 Ryland Hale
Ryland Hale Sami Sandakly
Sami Sandakly Janet Shipley
Janet Shipley Zoë Walters
Zoë Walters- 1Translational Epigenomics Team, Human Development and Health, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
- 2Sarcoma Molecular Pathology Team, Divisions of Molecular Pathology and Cancer Therapeutics, The Institute of Cancer Research, London, United Kingdom
Synovial Sarcomas (SS) are a type of Soft Tissue Sarcoma (STS) and represent 8–10% of all STS cases. Although SS can arise at any age, it typically affects younger individuals aged 15–35 and is therefore part of both pediatric and adult clinical practices. SS occurs primarily in the limbs, often near joints, but can present anywhere. It is characterized by the recurrent pathognomonic chromosomal translocation t(X;18)(p11.2;q11.2) that most frequently fuses SSX1 or SSX2 genes with SS18. This leads to the expression of the SS18-SSX fusion protein, which causes disturbances in several interacting multiprotein complexes such as the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex, also known as the BAF complex and the Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2). Furthermore, this promotes widespread epigenetic rewiring, leading to aberrant gene expression that drives the pathogenesis of SS. Good prognoses are characterized predominantly by small tumor size and young patient age. Whereas, high tumor grade and an increased genomic complexity of the tumor constitute poor prognostic factors. The current therapeutic strategy relies on chemotherapy and radiotherapy, the latter of which can lead to chronic side effects for pediatric patients. We will focus on the known roles of SWI/SNF, PRC1, and PRC2 as the main effectors of the SS18-SSX-mediated genome modifications and we present existing biological rationale for potential therapeutic targets and treatment strategies.
Introduction
Synovial sarcoma (SS) is a rare (1–3 cases per 1,000,000) aggressive high-grade malignancy mainly observed in adolescents and young adults, with a third of all SS cases occurring in patients under the age of 20. Pediatric SS predominantly occurs in the limbs (1), but can occur anywhere. This disease is characterized by the pathognomonic reciprocal t(X;18)(p11.2;q11.2) chromosome translocation, which leads to the fusion of the SS18 (formerly SYT) gene to one of three SSX genes (2–6). In two thirds of cases SS18 is fused to SSX1, with SSX2 as a fusion partner in most other cases, whilst a fusion with SSX4 occurs rarely. SS18 contains an SNH domain and a QPGY domain, both of which are present in the fusion protein. The SSX proteins contain a Krüppel associated box (KRAB) repression domain at their N-terminus that is excluded from the 79 C-terminal amino-acids (a.a.) fusing to SS18 (2–4, 7). SS18 functions as a co-activator of transcription, however the SSX proteins have been shown to moderate repression, allowing the oncoprotein the ability to activate and repress gene expression.
Other than the translocation, SS tumors are mutationally quiet. This is especially true in the pediatric setting (8). Furthermore, pediatric SS is associated with a better prognosis compared to its adult counterpart (9–16). Despite this, both patient populations face the same poor prognosis in metastatic context, which is associated with increased tumor genomic instability (8). A difference is seen in the rate of patients with metastases at diagnosis, reflected by 5–11% of pediatric patients (1, 17) compared to the 50% observed in newly diagnosed adult patients (1, 17, 18).
SS can be classified into three distinct histological subtypes (19). Monophasic SS is characterized by spindle cells of mesenchymal differentiation. Biphasic SS shows evidence of both epithelial and mesenchymal differentiation, resulting in epithelial-like structures among the spindle cells. The third subtype, undifferentiated SS, is characterized by a lack of differentiation. Currently, there is no clear evidence for a link between SSX fusion partner and histological subtype.
Like most other soft tissue sarcomas, systemic therapy for pediatric SS relies on heavy use of doxorubicin and ifosfamide associated with radiotherapy for local control (20), but surgical resection remains the only long-term curative technique. Low grade cases in pediatrics have shown no benefit from adjuvant chemotherapy and post-operative radiotherapy (21–25). There has been no progress in patient survival during the last three decades (26), which is linked to insufficient availability of therapeutics.
The hallmark SS chromosome translocation leads to the expression of a fusion protein consisting of the 379th N-terminal a.a. of SS18 to the 79th C-terminal a.a. of the given SSX fusion partner. These fusion proteins are widely considered to be the main driver of SS pathogenesis (27, 28), as their expression is sufficient to induce SS tumors in mice (29) and their silencing causes SS cells to revert to mesenchymal stem cell-like cells (30). However, neither fusion partner nor the fusion protein possess DNA binding domains (2, 3, 31). Regulation of gene expression by the fusion protein has been determined as indirect via interactions with protein complexes.
Recent evidence supporting epigenetic modifications driving sarcomagenesis (32) and the SS fusion acting as an epigenetic modifier (33, 34) has brought epigenetics to the forefront of the field for SS. Recent efforts have focused on unraveling the mechanism behind the SS18-SSX-mediated epigenetic rewiring.
In this paper, we summarize the evidence of the interplay between the SS fusion protein and the chromatin remodeling machinery with its associated epigenetic modifiers. We will discuss two key protein complex families, SWItch/Sucrose Non-Fermentable (SWI/SNF) and Polycomb Repressive Complexes (PRC) and how they can be targeted to improve current therapies and alleviate/avoid their harmful side-effects (35–38).
The SWI/SNF Complexes in SS
The SWI/SNF complexes, originally discovered in budding yeast (39–41), have been shown to be evolutionarily conserved, as seen by the presence of homologs of the complex components in Drosophila (42) and mammals (43–47). These complexes belong to the Trithorax Group (TrxG) proteins, and utilize ATP hydrolysis to regulate the chromatin state and control transcription (48–50). The SWI/SNF complexes have a canonically activating role, antagonistic to the one of the PRCs, whose description and link to SS is reviewed later.
There are two main mammalian SWI/SNF (mSWI/SNF) complex populations, BAF (BRG1 or BRM associated factors) and PBAF (Polybromo-associated BAF) (49, 51–54). The two populations contain either BRG1 or BRM, which are responsible for the catalytic ATPase activity of the complex, in addition to other core subunits (BAF155, BAF170, BAF47, BAF57) (55). The main difference between the two complexes is the inclusion of BAF250A/B or BAF200, in the BAF and PBAF complexes, respectively (56, 57). Moreover, only BAF complexes contain SS18 and have been shown to interact with the SS fusion proteins. The rest of the 12–14 complex subunits are tissue- and cell-type specific.
Historically, the first evidence for a link between the mSWI/SNF complexes and cancer was shown in Malignant Rhabdoid Tumors (MRT), with the hallmark biallelic inactivation of SMARCB1, the gene coding for the BAF47 protein, observed in 98% of tumors (58–60). This leads to a loss of function of the complexes thus promoting tumourigenesis. Later it was shown that up to 20% of all cancers show mutations in one of the subunits of the mSWI/SNF complexes, making it an extremely disrupted complex, with a key role in cancer (61–64). However, in SS the disruption of the BAF complex does not rely on a loss of function, but on a gain of function. The SS18-SSX fusion proteins have been shown to competitively replace the wild-type SS18 in the BAF complex and excluding BAF47 from it, leading to its proteasomal degradation (7, 65).
These oncogenic BAF complexes are subsequently retargeted to PRC repressed domains and have been shown to activate them. This is evidenced by the presence of BAF complexes on the SOX2 locus, and the decrease in PRC2 deposited tri-methylated lysine 27 on histone 3 (H3K27me3) marks on the same locus (65). This has been confirmed by McBride et al., showing a broad relocalization of the oncogenic BAF complexes in SS, specifically targeting PRC2 repressed domains and recruiting RNA Polymerase II to initiate transcription (66). Furthermore, the H3K27me3 levels increased at PRC2 repressed loci upon SS18-SSX knockdown, reverting back to a non-activated bivalent state (66), characterized by the presence of both activating H3K4me3 and repressive H3K27me3 marks (67). McBride et al. also described a targeting of the PBAF complexes to SS18-SSX activated genes (66), despite the complex not containing SS18, thus not incorporating the fusion protein. This suggests a downstream recruitment of PBAF complex following BAF relocalization.
The Role of the Polycomb Complexes in SS
The Polycomb Repressive Complexes 1 and 2 (PRC1 and PRC2) are members of a family of Polycomb group-proteins (PcG proteins). These multiprotein complexes are responsible for chromatin silencing of HOX genes (68, 69) through chromatin compaction (70, 71), mediated by their respective catalytic subunits. The current theory is that the SS18-SSX oncoprotein mediates its transcriptional silencing via interaction with PRC1 and PRC2, since studies have shown SS18-SSX to co-localize with the complexes (33, 72, 73). Figure 1 summarizes the important interplay between PRC2 and BAF for silencing and remodeling of chromatin, respectively.
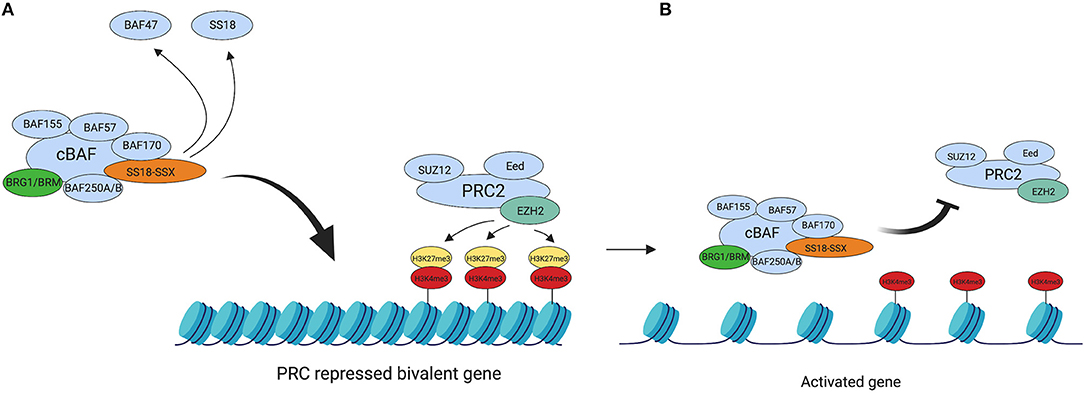
Figure 1. BAF complex retargeting in SS. (A) The oncogenic BAF complex, including one of the SS18-SSX fusion proteins is retargeted to PRC2 repressed domains. SS18-SSX inclusion in the BAF complex leads to the eviction of both SS18 and BAF47 from the complex. PRC2 mediates its transcriptional silencing activity via the catalytic subunit EZH2. EZH2 catalyses the tri-methylation of H3K27, which leads to chromatin compaction. (B) The oncogenic BAF complex evicts the PRC2 complex, thus repressing its activity and activating the previously bivalent promoter. This leads to an abnormal gene expression profile across the genome due to a widespread epigenetic rewiring and drives tumourigenesis. Created with BioRender.com.
PRC1 consists of two core subunits: RING1A/B and PCGF1-6. The canonical PRC1 (cPRC1), otherwise known as PRC1.2 or PRC1.4, contains the core subunits RING1A/B, with either polycomb group RING finger protein 2 or 4 (PCGF2 or PCGF4), respectively. These subunits are accompanied by Chromobox domains (Cbx) 2, 4, 6–8 and polyhomeotic homolog proteins 1–3 (HPH1-3) (74–77). There are also several heterogeneous non-canonical PRC1 complexes, however for the purpose of this review, we will be focussing mainly on cPRC1. The function of cPRC1 is the mono-ubiquitination of histone 2A at lysine 119 (H2AK119ub), controlled by the enzyme unit dRing that catalyses the E3 ligase activity (78, 79). This process also involves the Cbx proteins, which are thought to be the determinants for chromatin binding, since they interact with RING1A/B, to form a heterodimer with the core of the cPRC1 complex (74–76). The Cbx chromodomains have been found to preferably localize to H3K27me3 domains (77), which fits with the current hypothesis that PRC1 binds to trimethylated H3K27, in order to maintain suppression of transcription (69, 80–82).
The PCGF components are important for maintaining the protein-protein interactions that initiate chromatin silencing (83) and the knockdown of PCGF4 or either of the RING proteins, leads to a global reduction in H2AK119ub (69). Concurrent with this, Barco et al. also found that SS18-SSX2 influences PCGF4 by interacting with the polycomb complexes to downregulate PCGF4 and subsequently decrease the levels of H2AK119ub (84). This could indicate a method of reprieve from transcriptional silencing by PRC1, utilized by the modified BAF complex and further supports the idea that there is a sophisticated interplay between the different multiprotein complexes in SS.
The previous model suggested that PRC1 and PRC2 complemented each other when repressing chromatin, since many studies have identified that these PcG complexes commonly co-occupy many PcG target loci in Drosophila and mice (85–87). The theory was that PRC2 tri-methylated H3K27 and PRC1 maintained this state, due to the binding of a Cbx protein to H3K27me3 and the subsequent PRC1 binding (68, 88–90). However, compelling evidence shows that there may be PRC2 independent pathways for PRC1 chromatin silencing (91).
The Structure of PRC2 and Its Involvement in SS
PRC2 is another member of the PcG protein complexes. It exerts its chromatin silencing functions via its catalytic subunit Enhancer of Zeste 2 (EZH2) (92, 93), a histone methyltransferase. EZH2 catalyses di- or tri-methylation of lysine 27 on histone 3 (H3K27), to form H3K27me2 or H3K27me3, respectively. SS18-SSX and EZH2 mediate formation of H3K27me3, which leads to repression of tumor suppressor genes such as Early Growth Response 1 (EGR1) (94). The core PRC2 subunits of canonical PRC2 (cPRC2) are EZH2, SUZ12 and Eed (68, 90–95). There are also several PRC2 associated proteins (96, 97) but we will mainly focus on the core components of PRC2, since they are independently sufficient to perform di- and tri-methylation of H3K27 (98).
The previous hypothesis for the pathogenesis of SS was that the SS18-SSX oncoprotein completely knocked out the wild type BAF complex, which then disrupted the balance between the TrxG and PcG proteins (65, 99). It was thought that PRC2 activity was completely unopposed and therefore, EZH2 was able to catalyse over-production of the H3K27me3 motif. This chromatin repression also silences anti-tumourigenic proteins and EZH2 is able to maintain cells in a pluripotent state (100), which would fit with the neural stem-cell like landscape of SS (101, 102). Other cancers display an oncogenic dependency on the PcG complexes (55, 103), however McBride et al. have shown that EZH2 mediated activity may not be the driving factor for the pathogenesis of SS (66). There is currently no definitive theory for the mechanism of SS that accounts for the complex interplay between the TrxG and PcG complexes, therefore a better understanding of the effects and consequences of the expression of SS18-SSX fusion proteins is needed.
Novel Epigenetic Therapeutic Approaches
Due to the complexity of SS, its treatment will require multiple combined epigenetic therapies or combinations of targeted therapies, since single therapies have been trialed in the past and have only shown partial success (99, 104). A summary of potential therapeutic targets can be seen in Figure 2.
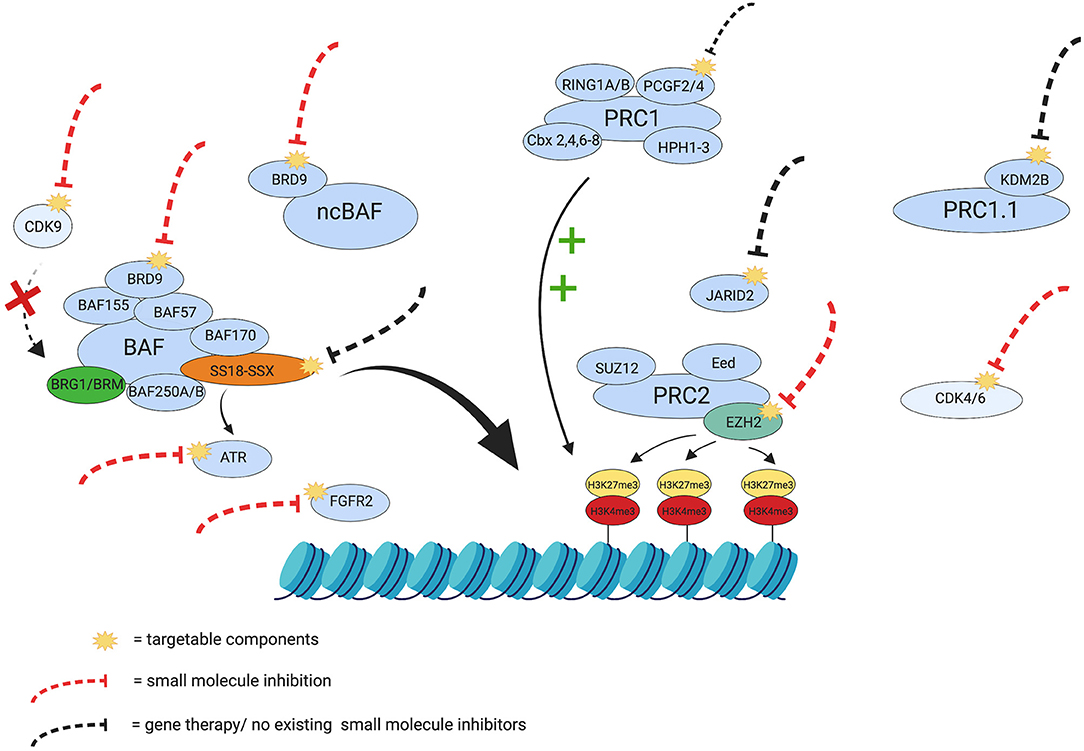
Figure 2. Therapeutic targets in Synovial Sarcoma. The yellow visuals represent the potentially targetable components discussed in this review. The red dashed lines indicate components in SS that already have small molecule inhibitors and the black dashed lines indicate potential targets that currently have no small molecule inhibitor or gene therapy. Certain targets in the figure (e.g., ATR and FGFR2) have been inhibited in other cancers and have shown potential synthetic lethality and growth reduction in SS, respectively, and have therefore been included for consideration as targets. The green plus signs indicate the maintenance of the repressive tri-methylation of H3K27, exerted by PRC1, and the red cross depicts the inhibition of BRG1 through dephosphorylation, should there be inhibition of CDK9. The figure also represents some of the interplay in SS that controls chromatin remodeling and silencing and emphasizes the need for a multifactorial approach to treatment and overcome potential treatment resistance. Created with BioRender.com.
SS have been shown to have high levels of EZH2 expression and subsequently high H3K27me3 motif expression (92, 99, 105). Moreover, EZH2 expression has been shown to correlate with poor prognosis in these tumors (105). Cho et al. also reported that in sarcomas, an increased expression of PRC2 and it's components were poor prognostic factors for overall survival (106). EZH2 catalyses the di- or tri-methylation of H3K27 via S-Adenosyl Methionine (SAM) and EZH2 expression was also found to be more commonly linked to metastatic SS (8, 106), which emphasizes the importance of EZH2 in metastatic SS. In hypomethylated prostate cancer, the addition of SAM showed a decrease in the proliferative and malignant potential of the cancer cells (107). This highlights the importance of SAM in cancer pathogenesis, therefore trialing a knockdown of SAM in SS should be considered for a combination therapy. Preclinical studies involving EZH2 inhibitors have shown some reduction in cell proliferation in SS cell lines (99, 104), which has prompted investigation in the clinical setting. Indeed, both pediatric and adult patients with refractory SS have been enrolled in phase I and II clinical trials testing the novel EZH2 inhibitor, tazemetostat (ClinicalTrials.gov Identifiers: NCT02601937, NCT02601950, and NCT02875548). Unfortunately, poor initial results in the adult setting were observed (108), which could be explained by the low levels of BAF47 present in SS (65, 109–111). This is further supported by the synthetic lethal effect exerted by combining BAF47 and PRC2 activity repression through EZH2 knockdown or inhibition with another EZH2 inhibitor (EPZ005687) (99). This result is coherent to what has previously been done in MRT with tazemetostat (103). BAF47 is an essential component of the complex under normal conditions (112, 113), but appears redundant for the activity of oncogenic BAF in SS. Instead, the complex relies on its gain of function through the SS18-SSX mediated specific retargeting (66), which explains the poor results shown in EZH2 inhibitor clinical trials.
JARID2 is a PRC2 associated protein (96, 97), which has DNA binding capabilities (114, 115) and has been shown to regulate the trimethyl mark of H3K27 in rhabdomyosarcomas in conjunction with PRC2 (116). Knockdown of JARID2 is linked to a global reduction of PRC2 binding to its target genes (117, 118), which indicates an interdependence for recruitment to target loci. An association between JARID2 and Eed suggests that Eed is the facilitator for the interaction between PRC2 and JARID2 (118), since knockdown of Eed in vitro and in Drosophila leads to global reduction in H3K27me3 (119). With a potential dependence on Eed, JARID2 has been shown to alter the methylation status of H3K27, thus implicating both Eed and JARID2 as potential targets.
SS differs from rhabdoid tumors and other BAF47 deficient tumors, since its key mechanism is the retargeting of the oncogenic BAF complexes through the 79th C-terminal a.a. of the SSX protein. This is supported by the fact that truncated SSX sequences disrupt the localization of the fusion proteins and inhibit SS proliferation (120), thus Figure 2 shows SS18-SSX to be a target for consideration.
Furthermore, the inhibition of a recently described non canonical BAF (ncBAF) complex (121), distinct from the two previously mentioned BAF and PBAF complexes, has been shown to be a potential synthetic lethal target for SS. The synthetic lethal approach relies on exploiting the vulnerability caused by the inclusion of the fusion genes in the cBAF complex, which becomes unable to perform its canonical role in the cells leading to an increased sensitivity to ncBAF inhibition. The depletion of the BRD9 subunit of the BAF and ncBAF complexes leads to cell death in both SS and MRT (122, 123). This is particularly interesting given the recent development of potent and specific BRD9 targeting compounds (124–128). Another synthetic lethal target for SS is the DNA damage response kinase ATR, which has been shown to impair growth of patient-derived SS xenografts (129). This study has indicated that SS has a dependence on ATR, which is apparent in other cancers and has been trialed in squamous non small cell lung cancer, small cell lung cancer, breast cancer and ovarian cancer (129, 130).
Another potential therapeutic target associated with the BAF complex, is cyclin-dependent kinase 9 (CDK9). It has recently been shown that its inhibition leads to the dephosphorylation of BRG1, one of the catalytic subunits of the BAF complex (131), which impairs its activity and activates silenced genes. The mechanism behind this is not completely understood but given the oncogenic relationship between the BAF complex and the SS fusion, this leads us to believe that it would be worth investigating the effect of CDK9 inhibition in a SS context.
Interestingly, another set of CDKs, namely CDK4 and CDK6, have been shown to be valuable therapeutic targets in SS as well. Targeting the cyclin D1-CDK4/6-Rb axis with the CDK4/6 inhibitor palbociclib leads to proliferation arrest and cell death in SS cell lines (132, 133). Palbociclib was approved for breast cancer treatment by the FDA in 2015 and is currently being trialed in various other cancers (134).
KDM2B is a histone demethylase and is a member of non-canonical PRC1, named PRC1.1 (77, 135). KDM2B is involved in the maintenance of embryonic stem cell (ESC) state in mouse cells (136) and is also implicated in multiple malignancies, such as gynecological, hematological, gastric, and pancreatic cancers (137–139). Its role in these cancers is linked to tumourigenesis and proliferation of the malignant cells, however more recently, KDM2B has been indicated for the maintenance of SS cell transformation (140).
SS18-SSX has also been shown to activate factors that influence lineage, most notably the FGF receptor gene (FGFR2) (101), which is important for inducing a neuronal lineage in stem cells (141). The effector proteins that are influenced by SS could provide another avenue for potential therapeutics. Another consideration for targeting is the protein-protein interaction that the PCGF proteins are responsible for maintaining, since this enables PRC1 mediated chromatin silencing and has not received much consideration previously. More insight into the different subtypes of SS could provide more targets or unknown synthetic lethal targets, however this would require more research into the genetic mechanisms involved in SS.
Conclusion
The growing evidence surrounding genetic abnormalities inducing a gain of function of the mSWI/SNF complexes, as opposed to the historically identified loss of function in MRT, makes a strong case for further investigation of the mechanism underlying the retargeting of the BAF complex. The interaction between the oncogenic BAF complex and the PcG complexes in SS is not fully understood either and this information may lead to the identification of novel therapeutic targets. There are multiple epigenetic targets discussed in this mini-review that, if targeted in combination, could provide a well-tolerated and safe therapy for pediatric and adult SS. However, a comprehensive understanding of the SS18-SSX induced epigenetic rewiring in SS is needed to allow the most influential epigenetic modifiers to be identified and incorporated into an effective therapy.
Author Contributions
RH and SS wrote the manuscript. All authors contributed to the manuscript and were involved in revisions and proof-reading. All authors approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005. Cancer. (2009) 115:3537–47. doi: 10.1002/cncr.24424
2. Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. (1994) 7:502–8. doi: 10.1038/ng0894-502
3. Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A, et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. (1995) 14:2333–40. doi: 10.1002/j.1460-2075.1995.tb07228.x
4. Shipley JM, Clark J, Crew AJ, Birdsall S, Rocques PJ, Gill S, et al. The t(X;18)(p11.2;q11.2) translocation found in human synovial sarcomas involves two distinct loci on the X chromosome. Oncogene. (1994) 9:1447–53. doi: 10.1016/0165-4608(94)90433-2
5. de Leeuw B, Balemans M, Olde Weghuis D, Geurts van Kessel A. Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum Mol Genet. (1995) 4:1097–9. doi: 10.1093/hmg/4.6.1097
6. Skytting B, Nilsson G, Brodin B, Xie Y, Lundeberg J, Uhlén M, et al. A novel fusion gene, SYT-SSX4, in synovial sarcoma. J Natl Cancer Inst. (1999) 91:974–5. doi: 10.1093/jnci/91.11.974
7. Thaete C, Brett D, Monaghan P, Whitehouse S, Rennie G, Rayner E, et al. Functional domains of the SYT and SYT-SSX synovial sarcoma translocation proteins and co-localization with the SNF protein BRM in the nucleus. Hum Mol Genet. (1999) 8:585–91. doi: 10.1093/hmg/8.4.585
8. Lagarde P, Przybyl J, Brulard C, Pérot G, Pierron G, Delattre O, et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol. (2013) 31:608–15. doi: 10.1200/JCO.2012.46.0147
9. Wright PH, Sim FH, Soule EH, Taylor WF. Synovial sarcoma. J Bone Joint Surg Am. (1982) 64:112–22. doi: 10.2106/00004623-198264010-00016
10. Singer S, Corson JM, Gonin R, Labow B, Eberlein TJ. Prognostic factors predictive of survival and local recurrence for extremity soft tissue sarcoma. Ann Surg. (1994) 219:165–73. doi: 10.1097/00000658-199402000-00008
11. Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. JCO. (1996) 14:1679–89. doi: 10.1200/JCO.1996.14.5.1679
12. Bergh P, Meis-Kindblom JM, Gherlinzoni F, Berlin Ö, Bacchini P, Bertoni F, et al. Synovial sarcoma. Cancer. (1999) 85:2596–607. doi: 10.1002/(SICI)1097-0142(19990615)85:12<2596::AID-CNCR16>3.0.CO;2-K
13. Spillane AJ, A'Hern R, Judson IR, Fisher C, Thomas JM. Synovial sarcoma: a clinicopathologic, staging, and prognostic assessment. JCO. (2000) 18:3794–803. doi: 10.1200/JCO.2000.18.22.3794
14. Spurrell EL, Fisher C, Thomas JM, Judson IR. Prognostic factors in advanced synovial sarcoma: an analysis of 104 patients treated at the Royal Marsden Hospital. Ann Oncol. (2005) 16:437–44. doi: 10.1093/annonc/mdi082
15. Brennan B, Stevens M, Kelsey A, Stiller CA. Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer. (2010) 55:85–90. doi: 10.1002/pbc.22453
16. Smolle MA, Parry M, Jeys L, Abudu S, Grimer R. Synovial sarcoma: do children do better? Eur J Surg Oncol. (2019) 45:254–60. doi: 10.1016/j.ejso.2018.07.006
17. Ferrari A, De Salvo GL, Oberlin O, Casanova M, De Paoli A, Rey A, et al. Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer. (2012) 48:1370–5. doi: 10.1016/j.ejca.2012.01.013
18. Bakri A, Shinagare AB, Krajewski KM, Howard SA, Jagannathan JP, Hornick JL, et al. Synovial sarcoma: imaging features of common and uncommon primary sites, metastatic patterns, and treatment response. Am J Roentgenol. (2012) 199:W208–15. doi: 10.2214/AJR.11.8039
20. Baldi GG, Orbach D, Bertulli R, Magni C, Sironi G, Casanova M, et al. Standard treatment and emerging drugs for managing synovial sarcoma: adult's and pediatric oncologist perspective. Expert Opin Emerg Drugs. (2019) 24:43–53. doi: 10.1080/14728214.2019.1591367
21. Baldini EH, Goldberg J, Jenner C, Manola JB, Demetri GD, Fletcher CD, et al. Long-term outcomes after function-sparing surgery without radiotherapy for soft tissue sarcoma of the extremities and trunk. J Clin Oncol. (1999) 17:3252–9. doi: 10.1200/JCO.1999.17.10.3252
22. Brecht IB, Ferrari A, Int-Veen C, Schuck A, Mattke AC, Casanova M, et al. Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer. (2006) 46:11–7. doi: 10.1002/pbc.20502
23. Cahlon O, Spierer M, Brennan MF, Singer S, Alektiar KM. Long-term outcomes in extremity soft tissue sarcoma after a pathologically negative re-resection and without radiotherapy. Cancer. (2008) 112:2774–9. doi: 10.1002/cncr.23493
24. Orbach D, Mc Dowell H, Rey A, Bouvet N, Kelsey A, Stevens MC. Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer. (2011) 57:1130–6. doi: 10.1002/pbc.23138
25. Ferrari A, De Salvo GL, Brennan B, van Noesel MM, De Paoli A, Casanova M, et al. Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol. (2015) 26:567–72. doi: 10.1093/annonc/mdu562
26. Wang S, Song R, Sun T, Hou B, Hong G, Mallampati S, et al. Survival changes in patients with Synovial Sarcoma, 1983-2012. J Cancer. (2017) 8:1759–68. doi: 10.7150/jca.17349
27. Limon J, Mrozek K, Mandant N, Nedoszytko B, Verhest A, Rys J, et al. Cytogenetics of synovial sarcoma: presentation of ten new cases and review of the literature. Genes Chromos Cancer. (1991) 3:338–45. doi: 10.1002/gcc.2870030504
28. Sandberg AA, Bridge JA. The Cytogenetics of Bone and Soft Tissue Tumors. Austin, TX: Landes (1994)
29. Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell. (2007) 11:375–88. doi: 10.1016/j.ccr.2007.01.016
30. Naka N, Takenaka S, Araki N, Miwa T, Hashimoto N, Yoshioka K, et al. Synovial sarcoma is a stem cell malignancy. Stem Cells. (2010) 28:1119–31. doi: 10.1002/stem.452
31. Lee Y-F, John M, Edwards S, Clark J, Flohr P, Maillard K, et al. Molecular classification of synovial sarcomas, leiomyosarcomas and malignant fibrous histiocytomas by gene expression profiling. Br J Cancer. (2003) 88:510–5. doi: 10.1038/sj.bjc.6600766
32. Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. (2016) 352:844–9. doi: 10.1126/science.aac7272
33. de Bruijn DRH, Allander SV, van Dijk AHA, Willemse MP, Thijssen J, van Groningen JJM, et al. The synovial-sarcoma-associated SS18-SSX2 fusion protein induces epigenetic gene (de)regulation. Cancer Res. (2006) 66:9474–82. doi: 10.1158/0008-5472.CAN-05-3726
34. Tamaki S, Fukuta M, Sekiguchi K, Jin Y, Nagata S, Hayakawa K, et al. SS18-SSX, the oncogenic fusion protein in synovial sarcoma, is a cellular context-dependent epigenetic modifier. PLoS ONE. (2015) 10:e0142991. doi: 10.1371/journal.pone.0142991
35. Skinner R, Sharkey IM, Pearson AD, Craft AW. Ifosfamide, mesna, and nephrotoxicity in children. J Clin Oncol. (1993) 11:173–90. doi: 10.1200/JCO.1993.11.1.173
36. Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. (2011) 21:440–6. doi: 10.1097/FPC.0b013e32833ffb56
37. De Angelis A, Urbanek K, Cappetta D, Piegari E, Ciuffreda LP, Rivellino A, et al. Doxorubicin cardiotoxicity and target cells: a broader perspective. Cardio-Oncology. (2016) 2:2. doi: 10.1186/s40959-016-0012-4
38. Ruysscher DD, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F. Radiotherapy toxicity. Nat Rev Dis Primers. (2019) 5:13. doi: 10.1038/s41572-019-0064-5
39. Neigeborn L, Carlson M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics. (1984) 108:845–58.
40. Peterson CL, Herskowitz I. Characterization of the yeast SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell. (1992) 68:573–83. doi: 10.1016/0092-8674(92)90192-F
41. Hirschhorn JN, Brown SA, Clark CD, Winston F. Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev. (1992) 6:2288–98. doi: 10.1101/gad.6.12a.2288
42. Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, et al. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2SWI2. Cell. (1992) 68:561–72. doi: 10.1016/0092-8674(92)90191-E
43. Khavari PA, Peterson CL, Tamkun JW, Mendel DB, Crabtree GR. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature. (1993) 366:170–4. doi: 10.1038/366170a0
44. Muchardt C, Yaniv M. A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J. (1993) 12:4279–90. doi: 10.1002/j.1460-2075.1993.tb06112.x
45. Kalpana G, Marmon S, Wang W, Crabtree G, Goff S. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science. (1994) 266:2002–6. doi: 10.1126/science.7801128
46. Muchardt C, Sardet C, Bourachot B, Onufryk C, Yaniv M. A human protein with homology to Saccharomyces cerevisiae SNF5 interacts with the potential helicase hbrm. Nucl Acids Res. (1995) 23:1127–32. doi: 10.1093/nar/23.7.1127
47. Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. (1996) 15:5370–82. doi: 10.1002/j.1460-2075.1996.tb00921.x
48. Côté J, Quinn J, Workman JL, Peterson CL. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science. (1994) 265:53–60. doi: 10.1126/science.8016655
49. Kwon H, Imbalzano AN, Khavari PA, Kingston RE, Green MR. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. (1994) 370:477–81. doi: 10.1038/370477a0
50. Imbalzano AN, Kwon H, Green MR, Kingston RE. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature. (1994) 370:481–5. doi: 10.1038/370481a0
51. Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. (1996) 10:2117–30. doi: 10.1101/gad.10.17.2117
52. Papoulas O, Beek SJ, Moseley SL, McCallum CM, Sarte M, Shearn A, et al. The Drosophila trithorax group proteins BRM, ASH1 and ASH2 are subunits of distinct protein complexes. Development. (1998) 125:3955–66.
53. Xue Y, Canman JC, Lee CS, Nie Z, Yang D, Moreno GT, et al. The human SWI/SNF-B chromatin-remodeling complex is related to yeast Rsc and localizes at kinetochores of mitotic chromosomes. Proc Natl Acad Sci USA. (2000) 97:13015–20. doi: 10.1073/pnas.240208597
54. Wang W. The SWI/SNF family of ATP-Dependent chromatin remodelers: similar mechanisms for diverse functions. In: Workman JL, editor. Protein Complexes that Modify Chromatin [Internet]. Berlin; Heidelberg: Springer Berlin Heidelberg (2003). p. 143–69. doi: 10.1007/978-3-642-55747-7_6
55. Middeljans E, Wan X, Jansen PW, Sharma V, Stunnenberg HG, Logie C. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE. (2012) 7:e33834. doi: 10.1371/journal.pone.0033834
56. Nie Z, Xue Y, Yang D, Zhou S, Deroo BJ, Archer TK, et al. A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol Cell Biol. (2000) 20:8879–88. doi: 10.1128/MCB.20.23.8879-8888.2000
57. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. (2013) 45:592–601. doi: 10.1038/ng.2628
58. Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. (1998) 394:203–6. doi: 10.1038/28212
59. Biegel JA, Zhou J-Y, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. (1999) 59:74–9.
60. Biegel JA, Fogelgren B, Zhou JY, James CD, Janss AJ, Allen JC, et al. Mutations of the INI1 rhabdoid tumor suppressor gene in medulloblastomas and primitive neuroectodermal tumors of the central nervous system. Clin Cancer Res. (2000) 6:2759–63.
61. Wilson BG, Roberts CWM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. (2011) 11:481–92. doi: 10.1038/nrc3068
62. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE. (2013) 8:e55119. doi: 10.1371/journal.pone.0055119
63. Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. (2014) 1:350–66. doi: 10.1002/ajmg.c.31410
64. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. (2015) 1:e1500447. doi: 10.1126/sciadv.1500447
65. Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. (2013) 153:71–85. doi: 10.1016/j.cell.2013.02.036
66. McBride MJ, Pulice JL, Beird HC, Ingram DR, D'Avino AR, Shern JF, et al. The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell. (2018) 33:1128–41.e7. doi: 10.1016/j.ccell.2018.05.002
67. Voigt P, Tee W-W, Reinberg D. A double take on bivalent promoters. Genes Dev. (2013) 27:1318–38. doi: 10.1101/gad.219626.113
68. Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. (2013) 20:1147–55. doi: 10.1038/nsmb.2669
69. Poynter ST, Kadoch C. Polycomb and trithorax opposition in development and disease. Wiley Interdisc Rev Dev Biol. (2016) 5:659–88. doi: 10.1002/wdev.244
70. Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. (2004) 306:1574–7. doi: 10.1126/science.1100576
71. Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. (2004) 38:413–43. doi: 10.1146/annurev.genet.38.072902.091907
72. Soulez M, Saurin AJ, Freemont PS, Knight JC. SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene. (1999) 18:2739. doi: 10.1038/sj.onc.1202613
73. Lubieniecka JM, de Bruijn DRH, Su L, van Dijk AHA, Subramanian S, van de Rijn M, et al. Histone deacetylase inhibitors reverse SS18-SSX–mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Cancer Res. (2008) 68:4303–10. doi: 10.1158/0008-5472.CAN-08-0092
74. Schoorlemmer J, Marcos-Gutiérrez C, Were F, Martínez R, García E, Satijn DP, et al. Ring1A is a transcriptional repressor that interacts with the Polycomb-M33 protein and is expressed at rhombomere boundaries in the mouse hindbrain. EMBO J. (1997) 16:5930–42. doi: 10.1093/emboj/16.19.5930
75. Bárdos JI, Saurin AJ, Tissot C, Duprez E, Freemont PS. HPC3 is a new human polycomb orthologue that interacts and associates with RING1 and Bmi1 and has transcriptional repression properties. J Biol Chem. (2000) 275:28785–92. doi: 10.1074/jbc.M001835200
76. Senthilkumar R, Mishra RK. Novel motifs distinguish multiple homologues of Polycomb in vertebrates: expansion and diversification of the epigenetic toolkit. BMC Genomics. (2009) 10:549. doi: 10.1186/1471-2164-10-549
77. Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell. (2012) 45:344–56. doi: 10.1016/j.molcel.2012.01.002
78. Cao R, Tsukada Y-I, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. (2005) 20:845–54. doi: 10.1016/j.molcel.2005.12.002
79. Elderkin S, Maertens GN, Endoh M, Mallery DL, Morrice N, Koseki H, et al. A phosphorylated form of Mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol Cell. (2007) 28:107–20. doi: 10.1016/j.molcel.2007.08.009
80. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. (2002) 298:1039–43. doi: 10.1126/science.1076997
81. Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. (2003) 17:1870–81. doi: 10.1101/gad.1110503
82. Min J, Zhang Y, Xu R-M. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. (2003) 17:1823–8. doi: 10.1101/gad.269603
83. Ishida A, Asano H, Hasegawa M, Koseki H, Ono T, Yoshida MC, et al. Cloning and chromosome mapping of the human Mel-18 gene which encodes a DNA-binding protein with a new ‘RING-finger' motif. Gene. (1993) 129:249–55. doi: 10.1016/0378-1119(93)90275-8
84. Barco R, Garcia CB, Eid JE. The synovial sarcoma-associated SYT-SSX2 oncogene antagonizes the polycomb complex protein Bmi1. PLoS ONE. (2009) 4:e5060. doi: 10.1371/journal.pone.0005060
85. Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. (2006) 441:349–53. doi: 10.1038/nature04733
86. Schwartz YB, Kahn TG, Nix DA, Li X-Y, Bourgon R, Biggin M, et al. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet. (2006) 38:700–5. doi: 10.1038/ng1817
87. Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. (2008) 4:e1000242. doi: 10.1371/journal.pgen.1000242
88. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. (2002) 16:2893–905. doi: 10.1101/gad.1035902
89. Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, et al. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat Genet. (2006) 38:694–9. doi: 10.1038/ng1792
90. Schwartz YB, Pirrotta V. A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet. (2013) 14:853–64. doi: 10.1038/nrg3603
91. Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. (2012) 148:664–78. doi: 10.1016/j.cell.2011.12.029
92. Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho Y-J, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. (2010) 18:316–28. doi: 10.1016/j.ccr.2010.09.006
93. Richly H, Aloia L, Di Croce L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. (2011) 2:e204. doi: 10.1038/cddis.2011.84
94. Ciarapica R, Miele L, Giordano A, Locatelli F, Rota R. Enhancer of zeste homolog 2 (EZH2) in pediatric soft tissue sarcomas: first implications. BMC Med. (2011) 9:63. doi: 10.1186/1741-7015-9-63
95. Liu PP, Xu YJ, Teng ZQ, Liu CM. Polycomb repressive complex 2: emerging roles in the central nervous system. Neuroscientist. (2017) 24:208–20. doi: 10.1177/1073858417747839
96. Smits AH, Jansen PWTC, Poser I, Hyman AA, Vermeulen M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. (2013) 41:e28. doi: 10.1093/nar/gks941
97. Vizán P, Beringer M, Ballaré C, Croce LD. Role of PRC2-associated factors in stem cells and disease. FEBS J. (2015) 282:1723–35. doi: 10.1111/febs.13083
98. Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. (2004) 15:57–67. doi: 10.1016/j.molcel.2004.06.020
99. Shen JK, Cote GM, Gao Y, Choy E, Mankin HJ, Hornicek FJ, et al. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci Rep. (2016) 6:25239. doi: 10.1038/srep25239
100. Gjerstorff MF, Relster MM, Greve KBV, Moeller JB, Elias D, Lindgreen JN, et al. SSX2 is a novel DNA-binding protein that antagonizes polycomb group body formation and gene repression. Nucleic Acids Res. (2014) 42:11433–46. doi: 10.1093/nar/gku852
101. Ishibe T, Nakayama T, Aoyama T, Nakamura T, Toguchida J. Neuronal differentiation of synovial sarcoma and its therapeutic application. Clin Orthop Relat Res. (2008) 466:2147–55. doi: 10.1007/s11999-008-0343-z
102. Garcia CB, Shaffer CM, Eid JE. Genome-wide recruitment to Polycomb-modified chromatin and activity regulation of the synovial sarcoma oncogene SYT-SSX2. BMC Genomics. (2012) 13:189. doi: 10.1186/1471-2164-13-189
103. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. (2013) 110:7922–7. doi: 10.1073/pnas.1303800110
104. Kawano S, Grassian AR, Tsuda M, Knutson SK, Warholic NM, Kuznetsov G, et al. Preclinical evidence of anti-tumor activity induced by EZH2 inhibition in human models of synovial sarcoma. PLoS ONE. (2016) 11:e0158888. doi: 10.1371/journal.pone.0158888
105. Changchien Y-C, Tátrai P, Papp G, Sápi J, Fónyad L, Szendroi M, et al. Poorly differentiated synovial sarcoma is associated with high expression of enhancer of zeste homologue 2 (EZH2). J Transl Med. (2012) 10:216. doi: 10.1186/1479-5876-10-216
106. Cho YJ, Kim SH, Kim EK, Han JW, Shin K-H, Hu H, et al. Prognostic implications of polycomb proteins ezh2, suz12, and eed1 and histone modification by H3K27me3 in sarcoma. BMC Cancer. (2018) 18:158. doi: 10.1186/s12885-018-4066-6
107. Schmidt T, Leha A, Salinas-Riester G. Treatment of prostate cancer cells with S-adenosylmethionine leads to genome-wide alterations in transcription profiles. Gene. (2016) 595:161–7. doi: 10.1016/j.gene.2016.09.032
108. Schoffski P, Agulnik M, Stacchiotti S, Davis LE, Villalobos VM, Italiano A, et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with synovial sarcoma (NCT02601950). J Clin Oncol. (2017) 35:11057. doi: 10.1200/JCO.2017.35.15_suppl.11057
109. Kohashi K, Oda Y, Yamamoto H, Tamiya S, Matono H, Iwamoto Y, et al. Reduced expression of SMARCB1/INI1 protein in synovial sarcoma. Mod Pathol. (2010) 23:981–90. doi: 10.1038/modpathol.2010.71
110. Rekhi B, Vogel U. Utility of characteristic ‘Weak to Absent' INI1/SMARCB1/BAF47 expression in diagnosis of synovial sarcomas. APMIS. (2015) 123:618–28. doi: 10.1111/apm.12395
111. Ito J, Asano N, Kawai A, Yoshida A. The diagnostic utility of reduced immunohistochemical expression of SMARCB1 in synovial sarcomas: a validation study. Hum Pathol. (2016) 47:32–7. doi: 10.1016/j.humpath.2015.09.010
112. Sen P, Luo J, Hada A, Hailu SG, Dechassa ML, Persinger J, et al. Loss of Snf5 induces formation of an aberrant SWI/SNF complex. Cell Rep. (2017) 18:2135–47. doi: 10.1016/j.celrep.2017.02.017
113. Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet. (2017) 49:1613–23. doi: 10.1038/ng.3958
114. Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. (2010) 24:368–80. doi: 10.1101/gad.1886410
115. Son J, Shen SS, Margueron R, Reinberg D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. (2013) 27:2663–77. doi: 10.1101/gad.225888.113
116. Walters ZS, Villarejo-Balcells B, Olmos D, Buist TWS, Missiaglia E, Allen R, et al. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene. (2014) 33:1148–57. doi: 10.1038/onc.2013.46
117. Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, et al. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell. (2009) 139:1303–14. doi: 10.1016/j.cell.2009.12.003
118. Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, et al. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. (2009) 139:1290–302. doi: 10.1016/j.cell.2009.12.002
119. Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, et al. Role of the polycomb protein Eed in the propagation of repressive histone marks. Nature. (2009) 461:762–7. doi: 10.1038/nature08398
120. Yoneda Y, Ito S, Kunisada T, Morimoto Y, Kanzaki H, Yoshida A, et al. Truncated SSX protein suppresses synovial sarcoma cell proliferation by inhibiting the localization of SS18-SSX fusion protein. PLoS ONE. (2013) 8:e77564. doi: 10.1371/journal.pone.0077564
121. Alpsoy A, Dykhuizen EC. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J Biol Chem. (2018) 293:3892–903. doi: 10.1074/jbc.RA117.001065
122. Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife. (2018) 7:e41305. doi: 10.7554/eLife.41305
123. Michel BC, D'Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. (2018) 20:1410. doi: 10.1038/s41556-018-0221-1
124. Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, et al. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J Med Chem. (2016) 59:1425–39. doi: 10.1021/acs.jmedchem.5b00256
125. Hohmann AF, Martin LJ, Minder JL, Roe J-S, Shi J, Steurer S, et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol. (2016) 12:672–9. doi: 10.1038/nchembio.2115
126. Martin LJ, Koegl M, Bader G, Cockcroft X-L, Fedorov O, Fiegen D, et al. Structure-based design of an in vivo active selective BRD9 inhibitor. J Med Chem. (2016) 59:4462–75. doi: 10.1021/acs.jmedchem.5b01865
127. Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo H-S, et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew Chem Int Ed Engl. (2017) 56:5738–43. doi: 10.1002/anie.201611281
128. Su J, Liu X, Zhang S, Yan F, Zhang Q, Chen J. Insight into selective mechanism of class of I-BRD9 inhibitors toward BRD9 based on molecular dynamics simulations. Chem Biol Drug Des. (2019) 93:163–76. doi: 10.1111/cbdd.13398
129. Jones SE, Fleuren EDG, Frankum J, Konde A, Williamson CT, Krastev DB, et al. ATR is a therapeutic target in synovial sarcoma. Cancer Res. (2017) 77:7014–26. doi: 10.1158/0008-5472.CAN-17-2056
130. Karnitz LM, Zou L. Molecular Pathways: targeting ATR in cancer therapy. Clin Cancer Res. (2015) 21:4780–5. doi: 10.1158/1078-0432.CCR-15-0479
131. Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell. (2018) 175:1244–58.e26. doi: 10.1016/j.cell.2018.09.051
132. Vlenterie M, Hillebrandt-Roeffen MHS, Schaars EWM, Flucke UE, Fleuren EDG, Navis AC, et al. Targeting cyclin-dependent kinases in synovial sarcoma: palbociclib as a potential treatment for synovial sarcoma patients. Ann Surg Oncol. (2016) 23:2745–52. doi: 10.1245/s10434-016-5341-x
133. Li X, Seebacher NA, Garbutt C, Ma H, Gao P, Xiao T, et al. Inhibition of cyclin-dependent kinase 4 as a potential therapeutic strategy for treatment of synovial sarcoma. Cell Death Dis. (2018) 9:446. doi: 10.1038/s41419-018-0474-4
134. Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front Oncol. (2018) 8:608. doi: 10.3389/fonc.2018.00608
135. Lagarou A, Mohd-Sarip A, Moshkin YM, Chalkley GE, Bezstarosti K, Demmers JAA, et al. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev. (2008) 22:2799–810. doi: 10.1101/gad.484208
136. He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat Cell Biol. (2013) 15:373–84. doi: 10.1038/ncb2702
137. Galbiati A, Penzo M, Bacalini MG, Onofrillo C, Guerrieri AN, Garagnani P, et al. Epigenetic up-regulation of ribosome biogenesis and more aggressive phenotype triggered by the lack of the histone demethylase JHDM1B in mammary epithelial cells. Oncotarget. (2017) 8:37091–103. doi: 10.18632/oncotarget.16181
138. Zhao X, Wang X, Li Q, Chen W, Zhang N, Kong Y, et al. FBXL10 contributes to the development of diffuse large B-cell lymphoma by epigenetically enhancing ERK1/2 signaling pathway. Cell Death Dis. (2018) 9:46. doi: 10.1038/s41419-017-0066-8
139. Kuang Y, Lu F, Guo J, Xu H, Wang Q, Xu C, et al. Histone demethylase KDM2B upregulates histone methyltransferase EZH2 expression and contributes to the progression of ovarian cancer in vitro and in vivo. Onco Targets Ther. (2017) 10:3131–44. doi: 10.2147/OTT.S134784
140. Banito A, Li X, Laporte AN, Roe J-S, Sanchez-Vega F, Huang C-H, et al. The SS18-SSX oncoprotein hijacks KDM2B-PRC1.1 to drive synovial sarcoma. Cancer Cell. (2018) 33:527–41.e8. doi: 10.1016/j.ccell.2018.01.018
Keywords: synovial sarcoma, SS18-SSX, epigenetics, therapeutic targets, SWI/SNF, PRC1, PRC2, chromatin remodeling
Citation: Hale R, Sandakly S, Shipley J and Walters Z (2019) Epigenetic Targets in Synovial Sarcoma: A Mini-Review. Front. Oncol. 9:1078. doi: 10.3389/fonc.2019.01078
Received: 23 July 2019; Accepted: 30 September 2019;
Published: 18 October 2019.
Edited by:
Karim Malik, University of Bristol, United KingdomReviewed by:
Scott C. Borinstein, Vanderbilt University, United StatesJean-François Michel Rual, University of Michigan, United States
Copyright © 2019 Hale, Sandakly, Shipley and Walters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoë Walters, ei5zLndhbHRlcnNAc290b24uYWMudWs=
†These authors have contributed equally to this work