 Valeria Ferla
Valeria Ferla Francesca Gaia Rossi1
Francesca Gaia Rossi1- 1Hematology Division, IRCCS Ca' Granda-Maggiore Policlinico Hospital Foundation, Milan, Italy
- 2University of Milan, Milan, Italy
Epstein–Barr virus (EBV) infection is correlated with several lymphoproliferative disorders, including Hodgkin disease, Burkitt lymphoma, diffuse large B-cell lymphoma (DLBCL), and post-transplant lymphoproliferative disorder (PTLD). The oncogenic EBV is present in 80% of PTLD. EBV infection influences immune response and has a causative role in the oncogenic transformation of lymphocytes. The development of PTLD is the consequence of an imbalance between immunosurveillance and immunosuppression. Different approaches have been proposed to treat this disorder, including suppression of the EBV viral load, reduction of immune suppression, and malignant clone destruction. In some cases, upfront chemotherapy offers better and durable clinical responses. In this work, we elucidate the clinicopathological and molecular-genetic characteristics of PTLD to clarify the biological differences of EBV(+) and EBV(–) PTLD. Gene expression profiling, next-generation sequencing, and microRNA profiles have recently provided many data that explore PTLD pathogenic mechanisms and identify potential therapeutic targets. This article aims to explore new insights into clinical behavior and pathogenesis of EBV(–)/(+) PTLD with the hope to support future therapeutic studies.
Introduction
The World Health Organization (WHO) classification of lymphoid malignancies considers four major diagnostic post-transplant lymphoproliferative disorder (PTLD) categories: early lesions, polymorphic PTLD that could be either polyclonal or monoclonal, Hodgkin lymphoma (HL), and monomorphic PTLD of which diffuse large B-cell lymphoma is most common (1).
PTLD can occur in 20% of hematopoietic stem cell (HSC) and solid organ transplant (SOT) recipients.
PTLD is associated with Epstein–Barr virus (EBV) infection in 60–80% of cases. In EBV infection in immunocompetent (IC) hosts, the virus forms an episome in latently infected B cells (2, 3). In post-transplant patients, immunosuppression causes T-cell inhibition with a consequent lack of T-cell modulation on B-cell proliferation. In particular, when an EBV(–) patient receives an EBV(+) transplant graft, immunosuppression causes uncontrolled proliferation of EBV-transformed B cells, which contributes to the development of PTLD (2, 4, 5).
The pathogenesis of EBV-PTLD is currently unclear; different hypotheses have been suggested as possible pathogenic mechanisms of these EBV-PTLD, such as chronic immune triggering by the graft, hit-and-run EBV infection (EBV induces chromosomal aberrations in cell genome and might be lost during malignant cell division), and other infectious agents [e.g., human herpesvirus 5, 6, or 8; (6–11)]. However, there is limited evidence supporting these hypotheses (Table 1).

Table 1. Major risk factors in the development of PTLD.
Clinically, there are differences between EBV(–) and EBV(+) PTLD. In particular, it has been described that EBV(–) PTLD arises later, after years of transplantation, whereas EBV(+) cases arise earlier, generally after months. Furthermore, EBV RNA is detected in early and polymorphic lesions, typical lesions early after transplantation.
In the literature, contradictory data are described regarding the diversity of prognosis between the EBV(+) and EBV(–) cases; in particular, the international multicenter prospective phase 2 PTLD-1 trial found no association with overall survival and EBV status [(22, 23); Table 2].

Table 2. Clinical aspects of EBV(+)/(–) PTLD.
From a therapeutic point of view, EBV(+) and EBV(–) PTLD have the same therapy; the only difference is regarding the EBV-specific adoptive immunotherapy.
Many studies have tried to investigate the genomic differences between the IC-DLBCL, EBV(+), and EBV(–) PTLD. What emerged was that EBV(–) PTLD has a genomic profile very similar to that of IC-DLBCL and a much greater biological complexity than EBV(+) PTLD (26–29).
Furthermore, it has been shown that EBV(+) PTLD, in addition to having a different genomic profile, has different genetic and tumor microenvironment alterations compared with those of EBV(–) PTLD (30–32).
Furthermore, EBV infection may alter the microRNA expression in B lymphocytes. MicroRNA is an important transcriptional and post-transcriptional regulator of gene expression.
In PTLD, EBV(+), B-cell lymphoma revealed different microRNA profiles, compared with normal B cells or EBV lymphoblastoid cell lines generated in vitro (33, 34).
These considerations seem to suggest that the pathogenesis of EBV(–) PTLD is to be considered much more similar to that of IC-DLBCL and that it is less influenced by post-transplantation factors. However, despite these differences, the fact that some EBV(–) PTLD respond well to reduction of immunosuppression similarly to EBV(+) PTLD remains to be clarified (35). Certainly, these studies seem to offer theoretical support for future therapeutic studies in EBV(+) and EBV(–) PTLD that appear to have a different pathogenesis.
The Genomic Landscape of Epstein–Barr Virus Positive and Negative Post-Transplant Lymphoproliferative Disorders
In this work, we want to illustrate the genomic complexity of EBV(+) and EBV(–) PTLD through the integration of different genomic approaches that have significantly improved our understanding of the genetic landscape of these disorders (Table 3).

Table 3. Genomic characterization of EBV(+) and EBV(–) PTLDs through different technologies approaches.
Molecular Characterization Through a Genomic Approach
Poirel et al. (36) studied PTLD cases with comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH). The overall incidence of chromosomal imbalances was described in half of PTLD cases, even in the polymorphic category. Latent EBV infection was found in the lesions of three quarters of cases. Non-random losses were 17p13; 1p36, 4q; and 17q23q25, Xp. The gains of 8q24, 3q27, 2p24p25, 5p, 9q22q34, 11, 12q22q24, 14q32, 17q, and 18q21 were the most frequent. Three amplifications −4p16, 9p22p24, and 18q21q23–were detected. FISH has confirmed the involvement of Bcl2 in this latter imbalance. Chromosomal imbalances tended to be more complex in EBV(–) cases than in EBV(+) cases. The identification of chromosomal regions non-randomly involved in lymphomagenesis supports the role of candidate genes to be identified by a combined approach using gene expression profiling (GEP) and CGH array.
In order to improve PTLD pathogenesis understanding, Rinaldi et al. studied recurrent lesions revealed by whole-genome profiling analysis (26). The most common gains in IC-DLBCL were chromosome 3q, 7q, 12, and 18q and in PTLD were chromosomes 5p and 11p. The most common losses in IC-DLBCL were chromosome 12p and in PTLD were 6q, 17p, 1p, and 9p. DNA loss did not always match with loss of heterozygosity (LOH), and uniparental disomy seems to target chromosome 10 in PTLD. They found small deletions and gains involving BCL2 and PAX5 and ZDHHC14 (known gene). These data show that PTLD, at a lower frequency, shares common genetic aspects with IC-DLBCL. 9p13 amplification supports the importance of PAX5 in PTLD pathogenesis. Different DNA copy number and LOH patterns support the hypothesis that uniparental disomy can have a role in lymphomagenesis.
High-density genome-wide single-nucleotide polymorphism (SNP)-based arrays were used by Rinaldi et al. (27) to compare PTLD with IC-DLBCL and to compare EBV(+) with EBV(–) PTLD. In PTLD, the more frequently deleted loci were small interstitial deletions targeting FRA1B, FRA2E, and FRA3B fragile sites. PTLD presents typical and different aberrations than does IC-DLBCL: the deletions at 2p16.1 (FRA2E), lack of del(13q14.3) (MIR15/MIR16), and copy neutral LOH affecting 6p MHC. EBV(+) PTLD presented less recurrent lesions than did EBV(–) PTLD, including a gain of 7p, del(4q25–q35), and gains of 7q, 11q24–q25.
Menter et al. (29) investigated PTLD through next-generation sequencing (NGS) using the Ion Torrent platform. Nuclear factor-κB pathway-related genes had fewer mutations in EBV(+) PTLD compared with IC-DLBCL. Moreover, in PTLD, compared with IC-DLBCL, TP53 was more frequently mutated, whereas ATM and B2M mutations were absent. TP53 mutations were more frequent in EBV(–) PTLD. Mutations in DNA damage control and immune-surveillance genes are different in PTLD with respect to IC-DLBCL. EBV seems to have a role in the different mutational pattern.
Molecular Characterization Through a Transcriptional Approach
Through gene expression analysis, Morscio et al. (38) and Craig et al. (30) showed that EBV(+) and EBV(–) PTLD have different microenvironment and gene expression profiles. They also demonstrated that EBV(–) PTLD and IC-DLBCL are biologically similar.
Through array comparative genome hybridization (aCGH) analysis, Ferreiro et al. (31) studied at genomic and transcriptomic levels EBV(+) PTLD, EBV(–) PTLD, and IC-DLBCL.
EBV(+) PTLD had a different CNA pattern as compared with EBV(–) PTLD and a lower genomic imbalance.
Moreover, EBV(+) PTLD showed distinct aCGH profiles with only one recurrent imbalance with EBV(–) PTLD. On the other hand, EBV(–) PTLD displayed similar recurrent aberrations (gain of 3/3q and 18q and loss of 6q23/TNFAIP3 and 9p21/CDKN2A) as compared with IC-DLBCL. These findings support the concept of a biological relationship between both conditions.
9p24.1 gain/amplification was the most frequent aberration in EBV(+) PTLD targeting PDCD1LG2/PDL2. These genes encode immunomodulatory programmed cell death ligands (39).
In lymphoproliferative disorder, particularly in primary mediastinal B-cell lymphoma, classical HL, and primary central nervous system lymphoma, 9p24.1 is a common copy number gain. The consequence of this alteration is an increase of PDL1 and PDL2 and their induction by JAK2 (40–43).
An upregulation of PDL1 was described in the majority of EBV(+) lymphomas, including PTLD (44–46). PDL1/2 signal regulates immune defenses against pathogens and T-cell tolerance/T-cell activation through the PD-1 receptor (47).
Green et al. (44) demonstrated an alternative activation mechanism of PDL1 in classical HL and EBV(+) lymphoma, in which EBV latent membrane protein 1 (LMP1) is involved in PDL1 upregulation. These results were also supported by Chen et al. (45), who demonstrated how EBV(+) lymphomas, including PTLD, express detectable PDL1. In lymphomas, genomic amplification or EBV infection causes the PD-1/PDL signaling pathway activation with the immune surveillance escape (Figure 1).
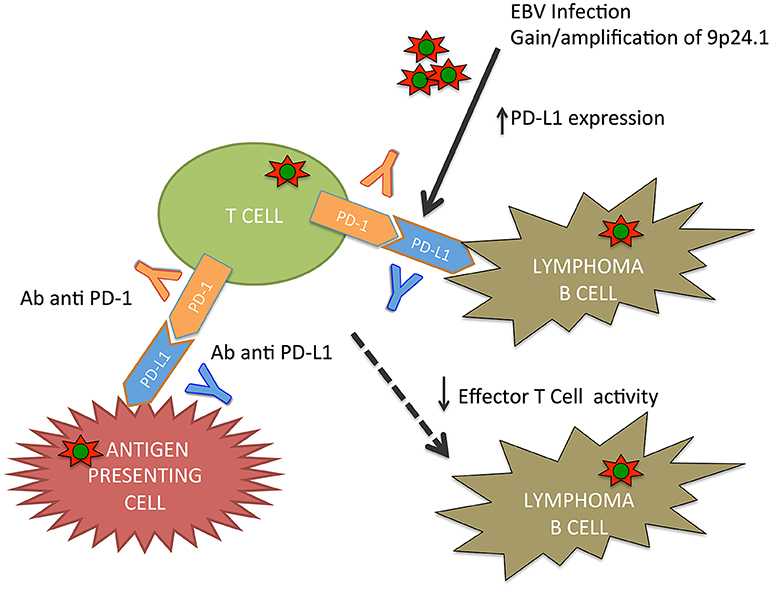
Figure 1. PD-1/PD-L1 pathways in EBV(+) PTDL.
The distinctive copy number alteration in EBV(+) PTLD was identified as a gain of 9p21 with respect to EBV(–) PTLD. Gain of 9p21 caused different CDKN2A expression. CDKN2A codes for cyclin-dependent kinase inhibitor 2A (p16INKA), an important regulator of the cell cycle; in particular, it decelerated cell progression through the G1 phase (48). In EBV(+) PTLD, immunohistochemistry (IHC) demonstrated that a gain of 9p21 was associated with exclusively cytoplasmic expression of the p16INKA protein. The p16INKA seems to be implicated in alternative oncogenic pathways and not as a tumor suppressor in EBV(+) PTLD (48, 49).
A gain of chromosome 3/3q was found in EBV(–) PTLD, and it was absent in EBV(+) PTLD. This alteration caused increased expression of FOXP1 in EBV(–) PTLD; these data were confirmed by QRT-PCR and IHC. FOXP1 encodes a transcriptional regulator implicated in different biological processes and in B-cell lymphomas pathogenesis; and it seems to play a critical role in the pathogenesis of EBV(–) PTLD. However, the connection between EBV infection and FOXP1 is uncertain because EBV downregulates FOXP1 in normal B cells (50–54).
IC-DLBCL has many points in common with EBV(–) PTLD. EBV(–) and EBV(+) PTLD demonstrated different genomic and gene expression profiles. In particular, GEP differences in EBV(+) and EBV(–) PTLD involve inflammation and immune response pathways (31), supporting the hypothesis that the EBV infection has a major impact on the gene expression and alterations in EBV(+) PTLD. On the other hand, the EBV(–) PTLD appears to be more similar to de novo lymphomas arising in transplanted patients.
Many studies support the role of cytokines in the pathogenesis of EBV(+) PTLD (55). This hypothesis is supported by the detection of IL-10 transcripts in PTLD biopsies. B-cell lymphomas isolated from EBV(+) PTLD produce IL-10 in a constitutive way and use it as an autocrine growth factor (56). For this reason, serum IL-10 has been proposed as an early marker of PTLD (57–60). It is unclear why IL-10 is altered in EBV(+) PTLD.
EBV infection modifies microRNA expression. Gene arrays demonstrate different microRNA profiles in EBV(+) B-cell lymphoma lines from patients with PTLD, as compared with in vitro generated EBV(+) lymphoblastoid cell lines or normal B cells. In particular, microRNA-194 (33) was found to be suppressed in EBV(+) PTLD. MicroRNA-194 overexpression increases apoptosis of EBV(+) B-cell lymphoma lines and attenuates IL-10 production. EBV seems to suppress microRNA-194 in order to increase IL-10 expression. Therefore, microRNA-194 may constitute a new approach to inhibiting proliferation of EBV(+) B-cell lymphomas in PTLD.
Conclusions and Prospective
In recent years, increasing understanding of the biologic and molecular PTLD pathogenesis has resulted in new therapeutic approaches and improved outcomes for these patients. Although the prognosis of EBV(+) in comparison with EBV(–) PTLD is not clear, frontline therapy in EBV(+) and EBV(–) PTLD is currently the same.
In this work, we reported much evidence that EBV(+) and EBV(–) PTLD have distinct genomic and transcriptomic landscape, although at the moment, clinical data do not completely support this hypothesis. EBV(–) PTLD and IC-DLBCL seem to be similar biological entities; for this reason, EBV(–) PTLD might be considered as a type of lymphoma that develops coincidentally in transplant recipients. Moreover, EBV(+) PTLD and EBV(+) DLBCL present many similarities, indicating that EBV both infection and reactivation have important consequence on their pathogenesis (30–32, 38).
PTLD therapy is a combination of reduction of immunosuppressive therapy, immunotherapy, and chemotherapy (23, 25). In this review, we summarize the clinical and biological differences of EBV(+) and EBV(–) PTLD, and we support a new therapeutic approach based on EBV status to improve outcomes of these patients.
The expression of viral antigens makes EBV(+) PTLD an attractive candidate for specific therapy. Unfortunately, latent EBV-infected B cells do not express EBV-thymidine kinase transcript/protein; and for this reason, they are unaffected by antiviral agents as purine nucleoside analog. Similarly, EBV-related lymphoproliferative disorders do not express viral protein kinase, and so monotherapy with nucleoside analogs failed to induce responses in EBV(+) PTLD. However, pharmacological induction of viral thymidine kinase by the administration of the histone deacetylase inhibitor arginine butyrate, followed by antiviral therapy, has shown promising results with an acceptable toxicity profile (61).
More recently, several studies demonstrated how immunomodulatory drugs such as lenalidomide or proteasome inhibitors, in particular bortezomib, can induce EBV lytic activation (62, 63).
The search for new antivirals is ongoing; in particular, a new antiviral agent hexadecyloxypropyl-cidofovir (HDP-CDV) exhibits a remarkable increase in antiviral activity in vitro against different double-stranded DNA viruses including EBV (64).
Constitutive activation of the PI3K/Akt/mTOR pathway was shown in in vitro EBV(+) PTLD cell lines. Inhibition of either Akt or PI3K, with specific inhibitors CAL-101 or MK-2206, respectively, suppresses EBV(+) PTLD cell growth; and the combination of rapamycin had a synergistic effect. The combination therapy with an Akt inhibitor, or a PI3K inhibitor, and rapamycin can be an efficacious treatment for EBV(+) PTLD (37).
Most results presented are based on in vitro data; further evaluation and prospective clinical trials are necessary before such agents can be used as a treatment for PTLD patients.
The upfront treatment of EBV(+) and EBV(–) PTLD is the same, except for the use of EBV-specific adoptive immunotherapy. Immune-based therapies are an effective approach because of EBV antigen expression. In particular, adoptive therapy is based on the high efficacy of unselected donor lymphocyte infusions in HSC transplantation PTLD (65). Attempts were made to isolate EBV-specific cytotoxic lymphocytes (CTLs) aiming to induce a strong EBV-specific cellular immune response without the risk of graft-vs.-host disease (GVHD). Both autologous and allogeneic [isolated from the donor itself or a partial human leukocyte antigen (HLA)-matched donor] CTLs, targeting specific immunogenic EBV antigens, can be used (66). In a large multicentric study, HSCT patients were treated with EBV-CTLs, either prophylactically or therapeutically (67). A Chinese prospective study in HSCT recipients demonstrated an increase in complete remission rates in patients treated with sequential administration of rituximab and EBV-CTLs (68).
Moreover, checkpoint inhibition seems to be a potential treatment option in EBV(+) PTLD. EBV infection/reactivation causes a cytotoxic T-cell dysfunction in lymphomas as PTLD and classical HL. EBV causes an upregulation of immune checkpoint markers. In classical HL, immune checkpoint inhibitors have demonstrated efficacy; and therefore, there has been an increasing interest in PTLD (69). Antigen-presenting cells express PD-L1 that bind the PD-1 receptor on T cells, thus inhibiting T-cell receptor functions. EBV plays a role in increasing PD-L1; these data support the role of checkpoint inhibition in PTLD (44). Kinch et al. demonstrated than PDL-1, PDL-2, and PD-1 were positive in more than half of PTLD cases following SOT (70). More clinical data are necessary to determine the safety, efficacy, and graft rejection risk or GVHD of immune checkpoint inhibitors in PTLD. Currently, a phase II trial (NCT03258567) of nivolumab in a cohort of patients—EBV(+) non-HLs including EBV(+) PTLD—is ongoing.
This review summarizes many steps that have been made in understanding the EBV(+)/(–) PTLD biology. The biological differences connected with the EBV status support the development of preventive/preventive strategies against EBV disease and implementation of existing therapies both in the frontline and in the setting of relapsed/refractory patients. Several molecular targeting agents including immunomodulatory agents, proteasome inhibitors, PI3K and Akt inhibitors, novel anti-CD20 monoclonal antibodies, and immune checkpoint inhibitors seem to have a therapeutic potential, providing a strong rationale for new clinical trials to improve the outcome of EBV post-transplant lymphoproliferative disorder.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LF declared a past co-authorship with author LB to the handling Editor.
References
1. Swerdlow S, Campo E, Harris NL, Jaffe ES, Pileri S, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC (2008).
2. Nourse JP, Jones K, Gandhi MK. Epstein-Barr Virus-related post-transplant lymphoproliferative disorders: pathogenetic insights for targeted therapy. Am J Transplant. (2011) 11:888–95. doi: 10.1111/j.1600-6143.2011.03499.x
3. Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol. (2012) 13:122–36. doi: 10.1007/s11864-011-0177-x
4. Paya CV, Fung JJ, Nalesnik MA, Kieff E, Green M, Gores G, et al. Epstein-Barr virus-induced posttransplant lymphoproliferative disorders. ASTS/ASTP EBV-PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting. Transplantation. (1999) 68:1517–25. doi: 10.1097/00007890-199911270-00015
5. McDonald RA, Smith JM, Ho M, Lindblad R, Ikle D, Grimm P, et al. Incidence of PTLD in pediatric renal transplant recipients receiving basiliximab, calcineurin inhibitor, sirolimus and steroids. Am J Transplant. (2008) 8:984–9. doi: 10.1111/j.1600-6143.2008.02167.x
6. Kapelushnik J, Ariad S, Benharroch D, Landau D, Moser A, Delsol G, et al. Post renal transplantation human herpesvirus 8-associated lymphoproliferative disorder and Kaposi's sarcoma. Br J Haematol. (2001) 113:425–8. doi: 10.1046/j.1365-2141.2001.02740.x
7. Shimizu N, Tanabe-Tochikura A, Kuroiwa Y, Takada K. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J Virol. (1994) 68:6069–73.
8. Jox A, Rohen C, Belge G, Bartnitzke S, Pawlita M, Diehl V, et al. Integration of Epstein-Barr virus in Burkitt's lymphoma cells leads to a region of enhanced chromosome instability. Ann Oncol. (1997) 8(Suppl. 2):131–5.
9. Ambinder RF. Gammaherpesviruses and “Hit-and-Run” oncogenesis. Am J Pathol. (2000) 156:1–3. doi: 10.1016/S0002-9440(10)64697-4
10. Capello D, Cerri M, Muti G, Berra E, Oreste P, Deambrogi C, et al. Molecular histogenesis of posttransplantation lymphoproliferative disorders. Blood. (2003) 102:3775–85. doi: 10.1182/blood-2003-05-1683
11. Capello D, Rossi D, Gaidano G. Post-transplant lymphoproliferative disorders: molecular basis of disease histogenesis and pathogenesis. Hematol Oncol. (2005) 23:61–7. doi: 10.1002/hon.751
12. Tsao L, Hsi ED. The clinicopathologic spectrum of posttransplantation lymphoproliferative disorders. Arch Pathol Lab Med. (2007) 131:1209–18. doi: 10.1043/1543-2165(2007)131[1209:TCSOPL]2.0.CO;2
13. Opelz G, Dohler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant. (2004) 4:222–30. doi: 10.1046/j.1600-6143.2003.00325.x
14. Nee R, Hurst FP, Dharnidharka VR, Jindal RM, Agodoa LY, Abbott KC. Racial variation in the development of posttransplant lymphoproliferative disorders after renal transplantation. Transplantation. (2011) 92:190–5. doi: 10.1097/TP.0b013e3182200e8a
15. Dharnidharka VR, Sullivan EK, Stablein DM, Tejani AH, Harmon WE, North American Pediatric Renal Transplant Cooperative S. Risk factors for posttransplant lymphoproliferative disorder (PTLD) in pediatric kidney transplantation: a report of the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). Transplantation. (2001) 71:1065–8. doi: 10.1097/00007890-200104270-00010
16. Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K. Posttransplant lymphoproliferative disorders after renal transplantation in the United States in era of modern immunosuppression. Transplantation. (2005) 80:1233–43. doi: 10.1097/01.tp.0000179639.98338.39
17. Opelz G, Henderson R. Incidence of non-Hodgkin lymphoma in kidney and heart transplant recipients. Lancet. (1993) 342:1514–6. doi: 10.1016/s0140-6736(05)80084-4
18. Landgren O, Gilbert ES, Rizzo JD, Socie G, Banks PM, Sobocinski KA, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. (2009) 113:4992–5001. doi: 10.1182/blood-2008-09-178046
19. Hartmann C, Schuchmann M, Zimmermann T. Posttransplant lymphoproliferative disease in liver transplant patients. Curr Infect Dis Rep. (2011) 13:53–9. doi: 10.1007/s11908-010-0145-9
20. Cockfield SM. Identifying the patient at risk for post-transplant lymphoproliferative disorder. Transpl Infect Dis. (2001) 3:70–8. doi: 10.1034/j.1399-3062.2001.003002070.x
21. Reshef R, Luskin MR, Kamoun M, Vardhanabhuti S, Tomaszewski JE, Stadtmauer EA, et al. Association of HLA polymorphisms with post-transplant lymphoproliferative disorder in solid-organ transplant recipients. Am J Transplant. (2011) 11:817–25. doi: 10.1111/j.1600-6143.2011.03454.x
22. Trappe R, Oertel S, Leblond V, Mollee P, Sender M, Reinke P, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. (2012) 13:196–206. doi: 10.1016/S1470-2045(11)70300-X
23. Crombie JL, LaCasce AS. Epstein Barr virus associated B-cell lymphomas and iatrogenic lymphoproliferative disorders. Front Oncol. (2019) 9:109. doi: 10.3389/fonc.2019.00109
24. Luskin MR, Heil DS, Tan KS, Choi S, Stadtmauer EA, Schuster SJ, et al. The impact of EBV status on characteristics and outcomes of posttransplantation lymphoproliferative disorder. Am J Transplant. (2015) 15:2665–73. doi: 10.1111/ajt.13324
25. Dierickx D, Tousseyn T, Gheysens O. How I treat posttransplant lymphoproliferative disorders. Blood. (2015) 126:2274–83. doi: 10.1182/blood-2015-05-615872
26. Rinaldi A, Kwee I, Poretti G, Mensah A, Pruneri G, Capello D, et al. Comparative genome-wide profiling of post-transplant lymphoproliferative disorders and diffuse large B-cell lymphomas. Br J Haematol. (2006) 134:27–36. doi: 10.1111/j.1365-2141.2006.06114.x
27. Rinaldi A, Capello D, Scandurra M, Greiner TC, Chan WC, Bhagat G, et al. Single nucleotide polymorphism-arrays provide new insights in the pathogenesis of post-transplant diffuse large B-cell lymphoma. Br J Haematol. (2010) 149:569–77. doi: 10.1111/j.1365-2141.2010.08125.x
28. Morscio J, Dierickx D, Tousseyn T. Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: what do we know so far? Clin Dev Immunol. (2013) 2013:150835. doi: 10.1155/2013/150835
29. Menter T, Juskevicius D, Alikian M, Steiger J, Dirnhofer S, Tzankov A, et al. Mutational landscape of B-cell post-transplant lymphoproliferative disorders. Br J Haematol. (2017) 178:48–56. doi: 10.1111/bjh.14633
30. Craig FE, Johnson LR, Harvey SA, Nalesnik MA, Luo JH, Bhattacharya SD, et al. Gene expression profiling of Epstein-Barr virus-positive and -negative monomorphic B-cell posttransplant lymphoproliferative disorders. Diagn Mol Pathol. (2007) 16:158–68. doi: 10.1097/PDM.0b013e31804f54a9
31. Ferreiro JF, Morscio J, Dierickx D, Vandenberghe P, Gheysens O, Verhoef G, et al. EBV-positive and EBV-negative posttransplant diffuse large B cell lymphomas have distinct genomic and transcriptomic features. Am J Transplant. (2016) 16:414–25. doi: 10.1111/ajt.13558
32. Marcelis L, Tousseyn T. The tumor microenvironment in post-transplant lymphoproliferative disorders. Cancer Microenviron. (2019) 12:3–16. doi: 10.1007/s12307-018-00219-5
33. Harris-Arnold A, Arnold CP, Schaffert S, Hatton O, Krams SM, Esquivel CO, et al. Epstein-Barr virus modulates host cell microRNA-194 to promote IL-10 production and B lymphoma cell survival. Am J Transplant. (2015) 15:2814–24. doi: 10.1111/ajt.13375
34. Fink SE, Gandhi MK, Nourse JP, Keane C, Jones K, Crooks P, et al. A comprehensive analysis of the cellular and EBV-specific microRNAome in primary CNS PTLD identifies different patterns among EBV-associated tumors. Am J Transplant. (2014) 14:2577–87. doi: 10.1111/ajt.12858
35. Reshef R, Vardhanabhuti S, Luskin MR, Heitjan DF, Hadjiliadis D, Goral S, et al. Reduction of immunosuppression as initial therapy for posttransplantation lymphoproliferative disorder(bigstar). Am J Transplant. (2011) 11:336–47. doi: 10.1111/j.1600-6143.2010.03387.x
36. Poirel HA, Bernheim A, Schneider A, Meddeb M, Choquet S, Leblond V, et al. Characteristic pattern of chromosomal imbalances in posttransplantation lymphoproliferative disorders: correlation with histopathological subcategories and EBV status. Transplantation. (2005) 80:176–84. doi: 10.1097/01.tp.0000163288.98419.0d
37. Sang AX, McPherson MC, Ivison GT, Qu X, Rigdon J, Esquivel CO, et al. Dual blockade of the PI3K/Akt/mTOR pathway inhibits posttransplant Epstein-Barr virus B cell lymphomas and promotes allograft survival. Am J Transplant. (2019) 19:1305–14. doi: 10.1111/ajt.15216
38. Morscio J, Dierickx D, Ferreiro JF, Herreman A, Van Loo P, Bittoun E, et al. Gene expression profiling reveals clear differences between EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am J Transplant. (2013) 13:1305–16. doi: 10.1111/ajt.12196
39. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
40. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. (2010) 116:3268–77. doi: 10.1182/blood-2010-05-282780
41. Steidl C, Telenius A, Shah SP, Farinha P, Barclay L, Boyle M, et al. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood. (2010) 116:418–27. doi: 10.1182/blood-2009-12-257345
42. Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, et al. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. (2011) 117:4056–64. doi: 10.1182/blood-2010-06-291310
43. Vandenberghe P, Wlodarska I, Tousseyn T, Dehaspe L, Dierickx D, Verheecke M, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin's lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol. (2015) 2:e55–65. doi: 10.1016/S2352-3026(14)00039-8
44. Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O'Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. (2012) 18:1611–8. doi: 10.1158/1078-0432.CCR-11-1942
45. Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. (2013) 19:3462–73. doi: 10.1158/1078-0432.CCR-13-0855
46. Veloza L, Teixido C, Castrejon N, Climent F, Carrio A, Marginet M, et al. Clinicopathological evaluation of the programmed cell death 1 (PD1)/programmed cell death-ligand 1 (PD-L1) axis in post-transplant lymphoproliferative disorders: association with Epstein-Barr virus, PD-L1 copy number alterations, and outcome. Histopathology. (2019) 75:799–812. doi: 10.1111/his.13857
47. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
48. LaPak KM, Burd CE. The molecular balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res. (2014) 12:167–83. doi: 10.1158/1541-7786.MCR-13-0350
49. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. (1996) 85:27–37. doi: 10.1016/s0092-8674(00)81079-x
50. Barrans SL, Fenton JA, Banham A, Owen RG, Jack AS. Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood. (2004) 104:2933–5. doi: 10.1182/blood-2004-03-1209
51. Wlodarska I, Veyt E, De Paepe P, Vandenberghe P, Nooijen P, Theate I, et al. FOXP1, a gene highly expressed in a subset of diffuse large B-cell lymphoma, is recurrently targeted by genomic aberrations. Leukemia. (2005) 19:1299–305. doi: 10.1038/sj.leu.2403813
52. Koon HB, Ippolito GC, Banham AH, Tucker PW. FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets. (2007) 11:955–65. doi: 10.1517/14728222.11.7.955
53. Price AM, Tourigny JP, Forte E, Salinas RE, Dave SS, Luftig MA. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-kappaB activation. J Virol. (2012) 86:11096–106. doi: 10.1128/JVI.01069-12
54. Rouhigharabaei L, Finalet Ferreiro J, Tousseyn T, van der Krogt JA, Put N, Haralambieva E, et al. Non-IG aberrations of FOXP1 in B-cell malignancies lead to an aberrant expression of N-truncated isoforms of FOXP1. PLoS ONE. (2014) 9:e85851. doi: 10.1371/journal.pone.0085851
55. Johannessen I, Perera SM, Gallagher A, Hopwood PA, Thomas JA, Crawford DH. Expansion in scid mice of Epstein-Barr virus-associated post-transplantation lymphoproliferative disease biopsy material. J Gen Virol. (2002) 83(Pt 1):173–8. doi: 10.1099/0022-1317-83-1-173
56. Beatty PR, Krams SM, Martinez OM. Involvement of IL-10 in the autonomous growth of EBV-transformed B cell lines. J Immunol. (1997) 158:4045–51.
57. Birkeland SA, Bendtzen K, Moller B, Hamilton-Dutoit S, Andersen HK. Interleukin-10 and posttransplant lymphoproliferative disorder after kidney transplantation. Transplantation. (1999) 67:876–81. doi: 10.1097/00007890-199903270-00015
58. Muti G, Klersy C, Baldanti F, Granata S, Oreste P, Pezzetti L, et al. Epstein-Barr virus (EBV) load and interleukin-10 in EBV-positive and EBV-negative post-transplant lymphoproliferative disorders. Br J Haematol. (2003) 122:927–33. doi: 10.1046/j.1365-2141.2003.04540.x
59. Baiocchi OC, Colleoni GW, Caballero OL, Vettore AL, Bulgarelli A, Dalbone MA, et al. Epstein-Barr viral load, interleukin-6 and interleukin-10 levels in post-transplant lymphoproliferative disease: a nested case-control study in a renal transplant cohort. Leuk Lymphoma. (2005) 46:533–9. doi: 10.1080/10428190400027837
60. Hinrichs C, Wendland S, Zimmermann H, Eurich D, Neuhaus R, Schlattmann P, et al. IL-6 and IL-10 in post-transplant lymphoproliferative disorders development and maintenance: a longitudinal study of cytokine plasma levels and T-cell subsets in 38 patients undergoing treatment. Transpl Int. (2011) 24:892–903. doi: 10.1111/j.1432-2277.2011.01282.x
61. Perrine SP, Hermine O, Small T, Suarez F, O'Reilly R, Boulad F, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood. (2007) 109:2571–8. doi: 10.1182/blood-2006-01-024703
62. Jones RJ, Iempridee T, Wang X, Lee HC, Mertz JE, Kenney SC, et al. Lenalidomide, thalidomide, and pomalidomide reactivate the Epstein-Barr virus lytic cycle through phosphoinositide 3-kinase signaling and ikaros expression. Clin Cancer Res. (2016) 22:4901–12. doi: 10.1158/1078-0432.CCR-15-2242
63. Granato M, Romeo MA, Tiano MS, Santarelli R, Gonnella R, Gilardini Montani MS, et al. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci Rep. (2017) 7:13052. doi: 10.1038/s41598-017-13533-7
64. Hostetler KY. Synthesis and early development of hexadecyloxypropylcidofovir: an oral antipoxvirus nucleoside phosphonate. Viruses. (2010) 2:2213–25. doi: 10.3390/v2102213
65. Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. (1994) 330:1185–91. doi: 10.1056/NEJM199404283301703
66. Bollard CM, Heslop HE. T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood. (2016) 127:3331–40. doi: 10.1182/blood-2016-01-628982
67. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. (2010) 115:925–35. doi: 10.1182/blood-2009-08-239186
68. Jiang X, Xu L, Zhang Y, Huang F, Liu D, Sun J, et al. Rituximab-based treatments followed by adoptive cellular immunotherapy for biopsy-proven EBV-associated post-transplant lymphoproliferative disease in recipients of allogeneic hematopoietic stem cell transplantation. Oncoimmunology. (2016) 5:e1139274. doi: 10.1080/2162402X.2016.1139274
69. Armand P. Immune checkpoint blockade in hematologic malignancies. Blood. (2015) 125:3393–400. doi: 10.1182/blood-2015-02-567453
Keywords: post-transplant lymphoproliferative disorders, Epstein–Barr virus, next-generation sequencing, microRNA, gene expression profile, tumor microenvironment
Citation: Ferla V, Rossi FG, Goldaniga MC and Baldini L (2020) Biological Difference Between Epstein–Barr Virus Positive and Negative Post-transplant Lymphoproliferative Disorders and Their Clinical Impact. Front. Oncol. 10:506. doi: 10.3389/fonc.2020.00506
Received: 21 November 2019; Accepted: 20 March 2020;
Published: 08 May 2020.
Edited by:
Francesco Maura, Cornell University, United StatesReviewed by:
Lucia Farina, National Cancer Institute Foundation (IRCCS), ItalyGuido Gini, Azienda Ospedaliero Universitaria Ospedali Riuniti, Italy
Copyright © 2020 Ferla, Rossi, Goldaniga and Baldini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Ferla, dmFsZXJpYS5mZXJsYUBwb2xpY2xpbmljby5taS5pdA==