 Virginia A. Jones
Virginia A. Jones Payal M. Patel
Payal M. Patel Frederick T. Gibson
Frederick T. Gibson Adriana Cordova
Adriana Cordova Kyle T. Amber
Kyle T. Amber- Skin Immunology Laboratory, Department of Dermatology, University of Illinois at Chicago, Chicago, IL, United States
Alterations in the extracellular matrix (ECM) likely facilitate the first steps of cancer cell metastasis and supports tumor progression. Recent data has demonstrated that alterations in collagen XVII (BP180), a transmembrane protein and structural component of the ECM, can have profound effects on cancer invasiveness. Collagen XVII is a homotrimer of three α1 (XVII) chains. Its intracellular domain contains binding sites for plectin, integrin β4, and BP230, while the extracellular domain facilitates interactions between the cell and the ECM. Collagen XVII and its shed ectodomain have been implicated in cell motility and adhesion and are believed to promote tumor development and invasion. A strong association of collagen XVII ectodomain shedding and tumor invasiveness occurs in squamous cell carcinoma (SCC). Aberrant expression of collagen XVII has been reported in many epithelial cancers, ranging from squamous cell carcinoma to colon, pancreatic, mammary, and ovarian carcinoma. Thus, in this review, we focus on collagen XVII's role in neoplasia and tumorigenesis. Lastly, we discuss the importance of targeting collagen XVII and its ectodomain shedding as a novel strategy to curb tumor growth and reduce metastatic potential.
Introduction
Cancer is a leading cause of death in developed countries (1). In the United States, cancer ranks as the second leading cause of overall mortality, with non-melanoma skin cancer (squamous cell carcinoma and basal cell carcinoma) as the most common form (2). Between the years 2007–2011, an estimated 4.9 million adults were treated for skin cancer, an increase of 1.5 million cases from 4 years prior (3). While incidence cannot be precisely determined, the data suggests that skin cancer incidence is likely increasing (2). Extensive research efforts are continuously underway to identify new targets for cancer treatment. Tumor microenvironment is of particular interest, particularly for invasiveness (4–6). The mechanism of tumor progression from the perspective of tumor stroma has provided a new outlook on cancer development. Following this model, studies have shown that extracellular matrix (ECM), historically considered a structural barrier to tumor cell migration, likely facilitates the first steps of cancer cell metastasis and supports tumor progression (6, 7).
Collagens within the tissue stroma represent a valuable target in cancer biology. At present, 28 triple helical collagen proteins with a variety of structures and functional properties have been discovered in humans (8). Here we will discuss collagen XVII (BP180), a type II hemidesmosomal transmembrane protein and a structural component of the dermoepidermal junction (9, 10). Given notable recent discoveries in COL17A1 expression in cancers, as well as the role of aberrant collagen XVII ectodomain shedding in squamous cell carcinoma, it appears that collagen XVII may play a particularly important role in epithelial cancer growth and invasiveness.
Cell biological analyses propose that collagen XVII functions as a cell-matrix adhesion molecule by stabilizing the hemidesmosome complex. Stabilization occurs by the projection of collagen XVII beneath hemidesmosomes in epithelia, particularly basal keratinocytes, thereby, mediating anchorage to the underlying basement membranes (10–13). Beyond its structural role, collagen XVII is presumed to play a role in skin inflammation, cell migration, and differentiation in physiologic and pathologic states (9, 14).
Structure of Collagen XVII, Interactions of its Subunits, and Function in Membrane Adhesion
Collagen XVII is a 180-kD type II transmembrane protein and structural component of the dermoepidermal anchoring complex, which facilitates adhesion of keratinocytes and certain other epithelial cells to the underlying basement membrane (10). Its gene, COL17A1, has been mapped to chromosome 10q24.3 (15, 16). Collagen XVII along with integrin α6β4, CD151, plectin, and BP230 are components of type I hemidesmosomes (HDs), which are responsible for cell-stromal adherence, cell polarization, and spatial organization of tissue architecture (10, 16). Collagen XVII and integrin α6β4 are transmembrane proteins that bind laminin-332 in the basement membrane (17). Collagen XVII acts as an anchor protein connecting extracellular and intracellular hemidesmosomal proteins (18). The importance of collagen XVII for the integrity of the dermoepidermal junction is demonstrated in inherited skin diseases caused by mutations in the COL17A1 gene, including junctional epidermolysis bullosa (JEB), and in numerous subepidermal blistering skin disorders with autoantibodies to collagen XVII (BP180), such as bullous pemphigoid (BP), linear IgA bullous dermatosis, lichen planus pemphigoides, and pemphigoid gestationis [Figure 1; (19, 20)].
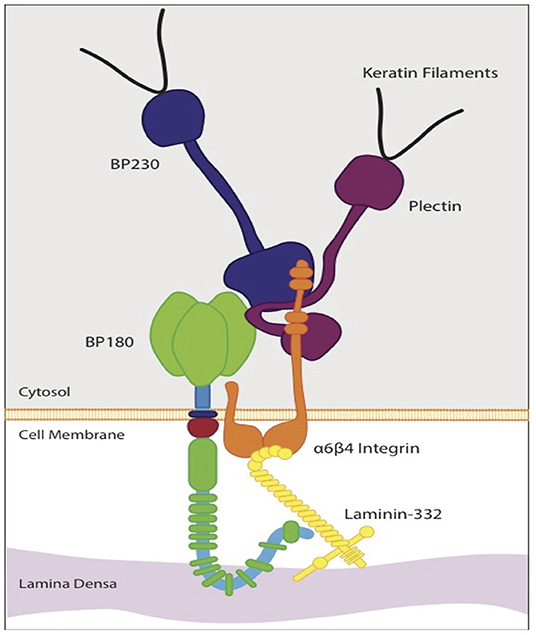
Figure 1. Schematic diagram of the molecular organization of a hemidesmosorne at the dermoepidermal basement membrane. The intracellular plaque is associated with transmembrane adhesion molecules, including integrins and transmembrane type XVII collagen (BP 180). It contains plectin and BP 230 (BPAG1). Type XVII collagen regulates expression and function of laminin- 332. The interaction between laminin-332 and the extracellular portions of a6β4 integrin stabilizes hemidesmosomes. This interaction is essential for hemidesmosome formation and epithelial adhesion.
Collagen XVII is a homotrimer of three α1 (XVII) chains, each with a globular cytosolic amino (N-) terminal domain, a short transmembrane stretch, and a flexible-rod extracellular carboxy (C-) terminal domain (10). The intracellular domain (ICD) of collagen XVII consists of 466 amino acids (aa) while the transmembrane domain (TD) and extracellular domain (ECD) are 23 and 1,008aa, respectively (21). The ICD lies in the outer plaque of the HD. The ECD extends into the lamina densa, an electron-dense zone, and loops back into the lamina lucida, an electron clear zone (22).
The ICD contains binding sites for plectin, integrin β4, and BP230 (16). The localization of collagen XVII into HDs has been shown to be dependent on its interaction with integrin β4, which has at least two binding sites in the ICD including one in the first 230aa stretch and another present in aa 231–401 [Figure 2; (23, 24)]. In the absence of integrin β4, HDs fail to form (24). Collagen XVII's ICD is also responsible for the recruitment and binding of BP230 and plectin, both of which are suggested to bind via a region of 280 aa residues (aa 180–460) (24). The recruitment and binding of BP230 and plectin mediate the linkage of HDs with the keratin cytoskeleton of epithelial cells, composed of keratin 5 and 14, and are necessary for the correct formation of HDs (24, 25).
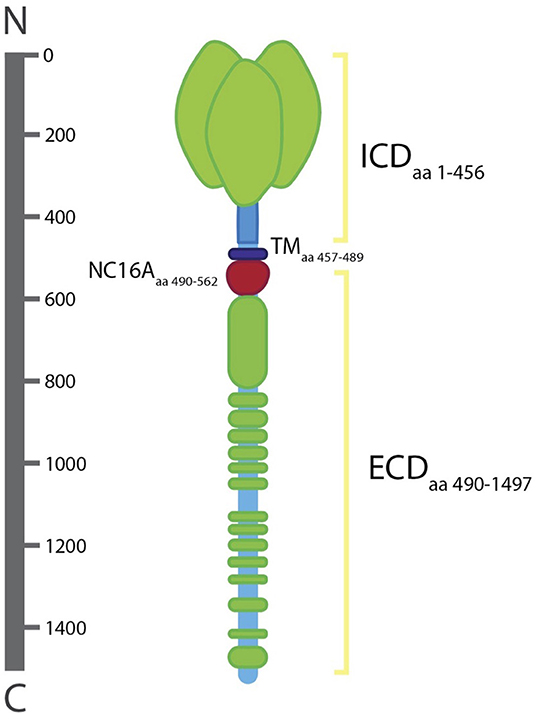
Figure 2. Structure of collagen XVII. The intracellular domain (lCD; aa 1–456), transmembrane domain (TD; aa 457–489), and extracellular domain (ECD; aa 490–1497). The ECD includ es the NC16A domain (aa 490–562). The ICD lies in the outer plaque of the HD and the ECD spans the lamina Iucida, extends to the lamina densa, and loops back into the lamina Iucida.
Collagen XVII's ECD facilitates interactions between the cell and the ECM by providing a link between the cytoplasmic structural components and the ECM (26). The ECD of 15 collagenous subdomains characterized by Gly-X-Y repeat sequences (COL1-COL15) separated by 16 non-collagenous subdomains (NC1-NC16) (10, 16). The ECD is constitutively shed from the cell surface leading to the formation of ~120-kD protein fragment, known as the ectodomain, which is integrated into the basement membrane. The ectodomain is essential for proper basement membrane formation and is suggested to play a role in cell motility, adhesion, and differentiation (27, 28).
Ectodomain Shedding
Under physiologic conditions, ectodomain shedding is catalyzed by the ADAMs (a disintegrin and metalloproteinase) sheddase family (28, 29). ADAMs are transmembrane proteins that promote ectodomain shedding of collagen XVII into the extracellular space by cleaving between the plasma membrane and the COL15 subdomain within the NC16A linker domain (aa 490–562), which contains sheddase recognition and cleavage sites (30).
The NC16A subdomain has an α-helical coiled-coil structure and has been identified as the likely site for association and initiation of triple helix formation in collagen XVII (31). Contrary to fibrillar collagen where coiled coils are most often found in the C-terminal propeptides and function to initiate trimerization and triple helix formation from the C to N terminus, collagen XVII, along with other membrane-associated collagens, contain coiled coils in the N-terminal non-collagenous domains, and thus, undergo triple helix formation from the N to C terminus (30, 32). The physiological cleavage sites are eight to eleven aa C-terminal from the coiled-coils within the region Leu524-Gly526 (30, 33).
ADAM9 and 10 have been identified as the major sheddases involved in ectodomain shedding (29). While ADAM17 was originally thought to be the major sheddase involved, it was shown that ADAM17 may play a more indirect role, as direct stimulation of ADAM17 activity did not lead to ectodomain shedding (29). The influence of sequence specificity, physical length of the substrate from proteolysis site, and substrate secondary structure on sheddase function and ectodomain shedding remains an area of controversy (34, 35). Ectodomain shedding can be increased by mutations in the coiled-coil motifs Val492-Ile505, which induces non-physiologic shedding C-terminally from the furin motif, a non-physiologic cleavage site (30). Likewise, deletion of the Ala528-Glu547, an area outside of the physiologic cleavage site, prevents ectodomain shedding (36). As such, structural motifs appear to play an important role in conformation-dependent susceptibility to sheddase related ectodomain cleavage.
The shed ectodomain interacts dynamically with the ECM including binding partners integrin α6 and laminin-332 (26). Integrin α6 and laminin-332 are both necessary for the localization of collagen XVII and stabilization of HDs (26, 37). Integrin α6 is proposed to bind to the juxtamembranous NC16 subdomain, and laminin-332 is proposed to bind near the extracellular carboxyl-terminal domain (37). The binding of autoantibodies with the ectodomain (ex. NC16A) can lead to the development of bullous pemphigoid, resulting in skin blistering and dermatitiss (20, 38, 39).
Functional Roles of Collagen XVII
Beyond its structural role in the hemidesmosome complex, collagen XVII is presumed to play a role in cell migration and differentiation in disease states (9). This postulation comes from studies that examine overexpression and knock-out models of collagen XVII in different cell lines and disease states.
The Role of Collagen XVII in Cell Differentiation
Ablation of COL17A1 has been proven to result in the loss of self-renewal and differentiation capacity of hair follicle stem cells (HFSCs) (13, 40). It has been demonstrated that COL17A1 is required for the maintenance of quiescence and immaturity, which thereby, maintains the self-renewing capabilities of HFSCs (13). In COL17A1- null mice, forced expression of COL17A1 in basal keratinocytes and HFSCs rescues melanocyte stem cells from premature differentiation while restoring TGF-β signaling (13). Additional studies have found that stem cell aging of HFSCs begins with DNA damage recruitment of neutrophil elastase and subsequent COL17A1 proteolysis, demonstrating that COL17A1 in HSFCs orchestrates stem-cell aging (40). Lastly, COL17A1 has been shown to play a major role in differentiation of hair-follicle associated pluripotent stem-cell markers (HAP)—which are nestin-expressing HFSC (41), into epidermal keratinocytes. This expression of COL17A1 is increased in HAP stem-cell markers only during periods of differentiation (42). Taken all together, these studies demonstrate the role of collagen XVII in cell differentiation in HFSC. Its role in regulating differentiation in other lines remains to be determined.
The Role of Collagen XVII in Cell Migration
Collagen XVII is also postulated to play a role in cell migration. Collagen XVII-deficient keratinocytes display increased spreading on laminin-332, increased expression of the integrin β4 subunit, and activated PI3K and Rac1, resulting in the formation of multiple unstable leading edges (lamellipodia) (17). These findings suggest collagen XVII is involved keratinocyte adhesion and directed motility coordination by interfering with integrin dependent PI3K activation (17, 43).
Furthermore, release of the ectodomain of collagen XVII from the cell surface has been associated with decreased keratinocyte motility, while addition of the ectodomain of collagen XVII inhibits motility in migrating cells (27, 44). It is postulated that an optimum adhesive strength is required for cell migration, with overly weak adhesions resulting in insufficient cell traction, while overly strong adhesions inhibit cell locomotion (44, 45). All together, these results demonstrate that collagen XVII inhibits keratinocyte migration, while collagen XVII's shed ectodomain leads to stabilization and cell immobilization (44).
This theory is proven in patients with junctional epidermolysis bullosa (JEB), a disorder characterized by mutations in the COL17A1 gene resulting in the absence of collagen XVII or expression of a structurally altered protein leading to subepidermal blistering and immature HDs (44, 46–48). The lack of a strong adhesion in these collagen XVII deficient keratinocytes is reflected by increased motility compared to controls, and is in line with the hypothesis of weakening basal keratinocytes adherence to the BM (44, 46, 47). Lastly, collagen XVII may coordinate keratinocyte migration during wound healing. Collagen XVII-deficient cells have proven to be hypermobile with faster healing of scratch wounds than controls (44). Conversely, wound closure is slower with the addition of exogenous ectodomain of collagen XVII (27).
The Role of Collagen XVII in Skin Inflammation
It is well-known that dysfunction of collagen XVII, either through genetic or autoantibody insult, leads to subepidermal blistering and skin inflammation. Nonetheless, a direct role of collagen XVII in skin inflammation is unclear. In a humanized NC16A mouse, mutation induced dysfunction of NC16A in basal keratinocytes (termed ΔNC16A), leads to development of spontaneous skin inflammatory disease (49). These mice exhibit severe pruritus, dysfunctional skin barrier, infiltrating immune cells, increased serum IgE levels, and high expression of thymic stromal lymphopoietin (TSLP) (49). Severe pruritus is not mediated by adaptive immunity or histamine, rather, it is dependent on increased expression of TSLP (49).
Involvement of Collagen XVII in Cancer Development
The malignant transformation of cancer is defined by its ability to invade normal tissues. As an essential component of the ECM, collagen XVII plays a significant role in forming a barrier against cell infiltration. Beyond its function as a cell-matrix adhesion molecule, collagen XVII's role in cancer biology has only recently been a point of investigation. The altered expression and regulation of collagen XVII, along with its effects on signaling cascades indicate its role in tumor development and invasion. Understanding this protein's role in tumor progression may be promising target in tumorigenesis.
Collagen XVII as It Pertains to Skin Cancer
Squamous Cell Carcinoma
In normal epidermis, collagen XVII expression ceases as keratinocytes migrate upwards and differentiate. SCC, however, demonstrates collagen XVII overexpression, suggesting de-differentiation (50, 51). Immunohistochemical analysis exhibited stronger immunoreaction of collagen XVII ectodomain in the primary SCC tumor and associated metastases (51). This is similarly noted in atypical keratinocytes of actinic keratoses and SCC in situ (51, 52). Viral mediated RNA interference knockdown of collagen XVII and/or β4 disrupted the migration and invasion of less aggressive SCC cells (52). Of note, no impact was observed on HSC-3, a highly invasive cell with upregulated collagen XVII (52).
Collagen XVII positivity has also been widely noted in head and neck SCC, especially within those originating from the oral cavity and larynx (53, 54). In a sample of tongue SCC samples, collagen XVII and laminin-332 were found to be co-localized at the invasive tumor fronts protruding into the surrounding tissue, and proteolytic shedding of collagen XVII enhanced its functioning as a chemotactic agent through its effects on transmigration of HSC-3 cells (50).
At the transcriptional level, overexpression of collagen XVII mRNA was found in the invasive tumors via reverse-transcriptase polymerase chain reaction (RT-PCR) at the epidermal level and within epithelial tissues. RT-PCR and northern hybridization confirmed the enhanced expression of collagen XVII in SCC (7, 55). Among oral keratinocytes stimulated with the tumor promoting phorbol ester (56), a 1.5-fold induction in collagen XVII mRNA expression was noted (53). In addition, quantitative PCR also confirmed COL17A1 expression and upregulation by transfection of miR-203a-3p inhibitor in oral SCC lines (57). The miRNA used in the analysis, miR-103a-3p, was found to bind human 3′UTRs of COL17A1 (58).
There is also a strong association with collagen XVII ectodomain shedding and tumor invasiveness in SCC (59). To investigate this role, a non-sheddable collagen XVII mutant with a deletion in the linker domain spanning over cleavage sites (60) was retrovirally introduced in SCC-25 cells, a cell used to assess aggregate size in SCC. Prevention of collagen XVII shedding resulted in smaller aggregate sizes, colony formation, and decreased Matrigel invasion, while collagen XVII re-expression restored tumorigenicity (59).
Basal Cell Carcinoma
Non-radioactive in situ hybridization revealed the presence of collagen XVII mRNA among basal keratinocytes of solid basal cell carcinoma (BCC) and the tumor islands of superficial BCC, while it was absent in the basal cells of healthy epidermis (9). Overall, immunostaining revealed decreased collagen XVII expression among the peripheral cells in solid and keratotic BCCs and the basal keratinocytes of invasive tumor fronts in superficial BCCs (9). A later study was unable to replicate this finding as collagen XVII immunostaining was mostly negative in the basal cell cancer islets, except in some dispersed spindle cells (51). Moreover, further evaluation is needed to confirm the role of collagen XVII in BCC progression.
Neural Crest Tumors
Collagen XVII expression was noted in the cells of neural crest origin and proliferating tissue melanocytes, but not in benign melanocytic tumors (55). All subtypes of melanoma were strongly positive whereas the nevus cells in nevoid melanoma were negative for significant staining (55). Furthermore, immunostaining of collagen XVII was statistically correlated with the invasive phenotype and the vertical “Breslow” thickness of melanomas (55). In vitro treatment of melanoma cells with aa 507–529 sequence specific antibody against collagen XVII endodomain promoted apoptosis, reduction of tumor cell proliferation, and cell adhesion (55). Sequencing of COL17A1 gene from melanoma cDNA detected several point mutations and in-frame deletions in the ectodomain coding region, suggesting the contribution of post-translational degradation in ectodomain deficiency (55).
In vivo models using skin specific-NC16A dysfunctional mice with B16 melanoma cells support the regulatory role of keratinocyte collagen XVII on melanoma tumorigenesis (61, 62). When compared to the controls, the experimental models showed increased levels of tumor volume and lymphatic metastasis (62). Collagen XVII dysfunction results in immediate inflammation, previously identified as a hallmark for cancer, through the influx of chemokines which further recruit myeloid derived suppressor cells (MDSCs) (61, 63). Decreasing the levels of MDSCs through treatment with specific antibodies resulted in the reduction of tumor volume, and the rate of metastatic development, demonstrating the role collagen XVII plays in curbing tumor invasion via regulation of MDSC infiltration (62). As some studies have noted an association between bullous pemphigoid and melanoma, particularly after immunotherapy, this remains a potentially relevant interaction (64, 65).
Collagen XVII as It Pertains to Other Cancers
In addition to squamous cell carcinoma and other skin cancers, dysregulation of collagen XVII appears to occur in numerous other cancers as outlined in Table 1.
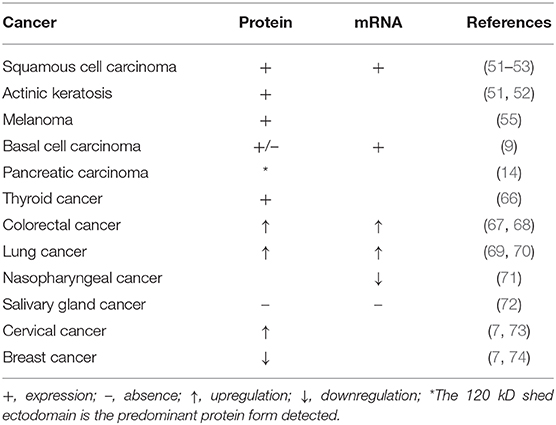
Table 1. Collagen XVII as it pertains to cancer development.
Pancreatic Cancer
Immunohistochemical visualization has demonstrated that mature type hemidesmosome 1 (collagen XVII + integrin α6β4; HD-1) can be found in the human pancreatic ductal epithelium, with subsequent disassembly during pancreatic carcinogenesis (14). The HD-1 breakdown is part of the phosphoinositide 3-kinase dependent induction of matrix-metalloprotease 9 (MMP-9), which cleaves collagen XVII and enables integrin α6β4 to promote cell migration and invasion (14, 52).
Colorectal Cancer
The role of collagen XVII as in colorectal cancer progression has been studied extensively. Overexpression of full-length collagen XVII and elevated mRNA levels have been found in samples of colorectal carcinoma (67, 68). Collagen XVII overexpression was associated with higher TNM staging and correlated with an infiltrative growth pattern, tumor budding, metastasis, and decreased survival rates (67). In vitro, the overexpression of murine collagen XVII promoted the invasiveness of colon cancer cells through the Matrigel (67). COL17A1 gene expression was upregulated via the PP2A-S727STAT3 mediated pathway (68). Inhibition of this pathway in colorectal cancer metastasis blocked suspension survival, sphere formation, tumor initiation, and metastasis, reinforcing the role of collagen XVII as an important prognostic factor in colorectal cancer patients (68).
Lung Cancer
In a sampling of non-small cell lung cancers, elevated expression of collagen XVII was seen in the stromal environment and was associated with increased metastatic potential (69). Microarray analysis revealed the upregulation of collagen XVII and elevated mRNA levels in spheroid cultures demonstrating its importance in the maintenance of the mesenchymal transition and metastatic ability in lung cancer like stem cells (70). Collagen XVII's shed ectodomain appears to stabilize laminin-332, in turn leading to activation of FAK/AKT/GSK3β. Ultimately, this leads to suppression of snail ubiquitination-degradation, propagating the epidermal-mesenchymal transition (70). Blockade of the signaling pathway also decreased the metastatic potential of lung metastases in vivo (70). Similar findings were demonstrated in A549 lung adenocarcinoma cell lines in adherent culture with induced collagen XVII overexpression (75). In these cells, the collagen XVII-laminin-332 pathway induced the FAK/AKT/GSK3β/β-catenin pathway leading to Oct4-HK2 activation and induction of lung cancer stem cells (CSCs)-like features (75). These include sphere formation, pluripotency marker expression, tumorigenic potential, metastasis, and metabolic reprogramming of CSC which is essential for CSC survival and maintenance (75). Notably, increased collagen XVII, Oct4, and HK2 are associated with poorer prognosis (75). A separate study examining lung tumorigenesis analyzed mice deficient for both Mob1A and Mob1B in bronchioalveolar epithelial cells as lung adenocarcinomas arise from bronchioalveolar stem cells (BASCs) (76). Mob1A/1B deficient adult mouse showed decreased mRNA levels of the COL17A1 gene in their lung epithelial cells resulting in subsequent suppression of tumor initiation (76).
Salivary Gland Cancer
Undifferentiated carcinoma (UDC) of the salivary gland, an uncommon disease responsible for <2% of major salivary gland tumors, carries histopathologic features similar to those noted in poorly differentiated nasopharyngeal carcinoma (NPC) (72, 77, 78). AMC-HN-9 cell lines are derived from UDC of the parotid gland and are arranged into sheets of atypical epithelial cells with an underlying fibrous stroma (72, 79). Collagen XVII is not expressed in AMC-HN-9 cells, and cytogenetics has not demonstrated gross deletions or rearrangements affecting collagen XVII gene loci (72). Previous studies have demonstrated that collagen XVII is expressed in normal salivary tissue epithelium (80). Therefore, the exact mechanism under which collagen XVII expression is lost in AMC-HN-9 cells remains uncertain (72).
Cervical Cancer
Aberrant expression of BP180 was first identified in the cervix immunohistochemically using a validated murine monoclonal antibody exhibiting specific reactivity to skin hemidesmosomes (73). A latter study confirmed collagen XVII overexpression in cervical tumors through immunohistochemistry, along with increased local dissemination and metastasis (7). Moreover, COL17A1 promoter was found to be hypomethylated in cervical carcinoma, head and neck squamous cell carcinoma, lung squamous cell carcinoma, and lung adenocarcinoma resulting in subsequent collagen XVII overexpression (7, 51, 55, 81, 82).
Breast Cancer
Immunohistochemistry on ductal breast cancers detected that collagen XVII is underexpressed (74). This underexpression is correlated with higher TNM staging, increased invasion, and postmenopausal status (7). The opposed direction of expression in breast cancer may be due to hypermethylation of the COL17A1 promoter (7). These findings suggest that the COL17A1 promoter methylation status dictates the direction of collagen XVII expression, with reduced expression leading to poorer prognosis (7).
Nasopharyngeal Cancer
Reduced mRNA expression of collagen XVII is observed in nasopharyngeal carcinoma cells (NPC) examined by RT-PCR (72). Previous studies have shown that downregulation of HD components (such as collagen XVII) can facilitate migration of carcinoma cells through detachment from the basement membrane (71, 83), suggesting that downregulation of HD protein expression may play a pivotal role in neoplastic progression to early invasion (84, 85). Downregulation of collagen XVII in NPC cell lines suggests that it is a shared feature of the malignant transformation of NPC (71).
Future Perspectives: Collagen XVII and Cancer Treatment
Collagen XVII assumes various roles in epithelial biology including but not limited to cell differentiation, cell migration, and skin inflammation. In a variety of tumors, genetic aberrations in COL17A1, full-length collagen XVII overexpression, or the accumulation of its proteolytic components have been linked to increased invasiveness of tumors (52, 62, 67, 69). For example, ectodomain shedding of COL XVII is catalyzed by ADAM9 and ADAM10 (27, 36, 86), and ectodomain release promotes tumor invasion (59). Selective blockage of collagen XVII ectodomain shedding was achieved in in vitro treatment of SCC-25 cells with the monoclonal NC16A-1 antibody raised against (aa 490–523) (87), and also demonstrated reduced tumor growth (59). Likewise, HSC-3 cell cultures treated with NC16A-1 antibody exhibited reduced invasion, confirming the role of shedding in SCC metastasis (59). In light of these findings, identifying therapeutic inhibitors of collagen XVII ectodomain shedding serves as a promising therapeutic avenue in SCC. While this in vivo and in vitro approach offers a potentially targeted means of inhibiting ectodomain shedding, several clinical studies have assessed protease inhibition, which presumably impacted ectodomain shedding.
MMP-9 is involved in the proteolytic cleavage of collagen XVII and is implicated in various forms of carcinomas including pancreatic, invasive cutaneous SCC, and mucosal SCC (14, 59, 88, 89). While MMP9 represents a tempting pharmacologic target for cancer therapy due to its role in facilitating extracellular matrix (ECM) degradation (90, 91), past RCTs from the 1990's to the early 2000's deemed MMP-9 inhibitors (MMPI) such as marimastat, unsuccessful in reducing tumor burden or mortality, and found increased rates of adverse effects (92–94). Previous generation MMPIs were flawed in their target to the MMP catalytic site, which is highly conserved throughout the MMP family (94), and resulted in drugs that blocked MMP-mediated proteolysis but could not selectively inhibit the specific pathologically overexpressed MMPs (94).
However, novel highly selective MMPIs offer the potential for more targeted therapy, with presumably fewer off target-related adverse events (95). Of these, Andecaliximab/GS-5745, an MMP-9 inhibitor prevents zymogen activation by binding MMP-9 functionally between the propeptide and catalytic domains (96), and also serves as an allosteric inhibitor (97). Andecaliximab has shown promise in phase I clinical trials of gastric adenocarcinoma (98). However, a phase II study failed to support the addition of andecaliximab to PD-1 inhibitor therapy (99), and a phase III study failed to support the addition of andecaliximab to the mFOLFOX6 regimen (100).
Given the importance of collagen XVII overexpression and ectodomain shedding in the tumorigenesis of SCC and other cancers, it serves as a promising treatment target. While more refined MMPIs may improve on previously ineffective treatments, multiple proteases may act on the same substrate, and there may additionally be compensation by untargeted metalloproteases. As such, targeted therapy directly blocking the substrate such as with NC16A-1 antibody may represent an innovative treatment. The absence of significant phenotype in mice unable to shed their collagen XVII ectodomain, suggests that inhibition of shedding may alternatively be a more tolerated therapeutic target (60).
Author Contributions
KA: conceptualization. VJ, PP, FG, AC, and KA: writing-original draft preparation. VJ and FG: figures. PP: table. VJ, PP, FG, and KA: writing-review and editing.
Funding
We acknowledge the Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago for financial support toward the open access publishing fee for this article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Tsuruta D, Kobayashi H, Imanishi H, Sugawara K, Ishii M, Jones JC. Laminin-332-integrin interaction: a target for cancer therapy? Curr Med Chem. (2008) 15:1968–75. doi: 10.2174/092986708785132834
2. PDQ® Adult Treatment Editorial Board. PDQ Skin Cancer Treatment. Bethesda, MD: National Cancer Institute (2019). Available online at: https://www.cancer.gov/types/skin/hp/skin-treatment-pdq (accessed February 11, 2020).
3. Guy GP Jr, Machlin SR, Ekwueme DU, Yabroff KR. Prevalence and costs of skin cancer treatment in the U.S., 2002-2006 and 2007-2011. Am J Prev Med. (2015) 48:183–7. doi: 10.1016/j.amepre.2014.08.036
4. Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. (2007) 8:221–33. doi: 10.1038/nrm2125
5. Garcia MG, Bayo J, Bolontrade MF, Sganga L, Malvicini M, Alaniz L, et al. Hepatocellular carcinoma cells and their fibrotic microenvironment modulate bone marrow-derived mesenchymal stromal cell migration in vitro and in vivo. Mol Pharm. (2011) 8:1538–48. doi: 10.1021/mp200137c
6. Fang M, Yuan J, Peng C, Li Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. (2014) 35:2871–82. doi: 10.1007/s13277-013-1511-7
7. Thangavelu PU, Krenacs T, Dray E, Duijf PH. In epithelial cancers, aberrant COL17A1 promoter methylation predicts its misexpression and increased invasion. Clin Epigenet. (2016) 8:120. doi: 10.1186/s13148-016-0290-6
8. Heino J. The collagen family members as cell adhesion proteins. Bioessays. (2007) 29:1001–10. doi: 10.1002/bies.20636
9. Parikka M, Kainulainen T, Tasanen K, Bruckner-Tuderman L, Salo T. Altered expression of collagen XVII in ameloblastomas and basal cell carcinomas. J Oral Pathol Med. (2001) 30:589–95. doi: 10.1034/j.1600-0714.2001.301003.x
10. Powell AM, Sakuma-Oyama Y, Oyama N, Black MM. Collagen XVII/BP180: a collagenous transmembrane protein and component of the dermoepidermal anchoring complex. Clin Exp Dermatol. (2005) 30:682–7. doi: 10.1111/j.1365-2230.2005.01937.x
11. Masunaga T, Shimizu H, Yee C, Borradori L, Lazarova Z, Nishikawa T, et al. The extracellular domain of BPAG2 localizes to anchoring filaments and its carboxyl terminus extends to the lamina densa of normal human epidermal basement membrane. J Invest Dermatol. (1997) 109:200–6. doi: 10.1111/1523-1747.ep12319337
12. Nievers MG, Schaapveld RQ, Sonnenberg A. Biology and function of hemidesmosomes. Matrix Biol. (1999) 18:5–17. doi: 10.1016/S0945-053X(98)00003-1
13. Tanimura S, Tadokoro Y, Inomata K, Binh NT, Nishie W, Yamazaki S, et al. Hair follicle stem cells provide a functional niche for melanocyte stem cells. Cell Stem Cell. (2011) 8:177–87. doi: 10.1016/j.stem.2010.11.029
14. Laval S, Laklai H, Fanjul M, Pucelle M, Laurell H, Billon-Gales A, et al. Dual roles of hemidesmosomal proteins in the pancreatic epithelium: the phosphoinositide 3-kinase decides. Oncogene. (2014) 33:1934–44. doi: 10.1038/onc.2013.146
15. Li KH, Sawamura D, Giudice GJ, Diaz LA, Mattei MG, Chu ML, et al. Genomic organization of collagenous domains and chromosomal assignment of human 180-kDa bullous pemphigoid antigen-2, a novel collagen of stratified squamous epithelium. J Biol Chem. (1991) 266:24064–9.
16. Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. (2015) 360:363–78. doi: 10.1007/s00441-014-2061-z
17. Loffek S, Hurskainen T, Jackow J, Sigloch FC, Schilling O, Tasanen K, et al. Transmembrane collagen XVII modulates integrin dependent keratinocyte migration via PI3K/Rac1 signaling. PLoS ONE. (2014) 9:e87263. doi: 10.1371/journal.pone.0087263
18. Liu Y, Li L, Xia Y. BP180 is critical in the autoimmunity of bullous pemphigoid. Front Immunol. (2017) 8:1752. doi: 10.3389/fimmu.2017.01752
19. Schmidt E, Wehr B, Wolf K, Sitaru C, Brocker EB, Zillikens D. Localisation of bullous pemphigoid antigen 180 (BP180) in cultured human keratinocytes: functionally relevant modification by calcium. Arch Dermatol Res. (2006) 298:283–90. doi: 10.1007/s00403-006-0690-0
20. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. (2018) 54:26–51. doi: 10.1007/s12016-017-8633-4
21. Franzke CW, Tasanen K, Schumann H, Bruckner-Tuderman L. Collagenous transmembrane proteins: collagen XVII as a prototype. Matrix Biol. (2003) 22:299–309. doi: 10.1016/S0945-053X(03)00051-9
22. Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. (2015) 360:529–44. doi: 10.1007/s00441-015-2216-6
23. Borradori L, Koch PJ, Niessen CM, Erkeland S, van Leusden MR, Sonnenberg A. The localization of bullous pemphigoid antigen 180 (BP180) in hemidesmosomes is mediated by its cytoplasmic domain and seems to be regulated by the beta4 integrin subunit. J Cell Biol. (1997) 136:1333–47. doi: 10.1083/jcb.136.6.1333
24. Koster J, Geerts D, Favre B, Borradori L, Sonnenberg A. Analysis of the interactions between BP180, BP230, plectin and the integrin alpha6beta4 important for hemidesmosome assembly. J Cell Sci. (2003) 116:387–99. doi: 10.1242/jcs.00241
25. Hopkinson SB, Jones JC. The N terminus of the transmembrane protein BP180 interacts with the N-terminal domain of BP230, thereby mediating keratin cytoskeleton anchorage to the cell surface at the site of the hemidesmosome. Mol Biol Cell. (2000) 11:277–86. doi: 10.1091/mbc.11.1.277
26. Nishie W, Kiritsi D, Nystrom A, Hofmann SC, Bruckner-Tuderman L. Dynamic interactions of epidermal collagen XVII with the extracellular matrix: laminin 332 as a major binding partner. Am J Pathol. (2011) 179:829–37. doi: 10.1016/j.ajpath.2011.04.019
27. Franzke CW, Tasanen K, Schacke H, Zhou Z, Tryggvason K, Mauch C, et al. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. (2002) 21:5026–35. doi: 10.1093/emboj/cdf532
28. Nishimura M, Nishie W, Shirafuji Y, Shinkuma S, Natsuga K, Nakamura H, et al. Extracellular cleavage of collagen XVII is essential for correct cutaneous basement membrane formation. Hum Mol Genet. (2016) 25:328–39. doi: 10.1093/hmg/ddv478
29. Franzke CW, Bruckner-Tuderman L, Blobel CP. Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J Biol Chem. (2009) 284:23386–96. doi: 10.1074/jbc.M109.034090
30. Nishie W, Jackow J, Hofmann SC, Franzke CW, Bruckner-Tuderman L. Coiled coils ensure the physiological ectodomain shedding of collagen XVII. J Biol Chem. (2012) 287:29940–8. doi: 10.1074/jbc.M112.345454
31. Van den Bergh F, Fu CL, Olague-Marchan M, Giudice GJ. The NC16A domain of collagen XVII plays a role in triple helix assembly and stability. Biochem Biophys Res Commun. (2006) 350:1032–7. doi: 10.1016/j.bbrc.2006.09.147
32. McAlinden A, Smith TA, Sandell LJ, Ficheux D, Parry DA, Hulmes DJ. Alpha-helical coiled-coil oligomerization domains are almost ubiquitous in the collagen superfamily. J Biol Chem. (2003) 278:42200–7. doi: 10.1074/jbc.M302429200
33. Nishie W, Lamer S, Schlosser A, Licarete E, Franzke CW, Hofmann SC, et al. Ectodomain shedding generates neoepitopes on collagen XVII, the major autoantigen for bullous pemphigoid. J Immunol. (2010) 185:4938–47. doi: 10.4049/jimmunol.1001524
34. Migaki GI, Kahn J, Kishimoto TK. Mutational analysis of the membrane-proximal cleavage site of L-selectin: relaxed sequence specificity surrounding the cleavage site. J Exp Med. (1995) 182:549–57. doi: 10.1084/jem.182.2.549
35. Black RA. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. (2002) 34:1–5. doi: 10.1016/S1357-2725(01)00097-8
36. Franzke CW, Tasanen K, Borradori L, Huotari V, Bruckner-Tuderman L. Shedding of collagen XVII/BP180: structural motifs influence cleavage from cell surface. J Biol Chem. (2004) 279:24521–9. doi: 10.1074/jbc.M308835200
37. Hopkinson SB, Findlay K, deHart GW, Jones JC. Interaction of BP180. (type XVII collagen) and alpha6 integrin is necessary for stabilization of hemidesmosome structure. J Invest Dermatol. (1998) 111:1015–22. doi: 10.1046/j.1523-1747.1998.00452.x
38. Wada M, Nishie W, Ujiie H, Izumi K, Iwata H, Natsuga K, et al. Epitope-dependent pathogenicity of antibodies targeting a major bullous pemphigoid autoantigen collagen XVII/BP180. J Invest Dermatol. (2016) 136:938–46. doi: 10.1016/j.jid.2015.11.030
39. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med. (2018) 5:201. doi: 10.3389/fmed.2018.00201
40. Matsumura H, Mohri Y, Binh NT, Morinaga H, Fukuda M, Ito M, et al. Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science. (2016) 351:aad4395. doi: 10.1126/science.aad4395
41. Hoffman RM. The pluripotency of hair follicle stem cells. Cell Cycle. (2006) 5:232–3. doi: 10.4161/cc.5.3.2397
42. Shirai K, Obara K, Tohgi N, Yamazaki A, Aki R, Hamada Y, et al. Expression of anti-aging type-XVII collagen (COL17A1/BP180) in hair follicle-associated pluripotent (HAP) stem cells during differentiation. Tissue Cell. (2019) 59:33–8. doi: 10.1016/j.tice.2019.06.001
43. de Pereda JM, Lillo MP, Sonnenberg A. Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J. (2009) 28:1180–90. doi: 10.1038/emboj.2009.48
44. Tasanen K, Tunggal L, Chometon G, Bruckner-Tuderman L, Aumailley M. Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am J Pathol. (2004) 164:2027–38. doi: 10.1016/S0002-9440(10)63762-5
45. Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. (1997) 385:537–40. doi: 10.1038/385537a0
46. Jonkman MF, de Jong MC, Heeres K, Pas HH, van der Meer JB, Owaribe K, et al. 180-kD bullous pemphigoid antigen (BP180) is deficient in generalized atrophic benign epidermolysis bullosa. J Clin Invest. (1995) 95:1345–52. doi: 10.1172/JCI117785
47. McGrath JA, Gatalica B, Christiano AM, Li K, Owaribe K, McMillan JR, et al. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet. (1995) 11:83–6. doi: 10.1038/ng0995-83
48. van Leusden MR, Pas HH, Gedde-Dahl TJr, Sonnenberg A, Jonkman MF. Truncated typeXVII collagen expression in a patient with non-herlitz junctional epidermolysis bullosa caused by a homozygous splice-site mutation. Lab Invest. (2001) 81:887–94. doi: 10.1038/labinvest.3780297
49. Zhang Y, Hwang BJ, Liu Z, Li N, Lough K, Williams SE, et al. BP180 dysfunction triggers spontaneous skin inflammation in mice. Proc Natl Acad Sci USA. (2018) 115:6434–9. doi: 10.1073/pnas.1721805115
50. Parikka M, Nissinen L, Kainulainen T, Bruckner-Tuderman L, Salo T, Heino J, et al. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration—a novel role for alphaIIb integrin and tirofiban. Exp Cell Res. (2006) 312:1431–8. doi: 10.1016/j.yexcr.2006.01.015
51. Stelkovics E, Korom I, Marczinovits I, Molnar J, Rasky K, Raso E, et al. Collagen XVII/BP180 protein expression in squamous cell carcinoma of the skin detected with novel monoclonal antibodies in archived tissues using tissue microarrays and digital microscopy. Appl Immunohistochem Mol Morphol. (2008) 16:433–41. doi: 10.1097/PAI.0b013e318162f8aa
52. Moilanen JM, Loffek S, Kokkonen N, Salo S, Vayrynen JP, Hurskainen T, et al. Significant role of collagen XVII and integrin beta4 in migration and invasion of the less aggressive squamous cell carcinoma cells. Sci Rep. (2017) 7:45057. doi: 10.1038/srep45057
53. Parikka M, Kainulainen T, Tasanen K, Vaananen A, Bruckner-Tuderman L, Salo T. Alterations of collagen XVII expression during transformation of oral epithelium to dysplasia and carcinoma. J Histochem Cytochem. (2003) 51:921–9. doi: 10.1177/002215540305100707
54. Tamas L, Szentkuti G, Eros M, Danos K, Brauswetter D, Szende B, et al. Differential biomarker expression in head and neck cancer correlates with anatomical localization. Pathol Oncol Res. (2011) 17:721–7. doi: 10.1007/s12253-011-9376-9
55. Krenacs T, Kiszner G, Stelkovics E, Balla P, Teleki I, Nemeth I, et al. Collagen XVII is expressed in malignant but not in benign melanocytic tumors and it can mediate antibody induced melanoma apoptosis. Histochem Cell Biol. (2012) 138:653–67. doi: 10.1007/s00418-012-0981-9
56. De Tata V, Ptasznik A, Cristofalo VJ. Effect of the tumor promoter phorbol 12-myristate 13-acetate. (PMA) on proliferation of young and senescent WI-38 human diploid fibroblasts. Exp Cell Res. (1993) 205:261–9. doi: 10.1006/excr.1993.1085
57. Yasukochi A, Kawakubo-Yasukochi T, Morioka M, Hazekawa M, Nishinakagawa T, Ono K, et al. Regulation of collagen type XVII expression by miR203a-3p in oral squamous cell carcinoma cells. J Biochem. (2019) 166:163–73. doi: 10.1093/jb/mvz024
58. Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. (2005) 120:21–4. doi: 10.1016/j.cell.2004.12.031
59. Galiger C, Loffek S, Stemmler MP, Kroeger JK, Mittapalli VR, Fauth L, et al. Targeting of cell surface proteolysis of collagen XVII impedes squamous cell carcinoma progression. Mol Ther. (2018) 26:17–30. doi: 10.1016/j.ymthe.2017.09.022
60. Jackow J, Schlosser A, Sormunen R, Nystrom A, Sitaru C, Tasanen K, et al. Generation of a functional non-shedding collagen XVII mouse model: relevance of collagen XVII shedding in wound healing. J Invest Dermatol. (2016) 136:516–25. doi: 10.1016/j.jid.2015.10.060
61. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
62. Hwang BJ, Zhang Y, Brozowski JM, Liu Z, Burette S, Lough K, et al. The dysfunction of BP180/collagen XVII in keratinocytes promotes melanoma progression. Oncogene. (2019) 38:7491–503. doi: 10.1038/s41388-019-0961-9
63. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. (2009) 182:4499–506. doi: 10.4049/jimmunol.0802740
64. Amber KT, Panganiban CM, Korta D, Feraudy S, Kelly KM, Grando SA. A case report of bullous pemphigoid associated with a melanoma and review of the literature. Melanoma Res. (2017) 27:65–7. doi: 10.1097/CMR.0000000000000307
65. Lopez AT, Khanna T, Antonov N, Audrey-Bayan C, Geskin L. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. (2018) 57:664–9. doi: 10.1111/ijd.13984
66. Nagle RB, Hao J, Knox JD, Dalkin BL, Clark V, Cress AE. Expression of hemidesmosomal and extracellular matrix proteins by normal and malignant human prostate tissue. Am J Pathol. (1995) 146:1498–507.
67. Moilanen JM, Kokkonen N, Loffek S, Vayrynen JP, Syvaniemi E, Hurskainen T, et al. Collagen XVII expression correlates with the invasion and metastasis of colorectal cancer. Hum Pathol. (2015) 46:434–42. doi: 10.1016/j.humpath.2014.11.020
68. Liu CC, Lin SP, Hsu HS, Yang SH, Lin CH, Yang MH, et al. Suspension survival mediated by PP2A-STAT3-Col XVII determines tumour initiation and metastasis in cancer stem cells. Nat Commun. (2016) 7:11798. doi: 10.1038/ncomms14027
69. Papay J, Krenacs T, Moldvay J, Stelkovics E, Furak J, Molnar B, et al. Immunophenotypic profiling of nonsmall cell lung cancer progression using the tissue microarray approach. Appl Immunohistochem Mol Morphol. (2007) 15:19–30. doi: 10.1097/01.pai.0000213143.32030.f5
70. Liu CC, Lin JH, Hsu TW, Hsu JW, Chang JW, Su K, et al. Collagen XVII/laminin-5 activates epithelial-to-mesenchymal transition and is associated with poor prognosis in lung cancer. Oncotarget. (2018) 9:1656–72. doi: 10.18632/oncotarget.11208
71. Lo AK, Yuen PW, Liu Y, Wang XH, Cheung AL, Wong YC, et al. Downregulation of hemidesmosomal proteins in nasopharyngeal carcinoma cells. Cancer Lett. (2001) 163:117–23. doi: 10.1016/S0304-3835(00)00683-2
72. Kim SH, Carey TE, Liebert M, Yoo SJ, Kwon HJ, Kim SY. Characterization of AMC-HN-9, a cell line established from an undifferentiated carcinoma of the parotid gland: expression of alpha6beta4 with the absence of BP180 and 230. Acta Otolaryngol. (2000) 120:660–6. doi: 10.1080/000164800750000513
73. Yamada T, Endo R, Tsukagoshi K, Fujita S, Honda K, Kinoshita M, et al. Aberrant expression of a hemidesmosomal protein, bullous pemphigoid antigen 2, in human squamous cell carcinoma. Lab Invest. (1996) 75:589–600.
74. Bergstraesser LM, Srinivasan G, Jones JC, Stahl S, Weitzman SA. Expression of hemidesmosomes and component proteins is lost by invasive breast cancer cells. Am J Pathol. (1995) 147:1823–39.
75. Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Li AF, et al. Involvement of collagen XVII in pluripotency gene expression and metabolic reprogramming of lung cancer stem cells. J Biomed Sci. (2020) 27:5. doi: 10.1186/s12929-019-0593-y
76. Otsubo K, Goto H, Nishio M, Kawamura K, Yanagi S, Nishie W, et al. MOB1-YAP1/TAZ-NKX2.1 axis controls bronchioalveolar cell differentiation, adhesion and tumour formation. Oncogene. (2017) 36:4201–11. doi: 10.1038/onc.2017.58
77. Batsakis JG, Luna MA. Undifferentiated carcinomas of salivary glands. Ann Otol Rhinol Laryngol. (1991) 100:82–4. doi: 10.1177/000348949110000115
78. Sheen TS, Tsai CC, Ko JY, Chang YL, Hsu MM. Undifferentiated carcinoma of the major salivary glands. Cancer. (1997) 80:357–63.
79. Kim SY, Chu KC, Lee HR, Lee KS, Carey TE. Establishment and characterization of nine new head and neck cancer cell lines. Acta Otolaryngol. (1997) 117:775–84. doi: 10.3109/00016489709113477
80. Aho S, Uitto J. 180-kD bullous pemphigoid antigen/type XVII collagen: tissue-specific expression and molecular interactions with keratin 18. J Cell Biochem. (1999) 72:356–67.
81. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. (2002) 21:5400–13. doi: 10.1038/sj.onc.1205651
82. Amenta PS, Hadad S, Lee MT, Barnard N, Li D, Myers JC. Loss of types XV and XIX collagen precedes basement membrane invasion in ductal carcinoma of the female breast. J Pathol. (2003) 199:298–308. doi: 10.1002/path.1303
83. Rabinovitz I, Mercurio AM. The integrin alpha 6 beta 4 and the biology of carcinoma. Biochem Cell Biol. (1996) 74:811–21.
84. Pauli BU, Schwartz DE, Thonar EJ, Kuettner KE. Tumor invasion and host extracellular matrix. Cancer Metastasis Rev. (1983) 2:129–52. doi: 10.1007/BF00048966
85. Bonkhoff H. Analytical molecular pathology of epithelial-stromal interactions in the normal and neoplastic prostate. Anal Quant Cytol Histol. (1998) 20:437–42.
86. Mauch C, Zamek J, Abety AN, Grimberg G, Fox JW, Zigrino P. Accelerated wound repair in ADAM-9 knockout animals. J Invest Dermatol. (2010) 130:2120–30. doi: 10.1038/jid.2010.60
87. Hofmann SC, Voith U, Schonau V, Sorokin L, Bruckner-Tuderman L, Franzke CW. Plasmin plays a role in the in vitro generation of the linear IgA dermatosis antigen LADB97. J Invest Dermatol. (2009) 129:1730–9. doi: 10.1038/jid.2008.424
88. Liu R, Gu J, Jiang P, Zheng Y, Liu X, Jiang X, et al. DNMT1-microRNA126 epigenetic circuit contributes to esophageal squamous cell carcinoma growth via ADAM9-EGFR-AKT signaling. Clin Cancer Res. (2015) 21:854–63. doi: 10.1158/1078-0432.CCR-14-1740
89. Oh ST, Stark A, Reichrath J. The disintegrin-metalloproteinases ADAM10 and ADAM17 are upregulated in cutaneous squamous cell carcinomas. Dermatoendocrinol. (2016) 8:e1228499. doi: 10.1080/19381980.2016.1228499
90. Jablonska-Trypuc A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzyme Inhib Med Chem. (2016) 31:177–83. doi: 10.3109/14756366.2016.1161620
91. Kapoor C, Vaidya S, Wadhwan V, Hitesh, Kaur G, Pathak A. Seesaw of matrix metalloproteinases (MMPs). J Cancer Res Ther. (2016) 12:28–35. doi: 10.4103/0973-1482.157337
92. Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JA. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer. (2002) 87:161–7. doi: 10.1038/sj.bjc.6600446
93. Sparano JA, Bernardo P, Stephenson P, Gradishar WJ, Ingle JN, Zucker S, et al. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: eastern cooperative oncology group trial E2196. J Clin Oncol. (2004) 22:4683–90. doi: 10.1200/JCO.2004.08.054
94. Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. (2018) 17:1147–55. doi: 10.1158/1535-7163.MCT-17-0646
95. Levin M, Udi Y, Solomonov I, Sagi I. Next generation matrix metalloproteinase inhibitors—novel strategies bring new prospects. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1927–39. doi: 10.1016/j.bbamcr.2017.06.009
96. Kumar V, Soni P, Garg M, Kamholz S, Chandra AB. Emerging therapies in the management of advanced-stage gastric cancer. Front Pharmacol. (2018) 9:404. doi: 10.3389/fphar.2018.00404
97. Marshall DC, Lyman SK, McCauley S, Kovalenko M, Spangler R, Liu C, et al. Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS ONE. (2015) 10:e0127063. doi: 10.1371/journal.pone.0127063
98. Shah MA, Starodub A, Sharma S, Berlin J, Patel M, Wainberg ZA, et al. Andecaliximab/GS-5745 alone and combined with mFOLFOX6 in advanced gastric and gastroesophageal junction adenocarcinoma: results from a phase I study. Clin Cancer Res. (2018) 24:3829–37. doi: 10.1158/1078-0432.CCR-17-2469
99. Shah MA, Metges J-P, Cunningham D, Shiu K-K, Wyrwicz L, Thai D, et al. A phase II, open-label, randomized study to evaluate the efficacy and safety of andecaliximab combined with nivolumab versus nivolumab alone in subjects with unresectable or recurrent gastric or gastroesophageal junction adenocarcinoma. J Clin Oncol. (2019) 37:75. doi: 10.1200/JCO.2019.37.4_suppl.75
100. Shah MA, Yanez Ruiz EP, Bodoky G, Starodub A, Cunningham D, Yip D, et al. A phase III, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of andecaliximab combined with mFOLFOX6 as first-line treatment in patients with advanced gastric or gastroesophageal junction adenocarcinoma (GAMMA-1). J Clin Oncol. (2019) 37:4. doi: 10.1200/JCO.2019.37.4_suppl.4
Keywords: collagen XVII, literature review, skin cancer, cancer, ectodomain shedding
Citation: Jones VA, Patel PM, Gibson FT, Cordova A and Amber KT (2020) The Role of Collagen XVII in Cancer: Squamous Cell Carcinoma and Beyond. Front. Oncol. 10:352. doi: 10.3389/fonc.2020.00352
Received: 16 December 2019; Accepted: 28 February 2020;
Published: 19 March 2020.
Edited by:
Takashi Hashimoto, Osaka City University, JapanReviewed by:
Wataru Nishie, Hokkaido University, JapanXinan Sheng, Peking University Cancer Hospital, China
Copyright © 2020 Jones, Patel, Gibson, Cordova and Amber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyle T. Amber, a3RhbWJlckB1aWMuZWR1