 Bruno Fattizzo1,2
Bruno Fattizzo1,2 Jessica Rosa1,2
Jessica Rosa1,2 Juri Alessandro Giannotta1,2
Juri Alessandro Giannotta1,2 Luca Baldini1,2Nicola Stefano Fracchiolla1*
Luca Baldini1,2Nicola Stefano Fracchiolla1*- 1Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico di Milano, Milan, Italy
- 2Dipartimento di Oncologia ed Oncoematologia, Università degli studi di Milano, Milan, Italy
T-cell acute lymphoblastic leukemia/lymphoma is an aggressive hematological neoplasm whose classification is still based on immunophenotypic findings. Frontline treatment encompass high intensity combination chemotherapy with good overall survival; however, relapsing/refractory patients have very limited options. In the last years, the understanding of molecular physiopathology of this disease, lead to the identification of a subset of patients with peculiar genetic profile, namely “early T-cell precursors” lymphoblastic leukemia, characterized by dismal outcome and indication to frontline allogeneic bone marrow transplant. In general, the most common mutations occur in the NOTCH1/FBXW7 pathway (60% of adult patients), with a positive prognostic impact. Other pathogenic steps encompass transcriptional deregulation of oncogenes/oncosuppressors, cell cycle deregulation, kinase signaling (including IL7R-JAK-STAT pathway, PI3K/AKT/mTOR pathway, RAS/MAPK signaling pathway, ABL1 signaling pathway), epigenetic deregulation, ribosomal dysfunction, and altered expression of oncogenic miRNAs or long non-coding RNA. The insight in the genomic landscape of the disease paves the way to the use of novel targeted drugs that might improve the outcome, particularly in relapse/refractory patients. In this review, we analyse available literature on T-ALL pathogenesis, focusing on molecular aspects of clinical, prognostic, and therapeutic significance.
Introduction
T-cell acute lymphoblastic leukemia/lymphoma (T-ALL/-LL) is an aggressive hematological tumor, driven by malignant transformation and expansion of T-cell progenitors. T-ALL and T-LL are distinguished by the presence of more or <20% marrow blasts, respectively (1, 2). The 2016 revision of WHO classification added a provisional entity called Early T-cell precursor (ETP) ALL. This subset is characterized by a unique immunophenotypic (reduced expression of T-cell markers, CD1a, CD8, and CD5) and genetic profile, indicating only limited early T-cell differentiation, with retention of some myeloid and stem cell characteristics (2).
Current treatment of T-ALL consists of high intensity combination chemotherapy, resulting in high overall survival, with the best outcomes observed in pediatric patients (3). Despite the high response rates after first-line therapy, about 20% of pediatric and 40% of adult patients will relapse (4). Differently from B-cell precursors ALL, where highly effective monoclonal antibodies as well as CD19 targeting chimeric antigen receptor (CAR) T-cells have been developed, in T-ALL only the purine nucleoside analog nelarabine is licensed for relapsed/refractory patients (1, 5). Relapsed/ refractory T-ALL treatment is therefore an unmet need and only new targeted drugs will have the potential to overturn the outcome of these patients.
The purpose of this review is to analyse available data on T-ALL pathogenesis, starting with a brief description of current T-ALL classification and treatment, and then focusing on molecular aspects of clinical, prognostic, and therapeutic significance.
Results
Snapshot on T-ALL Diagnosis, Classification, and Therapy
Diagnosis of T-cell ALL relies on a combination of morphology, immunophenotype, and cytogenetic features, many of which inform prognosis and treatment choices. The morphological distinction between L1 and L2 blasts has now lost clinical relevance since more precise immunophenotypic categories have been set. One of the most widely used is the European Group for the Immunological Characterization of Leukaemias subclassification based on the various stages of T-cell maturation (6). T-lymphoblasts are TdT+ and show positivity for cytoplasmic CD3, the only lineage specific marker. The variable expression of CD1a, CD2, CD4, CD5, CD7, and CD8 distinguishes pro-, pre-, cortical, and mature T-ALL. As regards the relationship between immunophenotype and prognosis, the best outcomes have been observed in the cortical T-cell ALL, while CD1a-negative patients show an increased relapse rate and a lower survival (7, 8). Noteworthy, ETP-ALL is a novel subcategory of T-ALL, characterized by a distinct gene expression profile and immunophenotype. ETP-ALL cells are tipically CD7+ but CD1a– and CD8–, CD5 weak, and express >1 myeloid or stem cell marker (i.e., CD34, CD13, or CD33). These cells originate from a subset of immature thymocytes directly derived from hematopoietic stem cells, thus able to differentiate into both T- and myeloid cells. ETP-ALL accounts for 15% of all T-cell ALL in children and about 35% in adult T-cell disease (9, 10).
As occurs in B-cell ALL, also in T-cell ALL prognosis is influenced by cytogenetics. In a large trial cytogenetic analysis displayed an abnormal karyotype in 72% of patients, with complex karyotypes (≥5 abnormalities) in about 8% of cases, significantly impacting on prognosis (5-year OS 19 vs. 51%, p = 0.006) (11). An increasing number of molecular abnormalities have been associated with T-cell ALL and will be discussed in a dedicated paragraph.
First Therapy Line
Regarding therapy, in the first-line setting, the standard of care for fit patients consists of ALL-based pediatric-inspired regimens, incorporating induction (combination of steroids, anthracyclines, and vincristine), consolidation, delayed intensification, and maintenance with central nervous system (CNS) prophylaxis (12, 13). Addition of the enzyme l-asparaginase, and more recently its pegylated E. coli-derived form (PEG-ASP), characterized by longer half-life and less anti-drug antibody formation, has been demonstrated to significantly improve response rates and OS both in pediatric (14) and adult patients (15, 16). As occurs in B-cell ALL, indication to allogenic hematopoietic stem cell transplant (alloHSCT) in T-ALL in first remission is based on high risk features at diagnosis and is more and more frequently MRD-driven (17). CNS involvement at diagnosis is more likely in T- than in B-cell ALL (9.6 vs. 4.4%; p = 0.001) and has been associated with inferior 5-year OS due to an increased risk of both systemic and CNS relapse (18). The most common prophylaxis employed is the combination of high-dose IV methotrexate and intrathecal chemotherapy (7, 11). A randomized trial stressed the importance of the use of 5 g/sq.m. in T-ALL, higher than those used in B-cell ALL (19). As regards ETP-ALL, a Spanish multicentre study showed the worse prognosis to be ascribed to a lower response to induction therapy than to an increased relapse rate, suggesting that use of different schedules, such as fludarabine, cytarabine, G-CSF, idarubicin (FLAG-IDA), and other more myeloid-oriented chemotherapies, or FLT3-targeted therapies, may play an advantage in this subcategory of patients (20). Current consolidation strategies comprise a delayed intensification including drugs used in induction phase, followed by a 2-year maintenance with 6-mercaptopurine and methotrexate, pulses of vincristine and steroids, and additional IT CNS prophylaxis. Molecular-based and flow cytometry-based techniques allow reliable assessment of minimal residual disease (MRD), whose monitoring at precise timepoints is the standard of care for ALL patients treated with curative intent. The molecular method consists of identifying clone-specific rearrangement with Sanger on next-generation sequencing into the immunoglobulin heavy chain gene or T-cell receptor genes by using a large panel of consensus primers, generating patient-specific real-time quantitative polymerase chain reaction assays for quantification in about 90% of cases, with a quantitative range of 10−4. Despite variable definitions of “early” assessment of MRD (from 6 to 10–16 weeks from the start of therapy), plenty of studies in ALL have confirmed that early MRD response is the most powerful predictor of long-term survival in adult patients with ALL (21–23). Finally, myeloablative alloHSCT should be considered for high-risk T-cell disease. Allocation to alloHSCT may vary among study groups, but generally speaking, failure to achieve CR after induction therapy, high white cell count at presentation, high risk cytogenetics/immunophenotype, and MRD persistence at defined timepoints can all be used to allocate to transplant (11, 24, 25). As regards the subcategory of ETP-ALL, two trials demonstrated improvement in survival in ETP-ALL patients transplanted early in case of treatment resistance (20). Considered its better prognosis, consolidation with alloHSCT is not considered necessary in T-LBL, unless suggested by an adverse course of the disease (26).
Relapsed Disease
About 80% of relapses occur within 2 years of diagnosis. With <7% of survival rate at 5 years (27), relapsed T-ALL has dismal outcome, and no standard strategies are available so far. Response rates using standard chemotherapy regimens such as FLAG-IDA are around 30–40%, with a median OS of 6 months in responders (28). Nelarabine is the only new agent specifically licensed for relapsed/refractory T-cell ALL/LBL. Used as single agent, this drug induced ORR of 14–55% in pediatric patients (29) and 41–46% in adults, with 1-year OS of 28% (30). Neurotoxicity is the major toxicity, affecting around 15% of patients, with more severe and irreversible cases in a minority of patients (31). Importantly, most of the patients obtaining a CR with nelarabine were able to proceed to alloHSCT.
Focus on the Molecular Pathways Involved in T-ALL Pathophysiology
T-ALL results from a multistep transformation process in which the accumulation of genetic alterations affects key oncogenic/tumor suppressors pathways, that are responsible for proliferation, survival and differentiation of T-cells (32, 33). The molecular steps involved in T-ALL pathogenesis encompass: transcriptional deregulation of oncogenes/oncosuppressors, NOTCH1 signaling, cell cycle deregulation, kinase signaling (including IL7R-JAK-STAT pathway, PI3K/AKT/mTOR pathway, RAS/MAPK signaling pathway, ABL1 signaling pathway), epigenetic deregulation, ribosomal dysfunction, and altered expression of oncogenic miRNAs or long non coding RNA (34) (Figure 1).
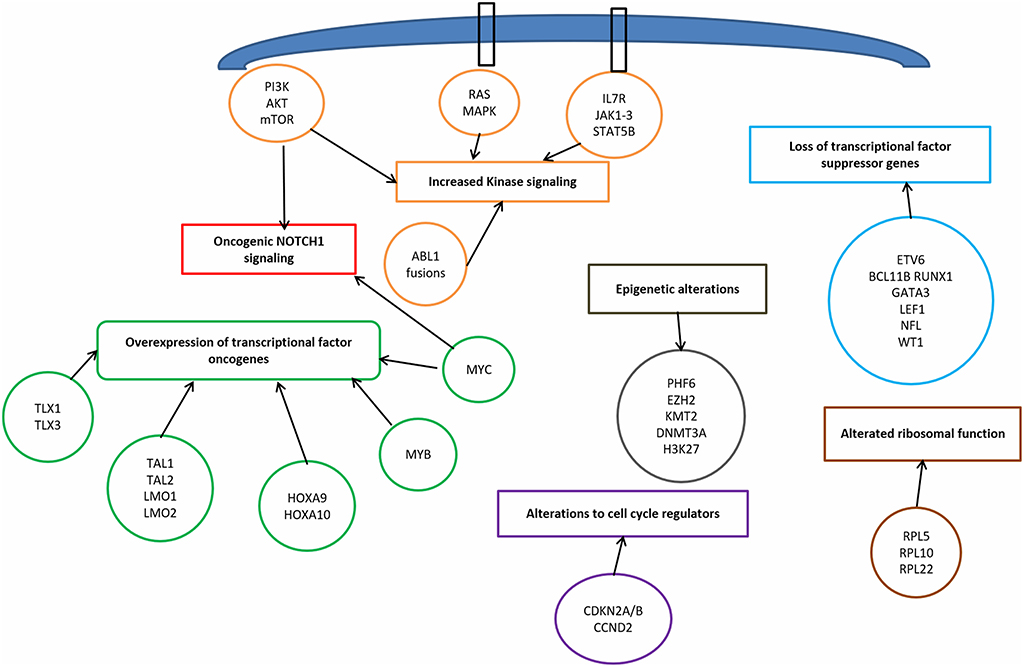
Figure 1. Signaling pathways involved in T-cell acute lymphoblastic leukemia pathophysiology.
Transcriptional Deregulation of Oncogenes and Oncosoppressors
Among the genetic abnormalities, chromosomal translocations of transcription factor oncogenes to regulatory regions of T-cell receptor (TCR) genes are characteristic of T-ALL (34). Approximately 50% of patients harbor chromosomal translocations involving 14q11 (TCR alfa and TCR delta) and 7q34 (TCR beta) (35). Other mechanisms involved are chromosomal rearrangements with other regulatory sequences, duplication/amplification, and mutations or small insertions generating novel regulatory sequences acting as enhancers (36).
Transcriptional factors belonging to bHLH, LMO, and HOX families are also implicated (Table 1). The largest subgroup, representing about 30–35% of T-ALL, is characterized by the abnormal expression of TAL1 (1p32), a bHLH member, which results from either t(1;14) (p32;q11), and t(1;7) (p32;q35) translocations, small insertions, mutations or 1p32 deletion (36, 37). TAL1 expression is associated with a late cortical thymocyte immunophenotype (CD1a-) (38), and correlates with favorable outcomes (35, 39).
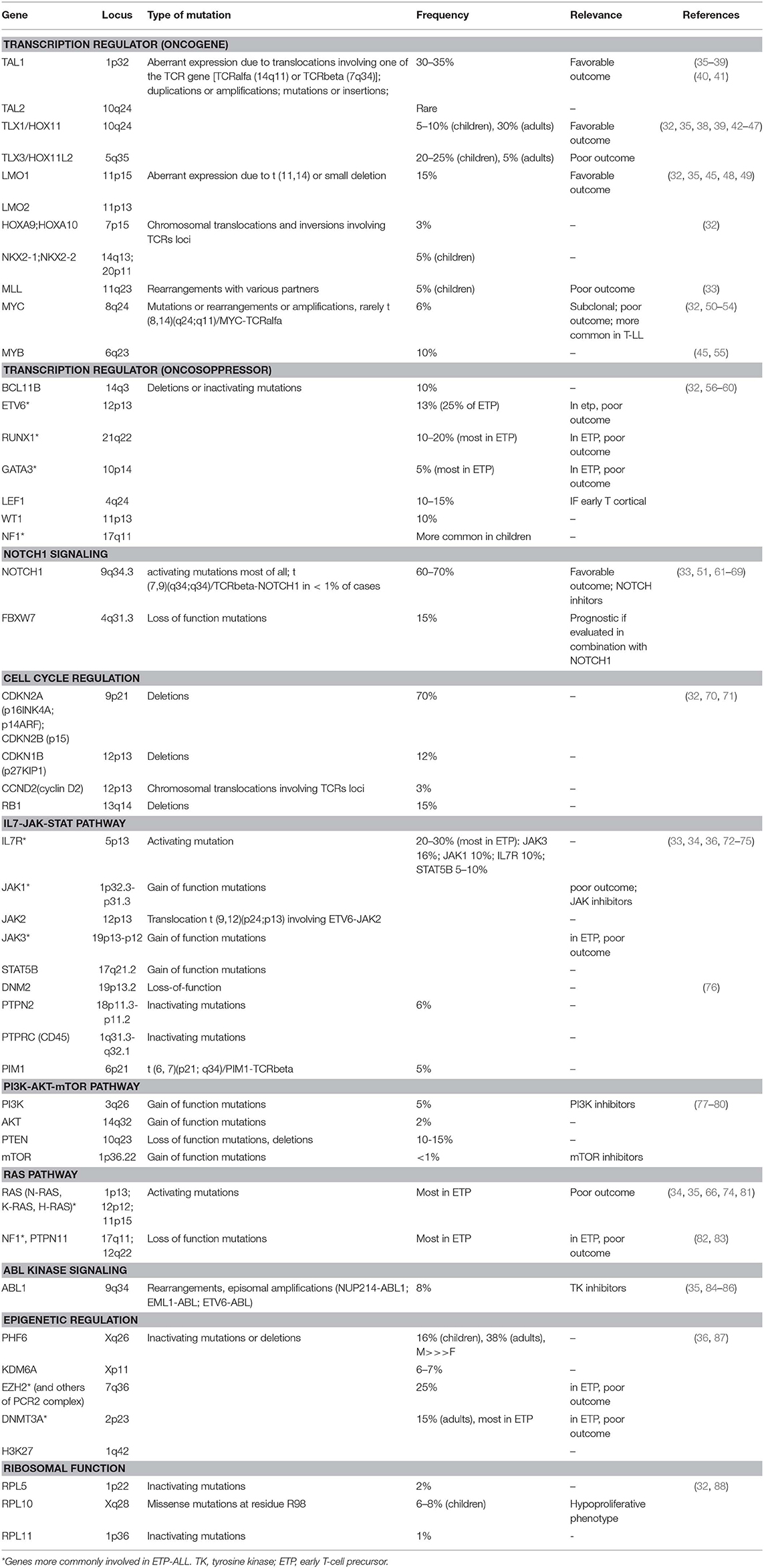
Table 1. Molecular pathways involved in T-ALL pathogenesis.
LMO1 (11p15) and LMO2 (11p13) are part of a transcriptional complex, and are aberrantly expressed at high levels in ~15% of T-ALL, due to both translocations to TCR loci and small chromosomal deletions (32, 45, 48, 49). Also these cases carry a favorable prognosis (35).
Among HOX genes family, TLX1 (10q24, formerly HOX11), and TLX3 (5q35) are over-expressed in T-ALL. TLX1+T-ALLs represent 30% of adult T-ALLs and result from the translocation t(10;14) (q24,q11); the latter involves the TCR locus (42, 43) and contributes to thymocyte arrest at the early cortical stage (CD1a+), conferring favorable outcome (32, 44, 45). On the contrary, TLX3 overexpression (20–25% of pediatric T-ALL) correlates with a poor outcome; it results from t(5;14) which places this oncogene under the control of T-cell regulatory sequences in the BCL11B locus (32, 35, 39, 46, 47).
Other Protoncogenes Involved: MLL, MYC, and MYB
MLL (11q23), originally described in pediatric acute myeloid leukemia, is also involved in T-ALL pathogenesis. The outcome of MLL-rearranged leukemias is generally unfavorable, however this relationship is less clear in T-ALL. MLL-MLLT1 rearrangement, present in 2–3% of T-ALL, has a better outcome, whereas PICALM-MLLT10 rearrangement (about 6–7% of cases) is linked to worse prognosis (39, 89, 90).
MYC (8q24) and MYB (6q23) are proto-oncogenes involved in the transcriptional deregulation observed in T-ALL. In early T-cell development, MYC plays an important role in the control of cell growth downstream NOTCH1 and TCR signaling (50). Moreover, rearrangements involving PI3K/AKT pathway often result in MYC overexpression (52). The translocation t(8;14), involving the TCR, is present in only 1% of MYC+ T-ALL (53), and other mechanisms occur: translocations involving others partners, duplications, amplifications, and reduced degradation (32). In a subgroup of about 6% of T-ALL, MYC translocations are secondary abnormalities, present in subclones, and are associated with induction failure, high rate of relapse, and with an aggressive clinical course (52). The genetic profile of these MYC- translocated T-ALL is characterized by concomitant abnormalities, including CDKN2A/B deletions, PTEN inactivation, and mutations typical of myeloid neoplasms, such as DNMT3A (54). Regarding MYB, it is activated in T-ALL harboring the t(6;7) translocation, which is common among children younger than 2 years of age, or as a result of duplications or amplification of 6q23 (45, 55).
In addition to oncogenes, tumor suppressors contribute to transcriptional deregulation in T-ALL, usually due to deletions or inactivating mutations. BCL11B (14q32), ETV6 (12p13), RUNX1 (21q22), GATA3 (10p14), LEF1 (4q24), WT1 (11p13), and NF1 (17q11) are the main oncosoppressors involved (32).
ETV6, RUNX1, and GATA3, described also in acute myeloid leukemia, are deleted or inactivated in ETP-ALL, and correlate with poor outcome: ETV6 (12p13) mutations account for ~25% of ETP-ALL (56), whilst RUNX1 (21q22), and GATA3 (10p14) mutations are less common. BCL11B (14q32) is mutated in 10% of T-ALL (57); LEF1 (4q24) in 10–15% and is associated with an early cortical thymocyte immunophenotype (58), and WT1 (11p13) in about 10% of cases (59). Monoallelic deletion of 17q12, involving the tumor suppressor NF1, is common in children, but it has been described also in adults (60).
NOTCH1 Pathway
NOTCH1 pathway is essential for T-cell lineage commitment and maturation of hematopoietic progenitors (61). Rarely, the t(7;9) (q34;q34.3) translocation leads to the expression of a constitutively active form of NOTCH1 (9q34.3) (62). However, in over 60% of T-ALLs, NOTCH1 aberrant expression results from activating mutations (63). These mutations lead to ligand-independent cleavage and activation of the intracellular NOTCH1 domain and to the stabilization of the active protein (33). Loss of function of negative regulators of NOTCH1 is an alternative mechanism. As a matter of fact, 10–15% of T-ALL, harbor mutations in FBXW7 (4q31.3), a protein that promotes NOTCH1 proteasomal degradation, and lead to increased NOTCH1 protein stability (64). In prognostic models, patients with NOTCH1 and FBXW7 mutations are defined as low risk cases (65, 66).
NOTCH1 pathway is also a central driver of T-cell metabolism and promotes leukemia cell growth via direct upregulation of anabolic pathways, including ribosome biosynthesis, protein translation and nucleotide and aminoacid metabolism. The effect on cell growth is enhanced by the upregulation of MYC (51, 67, 68). Furthermore, NOTCH1 activates mTOR/Akt pathway and increases the glucose uptake in maturating thymocytes. In summary, oncogenic Notch1 pathway is responsible for enhanced aerobic glycolysis and upregulation of anabolic pathway leading to increased proliferation (69).
Cell Cycle Deregulation
The loss of cell cycle control has a prominent role in the pathogenesis of T-ALL. Deletions of the cyclin-dependent kinase inhibitor 2A (CDKN2A encoding tumor suppressors p16INK4A and p14ARF) and 2B (CDKN2B encoding the tumor suppressor p15INK4B) loci on 9p21 are present in up to 70% of T-ALL, leading to abnormal proliferation control (70). Moreover, deletions in retinoblastoma 1 (RB1, locus on 13q14), a regulator of cell cycle progression, are found in 15% of T-ALL, and deletions involving the CDKN1B locus (12p13, encoding p27KIP1) are present in about 12% loci (32). Finally, high levels of cyclin D (CCND2) are present in 3% of T-ALLs, as a result of translocations with TCR loci (71).
Kinase Signaling Pathways
Kinase signaling pathways aberrantly activated in T-ALL include IL7R/JAK/STAT, PI3K/AKT/mTOR, RAS/MAPK, and ABL kinase signaling (34, 36).
IL7R/JAK/STAT pathway is essential for normal T-cell development and is triggered by the interaction between IL7 and its heterodimeric receptor. Upon ligand-binding, IL7R dimerizes and induces JAK1 and JAK3 phosphorylation, with consequent STAT5 activation. STAT5 dimerizes and translocates to the nucleus, where regulates many target genes, including BCL2 family members (72, 73). Activating mutations of IL7R (5p13), JAK1 (1p32), JAK3 (19p13), and/or STAT5B (17q21) are present in 20–30% of T-ALL cases, with a higher frequency in ETP-ALL patients (33, 74). JAK3 mutations are present in about 16% of T-ALL cases, and a strong association between JAK3 mutations and HOXA9 expression has been demonstrated (75). Furthermore, 6% of T-ALLs are characterized by haplo-insufficiency of negative regulators of this pathway, such as DNM2 (19p13), PTPN2 (18p11), and PTPRC (1q31) (76). The rare t(9,12) (p24;p13) translocation encodes a constitutively active kinase protein, ETV-JAK2, leading to aberrant JAK signaling (91). PIM1 is the ultimate target of the JAK/STAT downstream, and high PIM1 expression is a biomarker of activation of this pathway; PIM1 can be overexpressed also as a result of translocation t(6,7) (p21;q34), involving TCR beta (76).
PI3K/AKT/mTOR pathway is aberrantly activated in T-ALL, resulting in enhanced cell metabolism, proliferation, survival, differentiation, and impaired apoptosis (77). Hyperactivation of this oncogenic pathway is mainly caused by loss-of-function mutations/deletions of PTEN (10q23), occurring in about 10–15% of T-ALLs (78, 79). Additional mutations include gain-of-function mutations in regulatory and catalytic subunits of PI3K (3q26) (4,5% of cases), or in AKT (14q32) or mTOR (1p36) (2 and <1% of cases, respectively) (80).
RAS proteins, including H-RAS (11p15), N-RAS (1p13), and K-RAS (12p12), are fundamental signal transductors from cell surface to downstream effectors (34). RAS-MAPK signaling pathway is frequently hyperactivated in T-ALL, and RAS mutations are present in about 5–10% of cases, particularly in high risk ETP-ALL and in relapsing patients (35, 66, 74, 81). RAS pathway regulators may also be mutated: loss-of-function of NF1 (17q11) and PTPN11 (12q22) have been described in 3% of cases (82, 83).
Finally, ABL1 gene (9q34) is rearranged in 8% of cases, leading to constitutive kinase activity (84). The most frequent rearrangement is NUP214-ABL1 amplification (9q34 amplification), observed in 6% of patients (85), whilst EML1-ABL and ETV6-ABL1 are less common (35). NUP214-ABL is a secondary, subclonal alteration and has not been linked with poor prognosis (86).
Epigenetic Deregulation
Mutations in epigenetic factors are frequent in T-ALL: PHF6 (Xq26), SUZ12 (17q11), EZH2 (7q36), TET2 (4q24), H3F3A (1q42), KDM6A (Xp11), EED (11q14), SETD2 (3p21), and DNMT3A (2p23) mutations are the most common (32, 35). Considering the most frequent, PHF6 is a histone modifier, involved in transcriptional regulation, DNA damage response and cell cycle control. Loss-of-function mutations or deletions of this gene, exclusively found in male patients, are present in 16% of pediatrics and 38% of adults, and result in G2/M cell cycle arrest. Mutational loss of PHF6 is associated with the aberrant expression of the transcription factor oncogenes TLX1 and TLX3 (87). H3K27 regulates methylation, and together with the PRC2 complex (polycomb repressive complex 2, that includes EZH2, SUZ12, and EED) is mutated in up to 25% of T-ALLs (36).
Ribosomal Function
Ribosomes are cellular components required for protein synthesis, a crucial step in rapidly dividing leukemic cells. Ribosomal genes RPL5 (1p22), RPL10 (Xq28), and RPL11 (1p36) have been described to be mutated in T-ALL (32). RPL10 mutations are found in 6–8% of pediatrics, with the recurrent RPL10R98S mutant allele in most cases (32, 88). RPL10R98S mutant leukemia cells may increase the expression of anti-apoptotic protein BCL2. RPL10 R98S mutations are mutually exclusive with JAK/STAT mutations and are associated with a hypoproliferative phenotype (88).
Novel Therapeutic Strategies
Regarding therapy, T-ALL is an aggressive leukemia with limited options, particularly in the relapsed/refractory setting. A better understanding of T-ALL pathogenesis may allow the development of molecular targeted therapies (Table 2) (49). For instance, the high prevalence and prominent role of NOTCH1 mutations make it a promising therapeutic target. Clinical trials have explored the use of γ-secretase inhibitors (86), with limited efficacy and gastrointestinal toxicity (92) that can be reduced by the addition of steroids (93, 104). An example is PF-03084014 that has been tested in a clinical study of relapsed/refractory T-ALL/T-LL (A8641014), with one out of 8 patients experiencing complete response lasting about 3 months (94). Other options are NOTCH transcriptional complex inhibitors or antibodies against NOCTH1 (105). Cell cycle dysregulation by CDK4/CDK6 altered pathway is another potential target, and CDK4/CDK6 inhibitors (86) such as palbociclib recently entered clinical trials. The constitutive activation of PI3K/AKT/mTOR signaling pathway may also be targeted: several PI3K inhibitors showed anti-leukemic effects in T-ALL cell lines, whereas mTOR inhibitors seem to prolong survival in T-ALL cells (34). The most studied molecules were everolimus and temsirolimus (106), that induced variable responses (0–50%) in association to chemotherapy and in a small number of cases (94–96). The limited efficacy of mTOR inhibitors seems to be linked to the activation of compensatory signaling pathways (106). Furthermore, dual PI3K/mTOR inhibitors, NVP-BEZ235 and NVP-BKM120, have been studied. BEZ235 had antiproliferative and proapoptotic effect in T-ALL cell lines (97), and a clinical trial has been started (NCT01756118). BKM120/Buparlisib showed modest efficacy and was tolerable in advanced acute leukemia (only 1 patient with T-ALL) in a recent clinical trial (98). As regards cytokine signaling, JAK-STAT pathway is activated in T-ALL and about 5% of cases are driven by tyrosine kinase oncogene fusions, particularly the NUP214-ABL1 rearrangement (86). JAK inhibitors, such as Ruxolitinib and Tofacitinib, have been studied in preclinical models with activation of IL7R/JAK/STAT pathway (34, 86). In addition, imatinib, dasatinib, and nilotinib are all active against NUP214-ABL1-positive T-cells, with different ability to inhibit this kinase and induce apoptosis in preclinical studies (102). Finally, RPL10R98S mutant leukemia cells are potentially sensitive to Bcl2 inhibitor venetoclax (88). Venetoclax combined to chemotherapy induced a morphological remission in 60% of patients (including ETP-ALL) in a recent retrospective study (103).
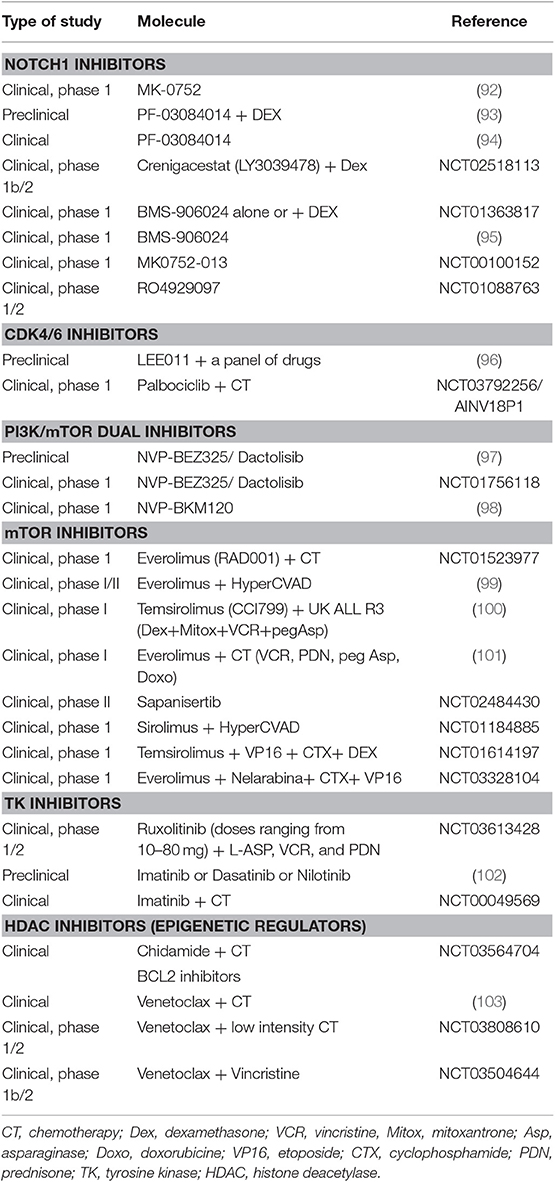
Table 2. Clinical and preclinical trials with target therapies in T-cell acute lymphoblastic leukemia.
Discussion and Future Perspectives
T-ALL is a genetically heterogeneous disease caused by a multistep process, involving cell growth, proliferation and differentiation of T-cells (36, 66). A better understanding of the molecular physiopathology may refine classification and prognostication. Regarding the former, molecular findings allowed the definition of the ETP-ALL subgroup, characterized by a distinct gene expression profile and immunophenotype (9). Moreover, high frequencies of FLT3, NRAS/KRAS, DNMT3A, IDH1, and IDH2 mutations have been found in ETP-ALL (107), similarly to what observed in myeloid leukemic stem cells. This new entity is associated with high levels of minimal residual disease after induction chemotherapy (10) and inferior long-term outcomes (25, 108). Beyond ETP-ALL, other recurrent mutations carry prognostic significance. Among them, the most common occur in the NOTCH1/FBXW7 pathway (60% of adult patients) (63), and confer a positive prognosis in most studies (65, 109, 110). A risk classification based on the presence or absence of NOTCH1/FBXW7, PTEN, or N/K-RAS mutations has been proposed (111). The good-risk group (significantly superior OS and inferior cumulative incidence of relapse) harbored mutations in the NOTCH1/FBXW7 pathway with no associated mutations in PTEN or N/K-RAS; mutated NOTCH1/FBXW7 genes plus mutations in PTEN or N/K-RAS were classified as poor risk with OS 44% and cumulative incidence of relapse 54%.
The study of genetic lesions involved in T-ALL pathogenesis may lead to the development of new targeting drugs. In particular, different inhibitors of NOTCH1 pathway are under active study, including γ-secretase inhibitors, blocking of NOTCH transcriptional complex, and antibodies against NOCTH1. Cell cycle blockers like palbociclib and PI3K-, mTOR- and dual inhibitors (everolimus and temsirolimus, NVP-BEZ235 and NVP-BKM120), showed promising anti-leukemic effect both in vitro and in vivo. Tyrosine kinase inhibitors targeting IL7R/JAK/STAT pathway (ruxolitinib and tofacitinib) and NUP214-ABL1-mutated ALL (imatinib, dasatinib and nilotinib) are all active against T-cell blasts. Finally, Bcl2 inhibitor venetoclax may have a role in RPL10R98S mutant ALL.
In conclusion, in the last years the better understanding of genetic lesions in T-ALL paved the way to novel target therapies, and many preclinical and clinical trials are ongoing. However, the rarity of the disease makes it hard to design specific trials, and the complexity of the molecular landscape may account for the limited efficacy of selective inhibitors in clinical studies. In this setting, differently from other leukemic contexts where chemo-free regimens are emerging (as observed for Ph+ B-ALL targeted with TK-inhibitors and bispecific antibodies), combination chemotherapy is still needed to establish a response. Nevertheless, the inhibition of more ancillary targets like Bcl2 seems to evoke better anti-leukemic effect and may lead the way for future studies and combinations.
Author Contributions
BF, JR, and JG wrote the paper and revised it for intellectual content. NF designed the study and revised the paper for intellectual content. LB revised the manuscript for intellectual content. All authors made substantial contributions to the conception or design of the work, revised it critically for important intellectual content provided approval for publication of the content, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hefazi M, Litzow MR. Recent advances in the biology and treatment of T-cell acute lymphoblastic leukemia. Curr Hematol Malig Rep. (2018) 13:265–74. doi: 10.1007/s11899-018-0455-9
2. Arber DA, Orazi A, Hasserjian R, Borowitz MJ, Le Beau MM, Bloomfield CD, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–406. doi: 10.1182/blood-2016-03-643544
3. Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol. (2012) 30:1663–9. doi: 10.1200/JCO.2011.37.8018
4. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. (2008) 371:1030–43. doi: 10.1016/S0140-6736(08)60457-2
5. Jabbour E, O'Bren S, Konopleva M, Kantarjian H. New insight into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer. (2015) 121:2517–28. doi: 10.1002/cncr.29383
6. Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, et al. European group for the immunological characterization of leukemias (EGIL). proposals for the immunological classification of acute leukemias. Leukemia. (1995) 9:1783–6.
7. Hoelzer D, Thiel E, Arnold R, Beck J. Successful subtype oriented treatment strategies in adult T-All; results of 744 patients treated in three consecutive GMALL studies. Blood. (2009) 114:324. doi: 10.1182/blood.V114.22.324.324
8. Vitale A, Guarini A, Ariola C, Mancini M, Mecucci C, Cuneo A, et al. Adult T-cell acute lymphoblastic leukemia: biologic profile at presentation and correlation with response to induction treatment in patients enrolled in the GIMEMA LAL 0496 protocol. Blood. (2006) 107:473–9. doi: 10.1182/blood-2005-04-1754
9. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukemia: a subtype of very high-risk acute lymphoblastic leukemia identified in two independant cohorts. Lancet Oncol. (2009) 10:147–56. doi: 10.1016/S1470-2045(08)70314-0
10. Wood BL, Winter SS, Dunsmore KP, Devidas M, Chen S, Asselin BL, et al. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in children's oncology group (COG) study AALL0434. Blood. (2014) 124:1–1. doi: 10.1182/blood.V124.21.1.1
11. Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. (2009) 114:5136–45. doi: 10.1182/blood-2009-08-231217
12. Huguet F, Leguay T, Raffoux E, Thomas X, Beldjord K, Delabesse E, et al. Pediatric-inspired therapy in adults with philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol. (2009) 27:911–8. doi: 10.1200/JCO.2008.18.6916
13. Ribera JM, Oriol A, Bethencourt C, Parody R, Hernández-Rivas JM, Moreno MJ, et al. Comparison of intensive chemotherapy, allogeneic or autologous stem cell transplantation as post-remission treatment for adult patients with high-risk acute lymphoblastic leukemia. results of the PETHEMA ALL-93 trial. Haematologica. (2005) 90:1346–56. doi: 10.1182/blood.V106.11.173.173
14. Kurtzberg J, Asselin B, Bernstein M, Buchanan GR, Pollock BH, Camitta BM. Polyethylene glycol-conjugated L-asparaginase versus native L-asparaginase in combination with standard agents for children with acute lymphoblastic leukemia in second bone marrow relapse: a children's oncology group study (POG 8866). J Pediatr Hematol Oncol. (2011) 33:610–6. doi: 10.1097/MPH.0b013e31822d4d4e
15. Rosen O, Müller HJ, Gökbuget N, Langer W, Peter N, Schwartz S, et al. Pegylated asparaginase in combination with high-dose methotrexate for consolidation in adult acute lymphoblastic leukaemia in first remission: a pilot study. Br J Haematol. (2003) 123:836–41. doi: 10.1046/j.1365-2141.2003.04707.x
16. Wetzler M, Sanford BL, Kurtzberg J, DeOliveira D, Frankel SR, Powell BL, et al. Effective asparagine depletion with pegylated asparaginase results in improved outcomes in adult acute lymphoblastic leukemia: cancer and leukemia group B study 9511. Blood. (2007) 109:4164–7. doi: 10.1182/blood-2006-09-045351
17. Zugmaier G, Gökbuget N, Klinger M, Viardot A, Stelljes M, Neumann S, et al. Long-term survival and T-cell kinetics in relapsed/refractory ALL patients who achieved MRD response after blinatumomab treatment. Blood. (2015) 10:2578–84. doi: 10.1182/blood-2015-06-649111
18. Lazarus HM, Richards SM, Chopra R, Litzow MR, Burnett AK, Wiernik PH, et al. Medical research council (MRC)/national cancer research institute (NCRI) adult leukaemia working party of the United Kingdom and the eastern cooperative oncology group. central nervous system involvement in adult acute lymphoblastic leukemia at diagnosis: results from the international ALL trial MRC UKALL XII/ECOG E2993. Blood. (2006) 108:465–72. doi: 10.1182/blood-2005-11-4666
19. Asselin BL, Devidas M, Wang C, Pullen J, Borowitz MJ, Hutchison R, et al. Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the children's oncology group (POG 9404). Blood. (2011) 118:874–83. doi: 10.1182/blood-2010-06-292615
20. Genescà E, Morgades M, Montesinos P, Barba P, Gil C, Guàrdia R, et al. Unique clinico-biological, genetic and prognostic features of adult early T-cell precursor acute lymphoblastic leukemia. Haematol Haematol. (2019). 2019:225078. doi: 10.3324/haematol.2019.225078
21. Vidriales M-B, Pérez JJ, López-Berges MC, Gutierrez NC. Minimal residual disease in adolescent (older than 14 years) and adult acute lymphoblastic leukemias: early immunophenotypic evaluation has high clinical value. Blood. (2003) 101:4695–700. doi: 10.1182/blood-2002-08-2613
22. Holowiecki J, Krawczyk-Kulis M, Giebel S, Jagoda K, Stella-Holowiecka B, Piatkowska-Jakubas B, et al. Status of minimal residual disease after induction predicts outcome in both standard and high-risk Ph-negative adult acute lymphoblastic leukaemia. the polish adult leukemia group ALL 4-2002 MRD Study. Br J Haematol. (2008) 142:227–37. doi: 10.1111/j.1365-2141.2008.07185.x
23. Bassan R, Spinelli O, Oldani E, Intermesoli T, Tosi M, Peruta B, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. (2009) 113:4153–62. doi: 10.1182/blood-2008-11-185132
24. Beldjord K, Chevret S, Asnafi V, Huguet F, Boulland ML, Leguay T, et al. Group for research on adult acute lymphoblastic leukemia (GRAALL). oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. (2014) 123:3739–49. doi: 10.1182/blood-2014-01-547695
25. Jain N, Lamb AV, O'Brien S, Ravandi F, Konopleva M, Jabbour E, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. (2016) 127:1863–9. doi: 10.1182/blood-2015-08-661702
26. Bassan R, Maino E, Cortelazzo S. Lymphoblastic lymphoma: an updated review on biology, diagnosis, and treatment. Eur J Haematol. (2016) 96:447–60. doi: 10.1111/ejh.12722
27. Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. (2007) 109:944–50. doi: 10.1182/blood-2006-05-018192
28. Specchia G, Pastore D, Carluccio P, Liso A, Mestice A, Rizzi R, et al. FLAG-IDA in the treatment of refractory/relapsed adult acute lymphoblastic leukemia. Ann Hematol. (2005) 84:792–5. doi: 10.1007/s00277-005-1090-9
29. Berg SL, Blaney SM, Devidas M, Lampkin TA, Murgo A, Bernstein M, et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the children's oncology group. J Clin Oncol. (2005) 23:3376–82. doi: 10.1200/JCO.2005.03.426
30. Gökbuget N, Basara N, Baurmann H, Beck J, Brüggemann M, Diedrich H, et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood. (2011) 118:3504–11. doi: 10.1182/blood-2011-01-329441
31. Kurtzberg J, Ernst TJ, Keating MJ, Gandhi V, Hodge JP, Kisoret DF, et al. Phase I study of 506U78 administered on a consecutive 5-day schedule in children and adults with refractory hematologic malignancies. J Clin Oncol. (2005) 23:3396–403. doi: 10.1200/JCO.2005.03.199
32. Belver L, Ferrando A. The genetics and mechanisms of T-cell acute lymphoblastic leukemia. Nat Rev Cancer. (2016) 16:494–507. doi: 10.1038/nrc.2016.63
33. Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica. (2015) 100:1301–10. doi: 10.3324/haematol.2015.130179
34. Bongiovanni D, Saccomani V, Piovan E. Aberrant signalling pathways in T-cell acute lymphoblastic leukemia. Int J Mol Sci. (2017) 18:1904. doi: 10.3390/ijms18091904
35. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. (2017) 35:975–83. doi: 10.1200/JCO.2016.70.7836
36. Girardi T, Vicente C, Colls J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. (2017) 129:1113–23. doi: 10.1182/blood-2016-10-706465
37. Chen Q, Ying-Chuan Yang C, Tsou Tsan J, Xia Y, Ragab AH, Peiper SC, et al. Coding sequences of the tal-1 gene are disrupted by chromosome translocation in human T-cell leukemia. J Exp Med. (1990) 172:1403–8. doi: 10.1084/jem.172.5.1403
38. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T-cell acute lymphoblastic leukemia. Cancer Cell. (2002) 1:75–87. doi: 10.1016/S1535-6108(02)00018-1
39. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. (2015) 125:3977–87. doi: 10.1182/blood-2015-02-580043
40. Kikuchi A, Hayashi Y, Kobayashi S, Hanada R, Moriwaki K, Yamamoto K, et al. Clinical significance of TAL1 gene alteration in childhood T-cell acute lymphoblastic leukemia and lymphoma. Leukemia. (1993) 7:933–8.
41. Bash RO, Crist WM, Shuster JJ, Link MP, Amylon M, Pullen J, et al. Clinical features and outcome of T-cell acute lymphoblastic leukemia in childhood with respect to alterations at the TAL1 locus: a Pediatric Oncology Group study. Blood. (1993) 81:2110–7.
42. Dube ID, Raimondi SC, Pi D, Kalousek DK. A new translocation, t(10;14) (q24;q11), in T-cell neoplasia. Blood. (1986) 67:1181–4. doi: 10.1182/blood.V67.4.1181.1181
43. Hatano M, Roberts CW, Mindem M, Crist WM, Korsmeyer SJ. Deregulation of a homeobox gene, HOX11, by the t(10;14) in T-cell leukemia. Science. (1991) 253:79–82. doi: 10.1126/science.1676542
44. Ferrando AA, Neuberg DS, Dodge RK, Paietta E, Larson RA, Wiernik PH, et al. Prognostic importance of TLX1 (HOX11) oncogene expressione in adults with T-cell acute lymphoblastic leukemia. Lancet. (2004) 363:535–6. doi: 10.1016/S0140-6736(04)15542-6
45. Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukemia. Br J Haematol. (2009) 144:147–56. doi: 10.1111/j.1365-2141.2008.07417.x
46. Bernard OA, Busson-LeConiat M, Ballerini P, Mauchauffé M, Della Valle V, Monni R, et al. A new recurrent and specific cripti trans location, t(5;14) (q35;q32), is associated with expressione of the Hox11L2 gene in T-cell acute lymphoblastic leukemia. Leukemia. (2001) 15:1495–504. doi: 10.1038/sj.leu.2402249
47. Bellerini P, Blaise A, Busson-Le Coniat M, Su XY, Zucman-Rossi J, et al. HOX11L2 expressione defines a clinical subtype of pediatric T-ALL associated with poor prognosis. Blood. (2002) 100:991–7. doi: 10.1182/blood-2001-11-0093
48. Van Vlierberghe P, van Grotel M, Beverloo HB, Lee C, Helgason T, Buijs-Gladdines J, et al. The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood. (2006) 108:3520–9. doi: 10.1182/blood-2006-04-019927
49. Van Vlierberghe P, Ferrando A. The molecular basis of T-cell acute lymphoblastic leukemia. J Clin Invest. (2012) 122:3398–406. doi: 10.1172/JCI61269
50. Dose M, Khan I, Guo Z, Kovalovsky D, Krueger A, von Boehmer H, et al. C-MYC mediates pre-TCR-induced proliferation but not developmental progression. Blood. (2006) 108:2669–77. doi: 10.1182/blood-2006-02-005900
51. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. (2006) 103:18261–6. doi: 10.1073/pnas.0606108103
52. La Starza R, Borga C, Barba G, Pierini V, Schwab C, Matteucci C, et al. Genetic profile of T-cell acute lymphoblastic leukemias with MYC translocations. Blood. (2014) 124:3577–82. doi: 10.1182/blood-2014-06-578856
53. Erikson J, Finger L, Sun L, ar-Rushdi A, Nishikura K, Minowada J, et al. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science. (1986) 232:884–6. doi: 10.1126/science.3486470
54. Tosello V, Milano G, Martines A, Macri N, Van Loocke W, Matthijssens F, et al. A novel t(8;14) (q24;q11) rearranged human cell line as a model for mechanistic and drug discovery studies of NOTCH1- indipendent human T-cell leukemia. Cells. (2018) 7:160. doi: 10.3390/cells7100160
55. Clappier E, Cuccuini W, Kalota A, Crinquette A, Cayuela J, Dik WA, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute lymphoblastic (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. (2007) 110:1251–61. doi: 10.1182/blood-2006-12-064683
56. Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T-cell leukemias. J Exp Med. (2011) 208:2571–9. doi: 10.1084/jem.20112239
57. Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. (2011) 118:4169–73. doi: 10.1182/blood-2010-11-318873
58. Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. (2010) 115:2845–51. doi: 10.1182/blood-2009-07-234377
59. Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S, et al. WT1 mutations in T-ALL. Blood. (2009) 114:1038–45. doi: 10.1182/blood-2008-12-192039
60. Gorello P, La Starza R, Varasano E, Chiaretti S, Elia L, Pierini V, et al. Combined interphase fluorescence in situ hybridization elucidates the genetic heterogeneity of T-cell acute lymphoblastic leukemia in adults. Haematologica. (2010) 95:79–86. doi: 10.3324/haematol.2009.010413
61. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, et al. Deficient T-cell fate specification in mice with an induced inactivation of NOTCH1. Immunity. (1999) 10:547–58. doi: 10.1016/S1074-7613(00)80054-0
62. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the drosophila NOTCH gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. (1991) 66:649–61. doi: 10.1016/0092-8674(91)90111-B
63. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T-cell acute lymphoblastic leukemia. Science. (2004) 306:269–71. doi: 10.1126/science.1102160
64. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J Exp Med. (2007) 204:1813–24. doi: 10.1084/jem.20070876
65. Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a group for research on adult acute lymphoblastic leukemia (GRAALL) study. Blood. (2009) 113:3918–24. doi: 10.1182/blood-2008-10-184069
66. Chiaretti S, Zini G, Bassan R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr J Hematol Infect Dis. (2014) 6:e2014073. doi: 10.4084/mjhid.2014.073
67. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. C-MYC is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. (2006) 20:2096–109. doi: 10.1101/gad.1450406
68. Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1- driven MYC enhancer promotes T-cell developmente, transformation and acute lymphoblastic leukemia. Nat Med. (2014) 20:1130–7. doi: 10.1038/nm.3665
69. Olivas-Aguirre M, Pottosin I, Dobrovinskaya O. Mytochondria as emerging targets for terapie against T-cell acute lymphoblastic leukemia. J Leukoc Biol. (2019) 105:935–46. doi: 10.1002/JLB.5VMR0818-330RR
70. Hebert J, Cayuela JM, Berkeley J, Sigaux F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. (1994) 84:4038–44. doi: 10.1182/blood.V84.12.4038.bloodjournal84124038
71. Clapper E, Cuccuini W, Cayuela JM, Vecchione D, Baruchel A, Dombret H, et al. Cyclin D2 dysregulation by chromosomal translocations to TCR loci in T-cell acute lymphoblastic leukemias. Leukemia. (2006) 20:82–6. doi: 10.1038/sj.leu.2404008
72. Jiang Q, Li WQ, Aiello FB, Mazzucchelli R, Asefa B, Khaled AR, et al. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev. (2005) 16:513–33. doi: 10.1016/j.cytogfr.2005.05.004
73. Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. (2007) 7:144–54. doi: 10.1038/nri2023
74. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukemia. Nature. (2012) 481:157–63.
75. De Bock CE, Cools J. JAK3 mutations and HOXA9 expression are important cooperating events in T-cell acute lymphoblastic leukemia. Mol Cell Oncol. (2018) 5:e1458014. doi: 10.1080/23723556.2018.1458014
76. La Starza R, Messina M, Gianfelici V, Pierini V, Matteucci C, Pierini T, et al. High PIM1 expression is a biomarker of T-cell acute lymphoblastic leukemia with JAK/STAT activation or t(6;7)(p21;q34)/TRB-PIM1 rearrangement. Leukemia. (2018) 2:1807–10. doi: 10.1038/s41375-018-0031-2
77. Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. (2003) 3:317–30. doi: 10.1038/nri1056
78. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. (2007) 13:1203–10. doi: 10.1038/nm1636
79. Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T-cell leukemia viability. J Clin Investig. (2008) 118:3762–74. doi: 10.1172/JCI34616
80. Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. (2009) 114:647–50. doi: 10.1182/blood-2009-02-206722. [Epub ahead of print].
81. Von Lintig FC, Huvar I, Law P, Diccianni MB, Yu AL, Boss GR. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res. (2000) 6:1804–10.
82. Balgobind BV, Van Vlierberghe P, Van Den Ouweland AMW, Beverloo HB, Terlouw-Kromosoeto JNR, Van Wering ER, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. (2008) 111:4322–8. doi: 10.1182/blood-2007-06-095075
83. Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. (2011) 208:1977–88. doi: 10.1084/jem.20110450
84. Hagemeijer A, Graux C. ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromo Cancer. (2010) 49:299–308. doi: 10.1002/gcc.20743
85. Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, et al. Fusion of NUP214 to ABL1 on amplified epitome in T-cell acute lymphoblastic leukemia. Nat Genet. (2004) 36:1084–9. doi: 10.1038/ng1425
86. Ferrando A. Can one target T-cell ALL? Best Pract Res Clin Haematol. (2018) 31:361–6. doi: 10.1016/j.beha.2018.10.001
87. Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymhoblastic leukemia. Nat Genet. (2010) 42:338–42. doi: 10.1038/ng.542
88. Kampen KR, Sulima SO, Verbelen B, Girardi T, Vereecke S, Rinaldi G, et al. The ribosomal RPL10 R98S mutation drives IRES-dependent BCL-2 translation in T-ALL. Leukemia. (2019) 33:319–32. doi: 10.1038/s41375-018-0176-z
89. Bond J, Marchand T, Touzart A, Cieslak A, Trinquand A, Sutton L, et al. An early thymic precursor phenotype predicts outcome exclusively in HOXA-overexpressing adult T-cell acute lymphoblastic leukemia: a group for research in adult acute lymphoblastic leukemia study. Haematologica. (2016) 101:732–40. doi: 10.3324/haematol.2015.141218
90. Bond J, Touzart A, Cieslak A, Trinquand A, Marchand T, Escoffre M, et al. NAP1L1- MLLT10 is a rare recurrent translocation that is associated with HOXA activation and poor treatment response in T-cell acute lymphoblastic leukemia. Br J Haematol. (2016) 174:470–3. doi: 10.1111/bjh.13772
91. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. (1997) 78:1309–12. doi: 10.1126/science.278.5341.1309
92. DeAngelo D, Stone RM, Silverman LB, Stock W, Attar EC, Fearen I, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol. (2006) 24:6585. doi: 10.1200/jco.2006.24.18_suppl.6585
93. Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, et al. Preclinical analysis of the γ-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. (2012) 11:1565–75. doi: 10.1158/1535-7163.MCT-11-0938
94. Papayannidis C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. (2015) 5:e350. doi: 10.1038/bcj.2015.80
95. Zweidler-McKay PA, DeAngelo DJ, Douer D, Dombret H, Ottmann OG, Vey N, et al. The safety and activity of BMS-906024, a Gamma Secretase Inhibitor (GSI) with Anti-Notch activity, in patients with relapsed T-Cell Acute Lymphoblastic Leukemia (T-ALL): initial results of a phase 1 trial. Blood. (2014) 124: 968.
96. Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, et al. Synergistic drug combinations with a CDK4/6 inhibitor in T-cell acute lymphoblastic leukemia. Clin Cancer Res. (2017) 23:1012–24. doi: 10.1158/1078-0432.CCR-15-2869
97. Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. (2010) 70:8097–107. doi: 10.1158/0008-5472.CAN-10-1814
98. Ragon BK, Kantarjian H, Jabbour E, Ravandi F, Cortes J, Borthakur G, et al. Buparlisib, a PI3K inhibitor, demonstrates acceptabletolerability and preliminary activity in a phase I trial ofpatients with advanced leucemia. Am J Hematol. (2017) 92:7–11. doi: 10.1002/ajh.24568
99. Daver N, Boumber Y, Kantarjian H, Ravandi F, Cortes J, Rytting ME, et al. A Phase I/II Study of the mTOR inhibitor everolimus in combination with hyperCVAD Chemotherapy in patients with relapsed/refractory acute lymphoblastic leukemia. Clin Cancer Res. (2015) 21:2704–14. doi: 10.1158/1078-0432.CCR-14-2888
100. Rheingold SR, Tasian SJ, Whitlock JA, Teachey DT, Borowitz MJ, Liu X, et al. A phase 1 trial of temsirolimus and intensive re-induction chemotherapy for 2nd or greater relapse of acute lymphoblastic leukaemia: a children's oncology group study (ADVL1114). Br J Haematol. (2017) 177:467–74. doi: 10.1111/bjh.14569
101. Place AE, Pikman Y, Stevenson KE, Harris MH, Pauly M, Sulis ML, et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. (2018) 65:e27062. doi: 10.1002/pbc.27062
102. Quintàs-Cardama A, Tong W, Manshouri T, Vega F, Lennon PA, Cools J, et al. Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T-cell malignancies. Leukemia. (2008) 22:1117–24. doi: 10.1038/leu.2008.80
103. Richard-Carpentier G, Jabbour E, Short NJ, Rausch CR, Savoy JM, Bose P, et al. Clinical experience with venetoclax in combination with chemotherapy for relapsed or refractory T-cell acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. (2019) 30:S2152–650. doi: 10.1016/j.clml.2019.09.608
104. Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T-ALL. Nat Med. (2009) 15:50–8. doi: 10.1038/nm.1900
105. Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. (2011) 25:83–90. doi: 10.1016/j.blre.2010.09.004
106. Evangelisti C, Chiarini F, McCubrey JA, Martelli AM. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: an update. Int J Mol Sci. (2018) 19:E1878. doi: 10.3390/ijms19071878
107. Neumann M, Coskun E, Fransecky L, Mochmann LH, Bartram I, Sartangi NF, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS ONE. (2013) 8:e53190. doi: 10.1371/journal.pone.0053190
108. Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood. (2013) 122:74–82. doi: 10.1182/blood-2013-03-491092
109. Clappier E, Collette S, Grardel N, Girard S, Suarez L, Brunie G, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. (2010) 24:2023–31. doi: 10.1038/leu.2010.205
110. Mansour MR, Sulis ML, Duke V, Foroni L, Jenkinson S, Koo K, et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J Clin Oncol. (2009) 27:4352–6. doi: 10.1200/JCO.2009.22.0996
111. Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, Lambert J, Beldjord K, Lengliné E, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a group for research in adult acute lymphoblastic leukemia study. J Clin Oncol. (2013) 31:4333–42. doi: 10.1200/JCO.2012.48.5292
Keywords: T-cell acute lymphoblastic leukemia, genome, molecular, target therapies, early T cell precursors acute lymphoblastic leukemia
Citation: Fattizzo B, Rosa J, Giannotta JA, Baldini L and Fracchiolla NS (2020) The Physiopathology of T- Cell Acute Lymphoblastic Leukemia: Focus on Molecular Aspects. Front. Oncol. 10:273. doi: 10.3389/fonc.2020.00273
Received: 16 December 2019; Accepted: 17 February 2020;
Published: 28 February 2020.
Edited by:
Basem M. William, The Ohio State University, United StatesReviewed by:
Giuseppe Gritti, Ospedale Papa Giovanni XXIII, ItalyLueder Hinrich Meyer, University of Ulm, Germany
Copyright © 2020 Fattizzo, Rosa, Giannotta, Baldini and Fracchiolla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Stefano Fracchiolla, bmljb2xhLmZyYWNjaGlvbGxhQHBvbGljbGluaWNvLm1pLml0