 Helen F. Titmarsh
Helen F. Titmarsh Richard O'Connor
Richard O'Connor Kevin Dhaliwal
Kevin Dhaliwal Ahsan R. Akram
Ahsan R. Akram- 1EPSRC and MRC CDT in Optical Medical Imaging, Universities of Edinburgh and Strathclyde, Edinburgh, United Kingdom
- 2Centre for Inflammation Research, Queen's Medical Research Institute, University of Edinburgh, Edinburgh Bioquarter, Edinburgh, United Kingdom
- 3Cancer Research UK Edinburgh Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, United Kingdom
Study of the c-Met-HGF axis in non-small cell lung cancer (NSCLC) has focused on the roles of c-MET signaling in neoplastic epithelial cells and the secretion of its ligand hepatocyte growth factor (HGF) by tumor stromal cells. However, there is increasing evidence that some leukocyte sub-sets also express c-MET raising the possibility of an immunomodulatory role for this axis. Consequently, the role of the c-MET- HGF axis in immunoncology is an active area of ongoing research. This review summarizes current knowledge of c-MET expression in NSCLC, the prognostic significance of these findings and the mechanisms by which the c-MET-HGF axis might act in NSCLC, focusing on the emerging evidence for an immunoregulatory role.
Introduction: Non-Immunological Roles For The c-Met-Hgf Axis In Non-Small Cell Lung Cancer
c-MET is a tyrosine kinase receptor with an extracellular α-chain and a transmembrane β-chain joined by disulphide bonds. In the steady state, the receptor is primarily found on epithelial cells where it regulates cell motility and proliferation (1). The ligand for c-MET is hepatocyte growth factor (HGF), which is produced by mesenchymal cells in an inactive form (1). Activation of HGF requires the action of a serine protease and this occurs in localized in areas of tissue injury (2). Abnormal c-MET signaling has been demonstrated in a number of human cancers as a result of c-MET receptor overexpression, receptor mutation or amplification, HGF overexpression or the formation of abnormal autocrine signaling (3). c-MET activation in cancer promotes; communication between mesenchymal cells and epithelial cells, tissue infiltration, cancer cell proliferation, and the induction of angiogenesis (1, 4–6). In addition selective inhibition or SiRNA knockdown of c-MET decreased the viability of non-small cell lung cancer (NSCLC) cells demonstrating the direct effects of c-MET in promoting tumor growth (7).
In lung adenocarcinomas various missense mutations and alternatively spliced products have been identified. These include Exon-14 splice mutations that result in reduced receptor degradation and prolonged activation of the c-MET receptor, and have been reported to occur in 3% of lung adenocarcinoma and 2.3% other lung tumors (8). In-vitro studies by Frampton et al., support the conclusion that Exon-14 splice mutated tumors will respond to anti c-MET therapies. Case reports from three patients also provides limited clinical data to support this conclusion (8). There are also selected reports of clinical responses to c-MET inhibitors in clinical settings in patients with splice mutations or amplification of the c-MET receptor (8–11). The frequency with which human NSCLC tumors express c-MET protein varies between studies, ranging from 25 to 100% and depends on the method of measuring c-MET, as well as and the authors' definition of c-MET positivity (Table 1). The association between tumor production of HGF and NSCLC survival is also summarized (Table 1). These observational studies are limited in their conclusions due to their retrospective nature and it is difficult to directly compare the different methodologies used, however, these studies highlight that there may be a trend toward increased c-MET expression or HGF production with poor patients outcomes. The importance of protein expression and gene amplification has been questioned, as clinical studies using c-MET targeting therapies recruiting patients with protein overexpression or gene amplification have produced disappointing results. This is well-reviewed by Hughes and Siemann (18), although early reports from a phase 1 clinical study of 40 patients [NCT00585195] suggest patients with high MET amplification may respond better to treatment with crizotinib (19). Identifying c-MET pathway activation may be a more appropriate selection criteria (18). With emerging evidence that c-MET may play a role in anti-cancer immune responses, understanding how c-MET may affect immunotherapy is another potential area to explore to see if these therapies could be used to greater effect.
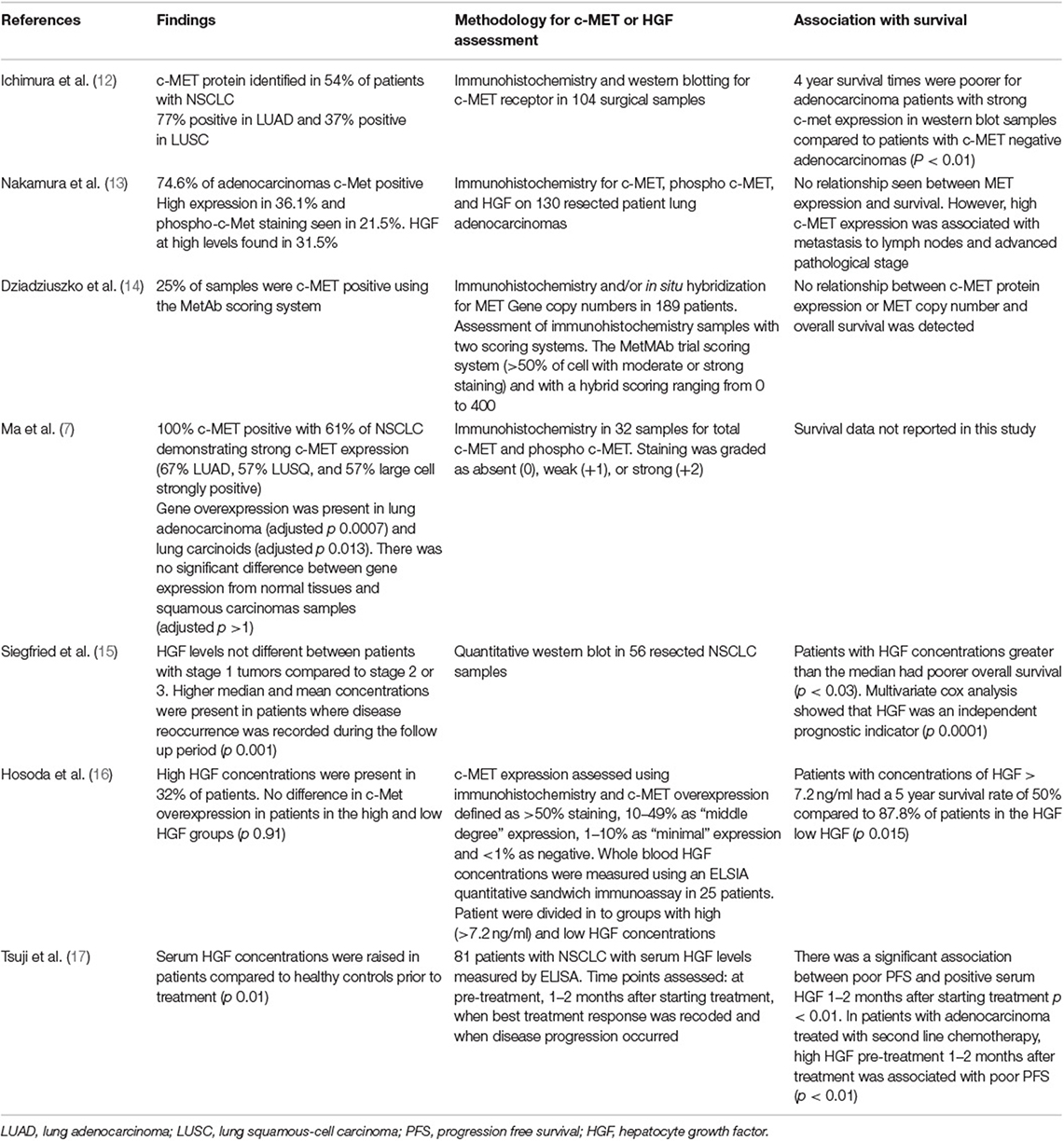
Table 1. Studies assessing c-MET and/or HGF in NSCLC, methods of assessment and correlation with survival.
c-MET and Immune Evasion
The ability to evade anti-tumor immune responses is a hallmark of cancer (20), however the discovery that checkpoint inhibition can overcome immunosuppression has established the anti-cancer potential of adaptive immune responses and ushered in the age of cancer immunotherapy (21). There is evidence that c-MET expression may be associated with expression of immunoregulatory molecules such as programmed cell death ligand (PD-L1) and Indoleamine-2,3-dixoygenase (IDO) in cancer cells. As a result, c-MET presents an attractive therapeutic target as there are clinically available inhibitors which may modulate the ability of cancer cells to evade anti-tumor immune responses. Programmed death 1 (PD-1) is a co-inhibitory molecule expressed on activated T-cells in response to antigenic stimulation. When the activating antigen is cleared PD-1 is down regulated, however where the antigen persists such as in cancer, PD-1 remains highly expressed on T-cells (22). As a means of preventing excessive immune response in the face of chronic antigen stimulation T-cell activity can be suppressed by binding of PD-1 to ligands on dendritic cells, B-cells and macrophage and cancer cells such as PD-L1 which are increased by inflammation or by some cancer mutations (22). PD-L1 binding with PD-1 on activated T-lymphocytes inhibits T-cell proliferation and cytokine release (22–24), thus inhibiting anti-tumor immune responses. PD-L1 expression occurs more frequently with MET activation in NSCLC (25), suggesting that besides its well-characterized direct anti-tumor effects inhibition of c-MET signaling may act indirectly to alleviate immunosuppression. Increasing PD-L1 expression correlated positively with MET gene amplification in 389 NSCLC samples (26). A separate study of 155 resected NSCLC tumor samples found MET activation was associated with PD-L1 expression and demonstrated that in NSCLC cell lines c-MET signaling directly induces PD-L1 expression (25). Aberrant c-MET activity can contribute to acquired tumor cell resistance to treatment with epidermal growth factor receptor (EGFR) targeting tyrosine kinases (TKIs) such as erlotinib (27, 28). This also includes the more recently available, third generation TKI osimertinib (29–31). There is some pre-clinical and clinical evidence that treating patients resistant to EGFR TKIs with c-MET inhibitors may improve outcomes. Treating erlotinib resistant cells with the c-MET inhibitor crizotinib decreased PD-L1 protein and gene expression, demonstrating a positive association between c-MET signaling and PD-L1 expression (32). Clinical reports also document responses to crizotinib in patients with osimertinib resistance (10). In addition, a marked response to a combination of erlotinib and crizotinib given concurrently in a patient with primary erlotinib resistance has also been reported (9). However, it is unknown from patient reports if crizotinib had any effect on PD-L1 expression. The results from Demuth et al. suggest that the c-MET axis may cause resistance to EGFR tyrosine kinases and increase the ability of the tumor to evade immune responses. If a relationship between c-MET inhibitors and PD-L1 expression is found in patient samples, then this could suggest that addition of c-MET inhibitors could also affect tumor immunity. This may be important in tumors with MET exon 14 alternations as they response more poorly to immune check point inhibitors. However, in a study where tumor samples with MET exon 14-alterations were available for assessment of PD-L1 status, outcomes with PD-1 blockade were poorer than for unselected patients (33). The authors also found treatment outcomes were not better in tumors with higher PD-L1 expression. The impact of the c-MET axis on PD-L1 related outcomes may relate to the type of abnormal c-MET activity present. c-MET may also affect inflammation induced PD-L1 expression.
IFN-γ induced upregulation of PD-L1 expression represents a negative control mechanism to limit the magnitude of T-cell responses (34, 35) and this pathway may also be modulated by c-MET inhibition. In response to IFN-γ produced by anti-tumor T-cells re-invigorated by checkpoint immunotherapy, increased tumor cell expression of PD-L1 can further raise the threshold for effective anti-tumor immunity (36). Although c-MET activation induces PD-L1 expression via a pathway distinct to that used by IFN-γ and independent of JAK/ STAT activation (25), inhibition of c-MET signaling blocks PD-L1 upregulation in response to both HGF and IFN-γ (25, 37). By blocking the capacity of IFN-γ to elevate PD-L1 expression c-MET inhibition may act as an effective co-treatment to increase the effectiveness of immune checkpoint blockade in patients with tumors with aberrant MET activity.
Production of Indoleamine-2,3-dixoygenase (IDO) by tumor cells is another pathway by which the c-MET-HGF axis may contribute to an immunosuppressive tumor microenvironment (38). IDO exerts immunosuppressive effects on T-cells and natural killer cells (NK cells) via regulation of kynurenic pathways and production of metabolites which deplete the extracellular environment of tryptophan required for T-cell function (39). Wang et al. performed experiments using the IDO expressing ovarian cancer cell line SKOV-3. Pharmacological inhibition of the c-MET receptor decreased IDO expression in a concentration dependent fashion. Additionally, when SKOV-3 cells were transfected to express the HGF variant NK4, which is a competitive antagonist for the c-MET receptor, IDO expression was inhibited. The variants expressing NK4 and therefore lacking IDO were more susceptible to the cytotoxic effects of NK cells suggesting that c-MET can indirectly influence NK effects on tumor cells. Murine experiments demonstrated that tumors expressing NK4 recruited more NK cells into the tumor stroma in-vivo and had reduced growth compared to tumors in the control group (38). Further work is required to see if increased IDO expression and therefore the function of NK or T-cells can be linked to c-MET expression in NSCLC cells.
The evidence above, indicates that role of aberrant c-MET activity on expression of immunoregulatory molecules by tumor cells is worthy of further investigation in both MET dependent and non-MET dependent NSCLC as is the potential combination of c-MET targeting therapies with immune check point inhibitors.
Leukocytes and c-MET Expression
The expression of c-MET is not limited to epithelial cells. The HGF-c-MET axis is involved in the binding, propagation, and survival of hematopoietic progenitor cells (40) and there is an emerging body of evidence that c-MET is expressed by mature leukocytes in murine and human tissues. The role of c-MET expressing leukocytes in inflammatory, autoimmune, and neoplastic disease is of increasing interest, although the role of these cells in human disease is still incompletely understood. The effect of c-MET on tumor associated leukocytes and cancer outcomes is worthy of further exploration as it may be possible to target leukocytes with c-MET inhibitors. Potential effects of the c-MET-HGF axis on leukocyte functions are summarized in Figure 1.
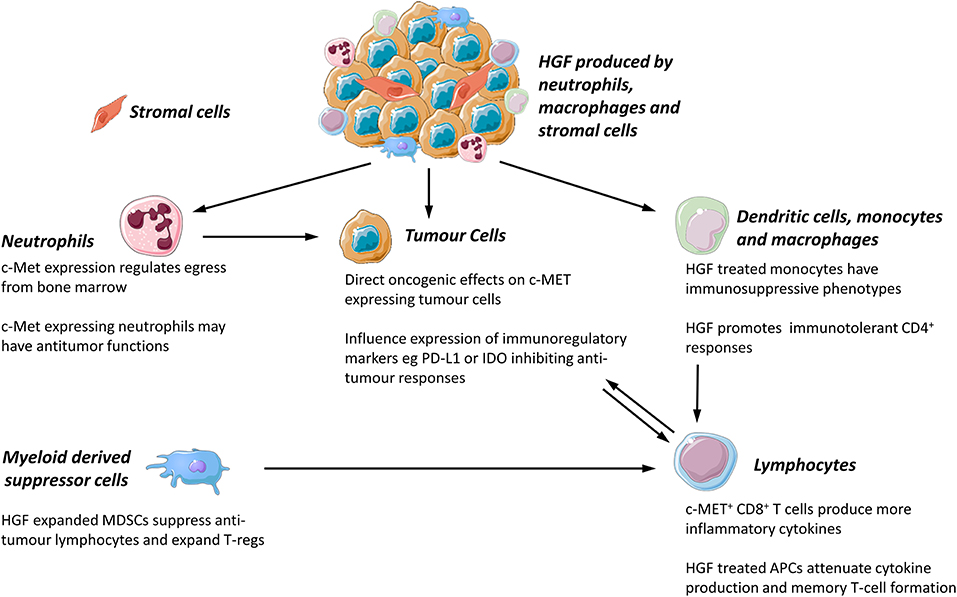
Figure 1. Proposed functions of c-MET/HGF axis in tumor associated leukocyte function. All images were sourced and modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com//.
Neutrophils are best understood for their role within the innate immune system, serving as short lived, phagocytic cells acting against pathogens such as bacteria (41). There is an increasing appreciation of the immunomodulatory role of neutrophils in shaping adaptive immune responses including anti-tumor immunity. The tumor microenvironment influences the phenotype and function of tumor-associated neutrophils (TANs) which can exert both pro- and anti-tumor effects. This has been reviewed elsewhere (42–44). The importance of better understanding the role of neutrophils in lung cancer is clear as they can comprise a significant proportion of the cellular tumor mass, making up between 5 and 25% of the infiltrating leukocytes (45, 46). Importantly neutrophil depletion studies in mice have demonstrated that neutrophils can limit the effectiveness of anti-PD-1 immunotherapy (47). This indicates factors influencing the recruitment and function of neutrophils within the tumor microenvironment may impact the efficacy of checkpoint inhibition (47). Given the use of PD-1 targeting therapies in NSCLC this may be important in the treatment of lung cancer patients. Evidence that c-MET targeting drugs may effect TAN functions and checkpoint inhibition efficacy is summarized below.
Sub-populations of murine and human neutrophils stimulated by inflammatory stimuli express c-MET (48). c-MET is required for the egress of neutrophils from the bone marrow and transendothelial migration of neutrophils into tissues in murine models of cancer (48, 49). c-MET silencing in neutrophils resulted in increased tumor growth in mice, demonstrating c-MET positive neutrophils have anti-tumor properties (48). Furthermore, in-vitro experiments showed c-MET knock out neutrophils have reduced nitric oxide-mediated cytotoxicity against neoplastic cells, whereas c-MET positive, HGF-stimulated neutrophils demonstrated increased nitric oxide (NO) release and enhanced cytotoxicity (48). TNF-α produced by tumor cells induced c-MET expression in neutrophils via activation of NF-kB (48), further illustrating how the prevailing cytokine milieu can ignite anti-tumor effector functions in neutrophils. However, besides its beneficial role in tumor cell killing, TAN production of NO may also suppress T-cell responses and limit the effectiveness of immunotherapy. In a murine melanoma model, adoptive T-cell transfer induced the recruitment of immunosuppressive c-MET positive neutrophils to the tumor and tumor draining lymph nodes (49). These TANs displayed increased expression of immunosuppressive genes including PD-L1, ARG1, and iNOS. The majority of TANs expressed PD-L1 and neutrophils from within the tumor and tumor-draining lymph nodes expressed higher levels of c-MET than those found in the spleen (49). Administering c-MET inhibitors alongside adoptive T-cell therapy prevented neutrophil recruitment, improved tumor eradication and increased survival in mouse tumor models (49). Cell-specific knockdown of c-MET expression in neutrophils also enhanced the effectiveness of adoptive T-cell therapy, reducing recruitment of reactive neutrophils and increasing survival of tumor reactive T-cells (49). The different pro and anti-tumor effects of c-Met expressing neutrophils reported in the Glodde and Finisguerra studies may be explained by differences in the inflammatory milieu of the tumor microenvironment. Following either adoptive T -cell therapy or checkpoint inhibition enhanced T-cell responses result in increased IFN-γ production which may promote suppressive effects. PD-L1 expression is readily induced in neutrophils in response to IFN-γ produced by activated T-cells (49). Thus, cytokines produced by both tumor cells [TNF-α promoting c-MET expression (50) and tumor reactive T-cells (via IFN-γ induced PD-L1 expression) (49)] co-operate in driving the recruitment and immunosuppressive phenotype of TANs. Limiting pro-tumor neutrophil mediated T-cell suppression may come at the expense of direct NO-mediated neutrophil killing of tumor cells. Therefore, whether there are anti-cancer benefits to inhibiting the c-MET dependent recruitment of TANs likely depends on the magnitude of the adaptive anti-tumor response. In the presence of a strong anti-tumor immune response or when used as an adjunct to checkpoint blockade or adoptive T-cell therapy, cytotoxic T-cells are likely to be the most efficient anti-tumor effector cells. However, as Glodde et al., where able to demonstrate a positive effect of c-MET silencing and inhibitors in mice with and without c-MET dependent tumors, the possibility of using c-MET inhibitors to improve checkpoint inhibitor associated outcomes in patients is worthy of investigation.
The role of TANs in c-MET associated patient outcomes has been investigated in treatment naive patients with hepatocellular carcinoma (HCC) (51). Although, tumor cell c-MET expression when considered alone was not predictive of outcome, MET expression by HCC cells was inversely associated with overall survival and disease free progression times in the subgroup of patients within this cohort with large numbers of neutrophils at the invading tumor margins (51) Further in-vitro work by this group, determined that exposing neutrophils to culture supernatants from HCC cell lines or primary HCC cells increased production of HGF by neutrophils. GM-CSF was found to be increased in culture supernatants from HCC cells and treatment of neutrophils with recombinant GM-CSF lead to increase in HGF, indicating GM-CSF is important in regulating neutrophil production of HGF. The possible pro-tumor effects of these HGF secreting neutrophils was also investigated. Conditioned media from HGF producing TANs promoted hepatoma cell migration, whereas the media from blood derived neutrophils, which produce less HGF did not. In addition, treatment of TANs with c-MET inhibitors reduced TAN associated hepatoma cell migration (51).
The studies by He et al., Finisguerra et al., and Glodde et al., suggest c-MET inhibition could become an adjunct therapy to established immunotherapies because they demonstrate a role for c-MET in the recruitment of TANs, show that tumor derived GM-CSF can stimulate increased HGF-production by neutrophils and that HGF can promote increased expression of PD-L1 and PD-L2 on neutrophils. Further work is needed to see if this could be applicable to the treatment of NSCLC. Currently, few studies have investigated expression of c-MET on TANs from human NSCLC. In four NSCLC patients, where neutrophils were isolated from tumor samples and from non-cancerous lung tissue, gene expression of MET in TANs was greater than in neutrophils from healthy tissues (48). However, in this study there was no assessment of c-MET protein expression or activation. Immunohistochemistry for HGF and the c-MET receptor has been investigated in eight patient bronchioaveolar lavage (BAL) samples and five paired tumor and adjacent non-tumor tissue samples from patients with bronchioaveloar carcinoma (BAC) (52). The c-MET receptor was absent on polymorphonuclear cells suggesting c-MET positive neutrophils are uncommon in BAC. Neutrophils were however a major source of HGF; HGF levels in the BAL fluid correlated with absolute BAL neutrophil counts and increasing BAL HGF concentrations were associated with shorter survival time. BAC, now termed adenocarcinoma spectrum disease, is often a more indolent cancer with a more favorable prognosis. Therefore, care should be taken when applying the results of this study to other types of NCSLC many of which will behave more aggressively.
Lymphocytes play a vital role in influencing immune responses to tumors. Whether lymphocytes mount an immune response or a promote immune tolerance toward tumor cells is influenced by a number of factors including, cytokines, tumor cell immunoregulatory molecules, and secreted proteins, as reviewed by Chraa et al. (53). In addition many recent advancements in NSCLC therapy act to alter T-cell responses. Although to our knowledge c-MET expressing lymphocytes have not be found in human NSCLC cancer there is evidence that subsets of lymphocytes may response to HGF in other types of human cancer. Therefore, if c-MET may have a direct effect on T-cells, potentially augmenting immunotherapy this could provide alternative uses for c-MET inhibitors in both MET dependent and non-MET dependent tumors.
Hepatocyte growth factor can attenuate CD8 positive responses in mice due to effects on antigen presenting cells (APCs)/generating tolerogenic dendritic cells (54, 55). c-Met expression has been identified in human melanoma samples (56) and a small percentage of murine cytotoxic T-cells express c-MET receptors in response to activation (56). c-MET positive CD8 positive T-cells are more cytotoxic than c-MET negative CD8 positive T-cells when assessed in vivo and in vitro (56). c-MET positive cytolytic T-cells expressed higher levels of granzyme B and perforin and showed a trend toward producing higher concentrations of IFN-γ and TNF-α. However, when these cells were exposed to HGF in vivo, cytolytic effects and cytokine production were reduced. This suggests HGF acts to restrain immune responses (56). c-MET inhibition may however, also act directly upon T-cells to enhance effector functions. The hypothesis is supported by experiments that indicate c-MET positive CD8 positive cells have increased ability to reduce melanoma pulmonary metastasis compared to c-MET negative CD8 positive cells, which can be abrogated by the addition of HGF (56). c-MET positive CD8 positive lymphocytes have been identified in human tumors; biopsy samples from four melanoma patients showed 8–35% of CD8 positive T-lymphocytes in close proximity to melanoma cells expressed c-MET (56). The prevalence of c-MET positive T-cells in other cancers, their functional capacities and the consequences of c-MET inhibition on T-cell function requires ongoing research.
c-MET positive B-lymphocytes in solid tumors have not, to the authors knowledge been reported. However, the HGF/c-Met axis has been shown to play a role in survival of mature murine B lymphocytes via the CD74 pathway (57), and is a paracrine factor involved with regulating B cell adhesion in lymphoid tissue (58, 59). High MET mRNA number has been associated with poor outcomes in patients with multiple myeloma (60). Additionally protein expression for c-MET has been found on the plasma cells of 40% of patients with multiple myeloma, but is absent in healthy donors (61). MET expression is documented in 30% of diffuse large B-cell lymphomas in one study (62) and is increased in 73.2% of patients in another study (63). Targeting c-MET has been suggested as a possible therapeutic option for this disease (63). Despite B lymphocytes being present at very low frequencies in NSCLC (46), tumor infiltrating B-lymphocytes have an important impact on CD4+ cells behavior and phenotype (64, 65). Therefore, the possibility that B-cells may express c-MET in the NSCLC microenvironment requires further investigation.
Lymphocyte responses can be influenced by other types of leukocytes such as dendritic cells. As there is some evidence of c-MET expressing dendritic cells targeting these cells may also provide another avenue via which c-MET inhibitors would be used to improve immunotherapy outcomes in cancer patients. Dendritic cells (DCs) are the primary antigen presenting cell population responsible for initiating T-cell activation and development of adaptive immune-responses. c-Met expression in dendritic cells has been best studied in inflammatory disease wherein it drives a tolerogenic rather than immunogenic phenotype. In a model of allergic asthma messenger RNA (mRNA) for c-MET was identified in murine DC cells and HGF suppressed the capability of DC to present antigens (66). HGF has also been shown to promote the development of tolerogenic DCs in experimental autoimmune encephalomyelitis (54). Besides the role of c-MET in promoting immunotolerance, Met-deficient skin DCs failed to migrate to skin draining lymph nodes following activation, resulting in reduced contact hypersensitivity responses (67). These results suggest that c-MET acts on DCs altering adaptive immune responses. Investigating the possible role of the c-MET-HGF axis in the regulation of anti-tumor immune responses is therefore an area worthy of investigation.
Monocytes are circulating precursors to macrophages and dendritic cells, which can have immunomodulatory effects on local T-cell responses (68). c-Met has been found on CD14 positive human monocytes during differentiation and in response to inflammatory cytokines such as lL-6, TNF-α, IFN-γ, IL-1B, and endotoxin (69–71). Activation of monocytes increases secretion of HGF and expression of the enzyme urokinase-type plasminogen activator (uPA) needed to convert pro-HGF to its active form, thus increasing both availability and bioactivity of HGF (70). In addition, HGF increases the transcription of cytokines including IL-6, GM-CSF and G-CSF (70). In contrast Chen et al. found HGF treated monocytes displayed an immunosuppressive phenotype and limited T-cell proliferation in an IL-10 dependent fashion (71). Similarly Rutella et al. found HGF-treated monocytes produced increased amounts of immunosuppressive IL-10 and lower levels of the immunostimulatory cytokine IL-12 p70 (72). Compared to GM-CSF/IL-4 matured monocytes, HGF treated cells were inefficient in driving T-cell proliferation and promoted expansion of suppressive Foxp3 positive regulatory T-cells (72). Gene profiling of HGF stimulated monocytes showed an increase in transcription of genes associated with immunotolerance and tumor progression such as IDO, compared to monocytes stimulated by GM-CSF and IL-4. Co-culturing CD4 positive T-cells and HGF stimulated monocytes with an IDO inhibitor partially reversed the hyporesponsiveness of CD4 positive cells to HGF exposed monocytes. This suggest that HGF may in part exert some immunosuppressive effects by affecting IDO expression. As discussed earlier in this review, the c-MET-HGF pathway has also been implicated in altered IDO expression by tumor cells (38). These studies suggest that HGF/c-MET axis may have indirect suppressive effects on T-cells as a consequence of altering the phenotype of antigen presenting cells favoring tolerogenic rather than immunogenic responses.
Myeloid derived suppressor cells (MDSCs) are a group of leukocytes which act to suppress T-cell effector functions and can be broadly divided into two populations myeloid (M-MDSCs) and polymorphonuclear (PMN-MDSCs) (73). M-MDSCs differ phenotypically from monocytes as they express no or very little of the T-cell ligand HLA-DR and can be recognized as CD11b+, HLA-DR−, CD14+CD15− cells (73). PMN-MDSCs can be classed as CD11b+CD15+CD14−/dim, HLA-DR− cells but are difficult to differentiate from neutrophils (73). Use of these markers as a standard to describe MDSCs has only recently been proposed and therefore literature which describe MDSCS as having a different phenotype are included in this review.
In human blood, CD14 negative leukocytes including MDSCs (CD14−, CD11b+ CD33+) can constitutively express low levels of c-Met (74). Culturing peripheral leukocytes with mesenchymal stem cells (MSCs) or HGF causes an increase in MDSCs. HGF-expanded MDSCs suppress allogeneic lymphocyte proliferation and expressed increased immunosuppressive/cytotoxic enzymes including iNOS and ARG-1 compared to at baseline (74). In addition the MDSCs cells were able to induce T-regs from stimulated peripheral blood leukocytes and the ability of MSCs to expand MDSCs populations was inhibited when c-MET was pharmacologically blocked or if HGF production was silenced in cells (74). This suggests that c-MET may be important in regulation of immunoregulatory processes. Indeed, an abstract from a pre-clinical study of a c-MET targeting therapeutic antibody ARGX-111 indicates that the drug can diminish c-MET positive MDSCs, suggesting a role of this drug in treating c-MET positive tumors and altering c-MET activity of cells in the tumor microenvironment (75).
Overall, there is currently little evidence that c-MET expressing leukocyte subsets exist in NSCLC tumors. However, evidence that the c-MET/HGF axis can influence leukocyte activity in the studies discussed above indicates that c-MET inhibitors may potentially be of benefit via their effects on immune cells. This raises the possibility that c-MET inhibitors could be used in patients with non-MET dependent tumors as well as those with tumor cells with aberrant activity.
Trials And Investigations Of c-Met Targeting Therapies And Immunotherapy In Met Dependent And Non-Dependent Tumors
The potential for c-MET to influence both immunoregulatory molecules in MET dependent cancers and leukocytes in either MET dependent or MET independent cancer raises the possibility that treatments targeting the c-MET axis could impact on the outcomes of anti-cancer immunotherapies. Current evidence suggest that immune check point inhibitors are less effective in tumors with driver mutations including c-MET (76). The conclusions of a report based on the IMMUNOTARGET registry, suggests where driver mutations are found, targeted therapies such as Tyrosine Kinase Inhibitors should be used before contemplating immunotherapy (76). However, a significant amount of pre-clinical investigation is required before this can be considered and this staged approach does not address the potential for concurrent therapy. The possibility of drug toxicity must also be considered. A clinical studying using crizotinib (prescribed for its effects on ALK rather than c-MET in this incidence), in patients given the PD1- PD-L1 targeting treatment nivolumab resulted in serious hepatic toxicity leading to the end of patient enrollment (77). Drugs and immunotherapies targeting c-MET have been developed for use in people. Early stage human trials using T-cells modified to identify the c-MET and PD-L1 proteins (CAR T-Cells) for the treatment of primary hepatocellular carcinoma are planned (78). The aim of this trial is to determine the efficacy of this therapy in increasing overall survival. The potential for these therapies to alter the immune cell populations as well as targeting neoplastic cells expressing c-MET and PD-L1 could be interesting in light of research suggesting c-MET has immunomodulatory functions. This may allow dual targeting of the c-MET and PD-L1 pathways without risking the toxicities seen when crizotinib was given to patients receiving immunotherapy (77).
The c-MET inhibitor crizotinib has been studied as a means of improving treatment outcomes, via an off target effect. Responses to some chemotherapy agents is partly due to causing “immunogenic cell death” which allows immune cells to be attracted to dying cancer cells and progression free and overall survival have been linked to the expression of receptors associated with immunogenic cell death. Crizotinib has been used to induce immunogenic cell death in a human NSCLC cell line (79). When cells treated with the chemotherapeutic agents and crizotinib were injected into mice more mice remained tumor free after 60 days than mice injected with tumor cells treated with chemotherapy alone. The in-vitro and in-vivo results suggest the c-MET inhibitor crizotinib increases immunogenic cell death and thus improves treatment outcomes. Additionally, these effects are related to T-lymphocyte infiltration and depletion of T-cells or blockade of IFN-γ abolishes these effects. As crizotinib effects appeared to be off target this again suggests a potential use of c-MET inhibitors in MET dependent and non-dependent tumors.
Summary
Although, the role of the HGF/c-MET axis as a regulator of immune function is incompletely understood it is emerging as a powerful influence in many aspects of immunity. The studies described above have illustrated its effects on first line responders such as neutrophils, in directing the migration and stimulatory capacity of dendritic cells and monocytes through to directly influencing the function of effector T-cells. These effects occur in concert with, and must be considered alongside, direct effects on tumor cells and stromal cells in the tumor microenvironment. As many novel anti-cancer therapies target immunoregulatory molecules or the c-MET receptor itself understanding the relationship between c-MET and immunological responses to tumors is critical in ensuring these drugs are used to their full potential benefit.
Author Contributions
HT performed the literature searches and wrote the first draft of the article. HT, RO'C, KD, and AA discussed and contributed to manuscript content. All authors reviewed the manuscript before submission.
Funding
HT was funded by EPSRC and MRC Center for Doctoral Training in Optical Medical Imaging (EP/L016559/1). AA was supported by a Cancer Research UK Clinician Scientist Fellowship (A24867).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Organ SL, Tsao M-S. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. (2011) 3(Suppl. 1):S7–S19. doi: 10.1177/1758834011422556
2. Miyazawa K. Hepatocyte growth factor activator (HGFA): a serine protease that links tissue injury to activation of hepatocyte growth factor. FEBS J. (2010) 277:2208–14. doi: 10.1111/j.1742-4658.2010.07637.x
3. Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. (2006) 12:3657–60. doi: 10.1158/1078-0432.CCR-06-0818
4. Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. (2012) 12:89. doi: 10.1038/nrc3205
5. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. (2013) 31:1089–96. doi: 10.1200/JCO.2012.43.9422
6. Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. (2018) 17:45. doi: 10.1186/s12943-018-0796-y
7. Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. (2005) 65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650
8. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. (2015) 5:850–9. doi: 10.1158/2159-8290.CD-15-0285
9. Gainor JF, Niederst MJ, Lennerz JK, Dagogo-Jack I, Stevens S, Shaw AT, et al. Dramatic response to combination erlotinib and crizotinib in a patient with advanced, EGFR-mutant lung cancer harboring de novo met amplification. J Thorac Oncol. (2016) 11:e83–5. doi: 10.1016/j.jtho.2016.02.021
10. Ou S-HI, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. (2016) 98:59–61. doi: 10.1016/j.lungcan.2016.05.015
11. Wang SXY, Zhang BM, Wakelee HA, Koontz MZ, Pan M, Diehn M, et al. Case series of MET exon 14 skipping mutation-positive non-small-cell lung cancers with response to crizotinib and cabozantinib. Anti Cancer Drugs. (2019) 30:537–41. doi: 10.1097/CAD.0000000000000765
12. Ichimura E, Maeshima A, Nakajima T, Nakamura T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn J Cancer Res. (1996) 87:1063–9. doi: 10.1111/j.1349-7006.1996.tb03111.x
13. Nakamura Y, Niki T, Goto A, Morikawa T, Miyazawa K, Nakajima J, et al. c-Met activation in lung adenocarcinoma tissues: an immunohistochemical analysis. Cancer Sci. (2007) 98:1006–13. doi: 10.1111/j.1349-7006.2007.00493.x
14. Dziadziuszko R, Wynes MW, Singh S, Asuncion BR, Ranger-Moore J, Konopa K, et al. Correlation between MET gene copy number by silver in situ hybridization and protein expression by immunohistochemistry in non-small-cell lung cancer. J Thorac Oncol. (2012) 7:340–7. doi: 10.1097/JTO.0b013e318240ca0d
15. Siegfried JM, Weissfeld LA, Singh-Kaw P, Weyant RJ, Testa JR, Landreneau R, et al. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. (1997) 57:433–9.
16. Hosoda H, Izumi H, Tukada Y, Takagiwa J, Chiaki T, Yano M, et al. Plasma hepatocyte growth factor elevation may be associated with early metastatic disease in primary lung cancer patients. Ann Thorac Cardiovasc Surg. (2012) 18:1–7. doi: 10.5761/atcs.oa.09.01522
17. Tsuji T, Sakamori Y, Ozasa H, Yagi Y, Ajimizu H, Yasuda Y, et al. Clinical impact of high serum hepatocyte growth factor in advanced non-small cell lung cancer. Oncotarget. (2017) 8:71805–16. doi: 10.18632/oncotarget.17895
18. Hughes VS, Siemann DW. Have clinical trials properly assessed c-Met inhibitors? Trends Cancer. (2018) 4:94–7. doi: 10.1016/j.trecan.2017.11.009
19. Camidge DR, Otterson GA, Clark JW, Ou SHI, Weiss J, Ades S, et al. (2018). Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): updated safety and efficacy findings from a phase 1 trial. J. Clin. Oncol. 36:9062. doi: 10.1200/JCO.2018.36.15_suppl.9062
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
21. Webb ES, Liu P, Baleeiro R, Lemoine NR, Yuan M, Wang Y, et al. Immune checkpoint inhibitors in cancer therapy. J Biomed Res. (2018) 32:317–26. doi: 10.7555/JBR.31.20160168
22. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. (2017) 18:153. doi: 10.1038/nri.2017.108
23. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. (2019) 18:10. doi: 10.1186/s12943-018-0928-4
24. Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, et al. PD-1/PD-L1 pathway: basic biology and role in cancer immunotherapy. J Cell Physiol. (2019) 234:16824–37. doi: 10.1002/jcp.28358
25. Saigi M, Alburquerque-Bejar JJ, Mc Leer-Florin A, Pereira C, Pros E, Romero OA, et al. MET-oncogenic and JAK2-inactivating alterations are independent factors that affect regulation of PD-L1 expression in lung cancer. Clin Cancer Res. (2018) 24:4579–87. doi: 10.1158/1078-0432.CCR-18-0267
26. Albitar M, Sudarsanam S, Ma W, Jiang S, Chen W, Funari V, et al. Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer. Oncotarget. (2018) 9:13682–93. doi: 10.18632/oncotarget.24455
27. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. (2007) 316:1039–43. doi: 10.1126/science.1141478
28. Morgillo F, Della Corte CM, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open. (2016) 1:e000060. doi: 10.1136/esmoopen-2016-000060
29. Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. (2015) 26:2073–8. doi: 10.1093/annonc/mdv319
30. Papadimitrakopoulou VA, Wu YL, Han JY, Ahn MJ, Ramalingam SS, John T, et al. LBA51Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. (2018) 29(suppl_8):mdy424.064. doi: 10.1093/annonc/mdy424.064
31. Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. LBA50Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol. (2018) 29(suppl_8):mdy424.063. doi: 10.1093/annonc/mdy424.063
32. Demuth C, Andersen MN, Jakobsen KR, Madsen AT, Sorensen BS. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget. (2017) 8:68221–9. doi: 10.18632/oncotarget.19920
33. Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. (2018) 29:2085–91. doi: 10.1093/annonc/mdy334
34. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. (2004) 64:1140–5. doi: 10.1158/0008-5472.CAN-03-3259
35. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
36. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
37. Martin V, Chiriaco C, Modica C, Acquadro A, Cortese M, Galimi F, et al. Met inhibition revokes IFNgamma-induction of PD-1 ligands in MET-amplified tumours. Br J Cancer. (2019) 120:527–36. doi: 10.1038/s41416-018-0315-3
38. Wang D, Saga Y, Sato N, Nakamura T, Takikawa O, Mizukami H, et al. The hepatocyte growth factor antagonist NK4 inhibits indoleamine-2,3-dioxygenase expression via the c-Met-phosphatidylinositol 3-kinase-AKT signaling pathway. Int J Oncol. (2016) 48:2303–9. doi: 10.3892/ijo.2016.3486
39. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara G, et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. (2002) 196:459–68. doi: 10.1084/jem.20020121
40. Weimar IS, Miranda N, Muller EJ, Hekman A, Kerst JM, de Gast GC, et al. Hepatocyte growth factor/scatter factor (HGF/SF) is produced by human bone marrow stromal cells and promotes proliferation, adhesion and survival of human hematopoietic progenitor cells (CD34+). Exp Hematol. (1998) 26:885–94.
41. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. (2018) 9:113. doi: 10.3389/fphys.2018.00113
42. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. (2016) 16:431. doi: 10.1038/nrc.2016.52
43. Grecian R, Walmsley SR, Whyte MKB. The role of neutrophils in cancer. Br Med Bull. (2018) 128:5–14. doi: 10.1093/bmb/ldy029
44. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. (2019) 16:601–20. doi: 10.1038/s41571-019-0222-4
45. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. (2014) 124:5466–80. doi: 10.1172/JCI77053
46. Kargl J, Busch SE, Yang GHY, Kim K-H, Hanke, ML, Metz HE, et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat Commun. (2017) 8:14381. doi: 10.1038/ncomms14381
47. Faget J, Groeneveld S, Boivin G, Sankar M, Zangger N, Garcia M, et al. Neutrophils and snail orchestrate the establishment of a pro-tumor microenvironment in lung cancer. Cell Rep. (2017) 21:3190–204. doi: 10.1016/j.celrep.2017.11.052
48. Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature. (2015) 522:349–53. doi: 10.1038/nature14407
49. Glodde N, Bald T, van den Boorn-Konijnenberg D, Nakamura K, O'Donnell JS, Szczepanski S, et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity. (2017) 47:789–802.e789. doi: 10.1016/j.immuni.2017.09.012
50. Bigatto V, De Bacco F, Casanova E, Reato G, Lanzetti L, Isella C, et al. TNF-α promotes invasive growth through the MET signaling pathway. Mol Oncol. (2015) 9:377–88. doi: 10.1016/j.molonc.2014.09.002
51. He M, Peng A, Huang X-Z, Shi D-C, Wang J-C, Zhao Q, et al. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology. (2016) 5:e1219828. doi: 10.1080/2162402X.2016.1219828
52. Wislez M, Rabbe N, Marchal J, Milleron B, Crestani B, Mayaud C, et al. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res. (2003) 63:1405–12. Available online at: https://cancerres.aacrjournals.org/content/63/6/1405.long
53. Chraa D, Naim A, Olive D, Badou A. T lymphocyte subsets in cancer immunity: friends or foes. J Leukoc Biol. (2019) 105:243–55. doi: 10.1002/JLB.MR0318-097R
54. Benkhoucha M, Santiago-Raber M-L, Schneiter G, Chofflon M, Funakoshi H, Nakamura T, et al. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+/Foxp3+regulatory T cells. Proc Natl Acad Sci USA. (2010) 107:6424–9. doi: 10.1073/pnas.0912437107
55. Benkhoucha M, Molnarfi N, Schneiter G, Walker PR, Lalive PH. The neurotrophic hepatocyte growth factor attenuates CD8+ cytotoxic T-lymphocyte activity. J Neuroinflamm. (2013) 10:919. doi: 10.1186/1742-2094-10-154
56. Benkhoucha M, Molnarfi N, Kaya G, Belnoue E, Bjarnadottir K, Dietrich PY, et al. Identification of a novel population of highly cytotoxic c-Met-expressing CD8(+) T lymphocytes. EMBO Rep. (2017) 18:1545–58. doi: 10.15252/embr.201744075
57. Gordin M, Tesio M, Cohen S, Gore Y, Lantner F, Leng L, et al. c-Met and its ligand hepatocyte growth factor/scatter factor regulate mature B cell survival in a pathway induced by CD74. J Immunol. (2010) 185:2020–31. doi: 10.4049/jimmunol.0902566
58. van der Voort R, Taher TE, Keehnen RM, Smit L, Groenink M, Pals S, et al. Paracrine regulation of germinal center B cell adhesion through the c-met-hepatocyte growth factor/scatter factor pathway. J Exp Med. (1997) 185:2121–31. doi: 10.1084/jem.185.12.2121
59. Skibinski G, Skibinska A, James K. The role of hepatocyte growth factor and its receptor c-met in interactions between lymphocytes and stromal cells in secondary human lymphoid organs. Immunology. (2001) 102:506–14. doi: 10.1046/j.1365-2567.2001.01186.x
60. Rocci A, Gambella M, Aschero S, Baldi I, Trusolino L, Cavallo F, et al. MET dysregulation is a hallmark of aggressive disease in multiple myeloma patients. Br J Haematol. (2014) 164:841–50. doi: 10.1111/bjh.12719
61. Wader KF, Fagerli U-M, Børset M, Lydersen S, Hov H, Sundan A, et al. Immunohistochemical analysis of hepatocyte growth factor and c-Met in plasma cell disease. Histopathology. (2012) 60:443–51. doi: 10.1111/j.1365-2559.2011.04112.x
62. Tjin EPM, Groen RWJ, Vogelzang I, Derksen PWB, Klok MD, Meijer HP, et al. Functional analysis of HGF/MET signaling and aberrant HGF-activator expression in diffuse large B-cell lymphoma. Blood. (2006) 107:760. doi: 10.1182/blood-2005-05-1929
63. Uddin S, Hussain AR, Ahmed M, Al-Dayel F, Bu R, Bavi P, et al. Inhibition of c-MET is a potential therapeutic strategy for treatment of diffuse large B-cell lymphoma. Lab Invest. (2010) 90:1346. doi: 10.1038/labinvest.2010.108
64. Bruno TC. Evaluating the antitumor role of B cells in patients with non-small cell lung cancer. J Clin Oncol. (2017) 35(7_suppl):75. doi: 10.1200/JCO.2017.35.7_suppl.75
65. Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, et al. Antigen-presenting intratumoral B cells affect CD4(+) TIL phenotypes in non-small cell lung cancer patients. Cancer Immunol Res. (2017) 5:898–907. doi: 10.1158/2326-6066.CIR-17-0075
66. Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, et al. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol. (2005) 175:4745–53. doi: 10.4049/jimmunol.175.7.4745
67. Baek J-H, Birchmeier C, Zenke M, Hieronymus T. The HGF receptor/Met tyrosine kinase is a key regulator of dendritic cell migration in skin immunity. J Immunol. (2012) 189:1699–707. doi: 10.4049/jimmunol.1200729
68. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349. doi: 10.1038/nri.2017.28
69. Chen Q, DeFrances MC, Zarnegar R. Induction of met proto-oncogene (hepatocyte growth factor receptor) expression during human monocyte-macrophage differentiation. Cell Growth Diff. (1996) 7:821–32.
70. Galimi F, Cottone E, Vigna E, Arena N, Boccaccio C, Giordano S, et al. Hepatocyte growth factor is a regulator of monocyte-macrophage function. J Immunol. (2001) 166:1241–7. doi: 10.4049/jimmunol.166.2.1241
71. Chen PM, Liu KJ, Hsu PJ, Wei CF, Bai CH, Ho LJ, et al. Induction of immunomodulatory monocytes by human mesenchymal stem cell-derived hepatocyte growth factor through ERK1/2. J Leukoc Biol. (2014) 96:295–303. doi: 10.1189/jlb.3A0513-242R
72. Rutella S, Bonanno G, Procoli A, Mariotti A, de Ritis DG, Curti A, et al. Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features. Blood. (2006) 108:218–27. doi: 10.1182/blood-2005-08-3141
73. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother. (2019) 68:687–97. doi: 10.1007/s00262-019-02302-2
74. Yen BL, Yen M-L, Hsu P-J, Liu K-J, Wang C-J, Bai C-H, et al. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Rep. (2013) 1:139–51. doi: 10.1016/j.stemcr.2013.06.006
75. Rolfo CD, Aftimos PG, Pallandre J-R, Morello V, Bouard A, Cazzanti M, et al. ARGX-111 shows activity in MET-amplified patients in a phase-I study and in preclinical models of myeloid-derived suppressor cell (MDSC) depletion in the tumor microenvironment. J Clin Oncol. (2016) 34(15_suppl):e14016. doi: 10.1200/JCO.2016.34.15_suppl.e14016
76. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. (2019) 30:1321–8. doi: 10.1093/annonc/mdz167
77. Spigel DR, Reynolds C, Waterhouse D, Garon EB, Chandler J, Babu S, et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation - positive advanced non-small cell lung cancer (CheckMate 370). J Thorac Oncol. (2018) 13:682–8. doi: 10.1016/j.jtho.2018.02.022
78. Guozhong J. Clinical Study on the Efficacy and Safety of c-Met/PD-L1 CAR-T Cell Injection in the Treatment of HCC. (2018). Available online at: https://clinicaltrials.gov/ct2/show/NCT03672305?cond=c-met$+$pd-l1andrank=1 (accessed July 16, 2019).
Keywords: c-MET, HGF, NSCLC, cancer immunology, immunotherapy
Citation: Titmarsh HF, O'Connor R, Dhaliwal K and Akram AR (2020) The Emerging Role of the c-MET-HGF Axis in Non-small Cell Lung Cancer Tumor Immunology and Immunotherapy. Front. Oncol. 10:54. doi: 10.3389/fonc.2020.00054
Received: 25 October 2019; Accepted: 13 January 2020;
Published: 06 February 2020.
Edited by:
Karen L. Reckamp, Cedars-Sinai Medical Center, United StatesReviewed by:
Joel W. Neal, Stanford University, United StatesTimothy F. Burns, University of Pittsburgh, United States
Copyright © 2020 Titmarsh, O'Connor, Dhaliwal and Akram. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen F. Titmarsh, aGVsZW4udGl0bWFyc2hAZWQuYWMudWs=; Ahsan R. Akram, YWhzYW4uYWtyYW1AZWQuYWMudWs=