 Aleksandr Klepinin1*
Aleksandr Klepinin1* Song Zhang2
Song Zhang2 Ljudmila Klepinina1
Ljudmila Klepinina1 Egle Rebane-Klemm1
Egle Rebane-Klemm1 Andre Terzic2
Andre Terzic2 Tuuli Kaambre1
Tuuli Kaambre1 Petras Dzeja2*
Petras Dzeja2*- 1Laboratory of Chemical Biology, National Institute of Chemical Physics and Biophysics, Tallinn, Estonia
- 2Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN, United States
A hallmark of cancer cells is the ability to rewire their bioenergetics and metabolic signaling circuits to fuel their uncontrolled proliferation and metastasis. Adenylate kinase (AK) is the critical enzyme in the metabolic monitoring of cellular adenine nucleotide homeostasis. It also directs AK→ AMP→ AMPK signaling controlling cell cycle and proliferation, and ATP energy transfer from mitochondria to distribute energy among cellular processes. The significance of AK isoform network in the regulation of a variety of cellular processes, which include cell differentiation and motility, is rapidly growing. Adenylate kinase 2 (AK2) isoform, localized in intermembrane and intra-cristae space, is vital for mitochondria nucleotide exchange and ATP export. AK2 deficiency disrupts cell energetics, causes severe human diseases, and is embryonically lethal in mice, signifying the importance of catalyzed phosphotransfer in cellular energetics. Suppression of AK phosphotransfer and AMP generation in cancer cells and consequently signaling through AMPK could be an important factor in the initiation of cancerous transformation, unleashing uncontrolled cell cycle and growth. Evidence also builds up that shift in AK isoforms is used later by cancer cells for rewiring energy metabolism to support their high proliferation activity and tumor progression. As cell motility is an energy-consuming process, positioning of AK isoforms to increased energy consumption sites could be an essential factor to incline cancer cells to metastases. In this review, we summarize recent advances in studies of the significance of AK isoforms involved in cancer cell metabolism, metabolic signaling, metastatic potential, and a therapeutic target.
Introduction
The significance of metabolism and metabolic signaling in human diseases is rapidly growing. New features and molecular players that are vital for cell homeostasis and function are being uncovered. Well-organized high-energy phosphoryl transfer systems are required to mediate intracellular communication between ATP-consuming and ATP-producing cellular compartments and thus to maintain normal growth and development of the cell (1–5). The main components of the cellular phosphotransfer system are AK, creatine kinase (CK), and glycolytic networks (1, 2). The significance of organized phosphotransfer was demonstrated by genetic manipulations in animal models, cellular systems, and alterations or mutations in separate phosphotransfer enzymes, which are associated with human diseases (6–16). Studies on Drosophila and mice model demonstrate that deletion of adenylate kinase 2 (AK2) is embryonically lethal, signifying the importance of AK phosphotransfer network in cell homeostasis (13, 17–19). In humans, mutations in the mitochondrial AK2 gene are associated with reticular dysgenesis characterized by immunodeficiency and sensorineural deafness, where processes of nucleotide signaling, cell differentiation, and motility are affected (15, 16, 20). So far, nine isoforms of adenylate kinase (AK1–AK9) and several subforms have been found and well characterized in mammalian cells (21). AK, which catalyzes reaction 2ADP↔AMP+ATP, is a recognized facilitator of AMP metabolic signaling, optimizing intracellular energetic communication, and local ATP supply (5, 22). Historically, the function of AK has been ascribed to de novo adenine nucleotide synthesis and cell energy economy through regulation of nucleotide ratios in different intracellular compartments and AMP-sensitive metabolic enzymes (14, 23, 24). The unique properties of AK lie on its ability to deliver γ- and β-phosphoryl groups of ATP, thereby doubling the ATP energetic potential. Moreover, the AK network provides an efficient mechanism for high-energy phosphoryl transport from mitochondria to ATP utilization sites (2). Evolutionary AK isoforms have been positioned to different subcellular compartments (21, 25). AK1, AK7, and AK8 are solely found in the cytosol; AK2, AK3, and AK4 are located in the mitochondria; and AK5 and AK9 can be found in either the cytosol or nucleus. Only AK1 and AK6 are known to be expressed in all tissues, whereas AK5 is expressed only in the brain (21). In the cytosol, the main isoform is AK1, which is predominantly expressed in high energy demand tissues such as the brain, heart, and skeletal muscles. AK2 is strategically located in the mitochondrial intermembrane and cristae space to facilitate high-energy phosphoryl exchange between mitochondria and cytosol (22). Two other AK isoforms, AK3 and AK4, are located in the mitochondrial matrix and are involved in the regulation of mitochondrial Krebs cycle and oxidative phosphorylation (OXPHOS), whereas AK5 and AK6 isoforms that are localized in the nucleus could serve to fulfill the energy needs of nuclear processes. In general, distinct intracellular localization and kinetic properties of AK isoforms favor energy support of specific cellular processes ranging from muscle contraction, electrical activity, cell motility, unfolded protein response, and mitochondrial/nuclear energetics (22). Importantly, reprogramming of energy metabolism has been proposed as one of the hallmarks of cancer (26), which is required to drive biosynthesis pathways necessary for rapid cell replication and proliferation. Cancer cells are believed to have a greater reliance on glycolytic phosphotransfer (27, 28). However, during the last decade, it was found that some tumors contain numerous mitochondria producing ATP predominantly via OXPHOS (29–31). The observed shift in hexokinase (HK) isoforms, upregulation of HK2 in cancer cells (32), indicates a closer integration of mitochondria with glycolytic phosphotransfer (see Figure 1). The association of HK2 with mitochondria and expression of pyruvate kinase PKM2 could promote effective yet uncontrolled energy distribution in cancer cells (27, 33, 34). Phosphotransfer enzymes such as CK and AK have been implicated in cancer cell proliferation (35, 36). However, it is not clear whether the redistribution of phosphotransfer enzymes, especially those which are localized in mitochondria, occurs during cancer formation. In this review, we focus on the significance of AK isoforms in the rewiring of cancer cell energy metabolism and AMP signaling. Specifically, we will overview how AK isoforms, localized in mitochondria (AK2 and AK4), and their main communication partners cytosolic AK (AK1 and AK6) are involved in cancer formation and metastasis.
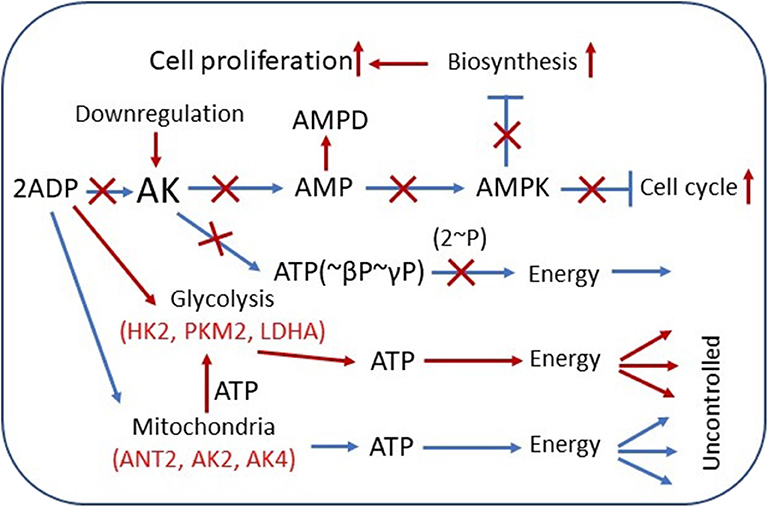
Figure 1. Overview of adenylate kinase (AK) isoform involvement in the rewiring of cancer cell metabolic signaling and energetic circuits. Increased competition for cytosolic ADP downregulates AK-mediated AMP signaling, reducing control over cell cycle and proliferation. AK expression is downregulated in several tumors. AMP can be consumed by AMPD and by 5′-NT, also overexpressed in some cancer cells. Augmented glycolytic metabolism, owing to higher affinity, scavenges cytosolic ADP, and uses mitochondrial ATP to drive glucose conversion to lactate. Overexpression of glycolytic HK2, PKM2, and LDHA and mitochondrial ANT2, AK2, AK4, and other genes in cancer cells promotes rewiring of energetic circuits resulting in unrestrained energy distribution. The result of these metabolic transformations is deficient AMP signaling and AMPK-mediated control of cellular katabolic and anabolic processes. Red color indicates the augmented pathways and gene expression in cancer cells. AMPD, AMP-deaminase; HK2, hexokinase 2; LDHA, lactate dehydrogenase A; ANT2, adenine nucleotide translocase 2; AMPK, AMP-activated protein kinase.
Adenylate Kinase 2 and Mitochondrial Creatine Kinase Interplay in Malignant Transformation
AK2 and mitochondrial CK (CKmit) are major phosphotransfer enzymes located in the intermembrane/cristae space in mitochondria (3, 14, 22). AK2 and CKmit provide nucleotide exchange and metabolic signaling capacity, allowing mitochondria to export ATP and reception of cytosolic feedback signals such as ADP, AMP, and creatine (22, 23, 37). Phosphotransfer enzymes CK and AK have been implicated in cancer cell proliferation (35, 36). In general, CK is involved in cancerous transformation, as CKB (brain-type CK) is upregulated in a variety of cancers to support growing energy needs (38). The elevation of creatine metabolites was noted in drug-resistant cancer cells (39). However, in other cancer types, the downregulation of CKB and rewiring of metabolism may play an important role in colon cancer progression (40). Moreover, several studies have demonstrated that in colorectal cancer (41), breast cancer (42), neuroblastoma (35), prostate cancer (43), and sarcoma (36, 44), the CKmit was downregulated. The reduction of CKmit in cancer cells was associated with the upregulation of adenylate kinase AK2 isoform in intermembrane space (36, 41, 42, 45, 46) (see Table 1). There is evidence that the expression of AK2 on the cell surface could facilitate nucleotide signaling and metastatic potential (60). It was found that Ak2 gene expression is upregulated in the metastatic pancreatic endocrine neoplasms (60), indicating the significance of nucleotide metabolic signaling in cancer invasion (61). Moreover, increased expression of the Ak2 on the surface of the metastatic F9DR murine terato-carcinoma cells compared with the nonmetastatic F9B9 cell line has been demonstrated (53). Furthermore, a recent study showed that AK2 has prognostic and therapeutic potential in lung adenocarcinoma (55). The knockdown of AK2 suppressed proliferation, migration, and invasion, as well as induced apoptosis and autophagy in human lung adenocarcinoma cells. In this regard, the AK2-FADD (Fas-associated protein with death domain) mediated apoptosis pathway was found to be defective in some tumor cells, which may contribute to tumor development by preventing apoptosis (62). A recent study indicates that AK2 and FADD are crucial for caspase-10 activation upon metabolic stress, and this activation is independent of death receptors and extrinsic pathway of apoptosis (63). Moreover, the deletion of the Ak2 gene or exit AK2 from mitochondria during apoptosis disrupts nucleotide exchange between mitochondria and cytosol, causing hyperpolarization of mitochondria and reactive oxygen species (ROS) production (20). It was found that the presence of AK2 in mitochondrial cristae nanochannels is critical for ATP export (2, 22). There are evidence that the AK2 upregulation could be used by cancer cells to support energy supply to biosynthetic processes and cellular growth (18, 64). These results, as well as studies on CK and AK knockout mice, demonstrate remarkable plasticity of cellular energetics and phosphotransfer systems, which could be used in cancer cells to promote uncontrolled cell growth (9, 17, 22, 65).
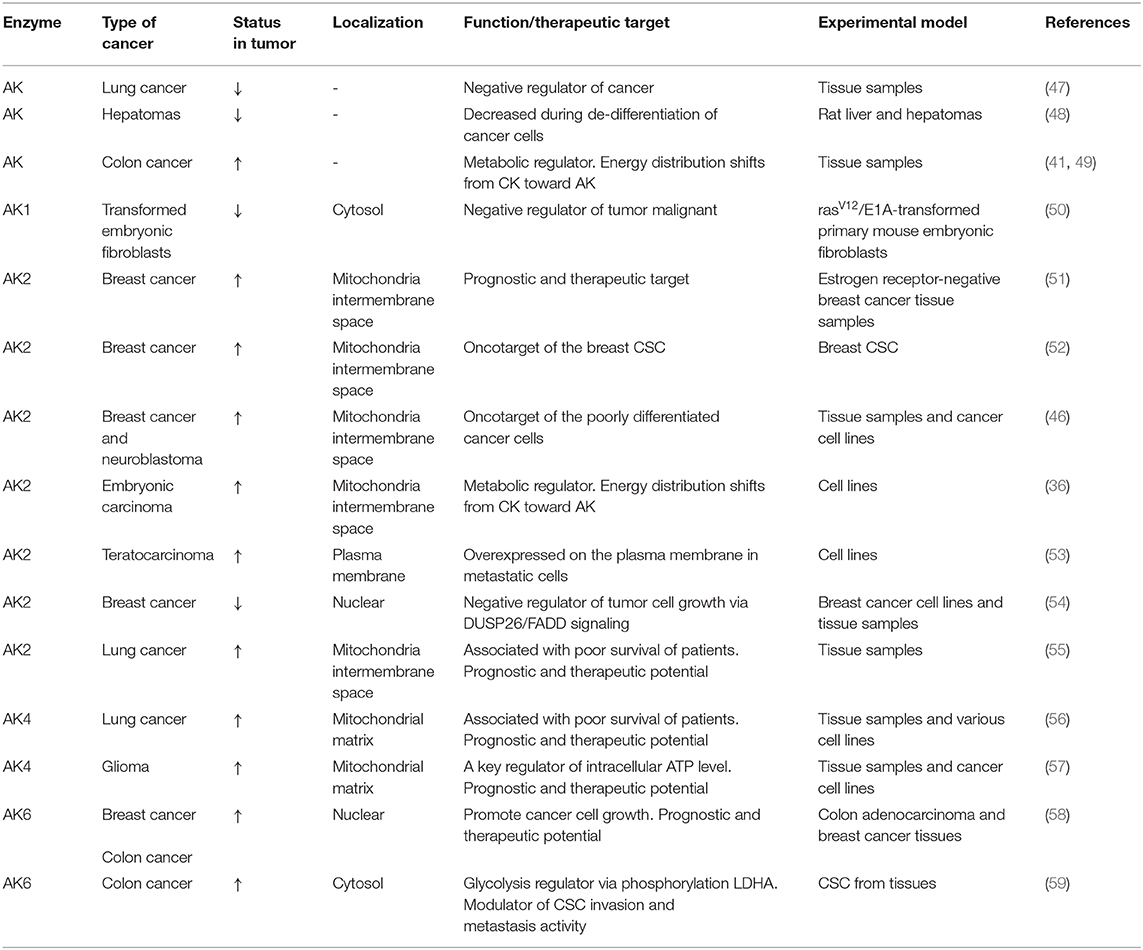
Table 1. Adenylate kinase isoforms in cancer.
Adenylate Kinase Modulate Tumor Cell Response to Survive Under Oxidative Stress
The ability to conduct metabolic signaling and rewire metabolism is critical for cell survival. The AK4 isoform increased expression has been associated with a poor clinical outcome marker for lung cancer (56) as well as for glioma patients (57) (see Table 1). It was found that AK4 expression is under tight control of noncoding RNA. The AK4 is negatively regulated by micro-RNA miR-556-3p and positively by circular RNA of ATP-binding cassette (ABC) subfamily B member 10, circ-ABCB10 (66). In same study was demonstrated that downregulation of AK4 restrained lung cancer progression and sensitized lung cancer cells to cisplatin (66). Moreover, new data indicate that AK4 was shown to be involved in the radioresistance of esophageal cancer cells (67) and in chemoresistance of other cancers (68, 69). Previously, it was suggested that overexpression of AK4 could protect cells against oxidative stress (70). Other studies on HeLa (68) and HEK293 cells (71) demonstrated that tumor cells respond to a hypoxic condition by upregulating the AK4. However, in HepG2 cells (71), it was found that under oxidative stress, AK4 oppositely was downregulated. Although AK4 might be downregulated, it can still regulate OXPHOS because it retains the nucleotide-binding capability, and it can interact with the mitochondrial adenine nucleotide translocase (ANT) (70). It was found that knockout of AK4 increased cellular ATP through raised OXPHOS activity as well as mitochondrial number (68). Fujisawa and colleagues in 2016 have proposed that there are two mechanisms how AK4 regulates mitochondrial respiration in cancer cells (68). First, in cancer cells, AK4 interacts with ANT, which forms with voltage-dependent anion channel (VDAC) and HK transmembrane complex AK4-ANT-VADC-HK (see Figure 1). Under the hypoxic conditions, the complex supports the high glycolytic activity of cancer cells. It allows efficient ADP recycling between mitochondrial ATP synthesis and glucose phosphorylation by HK, which interacts with the mitochondrial outer membrane (MOM) (68, 72). In addition, in hepatoma cells (73), it was found that up to 50% of ATP is provided by intramembrane space located AK2 through VDAC binding to HK. Thus, AK2 may also be a member of the metabolic circuit channeling ADP–ATP in and out of mitochondria. The second mechanism is related to the fact that AK4 and AK3 have highly homologous sequences; therefore, they compete with each other for their substrates (68). According to this mechanism, AK4 interferes with AK3 action in supplying of the GDP required for the conversion of succinyl-CoA to succinate. That is why overexpression of AK4 in HeLa cells induces a decrease in Krebs cycle metabolites such as succinate, fumarate, and malate while glutamine and glutamate are increased. In several tumors, it was shown that the predominate substrate for mitochondria is glutamine (74). However, further studies are needed to confirm the role of AK4 in mitochondria and Krebs cycle substrate metabolism.
Adenylate Kinase Network Role in Cancer Stem Cells
Traditional therapies against cancer, such as chemotherapy and radiotherapy, have many limitations. The limitation is due to systematic and local toxicity as well as drug resistance of small populations of tumor cells that have self-renewal properties. This small population of cells is called cancer stem cells (CSCs) (75). Previously, studies on CSC have shown that the cancer resistance for chemotherapy is related to increased OXPHOS in CSC. That is why a new generation of cancer chemotherapy could be targeted against pathways that interact with OXPHOS, such as the phosphotransfer system. Lamb and colleagues have shown on the breast cancer model that mitochondrial mass is a new biomarker of CSC, which have increased AK2 expression level (see Table 1) (52). In our previous study on neuroblastoma (NB) (46), which contains numerous CSC (76), and embryonal carcinoma cells (36), we also found that those cells have a high activity of AK2 (see Table 1). Moreover, another feature of CSC is that mitochondria are localized around the cell nucleus (77). There is evidence that AK2 can play an important role in communication between mitochondria and the nucleus (78). In another study using proteomic analysis of mouse teratocarcinoma cells (53), it was demonstrated that metastatic cancer cells have increased AK2 levels than have nonmetastatic cancer cells (see Table 1). As metastasis is related to cell motility, positioning of phosphotransfer enzymes to sites of increased energy consumption could be an important factor of tumor formation (59). The AK4 has been identified as a biomarker of metastasis in lung cancer (56, 79, 80). Overexpression of AK4 promoted lung cancer metastasis by enhancing hypoxia-inducible factor HIF-1 stability and epithelial-to-mesenchymal transition under hypoxia (79). Moreover, it was found that aferin-A could suppress AK4-HIF-1a signaling and may serve as a novel anti-metastatic agent in lung cancer (79). The AK4 was also implicated in breast and bladder cancers, where it promoted cell proliferation and invasion (81, 82). Furthermore, it was demonstrated that another AK isoform AK6 could affect colorectal cancer migration and invasion (59). Although significant progress has been made, at this time, the complete role of the AK system in cancer metastasis is still unclear. Moreover, the other reason why CSCs are drug resistant relates to the increased expression of ABC transporters in those cells (83, 84). The model for ABC transporters was proposed (85), which is based on 31P solid-state NMR spectroscopy, suggesting that intrinsic ATPase is coupled with AK activity where AK participates in ATP exchange. It is known that cytosolic and membrane-associated AK can regulate the activity of another ABC protein—K-ATP channel (86, 87). Nevertheless, the exact role of AK in supporting adenylate charge and function of ABC transporters in CSC remains unknown yet. In this respect, the ABC transporters are not unique proteins that possess both ATPase and AK activities; there are other proteins like AK6 (58), also known as transcription factor TAF9, human coilin interacting nuclear ATPase protein (hCINAP), and highly conserved DNA repair complex Rad50 (88).
Paradoxes Regarding the Role of Adenylate Kinase in Tumor Formation
Cancer is a very complex and diverse phenomenon, including tissue specificity and different phases. Enzymatic changes can be different in the initial and advanced stages of tumor growth (89, 90). There are some contradictory studies where it was found that in lung cancer and hepatoma, AK was downregulated compared with that in normal tissue (47, 48) (see Table 1), whereas a recent study has shown that high expression of AK2 correlates with a worse prognosis for lung cancer patients (55) (see Table 1). In mouse embryonic fibroblasts, it was demonstrated that during their transformation into tumor cells, a significant reduction of AK1 expression occurs (50). More recently, the existence of AK1 additional gene product AK1β has been reported, and it is known that the AK1β expression level is regulated by p53 (91). In some cancers, p53 is mutated or suppressed. In this context, experiments on mouse embryonic fibroblast (50) have shown that during their transformation into tumor cells, augmentation of AK1 might be related to the downregulation of AK1β (see Table 1). Also, Kim et al. have postulated that AK2 is a negative regulator of tumor growth (54) (see Table 1). They demonstrated that in some cells, the AK2 localized not only in mitochondria but also in the nucleus, where it interacted with dual-specificity phosphatase 26 (DUSP26). This protein complex can dephosphorylate FADD leading to suppressed cell growth. They also suggested that AK2 downregulation was associated with breast cancer formation. In contrast, Speers and colleagues have found that AK2 is overexpressed in ER-negative breast cancer (51) (see Table 1). They proposed that AK2 should be a novel target for the treatment of ER-negative breast cancer. Indeed, a diterpene lactone neoandrographolide from extracts of the traditional medicinal herb Andrographis paniculata has been suggested to inhibit AK2 and have strong anticancer properties (92). Nevertheless, studies on human breast cancer and colorectal cancer demonstrated another AK isoform AK6 was overexpressed during cancer formation (58) (see Table 1). These data correlate with our previous studies on colorectal and breast cancers (41, 46) (see Table 1). It was also shown, that in both colon and breast tissues, AK6 is located not only in nuclear but also in the cytosol. However, only in cytosolic compartmentalized AK6 did expression level increase during tumorogenesis of breast and colorectal cancer cells (58) (see Table 1). They have found that AK6's main function is to regulate ribosome assembly and, consequently, protein expression and cancer cell growth. Recently, it was demonstrated that hCINAP or AK6 is a potent modulator of metabolic reprogramming by phosphorylating LDHA, a key player in cancer glycolysis (59) (see Table 1). Thus, AK isoform role can be different depending on cancer cell type and development stage.
Adenylate Kinase-Mediated Amp Metabolic Signaling in Cancer Cells
In recent years, AK-mediated AMP signaling is emerging as one of the most versatile systems in the regulation of diverse cellular processes (5, 22, 93). Particularly, AMP signaling to AMP-activated protein kinase (AMPK) plays a critical role in adjusting ATP-producing and ATP-consuming processes (90, 94) (Figure 1). In several cancers, it has been demonstrated that AMPK, a master regulator of cellular energy homeostasis, possesses tumor suppressor function (95–97). In cells, AMPK activation/suppression is regulated via changes in cellular AMP levels. The principal activator of AMPK is the AK-catalyzed pathway, where it monitors cellular ATP–ADP balance and signals to AMPK by increased AMP cellular level. A recent study indicates that AK and AMPK cooperate to maintain cellular ATP levels (98). On the other end, AMP-deaminase (AMPD) and 5′-nucleotidase (5′-NT) suppress AMPK via decreasing AMP cellular levels (22, 99, 100). Moreover, the product of AMPD and 5'-NT reactions is adenosine, an immunosuppressive metabolite. At a high level in tumors, adenosine can promote cell growth, invasion, metastasis of cancer cells, and tumor immune evasion (101). Our previous work has demonstrated that in NB and heart adenocarcinoma cells HL-1, their mitochondrial permeability for AMP was increased than in healthy cells (46). It is known that AK2, which has unique localization in mitochondrial space, has a high affinity for AMP among AMP metabolizing enzymes. Therefore, it has been proposed that the AK2's primary function is to regulate intracellular AMP levels and to guard the cellular adenine nucleotide pool (22). Our study also suggested that cancer cells have a high level of AK2 (46) (Figure 1). Altogether, in cancer cells, most cellular AMP transport occurs via MOM where it is converted immediately to ADP and channeled into, maintaining a low cytosolic AMP concentration. Recent direct measurements of AK-mediated metabolic flux indicate that cancer cells have suppressed ATP β-phosphoryl energetics and AMP signaling, as indicated from 18O-labeling experiments demonstrating that highly aggressive breast cancer cells MDAMB231 have lower β-ATP[18O] turnover (AMP phosphorylation) than have the control MCF10A cells (Klepinin et al., in preparation). This could be due to the rewiring of energy metabolism and glycolytic takeover. Activated glycolysis usually suppresses AK metabolic flux apparently by scavenging ADP (102) (Figure 1). Suppression of AK phosphotransfer, AMP generation, and consequent signaling through AMPK could be the biggest culprit of a cancerous transformation of a cell (Figure 1). There is also evidence that other AMP removal pathway enzymes like AMPD2 as well as 5′-NT are upregulated in colorectal cancer (103, 104). In this regard, the 5′-NT expression in breast cancer depends on tumor estrogen receptor status, suggesting a coordinated network (105). Our previous work has shown that in several tumors, MOM permeability has also increased for ADP, which may be related with keeping an intracellular ADP level low (41, 42, 49, 106) (see Figure 1). It was found that not only AMP but also ADP can regulate the activity of AMPK (107). Further studies are needed to elucidate detailed mechanisms: (1) how increased MOM permeability for ADP and AMP and (2) raised expression of AMP metabolizing enzymes can regulate intracellular nucleotide levels and the activity of AMPK and (3) what the significance is of AMP metabolic signaling in cancer progression.
Conclusions
The present review is a snapshot from recent AK studies that focused on the significance of AK network in energetics and metabolic signaling in cancer cells. Of the nine AK isoforms (AK1–AK9), four of them (AK1, AK2, AK4, and AK6) are involved in the progression of malignant transformation. Studies indicate that AK isoforms (AK1, AK2, AK4, and AK6) have an important role in the regulation of cancer cell metabolism, metabolic signaling, and cell migration and invasion. Moreover, at the initial stage, suppression of AK phosphotransfer and AMP generation and consequently signaling through AMPK by a variety of factors could be the biggest culprit of the cancerous transformation of a cell. Downregulation of AK→ AMP→ AMPK signaling can lead to the loss of control of cell cycle, growth, and proliferation. In the later stages, as emerging data suggest, cancer cells may use the shift in AK isoforms and other phosphotransfer enzymes to rewire their energy supply circuits to support proliferation and metastasis. Knockdown of overexpressed AK2 in human lung adenocarcinoma cells suppressed proliferation, migration, and invasion as well as induced apoptosis and autophagy. In this regard, a diterpene lactone neoandrographolide from extracts of the traditional medicinal herb Andrographis paniculata has been suggested to inhibit AK2 and has strong anticancer properties. Further studies that involve all AK isoforms have the potential to bring new understanding and novel therapeutic strategies targeting the AK isoform network to suppress growth and metastasis of cancer cells.
Author Contributions
AK and SZ performed the study design, development of methodology, data analysis and interpretation, drafting of the manuscript, and critical revision. LK and ER-K analyzed and interpreted data and performed manuscript review. AT, TK, and PD performed the study conception, design, writing, and reviewing of the manuscript.
Funding
This work was supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (HL 85744-09) and U24DK100469 (Mayo Clinic Metabolomics Resource Core); Marriott Foundation and Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN, USA; Institutional Research Funding IUT23-1 of the Estonian Ministry of Education and Research; and Estonia national scholarship program Kristjan Jaak, which is funded and managed by Archimedes Foundation in collaboration with the Estonian Ministry of Education and Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AK, adenylate kinase; CK, creatine kinase; CKmit, mitochondrial creatine kinase; CKB, brain-type creatine kinase; MOM, mitochondrial outer membrane; ANT, adenine nucleotide translocase; AMPK, AMP-activated protein kinase; CSCs, cancer stem cells; NB, neuroblastoma; VDAC, voltage-dependent anion channel; ABC, ATP-binding cassette; FADD, Fas-associated protein with death domain; HIF, hypoxia-inducible factor; hCINAP, human coilin-interacting nuclear ATPase protein; DUSP26, dual-specificity phosphatase 26; OXPHOS, oxidative phosphorylation; AMPD, AMP-deaminase; 5′-NT, 5′-nucleotidase; LDHA, lactate dehydrogenase A; HK, hexokinase.
References
1. Dzeja P, Chung S, Terzic A. Integration of adenylate kinase, glycolytic and glycogenolytic circuits in cellular energetics. In: Saks V, editor. Molecular System Bioenergetics: Energy for Life. Weinheim: Wiley-VCH. (2007). p. 265–301. doi: 10.1002/9783527621095.ch8
2. Dzeja PP, Terzic A. Phosphotransfer networks and cellular energetics. J Exp Biol. (2003) 206:2039–47. doi: 10.1242/jeb.00426
3. Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, Wallimann T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol. (2006) 571:253–73. doi: 10.1113/jphysiol.2005.101444
4. Saks V, Monge C, Anmann T, Dzeja P. Integrated and organized cellular energetic systems: theories of cell energetics, compartmentation and metabolic channeling. In: Saks V, editor. Molecular System Bioenergetics: Energy for Life. Weinheim: Wiley-VCH. (2007). p. 59–109. doi: 10.1002/9783527621095.ch3
5. Zhang S, Nemutlu E, Terzic A, Dzeja P. Adenylate kinase isoform network: a major hub in cell energetics and metabolic signaling. system biology of metabolic and signaling networks. Springer Ser Biophys. (2014) 16:145–62. doi: 10.1007/978-3-642-38505-6_6
6. Dzeja PP, Bast P, Pucar D, Wieringa B, Terzic A. Defective metabolic signaling in adenylate kinase AK1 gene knock-out hearts compromises post-ischemic coronary reflow. J Biol Chem. (2007) 282:31366–72. doi: 10.1074/jbc.M705268200
7. Dzeja PP, Terzic A, Wieringa B. Phosphotransfer dynamics in skeletal muscle from creatine kinase gene-deleted mice. Mol Cell Biochem. (2004) 256-257:13–27. doi: 10.1023/B:MCBI.0000009856.23646.38
8. Janssen E, de Groof A, Wijers M, Fransen J, Dzeja PP, Terzic A, et al. Adenylate kinase 1 deficiency induces molecular and structural adaptations to support muscle energy metabolism. J Biol Chem. (2003) 278:12937–45. doi: 10.1074/jbc.M211465200
9. Janssen E, Terzic A, Wieringa B, Dzeja PP. Impaired intracellular energetic communication in muscles from creatine kinase and adenylate kinase (M-CK/AK1) double knock-out mice. J Biol Chem. (2003) 278:30441–9. doi: 10.1074/jbc.M303150200
10. Steeghs K, Benders A, Oerlemans F, de Haan A, Heerschap A, Ruitenbeek W, et al. Altered Ca2+ responses in muscles with combined mitochondrial and cytosolic creatine kinase deficiencies. Cell. (1997) 89:93–103. doi: 10.1016/S0092-8674(00)80186-5
11. van Deursen J, Heerschap A, Oerlemans F, Ruitenbeek W, Jap P, ter Laak H, et al. Skeletal muscles of mice deficient in muscle creatine kinase lack burst activity. Cell. (1993) 74:621–31. doi: 10.1016/0092-8674(93)90510-W
12. van Horssen R, Janssen E, Peters W, van de Pasch L, Lindert MM, van Dommelen MM, et al. Modulation of cell motility by spatial repositioning of enzymatic ATP/ADP exchange capacity. J Biol Chem. (2009) 284:1620–7. doi: 10.1074/jbc.M806974200
13. Horiguchi T, Fuka M, Fujisawa K, Tanimura A, Miyoshi K, Murakami R, et al. Adenylate kinase 2 deficiency limits survival and regulates various genes during larval stages of Drosophila melanogaster. J Med Invest. (2014) 61:137–50. doi: 10.2152/jmi.61.137
14. Noma T. Dynamics of nucleotide metabolism as a supporter of life phenomena. J Med Invest. (2005) 52:127–36. doi: 10.2152/jmi.52.127
15. Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, et al. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. (2009) 41:106–11. doi: 10.1038/ng.278
16. Pannicke U, Honig M, Hess I, Friesen C, Holzmann K, Rump EM, et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. (2009) 41:101–5. doi: 10.1038/ng.265
17. Dzeja PP, Chung S, Faustino RS, Behfar A, Terzic A. Developmental enhancement of adenylate kinase-AMPK metabolic signaling axis supports stem cell cardiac differentiation. PLoS ONE. (2011) 6:e19300. doi: 10.1371/journal.pone.0019300
18. Fujisawa K, Murakami R, Horiguchi T, Noma T. Adenylate kinase isozyme 2 is essential for growth and development of Drosophila melanogaster. Comp Biochem Physiol. (2009) 153:29–38. doi: 10.1016/j.cbpb.2009.01.006
19. Zhang S, Nemutlu E, Dzeja P. Metabolomic profiling of adenylate kinase AK1-/- and AK2+/- transgenic mice: effect of physical stress. Circulation. (2010) 122:A20435.
20. Ghaloul-Gonzalez L, Mohsen AW, Karunanidhi A, Seminotti B, Chong H, Madan-Khetarpal S, et al. Reticular dysgenesis and mitochondriopathy induced by adenylate kinase 2 deficiency with atypical presentation. Sci Rep. (2019) 9:15739. doi: 10.1038/s41598-019-51922-2
21. Panayiotou C, Solaroli N, Karlsson A. The many isoforms of human adenylate kinases. Int J Biochem Cell Biol. (2014) 49:75–83. doi: 10.1016/j.biocel.2014.01.014
22. Dzeja P, Terzic A. Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int J Mol Sci. (2009) 10:1729–72. doi: 10.3390/ijms10041729
23. Dzeja PP, Zeleznikar RJ, Goldberg ND. Adenylate kinase: kinetic behavior in intact cells indicates it is integral to multiple cellular processes. Int J Mol Sci. (1998) 184:169–82. doi: 10.1007/978-1-4615-5653-4_13
24. Noda L. Adenylate kinase. In: Boyer P, editor. The Enzymes, 3rd edition. New York, NY: Academic Press. (1973). p. 279-305. doi: 10.1016/S1874-6047(08)60068-2
25. Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther. (2000) 87:189–98. doi: 10.1016/S0163-7258(00)00048-6
26. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
27. Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol. (2010) 48:725–34. doi: 10.1016/j.yjmcc.2009.12.014
28. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer cell. (2006) 9:425–34. doi: 10.1016/j.ccr.2006.04.023
29. Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma? Biochim Biophys Acta. (2011) 1807:552–61. doi: 10.1016/j.bbabio.2010.10.012
30. Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. (2007) 274:1393–418. doi: 10.1111/j.1742-4658.2007.05686.x
31. Moreno-Sanchez R, Rodriguez-Enriquez S, Saavedra E, Marin-Hernandez A, Gallardo-Perez JC. The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? Biofactors. (2009) 35:209–25. doi: 10.1002/biof.31
32. Han CY, Patten DA, Lee SG, Parks RJ, Chan DW, Harper ME, et al. p53 Promotes chemoresponsiveness by regulating hexokinase II gene transcription and metabolic reprogramming in epithelial ovarian cancer. Mol Carcinog. (2019) 58:2161–74. doi: 10.1002/mc.23106
33. Masters C. Cellular differentiation and the microcompartmentation of glycolysis. Mech Ag Dev. (1991) 61:11–22. doi: 10.1016/0047-6374(91)90003-I
34. Zahra K, Dey T, Ashish Mishra SP, Pandey U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol. (2020) 10:159. doi: 10.3389/fonc.2020.00159
35. Klepinin A, Chekulayev V, Timohhina N, Shevchuk I, Tepp K, Kaldma A, et al. Comparative analysis of some aspects of mitochondrial metabolism in differentiated and undifferentiated neuroblastoma cells. J Bioenerg Biomembr. (2014) 46:17–31. doi: 10.1007/s10863-013-9529-5
36. Ounpuu L, Klepinin A, Pook M, Teino I, Peet N, Paju K, et al. 2102Ep embryonal carcinoma cells have compromised respiration and shifted bioenergetic profile distinct from H9 human embryonic stem cells. Biochim Biophys Acta Gen Subj. (2017) 1861:2146–54. doi: 10.1016/j.bbagen.2017.05.020
37. Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, Arandarcikaite O, et al. The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta. (2010) 1797:1018–27. doi: 10.1016/j.bbabio.2010.02.005
38. Li XH, Chen XJ, Ou WB, Zhang Q, Lv ZR, Zhan Y, et al. Knockdown of creatine kinase B inhibits ovarian cancer progression by decreasing glycolysis. Int J Biochem Cell Biol. (2013) 45:979–86. doi: 10.1016/j.biocel.2013.02.003
39. Dewar BJ, Keshari K, Jeffries R, Dzeja P, Graves LM, Macdonald JM. Metabolic assessment of a novel chronic myelogenous leukemic cell line and an imatinib resistant subline by H NMR spectroscopy. Metabolomics. (2010) 6:439–50. doi: 10.1007/s11306-010-0204-0
40. Mooney SM, Rajagopalan K, Williams BH, Zeng Y, Christudass CS, Li Y, et al. Creatine kinase brain overexpression protects colorectal cells from various metabolic and non-metabolic stresses. J Cell Biochem. (2011) 112:1066–75. doi: 10.1002/jcb.23020
41. Kaldma A, Klepinin A, Chekulayev V, Mado K, Shevchuk I, Timohhina N, et al. An in situ study of bioenergetic properties of human colorectal cancer: the regulation of mitochondrial respiration and distribution of flux control among the components of ATP synthasome. Int J Biochem Cell Biol. (2014) 55:171–86. doi: 10.1016/j.biocel.2014.09.004
42. Kaambre T, Chekulayev V, Shevchuk I, Karu-Varikmaa M, Timohhina N, Tepp K, et al. Metabolic control analysis of cellular respiration in situ in intraoperational samples of human breast cancer. J Bioenerg Biomembr. (2012) 44:539–58. doi: 10.1007/s10863-012-9457-9
43. Amamoto R, Uchiumi T, Yagi M, Monji K, Song Y, Oda Y, et al. The expression of ubiquitous mitochondrial creatine kinase is downregulated as prostate cancer progression. J Cancer. (2016) 7:50–9. doi: 10.7150/jca.13207
44. Patra S, Bera S, SinhaRoy S, Ghoshal S, Ray S, Basu A, et al. Progressive decrease of phosphocreatine, creatine and creatine kinase in skeletal muscle upon transformation to sarcoma. Febs Journal. (2008) 275:3236–47. doi: 10.1111/j.1742-4658.2008.06475.x
45. Lam YW, Yuan Y, Isaac J, Babu CV, Meller J, Ho SM. Comprehensive identification and modified-site mapping of S-nitrosylated targets in prostate epithelial cells. PLoS ONE. (2010) 5:e9075. doi: 10.1371/journal.pone.0009075
46. Klepinin A, Ounpuu L, Guzun R, Chekulayev V, Timohhina N, Tepp K, et al. Simple oxygraphic analysis for the presence of adenylate kinase 1 and 2 in normal and tumor cells. J Bioenerg Biomembr. (2016) 48:531–48. doi: 10.1007/s10863-016-9687-3
47. Balinsky D, Greengard O, Cayanis E, Head JF. Enzyme activities and isozyme patterns in human lung tumors. Cancer Res. (1984) 44:1058–62.
48. Criss WE, Litwack G, Morris HP, Weinhouse S. Adenosine triphosphate: adenosine monophosphate phosphotransferase isozymes in rat liver and hepatomas. Cancer Res. (1970) 30:370–5.
49. Chekulayev V, Mado K, Shevchuk I, Koit A, Kaldma A, Klepinin A, et al. Metabolic remodeling in human colorectal cancer and surrounding tissues: alterations in regulation of mitochondrial respiration and metabolic fluxes. Biochem Biophys Rep. (2015) 4:111–25. doi: 10.1016/j.bbrep.2015.08.020
50. Vasseur S, Malicet C, Calvo EL, Dagorn JC, Iovanna JL. Gene expression profiling of tumours derived from ras(V12)/EIA-transformed mouse embryonic fibroblasts to identify genes required for tumour development. Mol Cancer. (2005) 4:4. doi: 10.1186/1476-4598-4-4
51. Speers C, Tsimelzon A, Sexton K, Herrick AM, Gutierrez C, Culhane A, et al. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin Cancer Res. (2009) 15:6327–40. doi: 10.1158/1078-0432.CCR-09-1107
52. Lamb R, Bonuccelli G, Ozsvari B, Peiris-Pages M, Fiorillo M, Smith DL, et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget. (2015) 6:30453–71. doi: 10.18632/oncotarget.5852
53. Roesli C, Borgia B, Schhemann C, Gunthert M, Wunderli-Allenspach H, Giavazzi R, et al. Comparative analysis of the membrane proteome of closely related metastatic and nonmetastatic tumor cells. Cancer Res. (2009) 69:5406–14. doi: 10.1158/0008-5472.CAN-08-0999
54. Kim H, Lee HJ, Oh Y, Choi SG, Hong SH, Kim HJ, et al. The DUSP26 phosphatase activator adenylate kinase 2 regulates FADD phosphorylation and cell growth. Nat Commun. (2014) 5:4351. doi: 10.1038/ncomms4351
55. Liu H, Pu Y, Amina Q, Wang Q, Zhang M, Song J, et al. Prognostic and therapeutic potential of Adenylate kinase 2 in lung adenocarcinoma. Sci Rep. (2019) 9:17757. doi: 10.1038/s41598-019-53594-4
56. Jan YH, Tsai HY, Yang CJ, Huang MS, Yang YF, Lai TC, et al. Adenylate kinase-4 is a marker of poor clinical outcomes that promotes metastasis of lung cancer by downregulating the transcription factor ATF3. Cancer Res. (2012) 72:5119–29. doi: 10.1158/0008-5472.CAN-12-1842
57. Lanning NJ, Looyenga BD, Kauffman AL, Niemi NM, Sudderth J, DeBerardinis RJ, et al. A mitochondrial RNAi screen defines cellular bioenergetic determinants and identifies an adenylate kinase as a key regulator of ATP levels. Cell Rep. (2014) 7:907–17. doi: 10.1016/j.celrep.2014.03.065
58. Bai DM, Zhang JF, Li TT, Hang RL, Liu Y, Tian YL, et al. The ATPase hCINAP regulates 18S rRNA processing and is essential for embryogenesis and tumour growth. Nat Commun. (2016) 7:12310. doi: 10.1038/ncomms12310
59. Ji Y, Yang C, Tang Z, Yang Y, Tian Y, Yao H, et al. Adenylate kinase hCINAP determines self-renewal of colorectal cancer stem cells by facilitating LDHA phosphorylation. Nat Commun. (2017) 8:15308. doi: 10.1038/ncomms15308
60. Hansel DE, Rahman A, House M, Ashfaq R, Berg K, Yeo CJ, et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. (2004) 10(18 Pt 1):6152–8. doi: 10.1158/1078-0432.CCR-04-0285
61. Karhemo PR, Hyvonen M, Laakkonen P. Metastasis-associated cell surface oncoproteomics. Front Pharmacol. (2012) 3:192. doi: 10.3389/fphar.2012.00192
62. Lee HJ, Pyo JO, Oh Y, Kim HJ, Hong SH, Jeon YJ, et al. AK2 activates a novel apoptotic pathway through formation of a complex with FADD and caspase-10. Nat Cell Biol. (2007) 9:1303–10. doi: 10.1038/ncb1650
63. Kumari R, Deshmukh RS, Das S. Caspase-10 inhibits ATP-citrate lyase-mediated metabolic and epigenetic reprogramming to suppress tumorigenesis. Nat Commun. (2019) 10:4255. doi: 10.1038/s41467-019-12194-6
64. Oshima K, Saiki N, Tanaka M, Imamura H, Niwa A, Tanimura A, et al. Human AK2 links intracellular bioenergetic redistribution to the fate of hematopoietic progenitors. Biochem Biophys Res Commun. (2018) 497:719–25. doi: 10.1016/j.bbrc.2018.02.139
65. Dzeja PP, Hoyer K, Tian R, Zhang S, Nemutlu E, Spindler M, et al. Rearrangement of energetic and substrate utilization networks compensate for chronic myocardial creatine kinase deficiency. J Physiol. (2011) 589:5193–211. doi: 10.1113/jphysiol.2011.212829
66. Wu Z, Gong Q, Yu Y, Zhu J, Li W. Knockdown of circ-ABCB10 promotes sensitivity of lung cancer cells to cisplatin via miR-556-3p/AK4 axis. BMC Pulm Med. (2020) 20:10. doi: 10.1186/s12890-019-1035-z
67. Zang C, Zhao F, Hua L, Pu Y. The miR-199a-3p regulates the radioresistance of esophageal cancer cells via targeting the AK4 gene. Cancer Cell Int. (2018) 18:186. doi: 10.1186/s12935-018-0689-6
68. Fujisawa K, Terai S, Takami T, Yamamoto N, Yamasaki T, Matsumoto T, et al. Modulation of anti-cancer drug sensitivity through the regulation of mitochondrial activity by adenylate kinase 4. J Exp Clin Cancer Res. (2016) 35:48. doi: 10.1186/s13046-016-0322-2
69. Lei W, Yan C, Ya J, Yong D, Yujun B, Kai L. MiR-199a-3p affects the multi-chemoresistance of osteosarcoma through targeting AK4. BMC Cancer. (2018) 18:631. doi: 10.1186/s12885-018-4460-0
70. Liu RJ, Strom AL, Zhai JJ, Gal J, Bao SL, Gong WM, et al. Enzymatically inactive adenylate kinase 4 interacts with mitochondrial ADP/ATP translocase. Int J Biochem Cell B. (2009) 41:1371–80. doi: 10.1016/j.biocel.2008.12.002
71. Kong F, Binas B, Moon JH, Kang SS, Kim HJ. Differential expression of adenylate kinase 4 in the context of disparate stress response strategies of HEK293 and HepG2 cells. Arch Biochem Biophys. (2013) 533:11–7. doi: 10.1016/j.abb.2013.02.014
72. Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. (2007) 39:211–22. doi: 10.1007/s10863-007-9094-x
73. Nelson BD, Kabir F. Adenylate Kinase Is a Source of Atp for Tumor Mitochondrial Hexokinase. Biochimica Et Biophysica Acta. (1985) 841:195–200. doi: 10.1016/0304-4165(85)90021-2
74. Choi YK, Park KG. Targeting Glutamine Metabolism for Cancer Treatment. Biomol Ther (Seoul). (2018) 26:19–28. doi: 10.4062/biomolther.2017.178
75. Deshmukh A, Deshpande K, Arfuso F, Newsholme P, Dharmarajan A. Cancer stem cell metabolism: a potential target for cancer therapy. Mol Cancer. (2016) 15:555. doi: 10.1186/s12943-016-0555-x
76. Ross RA, Spengler BA. Human neuroblastoma stem cells. Semin Cancer Biol. (2007) 17:241–7. doi: 10.1016/j.semcancer.2006.04.006
77. Song IS, Jeong JY, Jeong SH, Kim HK, Ko KS, Rhee BD, et al. Mitochondria as therapeutic targets for cancer stem cells. World J Stem Cells. (2015) 7:418–27. doi: 10.4252/wjsc.v7.i2.418
78. Dzeja PP, Bortolon R, Perez-Terzic C, Holmuhamedov EL, Terzic A. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc Natl Acad Sci USA. (2002) 99:10156–61. doi: 10.1073/pnas.152259999
79. Jan YH, Lai TC, Yang CJ, Lin YF, Huang MS, Hsiao M. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1alpha to drive lung adenocarcinoma metastasis. J Hematol Oncol. (2019) 12:12. doi: 10.1186/s13045-019-0698-5
80. Jan YH, Lai TC, Yang CJ, Huang MS, Hsiao M. A co-expressed gene status of adenylate kinase 1/4 reveals prognostic gene signature associated with prognosis and sensitivity to EGFR targeted therapy in lung adenocarcinoma. Sci Rep. (2019) 9:12329. doi: 10.1038/s41598-019-48243-9
81. Xin F, Yao DW, Fan L, Liu JH, Liu XD. Adenylate kinase 4 promotes bladder cancer cell proliferation and invasion. Clin Exp Med. (2019) 19:525–34. doi: 10.1007/s10238-019-00576-5
82. Zhang J, Yin YT, Wu CH, Qiu RL, Jiang WJ, Deng XG, et al. AK4 promotes the progression of HER2-positive breast cancer by facilitating cell proliferation and invasion. Dis Markers. (2019) 2019:8186091. doi: 10.1155/2019/8186091
83. Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. (2008) 451:345–U11. doi: 10.1038/nature06489
84. Adams JM, Strasser A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. (2008) 68:4018–21. doi: 10.1158/0008-5472.CAN-07-6334
85. Kaur H, Lakatos-Karoly A, Vogel R, Noll A, Tampe R, Glaubitz C. Coupled ATPase-adenylate kinase activity in ABC transporters. Nat Commun. (2016) 7:13864. doi: 10.1038/ncomms13864
86. Dzeja PP, Terzic A. Phosphotransfer reactions in the regulation of ATP-sensitive K+ channels. Faseb J. (1998) 12:523–9. doi: 10.1096/fasebj.12.7.523
87. Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, et al. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA. (2001) 98:7623–8. doi: 10.1073/pnas.121038198
88. Bhaskara V, Dupre A, Lengsfeld B, Hopkins BB, Chan A, Lee JH, et al. Rad50 adenylate kinase activity regulates DNA tethering by Mre11/Rad50 complexes. Mol Cell. (2007) 25:647–61. doi: 10.1016/j.molcel.2007.01.028
89. Boissan M, Schlattner U, Lacombe ML. The NDPK/NME superfamily: state of the art. Lab Invest. (2018) 98:164–74. doi: 10.1038/labinvest.2017.137
90. Gonzalez A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: the yin and yang of cellular nutrient sensing and growth control. Cell Metab. (2020) 31:472–92. doi: 10.1016/j.cmet.2020.01.015
91. Collavin L, Lazarevic D, Utrera R, Marzinotto S, Monte M, Schneider C. wt p53 dependent expression of a membrane-associated isoform of adenylate kinase. Oncogene. (1999) 18:5879–88. doi: 10.1038/sj.onc.1202970
92. Sholihah MM, Indarto D, Pramana TY. The inhibitory effect ofAndrographis paniculataextract on proliferation of breast cancer cell line. Mat Sci Eng. (2019) 546:062029. doi: 10.1088/1757-899X/546/6/062029
93. Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. (2011) 39:1–13. doi: 10.1042/BST0390001
94. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev. (2007) 8:774–85. doi: 10.1038/nrm2249
95. Dasgupta B, Chhipa RR. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol Sci. (2016) 37:192–206. doi: 10.1016/j.tips.2015.11.007
96. Hardie DG, Alessi DR. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol. (2013) 11:36. doi: 10.1186/1741-7007-11-36
97. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. (2009) 9:563–75. doi: 10.1038/nrc2676
98. Takaine M, Imamura H, Yoshida S. AMP-activated protein kinase and adenylate kinase prevent the ATP catastrophe and cytotoxic protein aggregation. bioRxiv. (2019) 801738. doi: 10.1101/801738
99. Plaideau C, Liu JM, Hartleib-Geschwindner J, Bastin-Coyette L, Bontemps F, Oscarsson J, et al. Overexpression of AMP-metabolizing enzymes controls adenine nucleotide levels and AMPK activation in HEK293T cells. Faseb Journal. (2012) 26:2685–94. doi: 10.1096/fj.11-198168
100. Plaideau C, Lai YC, Kviklyte S, Zanou N, Lofgren L, Andersen H, et al. Effects of pharmacological amp deaminase inhibition and ampd1 deletion on nucleotide levels and AMPK activation in contracting skeletal muscle. Chem Biol. (2014) 21:1497–510. doi: 10.1016/j.chembiol.2014.09.013
101. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:57. doi: 10.1186/s40425-018-0360-8
102. Olson LK, Schroeder W, Robertson RP, Goldberg ND, Walseth TF. Suppression of adenylate kinase catalyzed phosphotransfer precedes and is associated with glucose-induced insulin secretion in intact HIT-T15 cells. J Biol Chem. (1996) 271:16544–52. doi: 10.1074/jbc.271.28.16544
103. Wang F, Liu Y, Jiang J, Qin Y, Ge X, Li J, et al. High expression of AMPD2 and obesity are associated with poor prognosis in colorectal cancer. Int J Clin Exp Pathol. (2018) 11:216–23.
104. Parr C, Jiang WG. Quantitative analysis of lymphangiogenic markers in human colorectal cancer. Int J Oncol. (2003) 23:533–9. doi: 10.3892/ijo.23.2.533
105. Spychala J, Lazarowski E, Ostapkowicz A, Ayscue LH, Jin A, Mitchell BS. Role of estrogen receptor in the regulation of ecto-5'-nucleotidase and adenosine in breast cancer. Clin Cancer Res. (2004) 10:708–17. doi: 10.1158/1078-0432.CCR-0811-03
106. Klepinin A, Ounpuu L, Mado K, Truu L, Chekulayev V, Puurand M, et al. The complexity of mitochondrial outer membrane permeability and VDAC regulation by associated proteins. J Bioenerg Biomembr. (2018) 50:339–54. doi: 10.1007/s10863-018-9765-9
Keywords: adenylate kinase, energy metabolism, phosphotransfer, mitochondria, cancer
Citation: Klepinin A, Zhang S, Klepinina L, Rebane-Klemm E, Terzic A, Kaambre T and Dzeja P (2020) Adenylate Kinase and Metabolic Signaling in Cancer Cells. Front. Oncol. 10:660. doi: 10.3389/fonc.2020.00660
Received: 15 February 2020; Accepted: 08 April 2020;
Published: 19 May 2020.
Edited by:
Stefano Falone, University of L'Aquila, ItalyReviewed by:
Daria Capece, Imperial College London, United KingdomPaul Dent, Virginia Commonwealth University, United States
Elisabetta Benedetti, University of L'Aquila, Italy
Copyright © 2020 Klepinin, Zhang, Klepinina, Rebane-Klemm, Terzic, Kaambre and Dzeja. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandr Klepinin, YWxla3NhbmRyLmtsZXBpbmluQGtiZmkuZWU=; Petras Dzeja, ZHplamEucGV0cmFzQG1heW8uZWR1