 Hui-Ching Wang
Hui-Ching Wang Leong-Perng Chan
Leong-Perng Chan Shih-Feng Cho
Shih-Feng Cho- 1Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
- 2Division of Hematology and Oncology, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
- 3Department of Otolaryngology-Head and Neck Surgery, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
- 4Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
Head and neck squamous cell carcinoma (HNSCC) is a highly aggressive solid tumor, with a 5-year mortality rate of ~50%. The development of immunotherapies has improved the survival of patients with HNSCC, but, the long-term prognosis of patients with recurrent or metastatic HNSCC remains poor. HNSCC is characterized by intratumoral infiltration of regulatory T cells, dysfunctional natural killer cells, an elevated Treg/CD8+ T cell ratio, and increased programmed cell death ligand 1 protein on tumor cells. This leads to an immunocompromised niche in favor of the proliferation and treatment resistance of cancer cells. To achieve an improved treatment response, several potential combination strategies, such as increasing the neoantigens for antigen presentation and therapeutic agents targeting components of the tumor microenvironment, have been explored and have shown promising results in preclinical studies. In addition, large-scale bioinformatic studies have also identified possible predictive biomarkers of HNSCC. As immunotherapy has shown survival benefits in recent HNSCC clinical trials, a comprehensive investigation of immune cells and immune-related factors/cytokines and the immune profiling of tumor cells during the development of HNSCC may provide more insights into the complex immune microenvironment and thus, facilitate the development of novel immunotherapeutic agents.
Introduction
Head and neck cancer, 90% of which is squamous cell carcinoma (HNSCC), is the sixth most common cancer globally (1). HNSCC is composed of a heterogeneous group of tumors developing from the mucosa of the nasal and oral cavity, oropharynx, hypopharynx, or larynx (2). The major risk factors for HNSCC are smoking and alcohol consumption. Other risk factors include high risk human papillomavirus (HPV) infection, which is associated with oropharyngeal cancer increasingly worldwide (3). The areca nut chewing is linked to development of oral cancer in south Asia, Taiwan, and Pacific islanders. Treatment of HNSCC involves a multidisciplinary approach composed of surgery, radiotherapy, chemotherapy, and targeted therapy. However, the prognosis of metastatic HNSCC remains extremely poor. A combination of cetuximab and chemotherapy (cisplatin and 5-fluorouracil) shows better clinical efficacy than conventional chemotherapy; however, the median overall survival time is ~10 months (4). In recent years, the introduction of immune checkpoint inhibitors (ICIs) targeting the programmed death 1-programmed death ligand 1 (PD1-PDL1) pathway has resulted in further improvements in the outcome of patients with metastatic HNSCC, but the results remain unsatisfactory when compared with other malignancies, like melanoma and lung cancer (5, 6).
Accumulating data suggest that the tumor microenvironment (TME) plays an important role in the pathogenesis and development of treatment resistance in a variety of malignancies, including HNSCC. Several cell subtypes, including regulatory T cells (Tregs), cancer-associated fibroblasts, and macrophages, together with non-cellular components, like extracellular matrix (ECM), have been shown to be associated with immunocompromised status and the dysfunction of normal immune cells, like cytotoxic T cells or dendritic cells in the TME of HNSCC (7). HPV infection status and smoking are also related with distinct immune TMEs (8, 9).
To achieve improved treatment responses and clinical outcomes in the immunotherapy era, it is important to understand the complex immune TME of HNSCC. In this review, we describe major cell subtypes and cellular components and discuss their function. In addition, we summarize potential strategies to overcome TME-mediated treatment resistance.
Tumor Microenvironment of HNSCC
The heterogeneity of molecular and cellular components has been reported in the TME of HNSCC (10, 11). However, the HNSCC TME is still characterized by some unique features, leading to immunosuppression and diminished anticancer immunity (Table 1). The TME is composed of stromal cells, immune cells, tumor cells, and cytokines, which mediate the interactions between these cells. HNSCC patients have decreased absolute T cell counts in the tumor and the circulation and the T cells have apoptotic features via the Fas/FasL signaling pathway and defective function (12, 16). The functional defects of tumor-infiltrating lymphocytes (TIL) include decreased expression of the CD3 zeta chain, decreased cytokine secretion, and loss of the ability to kill cancer cells (13–15). Tregs account for the major proportion of T cell components, which construct an immunosuppressive barrier, thus hindering the activity of effector T cells (Teffs) in the TME and interfering with the antitumor response to immunotherapy (26). A decrease in the number of immune cells with antigen-presenting machinery (such as dendritic cells) and in cytotoxic ability (such as natural killer cells) results in a profoundly immunodeficient tumor, which is common in HNSCC (16, 19, 27). Moreover, HNSCC tumors are characterized by desmoplastic stromal fibroblasts, which promote tumor invasion and progression via autocrine and paracrine factors (28, 29).
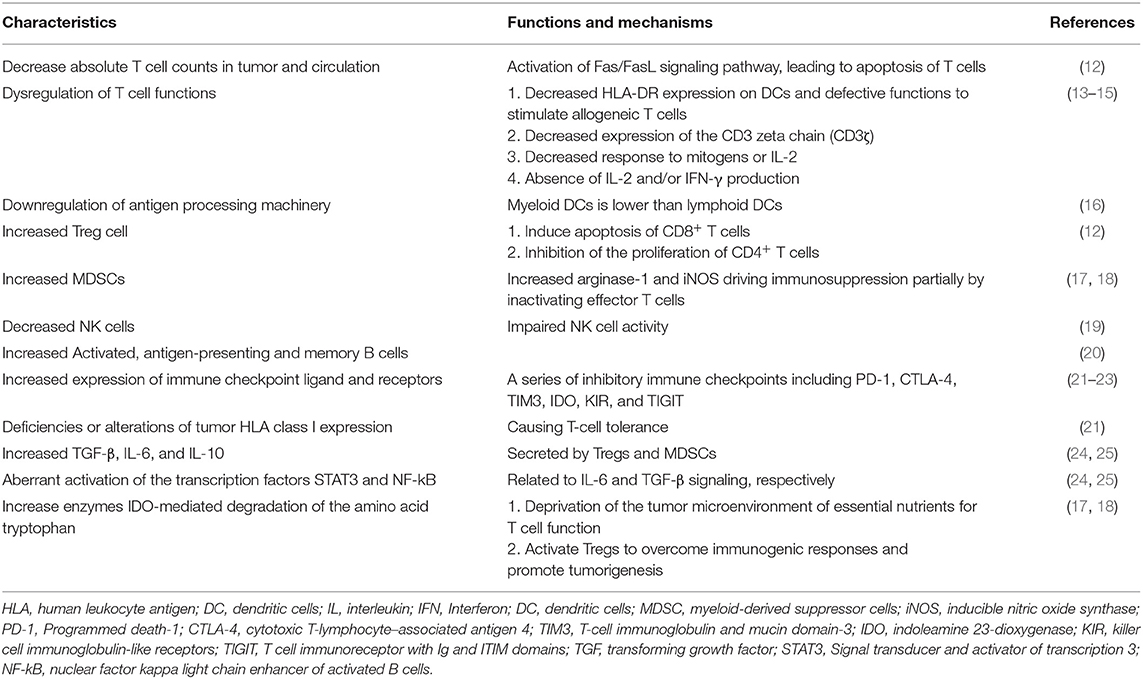
Table 1. Immune profilings of tumor microenvironment in HNSCC.
Communication within cancer cells, immune cells, and stromal cells via extracellular vesicles (EVs) is increasingly thought to be important (30). EVs not only deliver oncogenic proteins and non-coding RNA molecules to modulate tumor progression, but also modulate immune responses by inhibiting T cell proliferation and Th1 and Th17 differentiation (31). EVs promote suppressive immunity by activating Fas ligand (FasL), to induce CD8+ T cell apoptosis and the polarization of THP-1 to tumor-associated macrophages (TAMs) of the M2 phenotype (32, 33). Although several studies have analyzed the TME, it remains difficult to define HNSCC as an immune-inflamed, immune-excluded, or immune-desert tumor, due to diverse intratumor/peritumor expression patterns and the distribution of immune cells and cytokines (34, 35). The antitumor immune response to immunotherapy in the TME depends on the balance of stromal components, intratumoral Teffs, and immune-suppressive cell populations.
Cellular Component of the HNSCC TME
Regulatory T Cells
Tregs are a subset of T cells that contribute to the immunosuppressive TME in HNSCC (21). Treg recruitment is mediated by chemokines and associated receptors, such as CCL28-CCR10 and CXCL12-CXCR4 (36, 37). Tregs are characterized by specific markers, such as of CD4; CD25; and the transcription factor, forkhead box P3 (FOXP3) (22). Tregs express high levels of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which binds to CD80 and CD86 on antigen-presenting cells (APCs), leading to a reduced capacity to activate Teffs. Tregs exhibit their suppressor function by the consumption of interleukin-2 (IL-2), the secretion of granzyme and/or perforin to damage effector cells, and the production of immune-inhibitory cytokines and molecules, such as IL-10, IL-35, and transforming growth factor-β (38, 39). Tregs release large amounts of ATP and provide inhibitory signals to Teffs and APCs via the engagement of adenosine A2A receptor (A2AR) (40). In HNSCC, as in other malignancies, large numbers of Tregs infiltrate the TME. Intratumoral Tregs are more immunosuppressive than circulating Tregs, as evidenced by an increased expression of immune checkpoint molecules (23). A recent study identified a subset of Tregs with high levels of T-cell immunoglobulin and mucin domain-3 (TIM-3) expression from a population of CD4+CTLA-4+CD25high Treg cells. These high TIM-3-expressing Tregs are more immunosuppressive than Tregs with low levels of TIM-3 expression. After the administration of an anti-PD-1 monoclonal antibody, the expression of TIM-3 on this subgroup of T cells decreased (41). Another recent study demonstrated that Tregs are related to resistance to radiotherapy. The incorporation of an anti-CD25 antibody can overcome Treg-related treatment resistance (42). Several studies have demonstrated a negative prognostic impact of large numbers of Tregs in HNSCC (43, 44).
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) can be divided into three major subtypes, Ly6C+ monocytic MDSCs (M-MDSCs); Ly6G+ granulocytic polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs); and early stage e-MDSCs, which consist of the former two subsets deficient in myeloid lineage markers (45). The accumulation of MDSCs in the TME is associated with cancer progression and the inhibition of T cell activity and function (46). Various factors in the TME can induce the accumulation of MDSCs, including vascular endothelial growth factor (VEGF), IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (20). In addition, MDSCs regulate the TME by increasing the production of nitric oxide, reactive oxygen species, inducible NO synthase, and arginase-1; depleting various amino acids, such as L-arginine, L-tryptophan, and L-cystein; inducing pro-angiogenic factors; and elevating the expression of PD-L1 (17, 18). In HNSCC, a recent study demonstrated that a higher frequency of PMN-MDSCs is associated with poorer survival. Specifically, a subset of CD66b+/CD11b+/CD16+ mature PMN-MDSCs showed higher expression and activity of arginase I and demonstrated a greater suppressing effect on T cell proliferation and cytokine production than other MDSC subtypes. Moreover, high levels of CD11b+/CD16+ PMN-MDSCs, but not other PMN-MDSC subsets, are strongly correlated with adverse outcomes in HNSCC patients (47).
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) construct the stroma of the TME to promote the growth of cancer cells. CAFs possess different characteristics dependent on their status. For example, the active form of CAFs displays typical markers, such as α-smooth muscle actin and fibroblast activation protein and promotes tumor proliferation, invasion, and metastasis (48–50). CAFs regulate the TME via secretion of various cytokines and growth factors, such as VEGF, epidermal growth factor, C-X-C motif chemokine ligands, and C-C motif chemokine ligands (CCLs) (51, 52). Most importantly, CAFs secrete matrix-metalloproteinases (MMPs), which are crucial regulators of the TME and are responsible for degradation of the ECM (53). CAFs can be transformed from diverse progenitor cells, including endothelial cells, resting fibroblasts, and epithelial cells, via mesothelial-mesenchymal transition or epithelial-mesenchymal transition (EMT) (54, 55). In the TME of HNSCC, CAFs can promote the proliferation, migration, and invasion of tumor cells (29). CAFs also have a metabolic relationship with tumor cells. CAFs secrete hepatocyte growth factor (HGF), which then activates c-met to promote the progression of HNSCC (56). Additionally, HNSCCs secrete basic fibroblast growth factor (bFGF) which increases the phosphorylation of p44/42 mitogen-activated protein kinase, leading to the secretion of HGF from CAFs. Notably, the secretion of bFGF is also mediated by CAF-secreted HGF. Inhibition of c-met and the FGF receptor can reduce tumor volume. CAFs are also associated with the development of cancer stem cells, which is associated with treatment resistance (57). CAF secretes periostin, which promotes a cancer stem cell-like phenotype via interaction with protein tyrosine kinase 7 (58). Another study also showed that CAFs secrete several proteins that promote the expression of stemness-associated genes in HNSCC cells. Inhibition of these protein-associated pathways can suppress tumor growth (59).
Tumor-Associated Macrophages
TAMs have two distinct phenotypes, M1 and M2, with different morphological and biological characteristics (60, 61). The activated M1 phenotype promotes Th1 response and displays pro-inflammatory behaviors, whereas the activated M2 phenotype enhances Th2 response and mediates anti-inflammatory functions, which are more associated with tumor progression, invasion, metastasis, and the suppression of T cell immunity (61–63). Activated M2 macrophages demonstrate upregulated levels of IL-10, arginase-1, and peroxisome proliferator-activated receptor γ, which are known as markers of M2 TAMs (64–66). The M2 phenotype is induced by several cytokines, such as IL-4, IL-10, and IL-13. Activated M2 macrophages inhibit M1 TAMs and promote tissue remodeling through the production and secretion of anti-inflammatory cytokines, including IL-1 receptor antagonist, IL-10, transforming growth factor-β (TGF-β), VEGF, and tumor necrosis factor-α (TNF-α) (24, 25). In HNSCC, TAMs are recruited to the TME and directly contact SCC cells. A recent study showed that CCL18 derived from M2 macrophages is able to promote tumor metastasis by inducing EMT and stemness (67). Regarding clinical significance, a meta-analysis showed that high CD68+ and CD163+ TAM density is associated with poor cell differentiation and advanced disease status (68). Another meta-analysis showed that high stromal levels of CD163+ TAMs are associated with poorer overall and progression-free survival (69).
Other Cellular Subtypes
Human natural killer (NK) cells are important in the innate immune system and can be classified into two subgroups according to the surface expression of CD56 and CD16. CD56dim/CD16bright NK cells are predominantly responsible for natural cytotoxicity, whereas CD56bright/CD16dim NK cells regulate immune reactions through the secretion of cytokines, such as interferon-γ and TNF-α (70, 71). The activation of NK cells induces the apoptosis of target cancer cell, through the exocytosis of perforin and granzymes, FasL and TNF-related apoptosis-inducing ligand (TRAIL) activation, or antibody-dependent cellular cytotoxicity (ADCC) (72, 73). The natural killer group 2D (NKG2D) receptors on immune cells, including NK and several T cell subsets, play an important role in immunosurveillance. By identifying and engaging the NKG2D ligand (NKG2DL) on tumor cells, NK and T cells can exert anti-tumor effects. In HNSCC, high plasma levels of shed NKG2DLs correlate with NK cell inhibition and disease progression (74).
Neutrophils are involved in the adaptive immunity response. Tumor-associated neutrophils (TANs) exhibit both pro- and anti-tumor characteristics. Similar to TAMs, TANs are also divided into two subgroups, N1 and N2 (75). Neutrophils eradicate cancer cells by releasing the antimicrobial and cytotoxic contents of their granules or by secreting immune mediators to recruit other antitumor effector cells. However, other factors from the tumor can shift neutrophils into a pro-tumor phenotype (76). Neutrophils with the pro-tumor N2 phenotype possess CXCR4, VEGF, and MMP-9 markers, which facilitate tumorigenesis, promote tumor growth, stimulate angiogenesis, and mediate immunosuppression (75).
Non-cellular Components in the TME
The ECM contains large composites of non-cellular factors, including structural proteins, growth factors, proteoglycans, and glycoproteins, which form the main structure of the TME (77). MMPs, which are mainly produced by the ECM, are a large family of proteins and peptide hydrolases that mediate the degradation of the ECM and facilitate the migration of cancer cells (78). MMPs also activate bFGF, VEGF, and TGF-β and promote angiogenesis (79, 80). Fibronectin is the major glycoprotein in the ECM and it plays a crucial role in interactions between other molecules, such as integrins, collagens, and fibrin (81, 82). Increased levels of fibronectin are associated with tumor invasion, progression, and resistance to treatment (83, 84). Other molecules are also involved in cell adhesion and proliferation and assist in supporting the surrounding TME.
HPV Infection and Smoking Are Associated With a Distinct Immune TME
HPV Infection
HPV infection plays a pivotal role in the immune modulation of HNSCC. In general, HPV-positive HNSCCs demonstrate relatively inflamed immunity compared with HPV-negative HNSCCs (Table 2). A TME with a prolonged viral infection induces anti-tumor immunity via the expression of tumor-associated antigens (TAAs) and tumor-specific antigens in immune cells and tumor cells (8). After cytotoxic therapies (radiotherapy or chemotherapy), the antigen-processing machinery (APM) promotes the expression of major histocompatibility complex (MHC) class I molecules to present the antigen peptide from dying tumor cells to T cells (89). In addition, an increase in the infiltration of NK cells and T cells, including CD3+, CD4+, and CD8+ TILs, creates a vigorous TME that stimulates cellular immunity in HPV-positive HNSCCs (85, 86). Interestingly, HPV-positive oropharyngeal cancer demonstrates higher CD4+, higher CD8+, and lower CD4+/CD8+ ratio compared with HPV-negative HNSCC (85). Humoral immunity is also induced by the recruitment of CD19+/CD20+ B cells (87). Antigen presentation and cytotoxicity are promoted by gathering dendritic cells (DCs) and APCs (86). An increase in the number of intratumor and peritumor infiltrating immune cells results in a favorable prognosis and enhances the response to radiotherapy and immunotherapy (34). The interaction between HPV-negative oropharyngeal cancer cells and CAFs results in the secretion of chemokines via an IL-1/IL-1R-mediated mechanism, which is less prominent within the HPV-positive TME (88). Thus, the metabolic profiles are quite different between HPV-positive and HPV-negative HNSCCs.
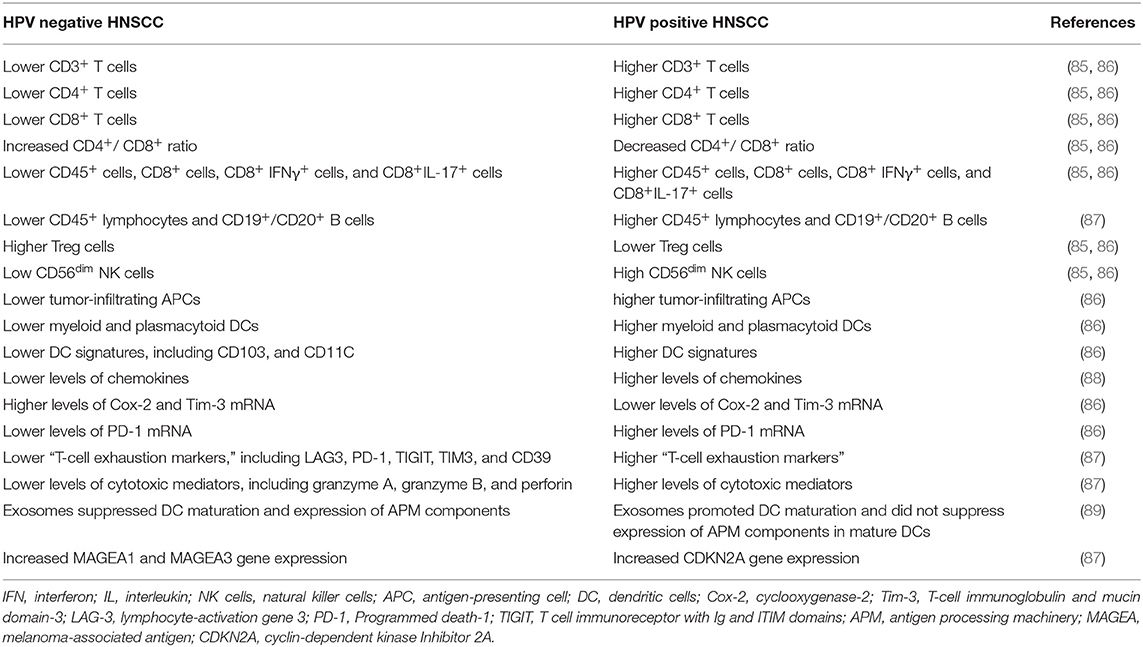
Table 2. Different immune modulations between HPV-negative and HPV-positive HNSCC.
The communication vesicles, EVs, also display different features depending on viral status. In HPV-positive cancers, exosomes carry viral proteins, genes, and TAAs (90, 91). However, these differences in EVs do not occur by influencing the T cell response. The functions of both CD4+ and CD8+ T cells are suppressed by these exosomes. The expression of co-stimulatory CD80 and CD83 molecules on immature DCs is up-regulated, but the expression of APM components is not suppressed in HPV-positive exosomes. In contrast, HPV(–) exosomes inhibit DC maturation and APM component expression (8). Moreover, HPV-negative tumors have a more active metabolic signature, with elevated expression of genes associated with glycolysis and oxidative phosphorylation (92). HPV-negative tumors are also characterized by increased MCT1 expression, which indicates that the regulation of lactate homoeostasis is more significant in promoting the invasion of HPV-negative HNSCCs (93).
Smoking
Smoking is a risk factor for the development of HNSCC and it promotes pro-inflammatory and immunosuppressive effects, which impact the TME of HNSCCs, to facilitate tumor development (94, 95). Smoking results in enrichment of immunogenic neoantigens which cause both pro- and anti-immunity effects in smoking-associated cancers, including lung cancer and HNSCC. In lung cancer, smoking leads to increased neoantigens and constructs an inflamed TME, which suggests higher response rates to ICIs in smokers. In contrast, the enhancement of immunogenic neoantigens by smoking forms a more immunosuppressive in TME in HNSCC by increased T cell apoptosis which is mediated through reactive oxygen and nitrogen species (94). In the TCGA database, enrichment scores from two Gene Expression Omnibus cohorts were higher in never-smoker and never-drinker (NSND) patients compared with smoker and drinker (SD) patients. To identify biological differences, gene set enrichment analysis of the TCGA dataset was performed and immunity-associated pathways were found to predominantly involve T-cell activation and differentiation in NSND patients. The TME in NSND patients is more immunoactive than the TME of SD patients, including an increased number of CD8+ TIL cells; increased INF-γactivation; overexpression of immune checkpoint ligands and receptors, such as indoleamine 23-dioxygenase 1 (IDO1) and PD-L1; and higher scores in the pembrolizumab-response signature (96). Tobacco smoking attenuates the cytotoxicity of the TME by repressing CD8+ T cells, NK cells, and DCs (9). Overall, smoking has a negative impact on immune responses, regardless of alcohol consumption.
Mechanisms of TME-Mediated Drug Resistance in HNSCC
The mechanisms of resistance to epidermal growth factor receptor (EGFR) inhibitors have been known for decades and they include nuclear localization of EGFR, activation of other ErbB family receptors, mutant forms of the receptor (EGFRvIII), or cross-talk with other signaling pathways (97, 98). However, issues of resistant mechanisms to immunotherapy have been gradually emphasized recently. These include a lack of production, editing, and presentation of neo-antigens; impaired intratumoral immune infiltration; impaired IFNγ signaling; immune factors within the TME; upregulation of alternative immune checkpoints; severe T-cell exhaustion; and T-cell epigenetic changes (99–102).
The downregulation of human leukocyte antigen (HLA) class I molecules and loss of β2-microglobulin expression interferes with antigen presentation to cytotoxic T cells (103). Specific oncogenic signaling pathways change the TME. Loss of the PTEN induces the expression of CCL2 and VEGF and blocks T-cell infiltration, leading to resistance to ICIs (104). Alterations in β-catenin/WNT signaling decrease CCL4 production and hinder the infiltration of DCs (105). During the development of ICI resistance, the TME shows an increase in the number of effector memory CD8 T cells (CCR7−CD45RA−), a lower CD4/CD8 ratio, and upregulation of TIM-3 on CD4 and CD8 T cells (100). Moreover, the major regulators of therapeutic response and resistance are Tregs and TAMs. In preclinical HNSCC mouse models, the Treg population is elevated during tumor rebound after combined treatment with ICI and radiation (26). Depletion of major histocompatibility complex class II-low TAMs increases chemotherapy-related DNA damage and apoptosis (106). Depletion of tumor-infiltrating Tregs using an anti-CD25 antibody, enhances the binding ability of activating Fc gamma receptors, increases Teff:Treg ratios, and improves the response to ICIs (107). High levels of alternative co-inhibitory receptors on T cells (e.g., CTLA-4, TIM-3, lymphocyte-activation gene 3, and V-domain Ig suppressor of T cell activation) and high levels of immune-suppressive cytokines or metabolites, causes T cell exhaustion, which also induces ICI resistance (108).
Potential Strategies to Overcome TME-Mediated Drug Resistance
Novel Therapeutic Agents or Combination Therapies
Due to the insufficient response elicited by immunotherapy alone, several mechanisms for the regulation of immunoresistant niches have been proposed, including defective immunorecognition, tumor insensitivity to T cell effector molecules, an immunosuppressive TME, and the compensatory regulation of multiple inhibitory and costimulatory immune checkpoints (Figure 1) (109). Combinations of diverse agents targeting distinct mechanisms have been investigated in recent years (Table 3).
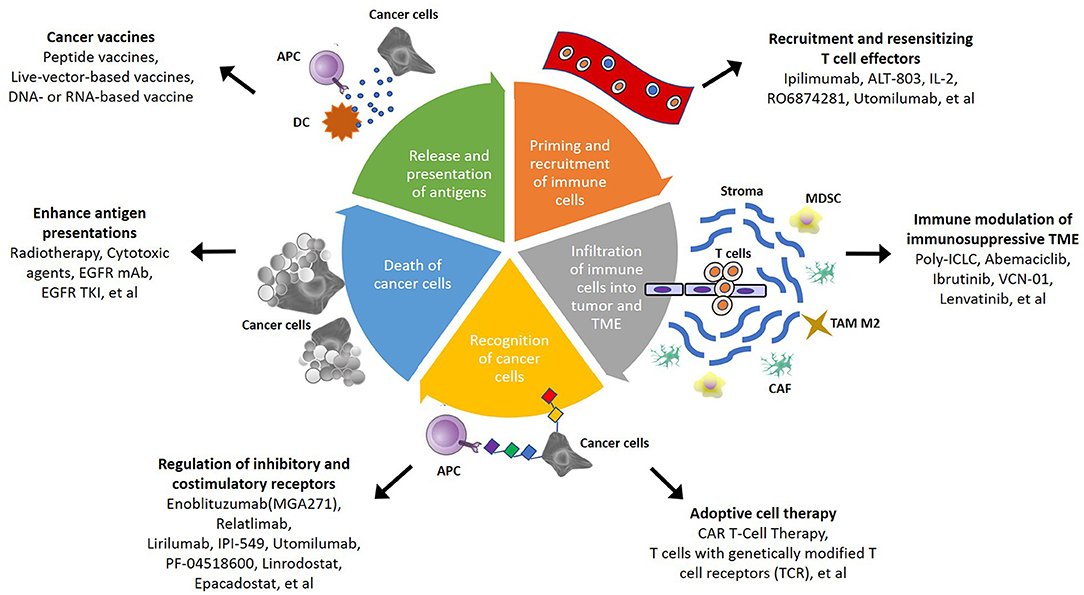
Figure 1. Schematic summary of potential strategies to overcome immunosuppressive TME in head and neck squamous cell carcinoma (HNSCC). In cancer-immunity cycle, there are several therapeutic strategies that can be applied to overcome TME-mediated treatment resistance. The steps of immune responses involve priming and recruitment of immune cells, infiltration of immune cells into tumor, and TME, recognition and death of cancer cells, then release and presentation of antigen from cancer cells. Targeting different mechanism of immune response become more potential therapeutic approach in the future.
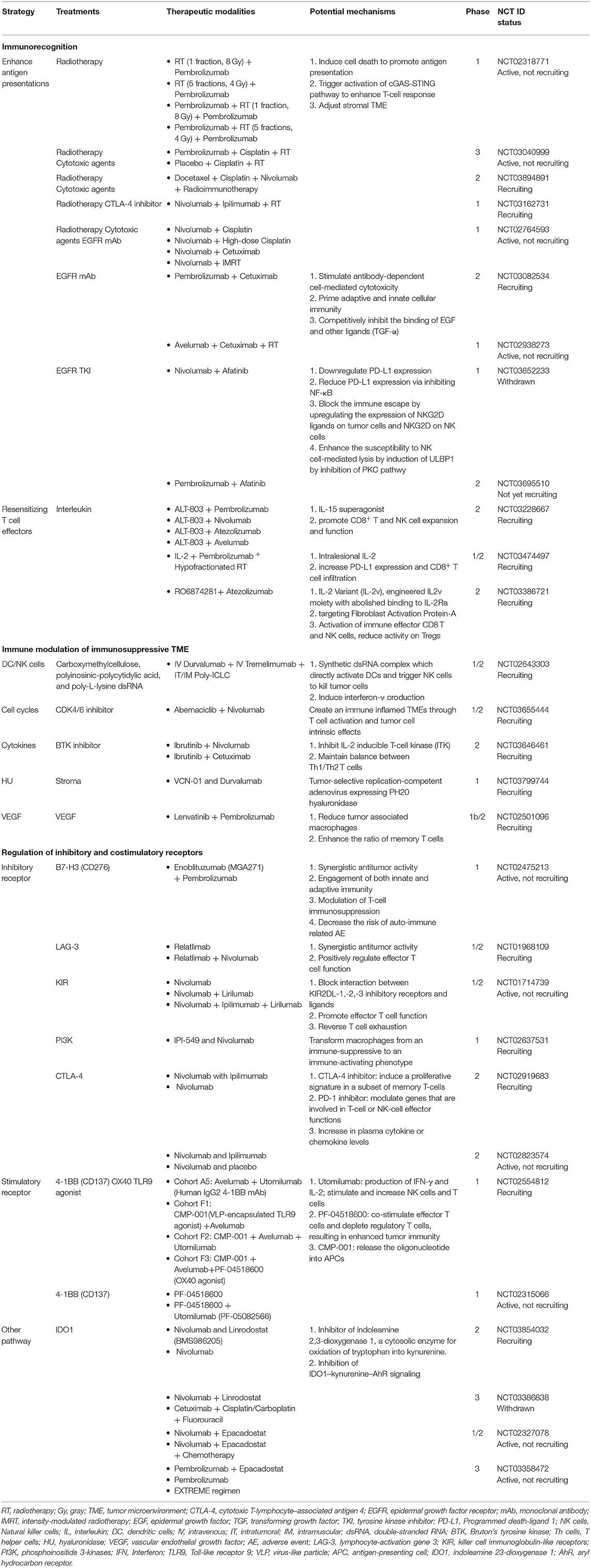
Table 3. Combination therapy to enhance PD-1/PD-L1-based treatment efficacy.
Defective immunorecognition involves dysfunction of antigen presentation in tumor cells, anergy of tumor-specific cytotoxic T lymphocyte, and immunoediting. Radiotherapy and cytotoxic therapy (NCT02318771, NCT03040999, NCT03894891, NCT03162731, NCT02764593, NCT02938273) induce cell death to promote antigen presentation and trigger activation of the cGAS-STING pathway to enhance the T-cell response. Moreover, radiation adjusts the stromal TME (110). Cetuximab (NCT02764593, NCT03082534, NCT02938273) binds to EGFR and to the CD16 receptor on NK cells and DCs, resulting in innate and adaptive immune responses, including ADCC and T cell priming (111). Afatinib (NCT03652233, NCT03695510), an EGFR tyrosine kinase inhibitor, downregulates PD-L1 expression via the inhibition of NF-κB. However, afatinib hinders immune escape by increasing the expression of NKG2D ligands on tumor cells and NKG2D on NK cells (112). ALT-803 (NCT03228667), an IL-15 superagonist, promotes CD8+ T cell and NK cell expansion and function and has demonstrated anti-tumor efficacy in preclinical models (113). Intralesional IL-2 (NCT03474497) increases PD-L1 expression and promotes CD8+ T cell infiltration (114). RO6874281 (NCT03386721), an engineered IL2v moiety, maintains its affinity for IL-2Rβγ, thus activating effector CD8 T cells and NK cells and reducing Treg activity (115).
The immunosuppressive TME also contributes to the low sensitivity of HNSCC to ICIs. Modulating different components of the TME improves the efficacy of ICIs and enhances self-immunity. Poly-ICLC (NCT02643303), a carboxymethylcellulose, polyinosinic-polycytidylic acid, and poly-L-lysine dsRNA, is a synthetic dsRNA complex that directly activates DCs, triggers NK cells, and induces interferon-γ production (116). Abemaciclib (NCT03655444), a CDK4/6 inhibitor, creates an immune inflamed TME through T cell activation and intrinsic tumor cell effects (117). Ibrutinib (NCT03646461), a Bruton's tyrosine kinase inhibitor, inhibits IL-2 inducible T-cell kinase (ITK), to strengthen specific anti-tumor responses (118). ITK plays a crucial role in maintaining the balance between Th1 and Th2 T cells. VCN-01 (NCT03799744), a selective oncolytic adenovirus encoding the human glycosylphosphatidylinositol-anchored enzyme, PH20 hyaluronidase, shows potential anti-tumor effects. Replication of the injected adenovirus in tumor cells results in cell death and the infection of adjacent tumor cells. Hyaluronidase also degrades hyaluronic acid (HA), which is abundant in the ECM and inhibits tumor cell growth and metastasis (119). Lenvatinib (NCT02501096), a multikinase inhibitor of VEGFR 1–3, fibroblast growth factor receptors (FGFR) 1–4, platelet-derived growth factor α receptors, RET, and KIT, reduces the number of TAMs and increases the ratio of memory T cells (120).
The regulation of inhibitory and costimulatory receptors synergically enhances the immunological anti-tumor effect. Inhibitory receptors, including B7-H3, LAG-3, killer cell immunoglobulin-like receptors (KIRs), phosphoinositide 3-kinases (PI3Ks), and CTLA-4, are applied in combination therapies. Enoblituzumab (NCT02475213), an Fc optimized, humanized IgG1 monoclonal antibody, promotes binding to activating FcγR and recognizes B7-H3, which is highly expressed in HNSCC. Combination therapy may contribute to synergistic antitumor activity (121). Relatlimab (NCT01968109), an anti-LAG-3 monoclonal antibody, shows an additive antitumor effect when administered with ICIs. LAG3 negatively regulates Teff function and is a marker of T cell exhaustion (122). IPI-549 (NCT02637531), a selective PI3K-γ inhibitor, transforms macrophages from an immune-suppressive to an immune-activating phenotype, which may help overcome resistance to ICIs (123). The well-known dual blockade therapy consisting of anti-CTLA-4 and anti-PD-1 antibodies (NCT02919683, NCT02823574), stimulates distinct immune cells and results in an inflammatory TME to overcome cancer cells (124). Similarly, cooperation with stimulatory receptors increases clinical benefits and treatment efficacy. Utomilumab (NCT01307267, NCT02315066), a 4-1BB/CD137 agonist, stimulates the activity and number of NK cells and T cells (125). PF-04518600 (NCT01307267, NCT02315066), a selective anti-OX40 antibody, activates OX40 and increases the proliferation of memory and effector T-lymphocytes (126). CMP-001 (NCT01307267), a Toll-like receptor 9 (TLR9) agonist, comprises a CpG-A oligodeoxynucleotide packaged in particles. It activates tumor-associated plasmacytoid DCs, which construct an interferon-rich TME and results in anti-tumor CD8+ T cell responses (127). IDO1, a major enzyme in tryptophan catabolism, is a target in clinical development, in combination with PD-1 ICIs (NCT03854032, NCT03386838, NCT02327078, NCT03358472). IDO1 converts tryptophan to kynurenine, which then activates aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, in Tregs, DCs, and NK cells. Activation of AhR induces subsequent cascades in three different cell types. In Tregs, AhR results in the nuclear translocation and enhancement of FoxP3 transcripts and IL10, eventually increasing Treg populations. In DCs, AhR promotes the production of IL-10 and inhibits IFNβ signaling. In NK cells, AhR induces the production of both IL-10 and IFNγ. The IDO1-kynurenine-AhR axis demonstrates a positive feedback loop. These effects on immune cells help establish an immunosuppressive TME. The inhibition of IDO reverses immunosuppression and enhances the response to ICIs (128).
Cancer Vaccines
Cancer vaccines targeting HPV antigens and tumor-associated antigens enhance the immune response in HNSCC. Therapeutic vaccines include peptide vaccines, live-vector-based vaccines, and DNA- or RNA-based vaccines. Peptide vaccines derived from HPV antigens are taken up by DCs and displayed by either MHC class I, class II, or both molecules, after which they induce a T-cell mediated immune response. Several trials have investigated such drugs, including DPX-E7 (NCT02865135), GL-0817/GL-0810 (NCT00257738), P16_37-63 peptide (NCT01462838, NCT02526316), and ISA 101(NCT02426892). However, the response rates have been variable in relatively small populations of patients (129, 130). Live-vector-based vaccines are more immunogenic and induce strong pathogen-derived CD8 epitopes (131). Recent cancer vaccine modalities include DNA and RNA vaccines encoding selected tumor antigens or synthetic long peptide (SLP) vaccines co-delivering CD4 and CD8 epitopes (132). DNA or peptide vaccines targeting HPV E6 and E7 oncoproteins have demonstrated specific clinical efficacy in precancerous lesions and have shown promise in the treatment of HPV-related HNSCC. However, the development of vaccines against HPV-independent HNSCC has been less successful due to the difficulty in identifying available targets (133). Additional vaccine modalities are required to overcome the immunosuppressive TME in HNSCC.
Cell-Based Therapy
T cells, including TILs, T cells with genetically modified T cell receptors (TCRs), and T cells transfected with chimeric antigen receptors (CAR), are the main types of cell-based therapy (134). Sufficient numbers of TILs overcome the immunosuppressive TME by removing other exhausted immune cells and inhibitory factors, such as cytokines. Adoptive immunotherapy using CAR T cells has displayed promising outcomes in hematological malignancies, such as leukemia and multiple myeloma. The process of CAR T cell therapy includes retrieving T cells from the patient's blood or tumor, training and stimulating their expansion in an in vitro system, and injecting the expanded cells back into the patient to promote cancer elimination. The development of tumor antigen-specific TCRs, for example HPV-targeted TCRs in genetically modify T cells, is another approach for adoptive immunotherapy. These modified T cells possess high levels of immune-signaling initiators and show rapid recognition of intracellular antigens, which can initiate an immune response against cancer cells. A phase I/II trial targeting the HLA-A*02:01-restricted epitope of E6 (E6 TCR T cells) enrolled patients with HPV-positive and HLA-A*02:01-positive metastatic epithelial cancers and showed that a dose up to 2 × 1011 cells was safe for patients. Partial responses in 2 of 12 patients (both with anal cancer) were reported (135). A phase I trial of T4 CAR T cell immunotherapy in HNSCC demonstrated safe intratumoral administration of T4 T-cells that co-express: (i) T1E28ζ, a CAR containing an ErbB ligand coupled to a CD28+CD3ζ endodomain and (ii) 4αβ, an IL-4-responsive chimeric cytokine receptor. Although a lymphopenia rate of 62% was observed, T4 manufacture was successful in 13/13 cases, yielding 2.5–7.5 Bn T cells (69 ± 13% transduced) (136). However, the development of adoptive cell therapy for HNSCC is still immature. There are still numerous difficulties and challenges including the identification of more specific peptide and genetic profiles of HNSCC cells. More precise knowledge of intracellular and extracellular neoantigens would help to identify potentially novel targets for cell therapy in HNSCC.
Potential Biomarkers in HNSCC Immunotherapy
Potential biomarkers in HNSCC have been discussed for many years, but there is still no consensus. Recent studies have tended to focus on specific biomarkers, including PD-L1 expression, HPV status, tumor immune infiltration, immune-associated signatures, gene expression profiles (GEPs), tumor mutational burden (TMB), the status of DNA mismatch repair, and smoking-related signatures. PD-L1 immunohistochemistry is the most frequently used marker in clinical practice. However, there are several challenges in the clinical application of these biomarkers. For example, PD-L1 is a heterogeneous marker with different intratumoral/temporal and primary/metastatic variations in expression (137). Different immunohistochemistry assays have been used, with different thresholds for positivity and different scoring criteria, including a tumor proportional score (TPS) and a combined proportional score (CPS) (138). HPV status also influences immunity within the TME and affects responses to immunotherapy (6). TILs, defined as CD8+ T cells and Tregs, have demonstrated a possible role in distinguishing ICI responders from ICI non-responders (139). GEP and TMB, analyzed by microarray or next-generation sequencing platforms, have been investigated as predictive biomarkers for biological phenotypes and clinical outcomes in HNSCC. Some analyses have shown that TMB, CPS, and GEP can serve as independent predictive biomarkers for responsiveness to anti-PD-1/PD-L1 antibodies (140). Tumors with more mutations influencing the DNA damage response, for example those with mismatch repair deficiency (dMMR), have a higher TMB and are more sensitive to ICIs. This contributed to the FDA approval of pembrolizumab for patients with dMMR or MSI-H tumors, regardless of histology (141–143). Overall, while the interactions between the tumor, the immune system, and the microenvironment are complex, more reliable predictive biomarkers are required to assess tumor responsiveness to immunotherapy.
Perspectives and Conclusions
As ICI monotherapy shows a durable response in only a small subset of patients, combination therapy with anti-PD-1/PD-L1 antibodies has emerged as an alternative and has shown encouraging results in the treatment of HNSCC. In addition, the anti-tumor effects of ICIs can be reinforced by increasing antigen presentation via radiation or chemotherapy/target therapy, modulating TME, or collaborating with costimulatory and inhibitory receptors on tumor cells or immune cells. The niches around cancer cells are crucial for interference with the efficacy of checkpoint inhibitors and they determine whether a tumor is “immunoactive” or “immunosuppressive.” Methods to overcome the immunotherapy resistance of the TME will become more crucial in the future. Multimodalities of treatment strategies aid in strengthening immunosurveillance and immunoediting. Studies to identify more specific targets for adoptive T cell therapies are ongoing. In addition, further studies designed to identify ideal biomarkers of individual tumors and to elucidate the mechanisms of immune escape are warranted.
Author Contributions
H-CW, L-PC, and S-FC substantially contributed to the conception, drafting, editing, and final approval of this manuscript.
Funding
The presented study was supported by Grant No. S10518-3 from the Kaohsiung Medical University Hospital, Kaohsiung, Taiwan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. (2005) 55:74–108. doi: 10.3322/canjclin.55.2.74
2. Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. (2008) 359:1143–54. doi: 10.1056/NEJMra0707975
3. Hsu WL, Yu KJ, Chiang CJ, Chen TC, Wang CP. Head and neck cancer incidence trends in Taiwan, 1980 ~ 2014. Int J Head Neck Sci. (2017) 1:180–9. doi: 10.6696/IJHNS.2017.0103.05
4. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. (2008) 359:1116–27. doi: 10.1056/NEJMoa0802656
5. Ferris RL, Blumenschein GR, Fayette J, Guigay J, Colevas AD, Licitra LF, et al. Further evaluations of nivolumab (nivo) versus investigator's choice (IC) chemotherapy for recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): CheckMate 141. Am Soc Clin Oncol. (2016) doi: 10.1200/JCO.2016.34.15_suppl.6009
6. Chow LQ, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. J Clin Oncol. (2016) 34:3838–45. doi: 10.1200/JCO.2016.68.1478
7. Peltanova B, Raudenska M, Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol Cancer. (2019) 18:63. doi: 10.1186/s12943-019-0983-5
8. Ludwig S, Sharma P, Theodoraki MN, Pietrowska M, Yerneni SS, Lang S, et al. Molecular and functional profiles of exosomes from HPV (+) and HPV (–) head and neck cancer cell lines. Front Oncol. (2018) 8:445. doi: 10.3389/fonc.2018.00445
9. Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol. (2009) 9:377–84. doi: 10.1038/nri2530
10. Keck MK, Zuo Z, Khattri A, Stricker TP, Brown CD, Imanguli M, et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin Cancer Res. (2015) 21:870–81. doi: 10.1158/1078-0432.CCR-14-2481
11. Hanna GJ, Liu H, Jones RE, Bacay AF, Lizotte PH, Ivanova EV, et al. Defining an inflamed tumor immunophenotype in recurrent, metastatic squamous cell carcinoma of the head and neck. Oral Oncol. (2017) 67:61–9. doi: 10.1016/j.oraloncology.2017.02.005
12. Duray A, Demoulin S, Hubert P, Delvenne P, Saussez S. Immune suppression in head and neck cancers: a review. Clin Dev Immunol. (2010) 2010:701657. doi: 10.1155/2010/701657
13. Reichert TE, Rabinowich H, Johnson JT, Whiteside TL. Mechanisms responsible for signaling and functional defects. J Immunother. (1998) 21:295–306. doi: 10.1097/00002371-199807000-00007
14. Whiteside TL. Immunobiology of head and neck cancer. Cancer Metastasis Rev. (2005) 24:95–105. doi: 10.1007/s10555-005-5050-6
15. Sakakura K, Chikamatsu K, Takahashi K, Whiteside TL, Furuya N. Maturation of circulating dendritic cells and imbalance of T-cell subsets in patients with squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. (2006) 55:151–9. doi: 10.1007/s00262-005-0697-y
16. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and-extrinsic factors. Immunity. (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
17. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
18. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
19. Echarri M, Lopez-Martin A, Hitt R. Targeted therapy in locally advanced and recurrent/metastatic head and neck squamous cell carcinoma (LA-R/M HNSCC). Cancers. (2016) 8:27. doi: 10.3390/cancers8030027
20. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. (2010) 185:2273–84. doi: 10.4049/jimmunol.1000901
21. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. (2007) 204:57–63. doi: 10.1084/jem.20061852
22. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
23. Jie H, Gildener-Leapman N, Li J, Srivastava R, Gibson S, Whiteside T, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. (2013) 109:2629–35. doi: 10.1038/bjc.2013.645
24. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
25. El-Rouby DH. Association of macrophages with angiogenesis in oral verrucous and squamous cell carcinomas. J Oral Pathol Med. (2010) 39:559–64. doi: 10.1111/j.1600-0714.2010.00879.x
26. Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin Cancer Res. (2018) 24:5368–80. doi: 10.1158/1078-0432.CCR-18-1038
27. Kather JN, Suarez-Carmona M, Charoentong P, Weis CA, Hirsch D, Bankhead P, et al. Topography of cancer-associated immune cells in human solid tumors. Elife. (2018) 7:e36967. doi: 10.7554/eLife.36967
28. Kunz-Schughart LA, Knuechel R. Tumor-associated fibroblasts (part I): active stromal participants in tumor development and progression? Histol Histopathol. (2002) 17:599–621. doi: 10.14670/HH-17.599
29. Wheeler SE, Shi H, Lin F, Dasari S, Bednash J, Thorne S, et al. Tumor associated fibroblasts enhance head and neck squamous cell carcinoma proliferation, invasion, and metastasis in preclinical models. Head Neck. (2014) 36:385–92. doi: 10.1002/hed.23312
30. Xie C, Ji N, Tang Z, Li J, Chen Q. The role of extracellular vesicles from different origin in the microenvironment of head and neck cancers. Mol Cancer. (2019) 18:83. doi: 10.1186/s12943-019-0985-3
31. Whiteside TL. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem Soc Trans. (2013) 41:245–51. doi: 10.1042/BST20120265
32. Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand–positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. (2005) 11:1010–20.
33. Al-Samadi A, Awad SA, Tuomainen K, Zhao Y, Salem A, Parikka M, et al. Crosstalk between tongue carcinoma cells, extracellular vesicles, and immune cells in in vitro and in vivo models. Oncotarget. (2017) 8:60123–34. doi: 10.18632/oncotarget.17768
34. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. (2016) 22:1865–74. doi: 10.1158/1078-0432.CCR-15-1507
35. Canning M, Guo G, Yu M, Myint C, Groves M, Byrd K, et al. Heterogeneity of the head and neck squamous cell carcinoma immune landscape and its impact on immunotherapy. Front Cell Dev Biol. (2019) 7:52. doi: 10.3389/fcell.2019.00052
36. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T reg cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
37. Zou L, Barnett B, Safah H, LaRussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+ CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. (2004) 64:8451–5. doi: 10.1158/0008-5472.CAN-04-1987
38. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
39. Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. (2006) 177:896–904. doi: 10.4049/jimmunol.177.2.896
40. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
41. Liu Z, McMichael EL, Shayan G, Li J, Chen K, Srivastava R, et al. Novel effector phenotype of Tim-3+ regulatory T cells leads to enhanced suppressive function in head and neck cancer patients. Clin Cancer Res. (2018) 24:4529–38. doi: 10.1158/1078-0432.CCR-17-1350
42. Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, et al. STAT3 Modulation of regulatory T cells in response to radiation therapy in head and neck cancer. J Natl Cancer Inst. (2019). doi: 10.1093/jnci/djz036. [Epub ahead of print].
43. Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+ CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. (2007) 13:6301–11. doi: 10.1158/1078-0432.CCR-07-1403
44. Al-Qahtani D, Anil S, Rajendran R. Tumour infiltrating CD25+ FoxP3+ regulatory T cells (Tregs) relate to tumour grade and stromal inflammation in oral squamous cell carcinoma. J Oral Pathol Med. (2011) 40:636–42. doi: 10.1111/j.1600-0714.2011.01020.x
45. Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol. (2010) 199:273–81. doi: 10.1007/s00430-010-0151-4
46. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
47. Lang S, Bruderek K, Kaspar C, Höing B, Kanaan O, Dominas N, et al. Clinical relevance and suppressive capacity of human myeloid-derived suppressor cell subsets. Clin Cancer Res. (2018) 24:4834–44. doi: 10.1158/1078-0432.CCR-17-3726
48. Lim KP, Cirillo N, Hassona Y, Wei W, Thurlow JK, Cheong SC, et al. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. J Pathol. (2011) 223:459–69. doi: 10.1002/path.2841
49. Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J Biol Chem. (1999) 274:36505–12. doi: 10.1074/jbc.274.51.36505
50. Wonganu B, Berger BW. A specific, transmembrane interface regulates fibroblast activation protein (FAP) homodimerization, trafficking and exopeptidase activity. Biochim Biophys Acta. (2016) 1858:1876–82. doi: 10.1016/j.bbamem.2016.05.001
51. Bello IO, Vered M, Dayan D, Dobriyan A, Yahalom R, Alanen K, et al. Cancer-associated fibroblasts, a parameter of the tumor microenvironment, overcomes carcinoma-associated parameters in the prognosis of patients with mobile tongue cancer. Oral Oncol. (2011) 47:33–8. doi: 10.1016/j.oraloncology.2010.10.013
52. Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med. (2013) 210:2851–72. doi: 10.1084/jem.20131195
53. Glentis A, Oertle P, Mariani P, Chikina A, El Marjou F, Attieh Y, et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat Commun. (2017) 8:924. doi: 10.1038/s41467-017-00985-8
54. Sandoval P, Jiménez-Heffernan JA, Rynne-Vidal Á, Pérez-Lozano ML, Gilsanz Á, Ruiz-Carpio V, et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J Pathol. (2013) 231:517–31. doi: 10.1002/path.4281
55. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. (2002) 110:341–50. doi: 10.1172/JCI0215518
56. Knowles LM, Stabile LP, Egloff AM, Rothstein ME, Thomas SM, Gubish CT, et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin Cancer Res. (2009) 15:3740–50. doi: 10.1158/1078-0432.CCR-08-3252
57. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. (2019) 18:70. doi: 10.1186/s12943-019-0994-2
58. Yu B, Wu K, Wang X, Zhang J, Wang L, Jiang Y, et al. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. (2018) 9:1082. doi: 10.1038/s41419-018-1116-6
59. Álvarez-Teijeiro S, García-Inclán C, Villaronga M, Casado P, Hermida-Prado F, Granda-Díaz R, et al. Factors secreted by cancer-associated fibroblasts that sustain cancer stem properties in head and neck squamous carcinoma cells as potential therapeutic targets. Cancers. (2018) 10:334. doi: 10.3390/cancers10090334
60. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
61. Lin CN, Chien CY, Chuang HC. Are friends or foes? New strategy for head and neck squamous cell carcinoma treatment via immune regulation. Int J Head Neck Sci. (2017) 1:105–13. doi: 10.6696/IJHNS.2017.0102.03
62. Weber M, Büttner-Herold M, Hyckel P, Moebius P, Distel L, Ries J, et al. Small oral squamous cell carcinomas with nodal lymphogenic metastasis show increased infiltration of M2 polarized macrophages–an immunohistochemical analysis. J Craniomaxillofac Surg. (2014) 42:1087–94. doi: 10.1016/j.jcms.2014.01.035
63. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. (2013) 229:176–85. doi: 10.1002/path.4133
64. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. (2013) 93:844–54. doi: 10.1038/labinvest.2013.69
65. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. (2005) 202:931–9. doi: 10.1084/jem.20050715
66. Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Ghassabeh GH, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. (2006) 108:525–35. doi: 10.1182/blood-2005-09-3777
67. She L, Qin Y, Wang J, Liu C, Zhu G, Li G, et al. Tumor-associated macrophages derived CCL18 promotes metastasis in squamous cell carcinoma of the head and neck. Cancer Cell Int. (2018) 18:120. doi: 10.1186/s12935-018-0620-1
68. Kumar AT, Knops A, Swendseid B, Martinez-Outschoorn UE, Harshyne L, Philp NJ, et al. Prognostic significance of tumor-associated macrophage content in head and neck squamous cell carcinoma: a meta-analysis. Front Oncol. (2019) 9:656. doi: 10.3389/fonc.2019.00656
69. Troiano G, Caponio VCA, Adipietro I, Tepedino M, Santoro R, Laino L, et al. Prognostic significance of CD68+ and CD163+ tumor associated macrophages in head and neck squamous cell carcinoma: a systematic review and meta-analysis. Oral Oncol. (2019) 93:66–75. doi: 10.1016/j.oraloncology.2019.04.019
70. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. (2001) 22:633–40. doi: 10.1016/S1471-4906(01)02060-9
71. Konjevic G, Jurisic V, Jovic V, Vuletic A, Martinovic KM, Radenkovic S, et al. Investigation of NK cell function and their modulation in different malignancies. Immunol Res. (2012) 52:139–56. doi: 10.1007/s12026-012-8285-7
72. Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. (2009) 128:7–15. doi: 10.1111/j.1365-2567.2009.03123.x
73. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. (2015) 6:368. doi: 10.3389/fimmu.2015.00368
74. Weil S, Memmer S, Lechner A, Huppert V, Giannattasio A, Becker T, et al. Natural killer group 2D ligand depletion reconstitutes natural killer cell immunosurveillance of head and neck squamous cell carcinoma. Front Immunol. (2017) 8:387. doi: 10.3389/fimmu.2017.00387
75. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta:“N1” versus “N2” TAN. Cancer cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
76. Zhang X, Zhang W, Yuan X, Fu M, Qian H, Xu W. Neutrophils in cancer development and progression: roles, mechanisms, and implications. Int J Oncol. (2016) 49:857–67. doi: 10.3892/ijo.2016.3616
77. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. (2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
78. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. (2002) 2:161–74. doi: 10.1038/nrc745
79. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. (2000) 2:737–44. doi: 10.1038/35036374
80. Tatti O, Vehviläinen P, Lehti K, Keski-Oja J. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp Cell Res. (2008) 314:2501–14. doi: 10.1016/j.yexcr.2008.05.018
81. Garcia AJ, Boettiger D. Integrin–fibronectin interactions at the cell-material interface: initial integrin binding and signaling. Biomaterials. (1999) 20:2427–33. doi: 10.1016/S0142-9612(99)00170-2
82. Brown AC, Dysart MM, Clarke KC, Stabenfeldt SE, Barker TH. Integrin α3β1 binding to fibronectin is dependent on the ninth type III repeat. J Biol Chem. (2015) 290:25534–47. doi: 10.1074/jbc.M115.656702
83. Lou X, Han X, Jin C, Tian W, Yu W, Ding D, et al. SOX2 targets fibronectin 1 to promote cell migration and invasion in ovarian cancer: new molecular leads for therapeutic intervention. OMICS. (2013) 17:510–8. doi: 10.1089/omi.2013.0058
84. Knowles LM, Gurski LA, Engel C, Gnarra JR, Maranchie JK, Pilch J. Integrin αvβ3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer Res. (2013) 73:6175–84. doi: 10.1158/0008-5472.CAN-13-0602
85. Matlung SE, Wilhelmina van Kempen PM, Bovenschen N, Baarle DV, Willems SM. Differences in T-cell infiltrates and survival between HPV+ and HPV-oropharyngeal squamous cell carcinoma. Future Sci OA. (2016) 2:FSO88. doi: 10.4155/fso.15.88
86. Partlová S, Bouček J, Kloudová K, Lukešová E, Zábrodský M, Grega M, et al. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV-associated compared to non-virally induced head and neck squamous cell carcinoma. Oncoimmunology. (2015) 4:e965570. doi: 10.4161/21624011.2014.965570
87. Lechner A, Schlößer HA, Thelen M, Wennhold K, Rothschild SI, Gilles R, et al. Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. Oncoimmunology. (2019) 8:1535293. doi: 10.1080/2162402X.2018.1535293
88. Al-Sahaf S, Hunter KD, Bolt R, Ottewell PD, Murdoch C. The IL-1/IL-1R axis induces greater fibroblast-derived chemokine release in human papillomavirus-negative compared to positive oropharyngeal cancer. Int J Cancer. (2019) 144:334–44. doi: 10.1002/ijc.31852
89. Bloy N, Garcia P, Laumont CM, Pitt JM, Sistigu A, Stoll G, et al. Immunogenic stress and death of cancer cells: contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev. (2017) 280:165–74. doi: 10.1111/imr.12582
90. Honegger A, Leitz J, Bulkescher J, Hoppe-Seyler K, Hoppe-Seyler F. Silencing of human papillomavirus (HPV) E6/E7 oncogene expression affects both the contents and the amounts of extracellular microvesicles released from HPV-positive cancer cells. Int J Cancer. (2013) 133:1631–42. doi: 10.1002/ijc.28164
91. Harden ME, Munger K. Human papillomavirus 16 E6 and E7 oncoprotein expression alters microRNA expression in extracellular vesicles. Virology. (2017) 508:63–9. doi: 10.1016/j.virol.2017.05.005
92. Fleming JC, Woo J, Moutasim K, Mellone M, Frampton SJ, Mead A, et al. HPV, tumour metabolism and novel target identification in head and neck squamous cell carcinoma. Br J Cancer. (2019) 120:356–67. doi: 10.1038/s41416-018-0364-7
93. Halestrap AP, Wilson MC. The monocarboxylate transporter family—role and regulation. IUBMB Life. (2012) 64:109–19. doi: 10.1002/iub.572
94. Desrichard A, Kuo F, Chowell D, Lee KW, Riaz N, Wong RJ, et al. Tobacco smoking-associated alterations in the immune microenvironment of squamous cell carcinomas. J Natl Cancer Inst. (2018) 110:1386–92. doi: 10.1093/jnci/djy060
95. Hernandez CP, Morrow K, Velasco C, Wyczechowska DD, Naura AS, Rodriguez PC. Effects of cigarette smoke extract on primary activated T cells. Cell Immunol. (2013) 282:38–43. doi: 10.1016/j.cellimm.2013.04.005
96. Foy JP, Bertolus C, Michallet MC, Deneuve S, Incitti R, Bendriss-Vermare N, et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann Oncol. (2017) 28:1934–41. doi: 10.1093/annonc/mdx210
97. Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. (2006) 12:5064–73. doi: 10.1158/1078-0432.CCR-06-0913
98. Hama T, Yuza Y, Suda T, Saito Y, Norizoe C, Kato T, et al. Functional mutation analysis of EGFR family genes and corresponding lymph node metastases in head and neck squamous cell carcinoma. Clin Exp Metastasis. (2012) 29:19–25. doi: 10.1007/s10585-011-9425-5
99. Bhatia S, Oweida A, Lennon S, Darragh LB, Milner D, Phan AV, et al. Inhibition of EphB4–Ephrin-B2 signaling reprograms the tumor immune microenvironment in head and neck cancers. Cancer Res. (2019) 79:2722–35. doi: 10.1158/0008-5472.CAN-18-3257
100. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
101. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515:577–81. doi: 10.1038/nature13988
102. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118:9–16. doi: 10.1038/bjc.2017.434
103. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
104. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
105. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
106. Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep. (2017) 19:101–13. doi: 10.1016/j.celrep.2017.03.038
107. Vargas FA, Furness AJ, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. (2017) 46:577–86. doi: 10.1016/j.immuni.2017.03.013
108. O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
109. Syn NL, Teng MW, Mok TS, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. (2017) 18:e731–41. doi: 10.1016/S1470-2045(17)30607-1
110. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol. (2018) 9:185. doi: 10.3389/fphar.2018.00185
111. Ferris RL, Lenz HJ, Trotta AM, García-Foncillas J, Schulten J, Audhuy F, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: Harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev. (2018) 63:48–60. doi: 10.1016/j.ctrv.2017.11.008
112. Liang H, Liu X, Wang M. Immunotherapy combined with epidermal growth factor receptor-tyrosine kinase inhibitors in non-small-cell lung cancer treatment. Onco Targets Ther. (2018) 11:6189–96. doi: 10.2147/OTT.S178497
113. Knudson KM, Hicks KC, Alter S, Schlom J, Gameiro SR. Mechanisms involved in IL-15 superagonist enhancement of anti-PD-L1 therapy. J Immunother Cancer. (2019) 7:82. doi: 10.1186/s40425-019-0551-y
114. Langan EA, Kümpers C, Graetz V, Perner S, Zillikens D, Terheyden P. Intralesional interleukin-2: a novel option to maximize response to systemic immune checkpoint therapy in loco-regional metastatic melanoma. Dermatol Ther. (2019) 32:e12901. doi: 10.1111/dth.12901
115. Soerensen MM, Ros W, Rodriguez-Ruiz ME, Robbrecht D, Rohrberg KS, Martin-Liberal J, et al. Safety, PK/PD, and anti-tumor activity of RO6874281, an engineered variant of interleukin-2 (IL-2v) targeted to tumor-associated fibroblasts via binding to fibroblast activation protein (FAP). Am Soc Clin Oncol. (2018) 36(Suppl. 15):e15155. doi: 10.1200/JCO.2018.36.15_suppl.e15155
116. Rady P, Cadet P, Bui T, Tyring S, Baron S, Stanton G, et al. Production of interferon gamma messenger RNA by cells of non-immune origin. Cytokine. (1995) 7:793–8. doi: 10.1006/cyto.1995.0095
117. Schaer D, Beckmann R, Dempsey J, Huber L, Forest A, Amaladas N, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. (2018) 22:2978–94. doi: 10.1016/j.celrep.2018.02.053
118. Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci USA. (2015) 112:E966–72. doi: 10.1073/pnas.1500712112
119. Hidalgo M, Gil M, Garcia-Carbonero R, Alvarez R, Laquente B, Moreno R, et al. First-in-human dose-escalation study of VCN-01, a selective oncolytic adenovirus with hyaluronidase activity in patients with advanced or metastatic cancer. Eur J Cancer. (2016) 69:S99–100. doi: 10.1016/S0959-8049(16)32895-7
120. Taylor MH, Rasco DW, Brose MS, Vogelzang NJ, Richey SL, Cohn AL, et al. A phase 1b/2 trial of lenvatinib plus pembrolizumab in patients with squamous cell carcinoma of the head and neck. Am Soc Clin Oncol. (2018) 36(Suppl. 15):6016. doi: 10.1200/JCO.2018.36.15_suppl.6016
121. Rizvi NA, Loo D, Baughman JE, Yun S, Chen F, Moore PA, et al. A phase 1 study of enoblituzumab in combination with pembrolizumab in patients with advanced B7-H3-expressing cancers. Am Soc Clin Oncol. (2016) 34(Suppl. 15):TPS3104. doi: 10.1200/JCO.2016.34.15_suppl.TPS3104
122. Lipson E, Long G, Tawbi H, Schadendorf D, Atkinson V, Maurer M, et al. 1302TiP CA224–047: a randomized, double-blind, phase II/III study of relatlimab (anti–LAG-3) in combination with nivolumab (anti–PD-1) versus nivolumab alone in previously untreated metastatic or unresectable melanoma. Ann Oncol. (2018) 29:mdy289.058. doi: 10.1093/annonc/mdy289.058
123. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. Am Soc Clin Oncol. (2018) 36(Suppl. 15):3013. doi: 10.1200/JCO.2018.36.15_suppl.3013
124. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti–CTLA-4 and anti–PD-1 leads to distinct immunologic changes in vivo. J Immunol. (2015) 194:950–9. doi: 10.4049/jimmunol.1401686
125. Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, et al. Structure of the 4–1BB/4–1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun. (2018) 9:4679. doi: 10.2210/pdb6mgp/pdb
126. Diab A, El-Khoueiry A, Eskens F, Ros W, Thompson J, Konto C, et al. A first-in-human (FIH) study of PF-04518600 (PF-8600) OX40 agonist in adult patients (pts) with select advanced malignancies. Ann Oncol. (2016) 27:359–78. doi: 10.1093/annonc/mdw378.08
127. Milhem M, Gonzales R, Medina T, Kirkwood JM, Buchbinder E, Mehmi I, et al. Abstract CT144: intratumoral toll-like receptor 9 (TLR9) agonist, CMP-001, in combination with pembrolizumab can reverse resistance to PD-1 inhibition in a phase Ib trial in subjects with advanced melanoma. Cancer Res. (2018) 78(Suppl. 13):CT144. doi: 10.1158/1538-7445.AM2018-CT144
128. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–kynurenine–aryl hydrocarbon axis. Clin Cancer Res. (2019) 25:1462–71. doi: 10.1158/1078-0432.CCR-18-2882
129. Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. (2019) 5:67–73. doi: 10.1001/jamaoncol.2018.4051
130. Schneider K, Grønhøj C, Hahn CH, von Buchwald C. Therapeutic human papillomavirus vaccines in head and neck cancer: a systematic review of current clinical trials. Vaccine. (2018) 36:6594–605. doi: 10.1016/j.vaccine.2018.09.027
131. Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. (2008) 8:108–20. doi: 10.1038/nrc2326
132. Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. (2002) 20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842
133. Wang C, Dickie J, Sutavani RV, Pointer C, Thomas GJ, Savelyeva N. Targeting head and neck cancer by vaccination. Front Immunol. (2018) 9:830. doi: 10.3389/fimmu.2018.00830
134. Qureshi HA, Lee SM. Immunotherapy approaches beyond PD-1 inhibition: the future of cellular therapy for head and neck squamous cell carcinoma. Curr Treat Options Oncol. (2019) 20:31. doi: 10.1007/s11864-019-0630-9
135. Hinrichs CS, Doran SL, Stevanovic S, Adhikary S, Mojadidi M, Kwong ML, et al. A phase I/II clinical trial of E6 T-cell receptor gene therapy for human papillomavirus (HPV)-associated epithelial cancers. Am Soc Clin Oncol. (2017) 35(Suppl. 15):3009. doi: 10.1200/JCO.2017.35.15_suppl.3009
136. Papa S, Adami A, Metoudi M, Achkova D, van Schalkwyk M, Parente Pereira A, et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). Am Soc Clin Oncol. (2018) 36(Suppl. 15):3046. doi: 10.1200/JCO.2018.36.15_suppl.3046
137. Okada S, Itoh K, Ishihara S, Shimada J, Kato D, Tsunezuka H, et al. Significance of PD-L1 expression in pulmonary metastases from head and neck squamous cell carcinoma. Surg Oncol. (2018) 27:259–65. doi: 10.1016/j.suronc.2018.04.007
138. Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol. (2017) 12:208–22. doi: 10.1016/j.jtho.2016.11.2228
139. Hanna GJ, Lizotte P, Cavanaugh M, Kuo FC, Shivdasani P, Frieden A, et al. Frameshift events predict anti–PD-1/L1 response in head and neck cancer. JCI Insight. (2018) 3:98811. doi: 10.1172/jci.insight.98811
140. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science. (2018) 362:eaar3593. doi: 10.1126/science.aar3593
141. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. (2017) 9:34. doi: 10.1186/s13073-017-0424-2
142. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
Keywords: head and neck cancer, microenvironment, biomarkers, immunotherapy, immunoresistance
Citation: Wang H-C, Chan L-P and Cho S-F (2019) Targeting the Immune Microenvironment in the Treatment of Head and Neck Squamous Cell Carcinoma. Front. Oncol. 9:1084. doi: 10.3389/fonc.2019.01084
Received: 23 August 2019; Accepted: 01 October 2019;
Published: 15 October 2019.
Edited by:
Cheng-Chia Yu, Chung Shan Medical University, TaiwanReviewed by:
Yi Feng Wu, Buddhist Tzu Chi General Hospital, TaiwanChih-Yen Chien, Kaohsiung Chang Gung Memorial Hospital, Taiwan
Copyright © 2019 Wang, Chan and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shih-Feng Cho, c2lmb25nOTZAZ21haWwuY29t