ, Newman BW, et al. Trastuzumab resistance induces EMT to transform HER2+ PTEN– to a triple negative breast cancer that requires unique treatment options. Sci Rep. (2015) 5:15821. doi: 10.1038/srep15821
119. De Cola A, Volpe S, Budani MC, Ferracin M, Lattanzio R, Turdo A, et al. miR-205-5p-mediated downregulation of ErbB/HER receptors in breast cancer stem cells results in targeted therapy resistance. Cell Death Dis. (2015) 6:e1823. doi: 10.1038/cddis.2015.192
120. Rodriguez CE, Berardi DE, Abrigo M, Todaro LB, Bal de Kier Joffe ED, Fiszman GL. Breast cancer stem cells are involved in Trastuzumab resistance through the HER2 modulation in 3D culture. J Cell Biochem. (2018) 119:1381–91. doi: 10.1002/jcb.26298
121. Notas G, Pelekanou V, Kampa M, Alexakis K, Sfakianakis S, Laliotis A, et al. Tamoxifen induces a pluripotency signature in breast cancer cells and human tumors. Mol Oncol. (2015) 9:1744–59. doi: 10.1016/j.molonc.2015.05.008
122. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. (2014) 16:864. doi: 10.1038/ncb3013
123. Mateo F, Arenas EJ, Aguilar H, Serra-Musach J, de Garibay GR, Boni J, et al. Stem cell-like transcriptional reprogramming mediates metastatic resistance to mTOR inhibition. Oncogene. (2016) 36:2737–49. doi: 10.1038/onc.2016.427
124. Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, et al. An integrin β3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol. (2014) 16:457–68. doi: 10.1038/ncb2953
125. He L, Gu J, Lim LY, Yuan ZX, Mo J. Nanomedicine-mediated therapies to target breast cancer stem cells. Front Pharmacol. (2016) 7:313. doi: 10.3389/fphar.2016.00313
126. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. (2013) 4:1795. doi: 10.1038/ncomms2766
127. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. (2015) 16:225–38. doi: 10.1016/j.stem.2015.02.015
128. Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. (2019) 24:41–53. doi: 10.1016/j.stem.2018.12.009
129. Juvekar A, Wulf GM. Closing escape routes: inhibition of IL-8 signaling enhances the anti-tumor efficacy of PI3K inhibitors. Breast Cancer Res. (2013) 15:308. doi: 10.1186/bcr3400
130. Todaro M, Turdo A, Bartucci M, Iovino F, Dattilo R, Biffoni M, et al. Erythropoietin activates cell survival pathways in breast cancer stem-like cells to protect them from chemotherapy. Cancer Res. (2013) 73:6393–400. doi: 10.1158/0008-5472.CAN-13-0248
131. Jia D, Li L, Andrew S, Allan D, Li X, Lee J, et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. (2017) 8:e2932. doi: 10.1038/cddis.2017.319
132. Tian J, Hachim MY, Hachim IY, Dai M, Lo C, Raffa FA, et al. Cyclooxygenase-2 regulates TGFbeta-induced cancer stemness in triple-negative breast cancer. Sci Rep. (2017) 7:40258. doi: 10.1038/srep40258
133. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. (2009) 14:3–9. doi: 10.1007/s10911-009-9109-9
134. Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol. (2012) 83:1084–103. doi: 10.1016/j.bcp.2012.01.002
135. Balaji SA, Udupa N, Chamallamudi MR, Gupta V, Rangarajan A. Role of the drug transporter ABCC3 in breast cancer chemoresistance. PLoS ONE. (2016) 11:e0155013. doi: 10.1371/journal.pone.0155013
136. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. (2014) 111:E5429–38. doi: 10.1073/pnas.1421438111
137. Li SY, Sun R, Wang HX, Shen S, Liu Y, Du XJ, et al. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J Control Release. (2015) 205:7–14. doi: 10.1016/j.jconrel.2014.11.011
138. Wu CP, Calcagno AM, Ambudkar SV. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr Mol Pharmacol. (2008) 1:93–105. doi: 10.2174/1874467210801020093
139. Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. (2013) 34:732–40. doi: 10.1038/aps.2013.27
140. Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, et al. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab Dispos. (2010) 38:1371–80. doi: 10.1124/dmd.109.031302
141. Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. (2007) 67:11012–20. doi: 10.1158/0008-5472.CAN-07-2686
142. Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. (2008) 68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499
143. Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR Jr, Chen X, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. (2009) 78:153–61. doi: 10.1016/j.bcp.2009.04.002
144. Sims JT, Ganguly SS, Bennett H, Friend JW, Tepe J, Plattner R. Imatinib reverses doxorubicin resistance by affecting activation of STAT3-dependent NF-kappaB and HSP27/p38/AKT pathways and by inhibiting ABCB1. PLoS ONE. (2013) 8:e55509. doi: 10.1371/journal.pone.0055509
145. Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Ther. (2016) 160:145–58. doi: 10.1016/j.pharmthera.2016.02.008
146. Yun J, Frankenberger CA, Kuo WL, Boelens MC, Eves EM, Cheng N, et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J. (2011) 30:4500–14. doi: 10.1038/emboj.2011.312
147. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. (2010) 120:485–97. doi: 10.1172/JCI39397
148. Singh JK, Farnie G, Bundred NJ, Simoes BM, Shergill A, Landberg G, et al. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res. (2013) 19:643–56. doi: 10.1158/1078-0432.CCR-12-1063
149. Schott AF, Goldstein L, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2 negative metastatic breast cancer (MBC). Clin Cancer Res. (2017) 23:5358–65. doi: 10.1158/1078-0432.CCR-16-2748
150. Yi T, Zhai B, Yu Y, Kiyotsugu Y, Raschle T, Etzkorn M, et al. Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc Natl Acad Sci USA. (2014) 111:E2182–90. doi: 10.1073/pnas.1404943111
151. Mukherjee S, Manna A, Bhattacharjee P, Mazumdar M, Saha S, Chakraborty S, et al. Non-migratory tumorigenic intrinsic cancer stem cells ensure breast cancer metastasis by generation of CXCR4(+) migrating cancer stem cells. Oncogene. (2016) 35:4937–48. doi: 10.1038/onc.2016.26
152. Graham NA, Graeber TG. Complexity of metastasis-associated SDF-1 ligand signaling in breast cancer stem cells. Proc Natl Acad Sci USA. (2014) 111:7503–4. doi: 10.1073/pnas.1405991111
153. Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. (2013) 15:677–87. doi: 10.1038/ncb2743
154. Lapteva N, Yang AG, Sanders DE, Strube RW, Chen SY. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. (2005) 12:84–9. doi: 10.1038/sj.cgt.7700770
155. Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. (2004) 64:8604–12. doi: 10.1158/0008-5472.CAN-04-1844
156. Pernas S, Martin M, Kaufman PA, Gil-Martin M, Gomez Pardo P, Lopez-Tarruella S, et al. Balixafortide plus eribulin in HER2-negative metastatic breast cancer: a phase 1, single-arm, dose-escalation trial. Lancet Oncol. (2018) 19:812–24. doi: 10.1016/S1470-2045(18)30147-5
157. Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res. (2011) 17:2074–80. doi: 10.1158/1078-0432.CCR-10-2636
158. Vater A, Klussmann S. Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer® therapeutics. Drug Discov Today. (2015) 20:147–55. doi: 10.1016/j.drudis.2014.09.004
159. Zboralski D, Hoehlig K, Eulberg D, Fromming A, Vater A. Increasing tumor-infiltrating t cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol Res. (2017) 5:950–6. doi: 10.1158/2326-6066.CIR-16-0303
160. Greenfield JP, Cobb WS, Lyden D. Resisting arrest: a switch from angiogenesis to vasculogenesis in recurrent malignant gliomas. J Clin Invest. (2010) 120:663–7. doi: 10.1172/JCI42345
161. Farnie G, Johnson RL, Williams KE, Clarke RB, Bundred NJ. Lapatinib inhibits stem/progenitor proliferation in preclinical in vitro models of ductal carcinoma in situ (DCIS). Cell Cycle. (2014) 13:418–25. doi: 10.4161/cc.27201
162. Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. (2000) 406:532–5. doi: 10.1038/35020106
163. Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. (2005) 19:1974–9. doi: 10.1101/gad.1310805
164. Rosenthal DT, Zhang J, Bao L, Zhu L, Wu Z, Toy K, et al. RhoC impacts the metastatic potential and abundance of breast cancer stem cells. PLoS ONE. (2012) 7:e40979. doi: 10.1371/journal.pone.0040979
165. Thomas P, Pranatharthi A, Ross C, Srivastava S. RhoC: a fascinating journey from a cytoskeletal organizer to a Cancer stem cell therapeutic target. J Exp Clin Cancer Res. (2019) 38:328. doi: 10.1186/s13046-019-1327-4
166. Simpson KJ, Dugan AS, Mercurio AM. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. (2004) 64:8694–701. doi: 10.1158/0008-5472.CAN-04-2247
167. Arpaia E, Blaser H, Quintela-Fandino M, Duncan G, Leong HS, Ablack A, et al. The interaction between caveolin-1 and Rho-GTPases promotes metastasis by controlling the expression of alpha5-integrin and the activation of Src, Ras and Erk. Oncogene. (2012) 31:884–96. doi: 10.1038/onc.2011.288
168. Luo M, Zhao X, Chen S, Liu S, Wicha MS, Guan JL. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. (2013) 73:5591–602. doi: 10.1158/0008-5472.CAN-13-1351
169. Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. (2009) 69:466–74. doi: 10.1158/0008-5472.CAN-08-3078
170. Thiagarajan PS, Sinyuk M, Turaga SM, Mulkearns-Hubert EE, Hale JS, Rao V, et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat Commun. (2018) 9:578. doi: 10.1038/s41467-018-02938-1
171. Nguyen K, Yan Y, Yuan B, Dasgupta A, Sun J, Mu H, et al. ST8SIA1 regulates tumor growth and metastasis in TNBC by activating the FAK-AKT-mTOR signaling pathway. Mol Cancer Ther. (2018) 17:2689–701. doi: 10.1158/1535-7163.MCT-18-0399
172. Kolev VN, Tam WF, Wright QG, McDermott SP, Vidal CM, Shapiro IM, et al. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget. (2017) 8:51733–47. doi: 10.18632/oncotarget.18517
173. Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. (2016) 29:255–69. doi: 10.1016/j.ccell.2016.02.006
174. Garrido-Castro AC, Goel S. CDK4/6 inhibition in breast cancer: mechanisms of response and treatment failure. Curr Breast Cancer Rep. (2017) 9:26–33. doi: 10.1007/s12609-017-0232-0
175. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. (2017) 548:471–5. doi: 10.1038/nature23465
176. Dutertre S, Descamps S, Prigent C. On the role of aurora-A in centrosome function. Oncogene. (2002) 21:6175–83. doi: 10.1038/sj.onc.1205775
177. Joukov V, De Nicolo A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci Signal. (2018) 11:eaar4195. doi: 10.1126/scisignal.aar4195
178. Li M, Gao K, Chu L, Zheng J, Yang J. The role of Aurora-A in cancer stem cells. Int J Biochem Cell Biol. (2018) 98:89–92. doi: 10.1016/j.biocel.2018.03.007
179. Zheng F, Yue C, Li G, He B, Cheng W, Wang X, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. (2016) 7:10180. doi: 10.1038/ncomms10180
180. Opyrchal M, Gil M, Salisbury JL, Goetz MP, Suman V, Degnim A, et al. Molecular targeting of the Aurora-A/SMAD5 oncogenic axis restores chemosensitivity in human breast cancer cells. Oncotarget. (2017) 8:91803–16. doi: 10.18632/oncotarget.20610
181. Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. (2010) 2:a000109. doi: 10.1101/cshperspect.a000109
182. Vazquez-Santillan K, Melendez-Zajgla J, Jimenez-Hernandez LE, Gaytan-Cervantes J, Munoz-Galindo L, Pina-Sanchez P, et al. NF-kappaBeta-inducing kinase regulates stem cell phenotype in breast cancer. Sci Rep. (2016) 6:37340. doi: 10.1038/srep37340
183. Ling L, Cao Z, Goeddel DV. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc Natl Acad Sci USA. (1998) 95:3792–7. doi: 10.1073/pnas.95.7.3792
184. Thu YM, Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. (2010) 21:213–26. doi: 10.1016/j.cytogfr.2010.06.002
185. Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, et al. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. (2001) 107:763–75. doi: 10.1016/S0092-8674(01)00599-2
186. Yamamoto M, Ito T, Shimizu T, Ishida T, Semba K, Watanabe S, et al. Epigenetic alteration of the NF-kappaB-inducing kinase (NIK) gene is involved in enhanced NIK expression in basal-like breast cancer. Cancer Sci. (2010) 101:2391–7. doi: 10.1111/j.1349-7006.2010.01685.x
187. Yang C, Atkinson SP, Vilella F, Lloret M, Armstrong L, Mann DA, et al. Opposing putative roles for canonical and noncanonical NFkappaB signaling on the survival, proliferation, and differentiation potential of human embryonic stem cells. Stem Cells. (2010) 28:1970–80. doi: 10.1002/stem.528
188. Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun. (2013) 4:2299. doi: 10.1038/ncomms3299
189. Zhang W, Tan W, Wu X, Poustovoitov M, Strasner A, Li W, et al. A NIK-IKKalpha module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell. (2013) 23:647–59. doi: 10.1016/j.ccr.2013.03.012
190. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. (2013) 501:338–45. doi: 10.1038/nature12625
191. Zare M, Bastami M, Solali S, Alivand MR. Aberrant miRNA promoter methylation and EMT-involving miRNAs in breast cancer metastasis: diagnosis and therapeutic implications. J Cell Physiol. (2017) 233:3729–44. doi: 10.1002/jcp.26116
192. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. (2013) 154:61–74. doi: 10.1016/j.cell.2013.06.005
193. Witt AE, Lee CW, Lee TI, Azzam DJ, Wang B, Caslini C, et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. (2017) 36:1707–20. doi: 10.1038/onc.2016.337
194. Rodriguez Bautista R, Ortega Gomez A, Hidalgo Miranda A, Zentella Dehesa A, Villarreal-Garza C, Avila-Moreno F, et al. Long non-coding RNAs: implications in targeted diagnoses, prognosis, and improved therapeutic strategies in human non- and triple-negative breast cancer. Clin Epigenet. (2018) 10:88. doi: 10.1186/s13148-018-0537-5
195. El Helou R, Pinna G, Cabaud O, Wicinski J, Bhajun R, Guyon L, et al. miR-600 acts as a bimodal switch that regulates breast cancer stem cell fate through WNT signaling. Cell Rep. (2017) 18:2256–68. doi: 10.1016/j.celrep.2017.02.016
196. Celia-Terrassa T, Liu DD, Choudhury A, Hang X, Wei Y, Zamalloa J, et al. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat Cell Biol. (2017) 19:711–23. doi: 10.1038/ncb3533
197. Deng L, Shang L, Bai S, Chen J, He X, Martin-Trevino R, et al. MicroRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res. (2014) 74:6648–60. doi: 10.1158/0008-5472.CAN-13-3710
198. Peng F, Li TT, Wang KL, Xiao GQ, Wang JH, Zhao HD, et al. H19/let-7/LIN28 reciprocal negative regulatory circuit promotes breast cancer stem cell maintenance. Cell Death Dis. (2017) 8:e2569. doi: 10.1038/cddis.2016.438
199. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene. (2017) 37:1062–74. doi: 10.1038/onc.2017.407
200. Deng J, Yang M, Jiang R, An N, Wang X, Liu B. Long Non-coding RNA HOTAIR regulates the proliferation, self-renewal capacity, tumor formation and migration of the Cancer Stem-Like Cell (CSC) subpopulation enriched from breast cancer cells. PLoS ONE. (2017) 12:e0170860. doi: 10.1371/journal.pone.0170860
201. Zeng L, Cen Y, Chen J. Long non-coding RNA MALAT-1 contributes to maintenance of stem cell-like phenotypes in breast cancer cells. Oncol Lett. (2018) 15:2117–22. doi: 10.3892/ol.2017.7557
202. Vidovic D, Huynh TT, Konda P, Dean C, Cruickshank BM, Sultan M, et al. ALDH1A3-regulated long non-coding RNA NRAD1 is a potential novel target for triple-negative breast tumors and cancer stem cells. Cell Death Differ. (2019). doi: 10.1038/s41418-019-0362-1. [Epub ahead of print].
203. Tu Z, Schmollerl J, Cuiffo BG, Karnoub AE. Microenvironmental regulation of long non-coding RNA LINC01133 promotes cancer-stem-cell-like phenotypic traits in triple-negative breast cancers. Stem Cells. (2019). doi: 10.1002/stem.3055. [Epub ahead of print].
204. Zhang Z, Sun L, Zhang Y, Lu G, Li Y, Wei Z. Long non-coding RNA FEZF1-AS1 promotes breast cancer stemness and tumorigenesis via targeting miR-30a/Nanog axis. J Cell Physiol. (2018) 233:8630–8. doi: 10.1002/jcp.26611
205. De Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J. (2018) 475:1611–34. doi: 10.1042/BCJ20170164
206. Sotgia F, Fiorillo M, Lisanti MP. Hallmarks of the cancer cell of origin: comparisons with “energetic” cancer stem cells (e-CSCs). Aging. (2019) 11:1065–8. doi: 10.18632/aging.101822
207. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. (2018) 24:1036–46. doi: 10.1038/s41591-018-0052-4
208. Wu T, Harder BG, Wong PK, Lang JE, Zhang DD. Oxidative stress, mammospheres and Nrf2 – new implication for breast cancer therapy? Mol Carcinogenesis. (2015) 54:1494–502. doi: 10.1002/mc.22202
209. Ryoo I-G, Choi B-H, Ku S-K, Kwak M-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. (2018) 17:246–58. doi: 10.1016/j.redox.2018.04.015
210. Kim D, Choi B-H, Ryoo I-G, Kwak M-K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. (2018) 9:896. doi: 10.1038/s41419-018-0903-4
211. Xu B, Wang S, Li R, Chen K, He L, Deng M, et al. Disulfiram/copper selectively eradicates AML leukemia stem cells in vitro and in vivo by simultaneous induction of ROS-JNK and inhibition of NF-κB and Nrf2. Cell Death Dis. (2017) 8:e2797. doi: 10.1038/cddis.2017.176
212. Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. (2008) 451:587–90. doi: 10.1038/nature06515
213. Chu F, Chou PM, Zheng X, Mirkin BL, Rebbaa A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer Res. (2005) 65:10183–7. doi: 10.1158/0008-5472.CAN-05-2002
214. Choi HK, Cho KB, Phuong NT, Han CY, Han HK, Hien TT, et al. SIRT1-mediated FoxO1 deacetylation is essential for multidrug resistance-associated protein 2 expression in tamoxifen-resistant breast cancer cells. Mol Pharm. (2013) 10:2517–27. doi: 10.1021/mp400287p
215. Heo J, Lim J, Lee S, Jeong J, Kang H, Kim Y, et al. Sirt1 regulates DNA methylation and differentiation potential of embryonic stem cells by antagonizing Dnmt3l. Cell Rep. (2017) 18:1930–45. doi: 10.1016/j.celrep.2017.01.074
216. Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. (2008) 2:241–51. doi: 10.1016/j.stem.2008.01.002
217. Ma W, Xiao GG, Mao J, Lu Y, Song B, Wang L, et al. Dysregulation of the miR-34a-SIRT1 axis inhibits breast cancer stemness. Oncotarget. (2015) 6:10432–44. doi: 10.18632/oncotarget.3394
218. Shi L, Tang X, Qian M, Liu Z, Meng F, Fu L, et al. A SIRT1-centered circuitry regulates breast cancer stemness and metastasis. Oncogene. (2018) 37:6299–315. doi: 10.1038/s41388-018-0370-5
219. O'Callaghan C, Vassilopoulos A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell. (2017) 16:1208–18. doi: 10.1111/acel.12685
220. Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA. (2012) 109:E187–96. doi: 10.1073/pnas.1105304109
221. Merino D, Whittle JR, Vaillant F, Serrano A, Gong JN, Giner G, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med. (2017) 9:eaam7049. doi: 10.1126/scitranslmed.aam7049
222. Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, et al. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. (2015) 5:12465. doi: 10.1038/srep12465
223. Ferrer I, Verdugo-Sivianes EM, Castilla MA, Melendez R, Marin JJ, Munoz-Galvan S, et al. Loss of the tumor suppressor spinophilin (PPP1R9B) increases the cancer stem cell population in breast tumors. Oncogene. (2016) 35:2777–88. doi: 10.1038/onc.2015.341
224. Sengupta S, Nagalingam A, Muniraj N, Bonner MY, Mistriotis P, Afthinos A, et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene. (2017) 36:5709–21. doi: 10.1038/onc.2017.164
225. Wang D, Xu J, Liu B, He X, Zhou L, Hu X, et al. IL6 blockade potentiates the anti-tumor effects of gamma-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. (2018) 25:330–9. doi: 10.1038/cdd.2017.162
226. Janghorban M, Xin L, Rosen JM, Zhang XH. Notch signaling as a regulator of the tumor immune response: to target or not to target? Front Immunol. (2018) 9:1649. doi: 10.3389/fimmu.2018.01649
227. Tominaga K, Shimamura T, Kimura N, Murayama T, Matsubara D, Kanauchi H, et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene. (2017) 36:1276–86. doi: 10.1038/onc.2016.293
228. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. (2009) 138:645–59. doi: 10.1016/j.cell.2009.06.034
229. Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. (2017) 9:1025–33. doi: 10.1038/nchem.2778
230. Huang X, Borgstrom B, Kempengren S, Persson L, Hegardt C, Strand D, et al. Breast cancer stem cell selectivity of synthetic nanomolar-active salinomycin analogs. BMC Cancer. (2016) 16:145. doi: 10.1186/s12885-016-2142-3
231. Taylor WR, Fedorka SR, Gad I, Shah R, Alqahtani HD, Koranne R, et al. Small-molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci Rep. (2019) 9:5926. doi: 10.1038/s41598-019-42251-5
232. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle. (2011) 10:3871–85. doi: 10.4161/cc.10.22.17976
233. Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. (2014) 32:2309–23. doi: 10.1002/stem.1746
Keywords: breast cancer stemness, chemoresistance, therapy failure, CSC-directed therapy, novel targets, plasticity, minimal residual disease
Citation: Sridharan S, Howard CM, Tilley AMC, Subramaniyan B, Tiwari AK, Ruch RJ and Raman D (2019) Novel and Alternative Targets Against Breast Cancer Stemness to Combat Chemoresistance. Front. Oncol. 9:1003. doi: 10.3389/fonc.2019.01003
Received: 26 June 2019; Accepted: 18 September 2019;
Published: 16 October 2019.
Edited by:
Seema Singh, Mitchell Cancer Institute, United StatesReviewed by:
Asfar S. Azmi, Wayne State University Karmanos Cancer Institute, United StatesKhalid El Bairi, Mohamed Premier University, Morocco
Vidya Sethunath, Baylor College of Medicine, United States
Copyright © 2019 Sridharan, Howard, Tilley, Subramaniyan, Tiwari, Ruch and Raman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dayanidhi Raman, ZGF5YW5pZGhpLnJhbWFuQHV0b2xlZG8uZWR1
†Co-first authors