 Kianna Robinson1
Kianna Robinson1 Venkataswarup Tiriveedhi1,2*
Venkataswarup Tiriveedhi1,2*- 1Department of Biological Sciences, Tennessee State University, Nashville, TN, United States
- 2Department of Pharmacology, Vanderbilt University, Nashville, TN, United States
Development of multidrug resistance (MDR) still remains a major obstacle to the long-term success of cancer therapy. P-glycoprotein (P-gp) is a well-identified membrane transporter with capability to efflux drug molecules out of the cancer cell leading to reduced efficiency of chemotherapy. Cancer cells upregulate P-gp expression as an adaptive response to evade chemotherapy mediated cell death. While several P-gp inhibitors have been discovered by in silico and pre-clinical studies, very few have successfully passed all phases of the clinical trials. Studies show that application of P-gp inhibitors in cancer therapy regimen following development of MDR achieved limited beneficial outcomes. While, the non-specific substrate binding to P-gp has made the drug-design a challenge, a bigger perplexing challenge comes from its role in tumor immunology. Expression of P-gp was noted immune cell phenotypes with apparently antagonistic functionality. Both pro-tumor MΦ2-macrophages and, anti-tumor NK-cell and Th17/CD4+T cell subsets have shown enhanced expression of P-gp. While drug based inhibition of P-gp in pro-tumor immune cell phenotypes could promote tumor elimination, however, it would not be a rational choice to exert inhibition of P-gp on anti-tumor immune cell phenotypes. This mutually exclusive paradigm of P-gp functionality requires a more comprehensive and detailed understanding of its role in tumor microenvironment with active interplay of cancer and immune cells in the tumor mileu. In this review, we focus on the current understanding of the role of P-gp in cancer cells and immune cells and finally attempt to highlight some caveats in the current understanding of its role in comprehensive tumor microenvironment along with challenges in the development of P-gp inhibitors toward anti-cancer therapy.
Introduction
Multidrug resistance (MDR) accounts for chemotherapeutic resistance in cancer cells (1). Three major proteins namely P-glycoprotein (P-gp, also referred to as MDR1), MDR-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP), were shown to play a critical in MDR (2). These three proteins belong to a family of 48 energy-dependent membrane transporter proteins called adenosine triphosphate (ATP)-binding cassette (ABC) efflux pumps (3, 4). This group of ABC transporters have a diverse epithelial cell surface expression including on gastrointestinal tract, hepatobiliary tract, renal tubules, adrenal cortex, placenta, and blood-brain barrier membranes (5). Under physiological conditions, ABC transporters are involved in efflux of lipids, sterols, small microbial peptides and toxins out of the cytoplasm (6). P-gp is most studied and well-characterized MDR transporter associated with resistance to cancer chemotherapy (7). Szackas et al. have previously tested 118 compounds with known putative mechanism of action on NCI-60 cancer cell lines (8). Their results have demonstrated that more than 95% of the compounds exerted a negative correlation between drug sensitivity and P-gp expression. Compounds such as geldanamycin, paclitaxel and its taxane analogs, doxorubicin, vinblastine, and bisantrene demonstrated a striking negative correlation, while compounds such as hydroxyurea, methotrexate, and 5-fluorouracil were found to have been invariably non-correlated or slightly positively correlated with drug sensitivity index. While some cancer cells (such as melonama and renal cancers) have an enhanced genetic and epigenetic modulators causing higher constitutive expression of P-gp, majority of other solid tumors induce expression of P-gp as a tumor resistance response following initiation of chemotherapy (9). Several P-gp inhibitors have been studied to improve the chemotherapeutic susceptibility of solid tumors (10). However, majority of these clinical trials have failed due to several reasons, an important reason being the high drug doses need to exert P-gp transporter inhibition. Currently, several new drug discovery projects have an array of novel pro-drug compounds in pipeline to bypass or exert a more sustained P-gp inhibition (11). Interestingly, over the past two and half decade accumulating evidence suggested that the expression of P-gp in inflammatory immune cell subset (12–15). This could exert a potential anti-cancer cytotoxic functionality. However, a detailed understanding of this apparently contrasting role of P-gp in cancer and immune cells in the context of tumor microenvironment is yet to evolve. In this review, we will briefly describe the molecular details of P-gp and prevailing understanding on its inhibitors. We will than focus on the current immunological evidence of P-gp in various immune cell phenotypes with potential future insights on tumor immunotherapy.
P-gp Genetics
The p-gp/abcb1 gene is located on chromosome 7q21.12 and contains 29 exons in a genomic region spanning 209.6 kb. The messenger RNA (mRNA) is 4872 bp in length, including the 5′ untranslated region (RefSeq accession NM_000927.3), which is expressed into a 141 KDa protein with 1,280 amino acids (16). To date (as of Nov 2019, NCBI-SNP view database), in the coding region alone, upto 1,200 single nucleotide polymorphisms (SNPs) have been reported with varied impact on protein expression and functionality. Of these the three most studied SNPs in the protein coding region of P-gp are rs1045642 (3435T>C, Ile1145Ile), rs2032582 (2677T>G/A, Ser893Ala/Thr), and rs1128503 (1236T>C, Gly412Gly) (17). Further, while 28% of the SNPs were reported in the transmembrane domain 72% of the SNPs were reported in intra- and extracellular regions of P-gp.
The synonymous mutation, C to T transition at position 3435 (rs1045642, 3435T>C) results in an unaltered amino acid sequence (Ile1145Ile) and could be expected not to change the protein functionality (18). In general, the 3435C allele occurs at 34–90% frequency across all populations with high expression 3435CC genotype in Africans compared to Caucasians or Japanese (19–21). Although this is a synonymous mutation, interestingly, it is not generally considered a silent mutation. Hoffmeyer et al. have demonstrated that 3435 TT genotype population demonstrated lower expression of P-gp in the epithelial cells of digestive tract (22). The 3435C allele showed higher mRNA transcript levels compared to the 3435T allele (23). This differential gene expression level is considered to be due to instability in the mRNA secondary structure which requires more time for mRNA folding/unfolding during translation process resulting in altered membrane insertion and tertiary structural orientation and thus leading to variations in the substrate affinity. For these reasons, the 3435CC genotype is correlated with a higher P-gp expression and function compared to either 3435CT or 3435TT genotypes (24, 25). In the context of tumor resistance, patients with 3435TT genotype might be expect to develop minimal resistance to chemotherapy compared to 3435CC genotype, and therefore requiring lower amount of drug for cancer cell elimination (25). Pharmacokinetic studies with cyclosporine have demonstrated that patients with 3435TT genotype had enhanced intracellular drug concentration compared to 3435CC genotype. Similarly, pharmacodynamic studies with tacrolimus and sirolimus have demonstrated that compared to 3435CC genotype, patients with 3435TT genotype had higher immunosuppression as evidenced by decreased circulating levels of inflammatory cytokine, interleukin-2 (IL-2) (26).
The rs2032582 SNP (2677T > G/A, Ser893Ala/ Thr), with three allelic variants, although well-studied have some discordant outcomes on the actual protein functionality. The frequency of 2677T allele coding for serine-893 varies as widely as 2–65% among various ethnicities (23). Interestingly, the frequency of homozygous 2677 GG genotype leading to 893-Ala/Ala P-gp is found to be as high as 81% in African populations, as compared to the frequency of only 10–32% in other demographics such as European, Mexican, Native America, Asian, and Indian populations. Along with these SNPs, another allele, 2677A bearing Thr-893 P-gp has been reported to be at lower frequency of only 0–17% across various populations. In spite of extensive studies on this non-synonymous mutation inducing SNP, the potential impact on the P-gp expression and functionality is unclear (27). The Ser-893 P-gp has shown to have apparently conflicting functional outcomes with all three (increase, decrease and no change) outcomes on the pharmacodynamics properties. Similarly, studies with Ala-893 vs. Ser-893 mutation have shown no difference in the treatment outcomes in inflammatory bowel diseases (Crohn's and ulcerative colitis) (28). Similarly, a third SNP, rs1128503 (1236T>C) bearing synonymous mutation leading to Gly412Gly P-gp is reported to have a wide frequency of 30 to 93% among various populations (29). However, the pharmacokinetic and pharmacodynamics differences between the genotypes, 1236CC/CT/TT, have not been confirmed (30).
P-gp: Protein Expression, Structure, and Function
Biedler et al. (31) for the first time suggested the potential existence of multidrug resistance (MDR) phenomena mediated by a cell surface protein (31). Riordan et al. (32) have first cloned the P-gp cDNA and expressed in mammalian cell lines to provide proof that MDR is indeed mediated by a membrane protein (32). Later, Schinkel et al. (33) using a murine abcb1 (P-gp) knock-out model demonstrated a 100-fold increased brain tissue concentration of antiparasitic medication, ivermectin, in these genetically engineered mice (33). For constitutive expression of this gene, there seems to be two transcriptional start regions in the proximal promoter region of exon 1 and intron 1. The mRNA transcript for this gene compromising of 5' untranslated region is 4,872 bp long and is translated into 1,280 amino acid P-gp protein. The secondary structure has twelve transmembrane domains (TMD) as also evidence by Kyte-Doolittle hydropathy plot (Figure 1). Several alternative transcripts and splice variants with undetermined significance were reported in the literature, however, not discussed in the current review. P-gp is post-translationally modified by differential phosphorylation and N-glycosylation which is thought to impact its final functionality (34). The serine residues of P-gp, S661 and S683 are phosphorylated by PKC and PKA, respectively (35). Additionally, phosphorylation of S683 by Pim-1 selectively on glycosylated P-gp is considered to induce multimerization and surface membrane stabilization (36). While two phosphorylation residues were shown to bind with tubulin, it has not been shown to be important in inducing downstream signaling and protein functionality (37, 38). The 12 TMDs form a hydrophobic pore-like-channel in the cell membrane to promote drug efflux of hydrophobic and amphipathic compounds (Figure 1). The two ATP-binding domains are located in the cytoplasmic intracellular side of the protein. The first high-resolution X-ray crystallography structure at a resolution of 3.8 Å of mouse P-gp, which has 87% homology with human P-gp, was reported in 2009 (39). Further studies with slightly improved (up to 3.3 Å) resolution also showed predominantly similar tertiary structural features (40, 41). The tertiary structure of P-gp protein exhibits high membrane flexibility to allow for multiple three-dimensional (3D) reorientations, possibly playing an important functional role in binding and efflux of a wide array of drug substrates (42). Interestingly, in silico structure activity relationship (SAR) studies demonstrated that P-gp had the capacity to differentially bind with stereoisomers of the same compound and also has multiple binding sites to allow binding and efflux of multiple drug substrates (43). While the initial SAR studies with P-gp have been challenging mainly due to its high hydrophobicity index making it insoluble in water and high tertiary structural flexibility, more recent studies by Alam et al. revealed a 3.5- Å resolution structure using reconstituted in lipidic nanodiscs allowing for much better SAR biochemical understanding (44).
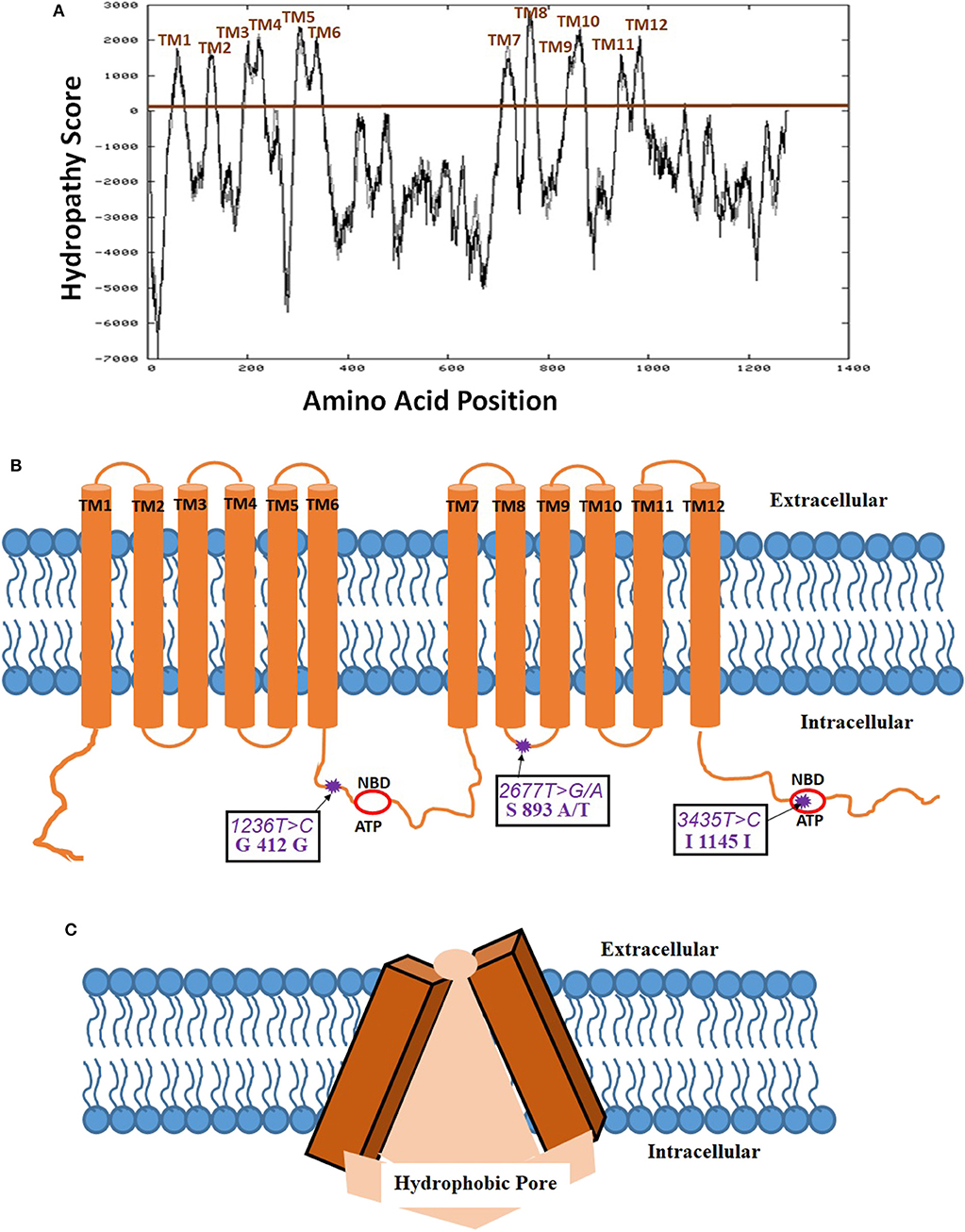
Figure 1. Membrane localization of P-glycoprotein (P-gp, PDB ID: 6QEX). (A) Kyte-Doolittle hydropathy plot determining the amino acid positions in the twelve transmembrane domains (TMD) of P-gp (https://embnet.vital-it.ch/cgi-bin/TMPRED_form_parser; EXPASy Bioinformatics resource portal); (B) Schematic of the membrane localization of 12 TMDs, 2 ATP- nucleotide binding domains (NBD), and 3 most common single nucleotide polymorphisms (SNP) on P-gp; (C) Tertiary three dimensional inverted V-shaped structure of P-gp.
The structure of P-gp displays the canonical ABC transporter fold consisting of two pseudo-symmetric transmembrane domains, with each half containing six transmembrane helices (TM) and one cytosolic domain has ATP-nucleotide binding functionality (NBD). The two NBD domains in P-gp, which are largely conserved in many ABC proteins, dimerize to bind and hydrolyze ATP at the interface. A 60–70 amino acid flexible linker with several phosphorylation sites connects the two pseudo-halves of P-gp (45). The cytoplasmic side of the protein encloses a 6000 Å3 large cavity (39). Drugs are thought to enter this cavity for binding through portals open on the cytoplasm and the inner leaflet of the membrane and exit out through the extracellular side which generally has a 70–200 Å3 pore size depending on the protein orientation (Figure 1). P-gp undergoes dynamic conformational changes to allow for an array of substrate binding and efflux which is associated with ATP binding and hydrolysis on the cytoplasmic side allowing unidirectional outward flow of the substrates. Thermodynamic studies have demonstrated that while inward V-conformation is energetically-feasible conformation and transient outward facing conformation is adopted at high-energy state with the consumption of ATP-derived energy (46). In spite of the controversies in the reported crystal structure regarding the location of ATP-binding domain due to the use of detergents, and the absence of nucleotides to obtain the crystal structure, however, it is well-documented that the two ATP-binding domains should be on the intracellular side as the cellular concentration of ATP (1–10 mM) far exceeds the domain binding constant (~0.01 mM) (47, 48). Regardless of the controversies on the P-gp tertiary membrane bound structure, the resolved crystal structure enables the in silico identification of the drug substrates and inhibitors for P-gp.
Several causes such as intrinsic cancer genomic instability, epigenetic mechanisms and inflammatory stressors in the tumor microenvironment have been implicated to play a critical role in the upregulation of P-gp expression (Figure 2) (49). Studies have demonstrated that gene rearrangements and tumor mutational burden are important mechanisms to control and modulate promoter region of abcb1 gene leading to its expression (50, 51). Oncogenes such as Ras, p53, c-Raf, etc. have been associated with the regulation of P-gp expression (52, 53). In various kinds of leukemias with enhanced P-gp expression, the promoter region of the gene was shown to be demethylated, suggesting the role of epigenetic modification toward activation of P-gp mediated drug resistance (9, 54). Studies have shown that, following cancer chemotherapy, there is an upregulation of acetyl-H3 and histone deacetylase activity (55, 56). The acetyl-H3 was shown to act at 968 bp upstream P-gp gene in the promoter region. Transcription factors such as CEBPβ have been shown to induce P-gp expression in MCF-7 breast cancer cell lines (57). Previous studies from our laboratory demonstrated that high salt-mediated osmotic stress (Δ0.05 mM NaCl) on MCF-7 and MDA-MB-231 breast cancer cells enhanced intracytoplasmic calcium concentration through activation of store operated calcium entry (SOCE) from endoplasmic reticulum (58). This hypertonic stress induced P-gp expression leading to paclitaxel drug resistance in these breast cancer cells. Further, our murine tumor studies demonstrated that orthotopic breast tumors with MCF-7 cells pre-cultured in hypertonic stress conditions, exerted higher tumor progression kinetics compared to the basal media cultured counterpart. These data suggest that osmotic stress in most solid organ tumors in itself induces P-gp mediated drug resistance.
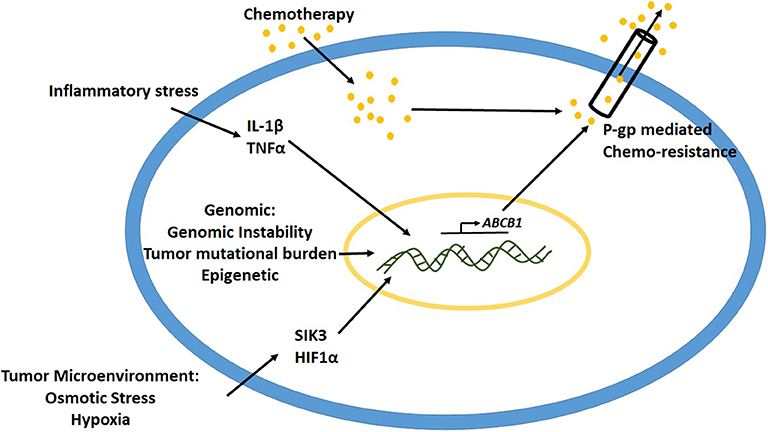
Figure 2. Mechanisms leading to upregulation of P-gp expression.
Studies from some laboratories have demonstrated that hypoxic stress enhances P-gp expression through interaction of HIF1α to P-gp promoter region (59). Sakata et al. have previous demonstrated that cells which developed hypoxia mediated drug resistance did not demonstrate significant increase in the mRNA of P-gp, thus suggesting that the mechanism of hypoxia mediated chemoresistance is different from P-gp pathway (60). Similarly, P-gp-independent MDR was also reported in osteosarcoma cells. Avnet et al. have demonstrated that short term changes in extracellular acidosis induced a reversal plasma membrane pH gradient along with decreased intracellular concentration of drugs such as doxorubicin and cisplatin, with no change P-gp functionality, thus suggesting a non-P-gp mediated MDR (61). Therefore, along with P-gp, other factors in the tumor microenvironment could also play a significant role in MDR.
P-gp Substrate and Drug Interactions
A large variety of molecules with divergent chemical structures (cyclic, linear, polar, non-polar, linear-hydrophobic, aromatic) and molecular weights (from 250 to 4,000 Da) are known to efflux through P-gp transporter (Table 1) (62). cyclosporine-A and verapamil were some of the first identified competitive inhibitors to P-gp. Crystallograhy studies (based on mouse P-gp) have shown that drug-binding motif in the inward V-orientation of P-gp is made up of both hydrophobic (and aromatic) residues to facilitate hydrophobic and van der Waals interactions, and also has few polar side chains (e.g., Q343, Q721, Q942, Q986, and S975) to facilitate the formation of hydrogen-bonds with ligands, thus explaining for the diverse substrate and inhibitor binding to P-gp (63, 64). A recent high-throughput screen of 10,804 compounds by Lee et al. has identified a total of 90 substrates of which 55 were novel. Among these, substrates for P-gp included anti-cancer small molecules such gedatolisib (PKI-587, phosphoinositide 3-kinase/mammalian target of rampamycin inhibitor), AT7159 (cyclin-dependent kinase inhibitor), AT9283 (Janus kinase 2/3 inhibitor), and ispinesib (kinesin spindle protein inhibitor) (65). Currently, although there is a lot of interest in the development of small molecule inhibitors of P-gp, to overcome multi-drug resistance (MDR) in cancer chemotherapy, this enthusiasm is curtailed by the fact that majority of previously discovered inhibitors could not succeed in passing FDA approved clinical phase trials. There are several reasons for this failure to pass clinical trials, but a major reason is that the tissue toxicity of the drugs at the high dose needed for P-gp inhibition (66). For example, verapamil and cyclosporine-A, some of the first discovered P-gp inhibitors tested in clinical trials, demonstrated low-affinity to P-gp requiring several micro-molar plasma concentration at which they have unacceptable cardiac and immunosuppressive side-effects, respectively (67, 68). The recently optimized P-gp inhibitors, tariquidar and zosuquidar, were designed for increased potency (10–100 nM) and enhanced specificity to P-gp (69–71). However, there seems to be some conflicting literature evidence on the role of tariquidar as an ATPase inhibitor or enhancer and if it is a substrate or inhibitor to P-gp (72, 73). Weidner et al., have recently reported that tariquidar is an inhibitor and nor substrate or both human and mouse P-gp (69).
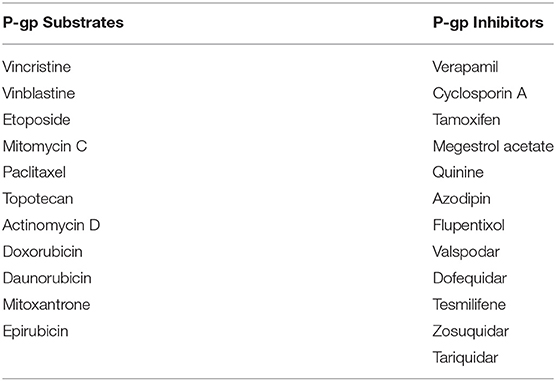
Table 1. List of the known P-gp substrates and inhibitors.
Unlike enzyme-substrate interactions, such as lock-and-key model or induced-fit model, the tertiary structure of P-gp does not have a well-defined ligand-binding pocket making it innately perplexing to design highly specific competitive inhibitors for P-gp (74, 75). However, development of uncompetitive inhibitors for drug-binding pockets, and also, as ATP-binding NBD is well-characterized development of competitive inhibitors for NBD is feasible. Some bivalent inhibitors such as reversible dimer of quetiapine (74) and prodrug dimer of paliperidone (74), with ability to bind with multiple interaction sites of P-gp have been designed with limited success to enable better inhibition compared to monovalent binding (76). A drug design strategy to modify the already identified and natural compounds with known P-gp interaction to enhance specificity and potency will minimize the dose needed for clinical human application. Epothilones which have a chemical structure similar to taxanes, a microtubule mediated cell-division inhibitors, have been suggested to be substrates of P-gp with poor specificity (77). An analog of epothilone B, ixabepilone (azaepothilone B), in combination with capecitabine, was demonstrated success in treating anthracycline and taxane resistant metastatic breast cancer (78, 79). Further ixabepilone demonstrated a 6–10 fold higher cancer cell cytotoxicity compared to epothilone B, against a panel of over 20 tumor cell lines which included both drug-sensitive and resistant P-gp overexpressing cancer phenotypes (43, 80). Similarly, semisynthetic analogs of taxanes have been utilized in the development of several novel compounds with significantly higher efficiency against paclitaxel-resistant cancers. These include cabazitaxel (FDA approved) and ortataxel which have been shown to be efficient in hormone refractory metastatic prostrate cancer (81, 82). Further, these compounds have been shown to be less amenable to efflux by P-gp. Along with taxanes extensive studies have been performed on synthesizing analogs of vinca alkaloids. Vinflunine, a fluorinated semisynthetic analog of vinblastine, displayed 2–13 fold diminished susceptibility to efflux by P-gp-mediated compared to vincristine and vinblastine (83). Consequently, vinflunine received approval in Europe (2009) as second-line therapeutic agent against urothelial cancers. Similarly, an isoindoline urea derivative of vinblastine (at the same chemical position C20) was shown to possess 100 fold higher cytotoxic potential against vinblastine- resistant cancer cell lines (84, 85). Along the lines, an aryl amide derivatives of vinblastine has also been demonstrated to be less sensitive to P-gp mediated efflux in cancer cell lines (86). However, the therapeutic efficiency of these drugs is yet be prove in clinical settings.
As P-gp is considered to recognize hydrophobic compounds for efflux, adding a polar moiety to the drug by chemical modification of the drug or conjugating the drug with polar ligand could be considered some of the possible strategies toward reduced P-gp mediated efflux. Various nano-sized carriers and drug-conjugates have been studied to treat P-gp mediated MDR. Liposome-mediated doxorubicin delivery has received FDA approval in as early as 1995 (87). Other ionic and block copolymer-based drug modifications are still under study. An albumin-bound paclitaxel, abraxane, has already received FDA approval for treatment of metastatic breast cancers (88). Opaxio/Xyotax, a poly-L-glutamic acid- paclitaxel, is currently under Phase III clinical trials for the treatment of ovarian and esophageal cancers (89). Cell-penetrating macromolecules (CPMs) and antibody-drug conjugates (ADCs) have been extensively utilized in targeted drug delivery and reduce side-effects (90, 91). Octaarginine-conjugated taxol has been extensively studied in resistant cancers. These conjugation techniques were primarily intended to enhance drug internalization (92). However, cytoplasmic drug concentration and eventual impact on lowering P-gp mediated efflux by these conjugation techniques is not convincing. Gemtuzumab-ozogamicin was approved by FDA for a brief time-period, but later the approval was withdrawn due to lack of improved overall survival profile (93). Similarly, CD33-conjugated maytansine is efficient in pre-clinical studies, however, clinical benefit is yet to be proven (94). Brentuximab-vedotin with a potency to evade P-gp-efflux seems to be one of the very few ADCs which received FDA approval for treatment of refractory hodgkin's lymphoma and systemic anaplastic large cell lymphoma (95). As the understanding of conjugation techniques improve more efficient compounds could designed.
P-gp Function in Tumor Immunity
The expression of P-gp on immune cells is shown to be correlated with immune cell activation, phenotype switch, and cytokine release. While expression of P-gp in peripheral circulating monocytes is extremely limited, however, its expression in tumor infiltrating anti-inflammatory MΦ2 tissue macrophages is extremely high (96). In dendritic cells, P-gp expression is correlated with their maturation and activation with enhanced professional antigen presenting functionality (97, 98). Blockade of P-gp with valspodar impaired DC maturation as shown by decreased expression of activation markers, CD80 and CD40 (98). Among all the innate immune cells, natural killer (NK) cells have been shown to have highest surface expression of P-gp, which is shown to correlate with the downstream cytotoxic functionality of these cells with enhanced Fas-mediated (Fas/FasL) surface binding of P-gp+NK cells to the target cells leading to release of inflammatory cytotoxic secretory granules leading to apoptotic death of target cell (99, 100).
The role of P-gp expression in adaptive immune cells varies with individual cell type. In B-cells, P-gp expression is correlated with cell migration and transitional phenotype in lymph nodes (101, 102). In CD4+T cells, P-gp is associated with inflammatory Th1/Th17 effector phenotype, while its expression is extremely limited in anti-inflammatory Treg phenotype (13, 103, 104). In CD8+T cells, the expression of P-gp is associated with memory (IL18Rα+CD161+CD62Llo) phenotype (105–107). These P-gp expressing CD8+ memory T cells in mucosal associated T-cells (such as in gastrointestinal tract) is associated with a bidirectional responses, with initial protective role to evade xenobiotic toxins, but later when normal microbiome is disrupted, could cause enhanced effector responses leading to autoimmune diseases such as crohn's disease and ulcerative colitis (107).
The immune cells in the patients with hematological malignancies, such as acute myeloid leukemia (AML), diffuse large B Cell lymphoma, multiple myeloma, and follicular lymphoma, demonstrate enhanced expression of P-gp, thus making these cancers resistant to chemotherapy (108, 109). Enhanced expression of P-gp in myeloid and lymphoid lineage cells of AML and B-cell lymphomas, respectively, is associated with upregulation of MAPkinase/ERK signaling (110). It is of interest to note that P-gp mediated chemoresistance could be overcome with monoclonal antibodies (mAb)-based anti-CD20 and anti-CD19 therapy, possibly because mAb could not be effluxed by P-gp (111).
To date there is very limited data from solid organ tumors showing P-gp expression in the infiltrating immune cells. Studies on human colorectal cancer demonstrated that there is enhanced frequency of P-gp expressing mucosal derived CD8+T cells in the tumor tissue specimens (112). However, the exact role of these CD8+T cells remains elusive. Further, the infiltration of the immune cells into the tumor could be skewed by the chemo-resistance of the cancer cells in the tumor microenvironment. In AML patients on long term chemotherapy there was enhanced CD4+CD161+P-gp+ T cells phenotype (113). Further this subset of CD4+helper-T-cells demonstrated diminished expression of T-cell exhaustion markers PD-1 and CTLA-4. The subsets of CD4+T-helper cells, Th17 and Th1 are known to induce anti-tumor effect through secretion of inflammatory cytotoxic cytokines such as IL-17, IFNγ, TNFα, and granzyme (114, 115). Interestingly, the tumor infiltrating P-gp-expressing CD4+T-cells (CD4+CD73+T cells) in breast and ovarian carcinomas were shown to exert enhanced secretion of these anti-cancer cytokines (116, 117). Importantly, chemical inhibition of P-gp inhibited vesicular secretion of these cytotoxic cytokines by these T-lymphocytes (14). Therefore, it will not be favorable to use P-gp-inhibitors in this scenario, as this might reduce the cytotoxic potential of these tumor infiltrating anti-cancer Th1 and Th17 CD4+T cell phenotypes (118). Further, as mentioned above P-gp expression is shown in pro-tumor MΦ2-macrophage phenotype and, anti-tumor NK-cell and Th17/CD4+T cell subsets, thus suggesting an apparently conflicting role of P-gp in tumor immunology. Therefore, the role of P-gp expression in tumor infiltrating immune cells should be more carefully studied in future to determine the potential application of combinatorial strategy of P-gp inhibitors with immune-checkpoint therapy (anti-CTLA4/anti-PD1) (119, 120).
Conclusion
Despite of implementing multi-drug regimens, cancer therapy is still a challenge as tumor cells quickly develop resistance. The role of P-gp in chemo-resistance is well-appreciated for over past three decades. However, development of P-gp-specific inhibitors requires a better understanding of the tissue distribution, cell type specificity, body distribution/toxicity, immune side-effects, and cell-specific cytotoxicity. Synthetic modification of current chemotherapeutic drugs to evade P-gp-mediated efflux seems to be a very difficult drug-discovery task. Advances in the understanding of the 3D-crystal structure of P-gp protein offered novel insights into the drug-design strategies. To make the matters more complicated, adopting a combinatorial therapeutic regimen with P-gp inhibitors could enhance tumor cell drug-sensitivity, but impair efficient infiltration of tumors with anti-tumor immune cells. These changes in tumor microenvironment require further in-depth research for efficient futuristic usage of P-gp inhibitors.
Author Contributions
KR and VT participated in manuscript drafting and revision of this review article.
Funding
This work was supported by NIH-5U54CA163066 (VT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. (2019) 575:299–309. doi: 10.1038/s41586-019-1730-1
2. Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. (2003) 22:7340–58. doi: 10.1038/sj.onc.1206938
3. Mohammad IS, He W, Yin L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed Pharmacother. (2018) 100:335–48. doi: 10.1016/j.biopha.2018.02.038
4. Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. (2001) 42:1007–17. doi: 10.1002/0471203076.emm0713
5. Gameiro M, Silva R, Rocha-Pereira C, Carmo H, Carvalho F, Bastos ML, et al. Cellular models and in vitro assays for the screening of modulators of P-gp, MRP1 and BCRP. Molecules. (2017) 22:600. doi: 10.3390/molecules22040600
6. Silva R, Vilas-Boas V, Carmo H, Dinis-Oliveira RJ, Carvalho F, de Lourdes Bastos M, et al. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. (2015) 149:1–123. doi: 10.1016/j.pharmthera.2014.11.013
7. Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Rep. (2018) 8:967. doi: 10.1038/s41598-018-19325-x
8. Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. (2004) 6:129–37. doi: 10.1016/j.ccr.2004.06.026
9. Henrique R, Oliveira AI, Costa VL, Baptista T, Martins AT, Morais A, et al. Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer. BMC Genomics. (2013) 14:898. doi: 10.1186/1471-2164-14-898
10. Amawi H, Sim HM, Tiwari AK, Ambudkar SV, Shukla S. ABC transporter-mediated multidrug-resistant cancer. Adv Exp Med Biol. (2019) 1141:549–80. doi: 10.1007/978-981-13-7647-4_12
11. Kumar A, Jaitak V. Natural products as multidrug resistance modulators in cancer. Eur J Med Chem. (2019) 176:268–91. doi: 10.1016/j.ejmech.2019.05.027
12. Wu YJ, Wang C, Wei W. The effects of DMARDs on the expression and function of P-gp, MRPs, BCRP in the treatment of autoimmune diseases. Biomed Pharmacother. (2018) 105:870–8. doi: 10.1016/j.biopha.2018.06.015
13. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. (2014) 211:89–104. doi: 10.1084/jem.20130301
14. Gupta S, Kim CH, Tsuruo T, Gollapudi S. Preferential expression and activity of multidrug resistance gene 1 product (P-glycoprotein), a functionally active efflux pump, in human CD8+ T cells: a role in cytotoxic effector function. J Clin Immunol. (1992) 12:451–8. doi: 10.1007/BF00918857
15. Zhang JC, Xie F, Yu XH, Deng ZY, Wang Y, Liang P, et al. Expression levels of P-glycoprotein in peripheral blood CD8+ T lymphocytes from HIV-1-infected patients on antiretroviral therapy. Int J Mol Med. (2014) 33:431–40. doi: 10.3892/ijmm.2013.1584
16. Bodor M, Kelly EJ, Ho RJ. Characterization of the human MDR1 gene. Aaps J. (2005) 7:E1–5. doi: 10.1208/aapsj070101
17. Tulsyan S, Mittal RD, Mittal B. The effect of ABCB1 polymorphisms on the outcome of breast cancer treatment. Pharmgenomics Pers Med. (2016) 9:47–58. doi: 10.2147/PGPM.S86672
18. Cizmarikova M, Wagnerova M, Schonova L, Habalova V, Kohut A, Linkova A, et al. MDR1 (C3435T) polymorphism: relation to the risk of breast cancer and therapeutic outcome. Pharmacogenomics J. (2010) 10:62–9. doi: 10.1038/tpj.2009.41
19. Schwab M, Eichelbaum M, Fromm MF. Genetic polymorphisms of the human MDR1 drug transporter. Annu Rev Pharmacol Toxicol. (2003) 43:285–307. doi: 10.1146/annurev.pharmtox.43.100901.140233
20. Dey S. Single nucleotide polymorphisms in human P-glycoprotein: its impact on drug delivery and disposition. Expert Opin Drug Deliv. (2006) 3:23–35. doi: 10.1517/17425247.3.1.23
21. Sakaeda T, Nakamura T, Okumura K. Pharmacogenetics of MDR1 and its impact on the pharmacokinetics and pharmacodynamics of drugs. Pharmacogenomics. (2003) 4:397–410. doi: 10.1517/phgs.4.4.397.22747
22. Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. (2000) 97:3473–8. doi: 10.1073/pnas.97.7.3473
23. Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta. (2009) 1794:860–71. doi: 10.1016/j.bbapap.2009.02.014
24. Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. (2007) 315:525–8. doi: 10.1126/science.1135308
25. Tryggvadottir H, Huzell L, Gustbee E, Simonsson M, Markkula A, Jirstrom K, et al. Interactions between ABCB1 genotype and preoperative statin use impact clinical outcomes among breast cancer patients. Front Oncol. (2018) 8:428. doi: 10.3389/fonc.2018.00428
26. Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet Genomics. (2005) 15:693–704. doi: 10.1097/01.fpc.0000178311.02878.83
27. Hung CC, Tai JJ, Lin CJ, Lee MJ, Liou HH. Complex haplotypic effects of the ABCB1 gene on epilepsy treatment response. Pharmacogenomics. (2005) 6:411–7. doi: 10.1517/14622416.6.4.411
28. Brant SR, Panhuysen CI, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. (2003) 73:1282–92. doi: 10.1086/379927
29. Aarnoudse AJ, Dieleman JP, Visser LE, Arp PP, van der Heiden IP, van Schaik RH, et al. Common ATP-binding cassette B1 variants are associated with increased digoxin serum concentration. Pharmacogenet Genomics. (2008) 18:299–305. doi: 10.1097/FPC.0b013e3282f70458
30. Sai K, Itoda M, Saito Y, Kurose K, Katori N, Kaniwa N, et al. Genetic variations and haplotype structures of the ABCB1 gene in a Japanese population: an expanded haplotype block covering the distal promoter region, and associated ethnic differences. Ann Hum Genet. (2006) 70:605–22. doi: 10.1111/j.1469-1809.2006.00260.x
31. Biedler JL, Riehm H. Cellular resistance to actinomycin D in Chinese hamster cells in vitro: cross-resistance, radioautographic, and cytogenetic studies. Cancer Res. (1970) 30:1174–84.
32. Riordan JR, Deuchars K, Kartner N, Alon N, Trent J, Ling V. Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature. (1985) 316:817–9. doi: 10.1038/316817a0
33. Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. (1994) 77:491–502. doi: 10.1016/0092-8674(94)90212-7
34. Lelong-Rebel IH, Cardarelli CO. Differential phosphorylation patterns of P-glycoprotein reconstituted into a proteoliposome system: insight into additional unconventional phosphorylation sites. Anticancer Res. (2005) 25:3925–35.
35. Czuba LC, Hillgren KM, Swaan PW. Post-translational modifications of transporters. Pharmacol Ther. (2018) 192:88–99. doi: 10.1016/j.pharmthera.2018.06.013
36. Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H, et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. (2008) 283:3349–56. doi: 10.1074/jbc.M707773200
37. Chambers TC, Pohl J, Glass DB, Kuo JF. Phosphorylation by protein kinase C and cyclic AMP-dependent protein kinase of synthetic peptides derived from the linker region of human P-glycoprotein. Biochem J. (1994) 299(Pt 1):309–15. doi: 10.1042/bj2990309
38. Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol. (2010) 78:310–8. doi: 10.1124/mol.109.061713
39. Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. (2009) 323:1718–22. doi: 10.1126/science.1168750
40. Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R, et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci USA. (2013) 110:13386–91. doi: 10.2210/pdb4ksd/pdb
41. Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, et al. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr D Biol Crystallogr. (2015) 71:732–41. doi: 10.1107/S1399004715000978
42. Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. (2014) 23:34–46. doi: 10.1002/pro.2387
43. Waghray D, Zhang Q. Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment. J Med Chem. (2018) 61:5108–5121. doi: 10.1021/acs.jmedchem.7b01457
44. Alam A, Kowal J, Broude E, Roninson I, Locher KP. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science. (2019) 363:753–756. doi: 10.1126/science.aav7102
45. Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang WK, et al. Structures of the multidrug transporter p-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J Biol Chem. (2017) 292:446–61. doi: 10.1074/jbc.M116.755884
46. Thonghin N, Collins RF, Barbieri A, Shafi T, Siebert A, Ford RC. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 A resolution. BMC Struct Biol. (2018) 18:17. doi: 10.1186/s12900-018-0098-z
47. Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, et al. Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol Pharmacol. (2016) 90:35–41. doi: 10.1124/mol.116.104190
48. Kim Y, Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science. (2018) 359:915–9. doi: 10.1126/science.aar7389
49. Wu J, Lin N, Li F, Zhang G, He S, Zhu Y, et al. Induction of P-glycoprotein expression and activity by Aconitum alkaloids: implication for clinical drug-drug interactions. Sci Rep. (2016) 6:25343. doi: 10.1038/srep25343
50. Huff LM, Wang Z, Iglesias A, Fojo T, Lee JS. Aberrant transcription from an unrelated promoter can result in MDR-1 expression following drug selection in vitro and in relapsed lymphoma samples. Cancer Res. (2005) 65:11694–703. doi: 10.1158/0008-5472.CAN-04-1349
51. Huff LM, Lee JS, Robey RW, Fojo T. Characterization of gene rearrangements leading to activation of MDR-1. J Biol Chem. (2006) 281:36501–9. doi: 10.1074/jbc.M602998200
52. Kopnin BP, Stromskaya TP, Kondratov RV, Ossovskaya VS, Pugacheva EN, Rybalkina EY, et al. Influence of exogenous ras and p53 on P-glycoprotein function in immortalized rodent fibroblasts. Oncol Res. (1995) 7:299–306.
53. Chen KG, Sikic BI. Molecular pathways: regulation and therapeutic implications of multidrug resistance. Clin Cancer Res. (2012) 18:1863–9. doi: 10.1158/1078-0432.CCR-11-1590
54. Chekhun VF, Lukyanova NY, Kovalchuk O, Tryndyak VP, Pogribny IP. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethylated targets. Mol Cancer Ther. (2007) 6:1089–98. doi: 10.1158/1535-7163.MCT-06-0663
55. Jin S, Scotto KW. Transcriptional regulation of the MDR1 gene by histone acetyltransferase and deacetylase is mediated by NF-Y. Mol Cell Biol. (1998) 18:4377–84. doi: 10.1128/MCB.18.7.4377
56. Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene. (2005) 24:8061–75. doi: 10.1038/sj.onc.1208955
57. Chen KG, Sale S, Tan T, Ermoian RP, Sikic BI. CCAAT/enhancer-binding protein beta (nuclear factor for interleukin 6) transactivates the human MDR1 gene by interaction with an inverted CCAAT box in human cancer cells. Mol Pharmacol. (2004) 65:906–16. doi: 10.1124/mol.65.4.906
58. Babaer D, Amara S, Ivy M, Zhao Y, Lammers PE, Titze JM, et al. High salt induces P-glycoprotein mediated treatment resistance in breast cancer cells through store operated calcium influx. Oncotarget. (2018) 9:25193–205. doi: 10.18632/oncotarget.25391
59. Wartenberg M, Ling FC, Muschen M, Klein F, Acker H, Gassmann M, et al. Regulation of the multidrug resistance transporter P-glycoprotein in multicellular tumor spheroids by hypoxia-inducible factor (HIF-1) and reactive oxygen species. Faseb J. (2003) 17:503–5. doi: 10.1096/fj.02-0358fje
60. Sakata K, Kwok TT, Murphy BJ, Laderoute KR, Gordon GR, Sutherland RM. Hypoxia-induced drug resistance: comparison to P-glycoprotein-associated drug resistance. Br J Cancer. (1991) 64:809–14. doi: 10.1038/bjc.1991.405
61. Avnet S, Lemma S, Cortini M, Pellegrini P, Perut F, Zini N, et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget. (2016) 7:63408–23. doi: 10.18632/oncotarget.11503
62. Sharom FJ. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: its role in modulating protein function. Front Oncol. (2014) 4:41. doi: 10.3389/fonc.2014.00041
63. Pluchino KM, Hall MD, Moen JK, Chufan EE, Fetsch PA, Shukla S, et al. Human-mouse chimeras with normal expression and function reveal that major domain swapping is tolerated by P-glycoprotein (ABCB1). Biochemistry. (2016) 55:1010–23. doi: 10.1021/acs.biochem.5b01064
64. Kim IW, Booth-Genthe C, Ambudkar SV. Relationship between drugs and functional activity of various mammalian P-glycoproteins (ABCB1). Mini Rev Med Chem. (2008) 8:193–200. doi: 10.2174/138955708783744100
65. Lee TD, Lee OW, Brimacombe KR, Chen L, Guha R, Lusvarghi S, et al. A high-throughput screen of a library of therapeutics identifies cytotoxic substrates of P-glycoprotein. Mol Pharmacol. (2019) 96:629–40. doi: 10.1124/mol.119.115964
66. Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, et al. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genomics. (2011) 21:152–61. doi: 10.1097/FPC.0b013e3283385a1c
67. Hollt V, Kouba M, Dietel M, Vogt G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem Pharmacol. (1992) 43:2601–8. doi: 10.1016/0006-2952(92)90149-D
68. List AF, Kopecky KJ, Willman CL, Head DR, Persons DL, Slovak ML, et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood. (2001) 98:3212–20. doi: 10.1182/blood.V98.12.3212
69. Weidner LD, Fung KL, Kannan P, Moen JK, Kumar JS, Mulder J, et al. Tariquidar is an inhibitor and not a substrate of human and mouse P-glycoprotein. Drug Metab Dispos. (2016) 44:275–82. doi: 10.1124/dmd.115.067785
70. Kemper EM, Cleypool C, Boogerd W, Beijnen JH, van Tellingen O. The influence of the P-glycoprotein inhibitor zosuquidar trihydrochloride (LY335979) on the brain penetration of paclitaxel in mice. Cancer Chemother Pharmacol. (2004) 53:173–8. doi: 10.1007/s00280-003-0720-y
71. Tang R, Faussat AM, Perrot JY, Marjanovic Z, Cohen S, Storme T, et al. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer. (2008) 8:51. doi: 10.1186/1471-2407-8-51
72. Loo TW, Clarke DM. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem Pharmacol. (2014) 92:558–66. doi: 10.1016/j.bcp.2014.10.006
73. Pajeva IK, Wiese M. Structure-activity relationships of tariquidar analogs as multidrug resistance modulators. Aaps J. (2009) 11:435–44. doi: 10.1208/s12248-009-9118-z
74. Emmert D, Campos CR, Ward D, Lu P, Namanja HA, Bohn K, et al. Reversible dimers of the atypical antipsychotic quetiapine inhibit p-glycoprotein-mediated efflux in vitro with increased binding affinity and in situ at the blood-brain barrier. ACS Chem Neurosci. (2014) 5:305–17. doi: 10.1021/cn4002329
75. Jagodinsky JC, Akgun U. Characterizing the binding interactions between P-glycoprotein and eight known cardiovascular transport substrates. Pharmacol Res Perspect. (2015) 3:e00114. doi: 10.1002/prp2.114
76. Pires MM, Hrycyna CA, Chmielewski J. Bivalent probes of the human multidrug transporter P-glycoprotein. Biochemistry. (2006) 45:11695–702. doi: 10.1021/bi0608109
77. Nobili S, Landini I, Mazzei T, Mini E. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med Res Rev. (2012) 32:1220–62. doi: 10.1002/med.20239
78. Shen H, Lee FY, Gan J. Ixabepilone, a novel microtubule-targeting agent for breast cancer, is a substrate for P-glycoprotein (P-gp/MDR1/ABCB1) but not breast cancer resistance protein (BCRP/ABCG2). J Pharmacol Exp Ther. (2011) 337:423–32. doi: 10.1124/jpet.110.175604
79. Egerton N. Ixabepilone (ixempra), a therapeutic option for locally advanced or metastatic breast cancer. P T. (2008) 33:523–31.
80. Rivera E, Gomez H. Chemotherapy resistance in metastatic breast cancer: the evolving role of ixabepilone. Breast Cancer Res. (2010) 12(Suppl 2):S2. doi: 10.1186/bcr2573
81. Tsao CK, Cutting E, Martin J, Oh WK. The role of cabazitaxel in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Urol. (2014) 6:97–104. doi: 10.1177/1756287214528557
82. Flores JP, Saif MW. Novel oral taxane therapies: recent Phase I results. Clin Investig. (2013) 3:333–41. doi: 10.4155/cli.13.18
83. Hainsworth JD, Meluch AA, Lane CM, Spigel DR, Burris HA 3rd, Gandhi JG, et al. Single agent vinflunine in the salvage treatment of patients with castration-resistant prostate cancer: a phase II trial of the Sarah Cannon research consortium. Cancer Invest. (2010) 28:275–9. doi: 10.3109/07357900902918460
84. Barker TJ, Duncan KK, Otrubova K, Boger DL. Potent vinblastine C20' ureas displaying additionally improved activity against a vinblastine-resistant cancer cell line. ACS Med Chem Lett. (2013) 4:985–8. doi: 10.1021/ml400281w
85. Carney DW, Lukesh JC, Brody DM, Brutsch MM, Boger DL. Ultrapotent vinblastines in which added molecular complexity further disrupts the target tubulin dimer-dimer interface. Proc Natl Acad Sci USA. (2016) 113:9691–8. doi: 10.1073/pnas.1611405113
86. Lukesh JC, Carney DW, Dong H, Cross RM, Shukla V, Duncan KK, et al. Vinblastine 20' amides: synthetic analogues that maintain or improve potency and simultaneously overcome Pgp-derived efflux and resistance. J Med Chem. (2017) 60:7591–604. doi: 10.1021/acs.jmedchem.7b00958
87. Uziely B, Jeffers S, Isacson R, Kutsch K, Wei-Tsao D, Yehoshua Z, et al. Liposomal doxorubicin: antitumor activity and unique toxicities during two complementary phase I studies. J Clin Oncol. (1995) 13:1777–85. doi: 10.1200/JCO.1995.13.7.1777
88. De Luca R, Profita G, Cicero G. Nab-paclitaxel in pretreated metastatic breast cancer: evaluation of activity, safety, and quality of life. Onco Targets Ther. (2019) 12:1621–7. doi: 10.2147/OTT.S191519
89. Pooja D, Kulhari H, Kuncha M, Rachamalla SS, Adams DJ, Bansal V, et al. Improving efficacy, oral bioavailability, and delivery of paclitaxel using protein-grafted solid lipid nanoparticles. Mol Pharm. (2016) 13:3903–12. doi: 10.1021/acs.molpharmaceut.6b00691
90. Shin MC, Zhang J, Min KA, Lee K, Byun Y, David AE, et al. Cell-penetrating peptides: achievements and challenges in application for cancer treatment. J Biomed Mater Res A. (2014) 102:575–87. doi: 10.1002/jbm.a.34859
91. Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. (2019) 394:793–804. doi: 10.1016/S0140-6736(19)31774-X
92. Rompicharla SVK, Kumari P, Ghosh B, Biswas S. Octa-arginine modified poly(amidoamine) dendrimers for improved delivery and cytotoxic effect of paclitaxel in cancer. Artif Cells Nanomed Biotechnol. (2018) 46:847–59. doi: 10.1080/21691401.2018.1470527
93. Jen EY, Ko CW, Lee JE, Del Valle PL, Aydanian A, Jewell C, et al. FDA Approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res. (2018) 24:3242–6. doi: 10.1158/1078-0432.CCR-17-3179
94. Lopus M. Antibody-DM1 conjugates as cancer therapeutics. Cancer Lett. (2011) 307:113–8. doi: 10.1016/j.canlet.2011.03.017
95. Donato EM, Fernandez-Zarzoso M, Hueso JA, de la Rubia J. Brentuximab vedotin in Hodgkin lymphoma and anaplastic large-cell lymphoma: an evidence-based review. Onco Targets Ther. (2018) 11:4583–90. doi: 10.2147/OTT.S141053
96. Cory TJ, He H, Winchester LC, Kumar S, Fletcher CV. Alterations in P-glycoprotein expression and function between macrophage subsets. Pharm Res. (2016) 33:2713–21. doi: 10.1007/s11095-016-1998-x
97. van de Ven R, de Jong MC, Reurs AW, Schoonderwoerd AJ, Jansen G, Hooijberg JH, et al. Dendritic cells require multidrug resistance protein 1 (ABCC1) transporter activity for differentiation. J Immunol. (2006) 176:5191–8. doi: 10.4049/jimmunol.176.9.5191
98. Lloberas N, Rama I, Llaudo I, Torras J, Cerezo G, Cassis L, et al. Dendritic cells phenotype fitting under hypoxia or lipopolysaccharide; adenosine 5'-triphosphate-binding cassette transporters far beyond an efflux pump. Clin Exp Immunol. (2013) 172:444–54. doi: 10.1111/cei.12067
99. Klimecki WT, Taylor CW, Dalton WS. Inhibition of cell-mediated cytolysis and P-glycoprotein function in natural killer cells by verapamil isomers and cyclosporine A analogs. J Clin Immunol. (1995) 15:152–8. doi: 10.1007/BF01543107
100. N'Cho M, Hobbs JA, Brahmi Z. Over-expression of multidrug resistance P-glycoprotein inhibits NK granule-mediated lytic ability without affecting the Fas lytic pathway. Hum Immunol. (1999) 60:223–30. doi: 10.1016/S0198-8859(98)00112-8
101. Wirths S, Lanzavecchia A. ABCB1 transporter discriminates human resting naive B cells from cycling transitional and memory B cells. Eur J Immunol. (2005) 35:3433–41. doi: 10.1002/eji.200535364
102. Tsujimura S, Adachi T, Saito K, Kawabe A, Tanaka Y. Relevance of P-glycoprotein on CXCR4(+) B cells to organ manifestation in highly active rheumatoid arthritis. Mod Rheumatol. (2018) 28:276–86. doi: 10.1080/14397595.2017.1341458
103. Pendse SS, Briscoe DM, Frank MH. P-glycoprotein and alloimmune T-cell activation. Clin Appl Immunol Rev. (2003) 4:3–14. doi: 10.1016/S1529-1049(03)00007-2
104. Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. (2010) 70:4850–8. doi: 10.1158/0008-5472.CAN-10-0283
105. Kooij G, Kroon J, Paul D, Reijerkerk A, Geerts D, van der Pol SM, et al. P-glycoprotein regulates trafficking of CD8(+) T cells to the brain parenchyma. Acta Neuropathol. (2014) 127:699–711. doi: 10.1007/s00401-014-1244-8
106. Freeman BE, Hammarlund E, Raue HP, Slifka MK. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci USA. (2012) 109:9971–6. doi: 10.1073/pnas.1203543109
107. Chen ML, Sundrud MS. Cytokine networks and T-cell subsets in inflammatory bowel diseases. Inflamm Bowel Dis. (2016) 22:1157–67. doi: 10.1097/MIB.0000000000000714
108. Gerrard G, Payne E, Baker RJ, Jones DT, Potter M, Prentice HG, et al. Clinical effects and P-glycoprotein inhibition in patients with acute myeloid leukemia treated with zosuquidar trihydrochloride, daunorubicin and cytarabine. Haematologica. (2004) 89:782–90.
109. Yoshimori M, Takada H, Imadome K, Kurata M, Yamamoto K, Koyama T, et al. P-glycoprotein is expressed and causes resistance to chemotherapy in EBV-positive T-cell lymphoproliferative diseases. Cancer Med. (2015) 4:1494–504. doi: 10.1002/cam4.494
110. Shen H, Xu W, Luo W, Zhou L, Yong W, Chen F, et al. Upregulation of mdr1 gene is related to activation of the MAPK/ERK signal transduction pathway and YB-1 nuclear translocation in B-cell lymphoma. Exp Hematol. (2011) 39:558–69. doi: 10.1016/j.exphem.2011.01.013
111. Pop I, Pop L, Vitetta ES, Ghetie MA. Generation of multidrug resistant lymphoma cell lines stably expressing P-glycoprotein. Oncol Rep. (2008) 19:889–95. doi: 10.3892/or.19.4.889
112. Ling L, Lin Y, Zheng W, Hong S, Tang X, Zhao P, et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci Rep. (2016) 6:20358. doi: 10.1038/srep20358
113. Alsuliman A, Muftuoglu M, Khoder A, Ahn YO, Basar R, Verneris MR, et al. A subset of virus-specific CD161(+) T cells selectively express the multidrug transporter MDR1 and are resistant to chemotherapy in AML. Blood. (2017) 129:740–58. doi: 10.1182/blood-2016-05-713347
114. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. (2009) 31:787–98. doi: 10.1016/j.immuni.2009.09.014
115. Disis ML. Immune regulation of cancer. J Clin Oncol. (2010) 28:4531–8. doi: 10.1200/JCO.2009.27.2146
116. Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Hasko G. Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer. (2016) 2:95–109. doi: 10.1016/j.trecan.2016.01.003
117. Frank MH, Denton MD, Alexander SI, Khoury SJ, Sayegh MH, Briscoe DM. Specific MDR1 P-glycoprotein blockade inhibits human alloimmune T cell activation in vitro. J Immunol. (2001) 166:2451–9. doi: 10.4049/jimmunol.166.4.2451
118. Kooij G, Backer R, Koning JJ, Reijerkerk A, van Horssen J, van der Pol SM, et al. P-glycoprotein acts as an immunomodulator during neuroinflammation. PLoS ONE. (2009) 4:e8212. doi: 10.1371/journal.pone.0008212
119. Liu S, Chen S, Yuan W, Wang H, Chen K, Li D, et al. PD-1/PD-L1 interaction up-regulates MDR1/P-gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. (2017) 8:99901–12. doi: 10.18632/oncotarget.21914
Keywords: P-glycoprotein, cancer, tumor immunology, chemotherapy, metastasis
Citation: Robinson K and Tiriveedhi V (2020) Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 10:265. doi: 10.3389/fonc.2020.00265
Received: 26 November 2019; Accepted: 17 February 2020;
Published: 05 March 2020.
Edited by:
Sungpil Yoon, Sungkyunkwan University, South KoreaReviewed by:
Andaleeb Sajid, Yale University, United StatesSilvia Lemma, Rizzoli Orthopaedic Institute (IRCCS), Italy
Copyright © 2020 Robinson and Tiriveedhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Venkataswarup Tiriveedhi, dnRpcml2ZWVAdG5zdGF0ZS5lZHU=