 Sherly Mardiana1,2
Sherly Mardiana1,2 Saar Gill1,2*
Saar Gill1,2*- 1Center for Cellular Immunotherapies, University of Pennsylvania School of Medicine, Philadelphia, PA, United States
- 2Division of Hematology-Oncology and Center for Cellular Immunotherapies, University of Pennsylvania, PA, United States
Relapse after conventional chemotherapy remains a major problem in patients with myeloid malignancies such as acute myeloid leukemia (AML), and the major cause of death after diagnosis of AML is from relapsed disease. The only potentially curative treatment option currently available is allogeneic hematopoietic stem cell transplantation (allo-HSCT), which through its graft-vs.-leukemia effects has the ability to eliminate residual leukemia cells. Despite its long history of success however, relapse following allo-HSCT is still a major challenge and is associated with poor prognosis. In the field of adoptive therapy, CD19-targeted chimeric antigen receptor (CAR) T cells have yielded remarkable clinical success in certain types of B-cell malignancies, and substantial efforts aimed at translating this success to myeloid malignancies are currently underway. While complete ablation of CD19-expressing B cells, both cancerous and healthy, is clinically tolerated, the primary challenge limiting the use of CAR T cells in myeloid malignancies is the absence of a dispensable antigen, as myeloid antigens are often co-expressed on normal hematopoietic stem/progenitor cells (HSPCs), depletion of which would lead to intolerable myeloablation. This review provides a discussion on the current state of CAR T cell therapy in myeloid malignancies, limitations for clinical translation, as well as the most recent approaches to overcome these barriers, through various genetic modification and combinatorial strategies in an attempt to make CAR T cell therapy a safe and viable option for patients with myeloid malignancies.
Introduction
Myeloid malignancies are clonal diseases of the hematopoietic stem or progenitor cells (HSPCs) that arise from genetic and/or epigenetic changes, resulting in deleterious effects on critical pathways such as cell differentiation, proliferation, and self-renewal. These malignancies can be categorized into myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), MDS/MPN such as chronic myelomonocytic leukemia (CMML), and acute myeloid leukemia (AML) (1). Currently, the only therapeutic modality that is potentially curative in hematological malignancies is allogeneic hematopoietic stem cell transplantation (allo-HSCT). In addition to anti-tumor effects from chemotherapy and/or total body irradiation given to patients prior to allo-HSCT, the overall therapeutic effect of allo-HSCT also relies on the graft-vs.-leukemia (GvL) phenomenon, that occurs when donor T cells recognize foreign antigens on the host's hematopoietic tissues and subsequently mediate tumor elimination (2). This GvL phenomenon demonstrates the responsiveness of myeloid diseases to T cells. However, despite the successful history of HSCT with ~20,000 people undergoing HSCT annually in the US alone (3), relapse following allo-HSCT is still a major challenge and is associated with poor prognosis.
Over the years, a plethora of immunotherapy-based treatment approaches for hematological malignancies have been developed and tested in both pre-clinical and clinical settings. Among these, the most striking progress has occurred in B-lymphoid malignancies such as acute lymphocytic leukemia (ALL) and certain lymphomas, where immunotherapy agents such as chimeric antigen receptor (CAR) T cells, bispecific T cell engagers (BiTEs), and immune checkpoint blockade (ICB) particularly in Hodgkin lymphoma, have shown robust clinical responses (4–7). Building on these successes, substantial efforts have been directed toward translating these approaches to treating myeloid malignancies. BiTEs recognizing the T cell molecule CD3 and a myeloid cell antigen such as CLL-1, CD123, or CD33 are under investigation (8). The use of ICB as monotherapy in AML and MDS has unfortunately shown only modest clinical responses, and ICB is now being tested in combination with hypomethylating agents (HMAs), which are thought to have immunomodulatory effects including enhancing tumor cell antigen presentation and co-stimulatory molecule expression (9). Indeed, HMAs such as decitabine and azacitidine have been reported to upregulate the expression of several cancer testis antigens such as NY-ESO-1 and MAGE (10, 11).
In the context of cellular therapy, the US Food and Drug Administration (FDA) approval of two CD19-directed CAR T cells products, KymriahTM and YescartaTM, revolutionized the field of immunotherapy and ignited our enthusiasm for translating this promising technology to the treatment of myeloid malignancies. However, unlike B cell malignancies that express several antigens exclusive to the B cell lineage such as CD19, CD20, CD22, or BCMA in the case of myeloma, most tumor antigens targetable in myeloid malignancies are shared with a wide range of healthy cells including HSPCs. Moreover, whereas B cell aplasia that occurs as a result of B cell antigen-specific CAR T cell therapy is clinically benign and can be managed with infusions of intravenous immunoglobulin, prolonged myeloablation is not feasible due to a risk of infection and transfusion dependence. Herein, we provide an overview of the current state as well as future prospects of CAR T cell therapy options for patients with myeloid malignancies, specifically focusing on AML.
The Current State of CAR T Cell Therapy in AML
The first reported clinical trial that demonstrated biological activity of CAR T cells in AML was published in 2013 by Ritchie et al. utilizing a second generation CD28-ζ CAR directed against the Lewis Y antigen. Although only limited efficacy was observed, this was an important study given its demonstration of CAR T cell biological activity in AML patients in the absence of overt hematopoietic toxicity (12). Currently, there are more than twenty CAR T cell clinical trials enrolling patients with AML, mostly targeting CLL-1, CD33, or CD123 (13). CD33 and CD123 are both appealing target antigens, given they are both almost ubiquitously expressed on AML blasts, though they are also expressed by healthy HSPCs (14). While mature clinical data have yet to be published, there have been a number of case reports and pilot studies reporting the use of CAR T cells in AML (15–18). For example, a partial response to CD33-specific CAR T cells was described in a case report of a 41-year-old male. The patient had a marked transient reduction of AML blasts in the marrow before the disease progressed at 9 weeks post CAR T cell treatment. A significant cytokine release syndrome (CRS) was observed in this patient (15).
CLL-1 (also known as CLEC12A) is also an attractive target for CAR T cells owing to its high expression in AML and reported absence in healthy HSPCs, though in our hands CLL-1 is detected on all monocytes as well as on some early hematopoietic cells (19). CLL-1 is also known to be rarely expressed on non-hematological cells (20). It has been reported that more than 60% AML samples with CD33 expression also express CLL-1 (21), leading to the idea of creating dual-specific CAR T cells for both antigens. Supported by pre-clinical data showing specificity and anti-tumor potency of dual CD33-CLL-1 CAR T cells, a first-in-human trial using these dual CAR T cells was conducted. A preliminary report by Liu et al. described that two patients with refractory/relapsed (R/R) AML were treated with the dual CAR T cells following fludarabine and cyclophosphamide preconditioning. Both of these patients experienced measurable residual disease (MRD)−ve complete remission accompanied by pancytopenia within 3 weeks of CAR T cell infusion. These patients later underwent anti-thymocyte globulin (ATG)-based HSCT with subsequent hematopoietic recovery (18, 20).
At the University of Pennsylvania, a clinical trial of CD123-specific CAR T cells manufactured via mRNA electroporation was recently completed. This method, rather than lentiviral transduction, was chosen to prevent long-term CAR T cell persistence thus avoiding the risk of severe myeloablation. Patients with R/R AML received lymphodepleting chemotherapy prior to treatment with transiently acting CAR T cells. In this study, although no measurable anti-tumor responses were observed, some level of CAR T cell bioactivity was found, as demonstrated by the cytokine release syndrome (CRS) and/or fever experienced by all treated patients. As expected with mRNA-based CAR T cells, only transient CAR T cell detection in vivo was observed. Of note, no overt vascular, hematologic or neurologic toxicity was reported despite expression of the target antigen on healthy hematopoietic tissues and some small-caliber blood vessels (17). This favorable safety profile supported the development of a clinical trial using a lentiviral transduction system (CD123-4-1BB-ζ), which is currently open (clinicaltrials.gov identifier: 03766126). In addition, early results from an ongoing trial at the City of Hope Medical Center (clinicaltrials.gov identifier: 02159495) were also recently reported. This study employed second generation CD28-ζ CAR T cells targeting CD123 manufactured by lentiviral transduction. Six patients with refractory AML were treated with either 50 or 200 million CAR T cells after being preconditioned with chemotherapy. Results revealed one of the two patients treated with the lower dose of CAR T cells experienced a transient morphologic leukemia-free state. This patient then received a subsequent infusion of CAR T cells 3 months later and achieved blast reduction from 77.9 to 0.9%. Encouragingly, two of the four patients receiving a higher dose of 200 million CAR T cells experienced complete remission and went on to HSCT. The other two patients had transient partial responses. Grade 1 or 2 CRS was reported in most patients, but no dose limiting toxicity including cytopenia was observed at the time of the report (22). As of February 2019, 24 patients were reported to have been enrolled (23), and further results on additional patients from this study are eagerly awaited.
Limitations of CAR T Cell Therapy in AML and Strategies to Overcome Them
Lack of a Leukemia-Specific Antigen for Use as Target for CAR T Cells
The fundamental biological barrier limiting the application of CAR T cell therapy in AML is the absence of an AML-specific antigen. AML cells express various cell surface antigens including CD123, CD34, CD33, and many others. However, these same antigens are also shared by healthy HSPCs and their myeloid and/or lymphoid progenitors (24). CD123-directed and CD33-directed CAR T cells have both shown highly potent anti-tumor activity in pre-clinical models (25–27). However, CAR T cells are unable to differentiate between normal and cancerous cells. Unlike in the case of CD19 CAR T cells whereby complete elimination of both normal and cancerous B cells is clinically benign, in stark contrast, prolonged myeloablation as a result of CAR T cells targeting myeloid antigens shared with normal myeloid progenitors is ultimately fatal due to neutropenic infections and bleeding complications. In order to mitigate such toxicities, substantial efforts have been invested in developing and testing a variety of solutions.
Limiting CAR T Cell Persistence to Prevent Protracted Myeloablation
One potential strategy to prevent the risk of bone marrow failure following CAR T cells that target the highly expressed but non-leukemia specific antigens such as CD33 and CD123 is to limit long-term CAR T cell persistence in vivo. Numerous approaches have been tested both in pre-clinical and clinical studies to incorporate a “safety switch” into T cells, allowing for elimination of T cells in vivo if needed. A suicide gene that has long been utilized in T cell therapy is the herpes simplex virus-thymidine kinase (HSV-tk), which allows for selective depletion of expressing cells upon administration of a prodrug. In this case, HSV-tk is able to turn the prodrug into a toxic compound that halts DNA replication, hence resulting in cell death (28). The use of HSV-tk however is limited by immunogenicity of the viral enzyme and the relatively long latency to activation, which is not suitable for managing toxicity that requires immediate termination (29). A more advanced suicide system employs the co-expression of inducible caspase 9 (iCasp9) in T cells. This construct fuses the intracellular domain of caspase 9, a known pro-apoptotic protein, to a drug-binding domain from FK506-binding protein. Administration of a synthetic molecule drug called AP1903 leads to dimerization of the fusion proteins and ultimately rapid ablation of T cells (30, 31). The iCasp9 suicide system was tested clinically in the setting of haploidentical stem cell transplantation (32), and has also been explored in the setting of CAR T cell therapy in pre-clinical study by Hoyos et al. (33). Subsequently, the iCasp9 suicide system has been incorporated in the CAR construct of various clinical trials (clinicaltrials.gov identifier: 02992210, 01822652) (29), however to our knowledge AP1903 administration has never been required and hence there is no formal proof of its efficacy in the CAR T cell setting.
Another option for a safety switch is to engineer T cells to co-express a truncated well-characterized surface antigen against which clinically approved monoclonal antibodies exist (for example truncated EGFR targetable by cetuximab, and truncated CD20 targetable by rituximab). This platform could allow antibody-based elimination of the adoptively transferred T cells by complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity (ADCC) (34). There are also non-specific drugs that eliminate the transferred T cells as well as endogenous T cells, including anti-thymocyte globulin (ATG), or the anti-CD52 monoclonal antibody alemtuzumab (35). A completely different approach to limit CAR T cell persistence that does not require administration of exogenous antibodies is to use mRNA electroporation for incorporation of the CAR into T cells, whereby CAR T cell function is inherently limited due to degradation of the mRNA (17).
It is increasingly apparent that the choice of co-stimulatory domain in the CAR constructs could affect the CAR T cell ability to persist in vivo. Comparison between CD28 and 4-1BB has revealed superior persistence of CAR T cells incorporating 4-1BB signaling domain over those with CD28 (36). Although never directly compared in the context of anti-AML CAR T cells, the use of CD28 co-stimulatory domain may lead to shorter CAR T cell persistence and could therefore impact the duration of CAR T cell-induced myeloablation. Notably, accumulating evidence from CD19-directed CAR T cell trials in B-cell leukemia suggests that durable clinical response appears to be associated with CAR T cell expansion and persistence, and failure to persist seems to correlate with increased risk of relapse (37, 38). The optimal duration of CAR T cell persistence for disease response, at least for CD19 CAR T cells, is predicted to be at least 3–6 months (4, 39). Therefore, placing a limit on CAR T cell persistence will likely create a new problem of limiting CAR T cell therapeutic potential and consequently increasing the risk of disease relapse.
Creating an Artificial “AML-Specific” Antigen by Genetic Editing of Allograft
A novel strategy to allow for CAR T cell long-term persistence without the unwanted prolonged myeloablation effect is to edit out the CAR target antigen, for example CD33, from a donor allograft. The idea is to transplant CD33−/− HSPCs into the patient. Once engrafted, CD33-specific CAR T cells manufactured from the same donor may then be given to the patient, with the goal of allowing for normal hematopoiesis by the CD33−/− allograft in the presence of continued CAR T cell persistence. Indeed, our group has evaluated this approach in both in vitro and in vivo studies. Using the CRISPR/Cas9 technology, we demonstrated that CD33−/− HSPCs and their progeny were resistant to CD33-directed CAR T cells in murine xenograft. Importantly, such CD33 deletion did not impair the hematopoietic and immunological function of the HSPCs and their progeny in murine xenograft and in non-human primate models (26). A clinical trial involving the use of allogeneic CD33−/− HSCT prior to CAR T cell infusion is currently being devised at the University of Pennsylvania for patients with R/R AML. During the conduct of the trial, careful assessment of potential side effects will include off-target editing in HSPCs, clinical consequences of CD33 deletion in the bone marrow, as well as the effect of CAR T cells on healthy tissues that may express CD33.
Another potential antigen that may be edited using a similar approach is CD123. However, since CD123 serves a function as the alpha subunit of the IL-3 receptor, complete removal of CD123 in the hematopoietic system is predicted to have a wide range of deleterious effects, given that IL-3 is a pleiotropic cytokine involved in hematopoietic development (40). Thus, an alternative approach could include targeted removal of the epitope on the CD123 molecule that is recognized by the CAR T cells, or to knockdown (instead of completely knockout) CD123 expression in donor HSPCs to a level below the CAR T cell activation threshold, but is still sufficient to preserve normal CD123 signaling and hematopoiesis. This approach is currently under investigation.
Identifying Leukemia-Specific Neoantigens
Designing a potent yet specific treatment that is able to facilitate tumor eradication whilst sparing normal cells is considered the “holy grail” in cellular therapy. The majority of CAR T cell target antigens to date are those overexpressed on tumor cells but also expressed at lower levels on normal tissues. While such antigens, for example GD2, Lewis Y and CEA may serve as relatively safe targets for CAR T cells (provided that their expression levels on healthy vs. malignant cells are sufficiently distinguishable) (41), this kind of differential expression unfortunately does not exist for most myeloid antigens, hence prompting the search for truly tumor-specific antigens. A newly formed antigen that results specifically from a disease-causing or disease-associated mutation would be the preferred target for CAR T cells as it should be expressed by malignant cells but not healthy cells (42). The Cancer Genome Atlas Research Network has conducted a comprehensive study to examine the mutational composition of de novo AML, and a number of recurrent mutations contributing to leukemogenesis have been identified (43, 44). AML genomes are amongst those with the lowest mutational burden, and presumably present very few neoantigens (45). Nonetheless, there are some neoantigens that have been described in AML, including mutations in the metabolic enzymes IDH1 and IDH2, which are present in ~20% of de novo AML cases (46, 47). Immune recognition of these IDH1/2 mutants have been demonstrated, suggesting endogenous processing and presentation of the mutant epitopes (48). In addition, mutation in NPM1 gene is also one of the most frequent genetic alterations in patients with AML, with up to 60% patients with normal cytogenetics reportedly having NPM1 mutation (49). Similar to IDH1/2 mutants, the NPM1 mutant epitopes have also been shown to be immunogenic, inducing both CD8+ and CD4+ T cell responses (49). The existence of such neoantigens in AML, albeit at relatively low frequency, makes them an attractive target for T cell therapy. However, the proteins encoded by these disease-associated mutations are expressed intracellularly, and therefore are not accessible to the CAR.
Dysregulated splicing can also be a source of neoantigens, if it results in alternative isoforms that are distinguishable from their wildtype counterpart. A recent study reported that about a third of expressed genes in AML undergo differential RNA splicing. This may result in splice variants and therefore potential neoantigens (50). In a subsequent study from the same group, expression of two novel splice variants, one for Flt3 and another for NOTCH2, was reported to be found in 50 and 73% of AML cases, respectively, but was absent in healthy donors (51). Additionally, another AML-specific isoform was described to arise from CD44, called the CD44v6 variant. In one study, more than 60% of the AML cases showed expression of CD44v6, which was not present in normal HSPC. Unlike other disease-associated mutations that are expressed intracellularly, CD44v6 is expressed on the cell surface, making it accessible to the CAR. Indeed, CAR T cells targeting the CD44v6 variant were generated and reported to exert robust anti-tumor responses against primary AML cells, apparently whilst preserving the HSPC compartment (52).
Although CARs can only recognize antigens expressed on the cell surface, notably a recent innovative study by Rafiq et al. has demonstrated that generating a CAR that recognizes an intracellular antigen is in fact possible. In this study, a T cell receptor (TCR)-mimic CAR, whose specificity was directed toward the intracellular onco-protein WT1 that was presented on the cell surface in the context of MHC, therefore targeting a peptide-MHC complex (53). This study provided a proof-of-concept for the possibility of expanding the range of CAR recognition beyond extracellular antigens, and that intracellular tumor-specific mutant epitopes could be harnessed for CAR T cell therapy. Although not yet tested clinically in the context of CAR T cells, the clinical use of WT-1-directed T cells was recently reported in a study by Chapuis et al. in a TCR-based adoptive therapy setting (54). This study sought to investigate whether AML relapse risk following HSCT could be decreased by prophylactically giving the patients WT-1-directed T cells. Remarkably, all twelve patients who received WT-1-directed T cells post HSCT had 100% relapse-free survival rate at a median of 44 months post transfer, which was significantly higher compared to the 54% relapse-free rate observed in the accompanying comparative group of 88 patients who had similar risk AML (54).
Challenges in Manufacturing CAR T Cells
The manufacture of CAR T cells in patients with active AML may prove to be problematic. It has been previously reported that AML cells secrete soluble factors that inhibit T cell proliferation (55). A study from our group has demonstrated that the presence of AML blasts could indeed be detrimental to the ability of T cells to expand and thus negatively affect CAR T cell production (56). In our recent clinical trial experience for the treatment of R/R AML, we encountered some CD123-CAR T cell manufacturing problems, resulting in ~40% of the planned doses failing to be administered (17). It is important to note however, that another group at the City of Hope did not encounter similar difficulties (22), the difference of which may be attributable to the different CAR T cell production methods used—our group used mRNA electroporation to deliver the CAR, whereas the other group used lentiviral transduction. Another potential issue that may give rise to challenges in the generation of CAR T cells is the fact that patients with AML that are candidates for CAR T cell therapy will have most likely undergone heavy and intense treatments that may make it harder to obtain good quality T cell product for CAR T cell manufacture. It is possible that careful selection of patients may potentially address this issue, perhaps by selecting patients relatively early in their treatment course but who are likely to be chemo-refractory based on accepted prognostic markers (57). Alternatively, many groups are now evaluating the use of allogeneic product from healthy donors as the source for CAR T cell manufacture, though it comes with its own risks of graft vs. host disease (GvHD) and/or rejection of the transferred CAR T cells (58).
A number of solutions to circumvent the risks associated with allo-CAR T cell therapy have been examined. For instance, intensifying the pre-conditioning regimen may be sufficient to prevent CAR T cell rejection. In an attempt to alleviate GvHD, data from various pre-clinical studies point to a new approach involving genetic disruption of TCR from CAR T cells (59, 60). As a result, this strategy has entered the clinic, and in 2017 it was reported that two children with R/R B-ALL were successfully treated with allogeneic TCR-deficient CD19-CAR T cells (61). Additional clinical trials using these so-called universal CAR T cells are ongoing (clinicaltrials.gov identifier: 03166878, 03229876). While this approach is promising, it is important to note that there still exists a risk of GvHD caused by even very small percentage of TCR+ cells that may remain in the T cell population (62). Optimization of TCR deletion during CAR T cell manufacture would therefore be a crucial step for the clinical application of universal CAR T cells.
Numerous studies have also assessed potential alternatives to CAR T cells that may help reduce the risk of GvHD. To date, most of the CAR T cells used in the clinic are made from unselected T cells, consisting primarily of αβ T cells. An attractive option is to select a T cell subset that is less likely to induce GvHD for the manufacture of CAR T cells. As such, studies have explored the possibility of utilizing γδ T cells, which unlike αβ T cells, are not alloreactive and do not induce GvHD (63). The incorporation of CAR into γδ cells was first reported in 2004 using a first-generation CAR specific for GD2. The study reported that elevated antigen-specific tumor reactivity was observed (64). Although γδ cells represent only a small percentage of up to 5% circulating T cells, in another study using GD2-directed CAR T cells, it was demonstrated that γδ cells (both Vδ1 and Vδ2 subsets) could be successfully expanded and transduced to sufficient numbers for use in clinical studies. CAR expression by these γδ cells was shown to augment their innate cytotoxicity in a CAR-antigen specific manner (65). The clinical use of CAR γδ cells has not been reported to date, however there have been numerous clinical trials evaluating the safety and activity of adoptively transferred Vδ2 T cells (with no CAR manipulation) for multiple cancer types (66). Accruing data suggest that γδ cells may offer a safer alternative to the conventional αβ T cells for allogeneic use of CAR T cells. However, there may be differential anti-tumor and pro-tumor functions in different γδ cell subtypes (67), suggesting that careful selection of γδ T cell subsets to use for CAR manipulation will be of high importance.
Alternatively, other effector cells such as NKT and NK cells may also be used for adoptive therapy to mitigate GvHD risks. CAR incorporation into NKT cells has been done in some pre-clinical studies against several tumor antigens (68, 69). Similarly, NK cells engineered to express CAR (CAR-NK) have also been tested in various in vivo models, using both primary human NK cells and the NK-92 cell line, with the goal of augmenting their cytotoxic function while exploiting their innate mechanisms for detecting and killing cancerous cells (70, 71). A number of CAR-NK cells are currently being tested in phase I/II clinical trials, mostly targeting CD19 but also other antigens including CD7, CD33, MUC-I, and Her2 (72). Of note, insufficient numbers of NK cells that can be used for therapy, as well as their lack of in vivo persistence, have long posed a significant limitation on NK cell-based therapeutic approaches. A phase I/II dose escalation study of CAR-NK cells has been initiated to address this issue in patients with B-cell malignancies (clinicaltrials.gov identifier: 03056339). At present, increasing NK cell expansion and persistence remains an active area of study. Encouragingly, a recent publication reported that inclusion of IL-15 transgene can lead to successful expansion of cord blood-derived CAR123-NK cells (71).
Immunosuppression Induced by AML
Though AML has long been known to be immune-responsive, as highlighted by the graft vs. leukemia effects following allo-HSCT (2), accumulating evidence has suggested AML to also be an immunosuppressive and/or immune-evasive disease (73). A wide range of mechanisms by which AML causes immune-evasion have been reported, the most compelling of which is acquired partial uniparental disomy (UPD) of chromosome 6p, which has been reported in AML patients relapsing after haploidentical HSCT. UPD of chromosome 6p, where the human leukocyte antigen (HLA) is encoded, leads to the loss of the mismatched HLA haplotype, rendering the host's leukemia unrecognizable by donor T cells (74, 75). Notably, downregulation of MHC class I and/or MHC class II has also been observed in AML patients (76, 77), and a recent study by Christopher et al. reported MHC class II downregulation to be associated with AML relapse after transplantation (77). While these specific mechanisms are not likely to be relevant for post-CART cell relapse, they are reminiscent of recent observations regarding loss of the target epitope after anti-CD19 CART cell therapy (78). Various other immunosuppressive mechanisms have also been described for AML, most of which are based on in vitro and pre-clinical observation. It is important to identify the immunosuppressive axes responsible for AML immune evasion in order to uncover potential therapeutic targets and thus develop appropriate therapies. Discussed below are some of the known pathways through which AML can induce immunosuppression (Figure 1).
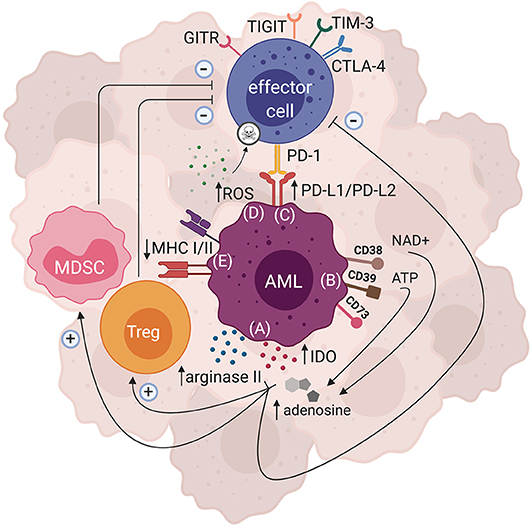
Figure 1. Mechanisms of AML-induced immunosuppression. There are several immunosuppressive pathways that have been described for AML. Both direct and indirect mechanisms can ultimately lead to AML immune escape. (A) Enzymes such as arginase II and IDO can be expressed by AML blasts, leading to production of metabolites that hinder effector cell function and proliferation, while polarizing the tumor microenvironment to become more immunosuppressive by favoring Treg and MDSC expansion. (B) AML blasts can express ectonucleotideases such as CD38, CD39, and CD73 that are involved in the breakdown of ATP and NAD+ to adenosine, which subsequently dampens effector cell function and enhances the activity of immunosuppressive cells. (C) Inhibitory ligands such as PD-L1 and PD-L2 can be expressed by AML blasts, and upon binding with their cognate PD-1 receptor may lead to effector cell suppression. Expression of other inhibitory receptors such as GITR, TIGIT, TIM-3, and CTLA-4 have also been shown in AML. (D) AML blasts are able to produce large amounts of ROS that subsequently trigger apoptosis of effector cells. (E) AML blasts can also downregulate their MHCI and/or MHCII expression, thus impairing their antigen presentation resulting in immune evasion.
Secretion of Immunosuppressive Soluble Factors
T cell dysfunction observed in the presence of AML blasts may be attributable, in part, to the immunosuppressive soluble factors secreted by AML blasts (55, 79, 80). For example, the activity of arginase II enzyme has been shown to be significantly elevated in the plasma of patients with AML in comparison to normal donors. When T cells are cultured with the plasma from these patients, a decrease in T cell proliferation was observed, and this could be rescued by replacement of arginine (80). In this study, the production of arginase II by AML blasts was reported to not only directly dampen T cell activity, but also polarize the monocyte population toward an immunosuppressive phenotype (80). Arginase inhibitors are in clinical development for advanced solid malignancies (clinicaltrials.gov identifier: 02903914) (81).
Another enzyme that has attracted interest in AML and numerous other malignancies is indoleamine 2,3 dioxygenase (IDO). This enzyme is involved in the oxidation of tryptophan to N-formylkynurenine, and is expressed at high levels in activated dendritic cells (DCs) and macrophages. The breakdown of tryptophan is known to hinder T cell proliferation and differentiation (82). In addition, the activity of IDO enzyme has also been reported to promote Treg conversion and enhance their immunosuppressive function (83–85). Increased levels of kynurenine are detected in patients with AML and correlated with diminished overall survival (86). AML blasts may be the source of the elevated levels of kynurenine seen in patients, given that they express IDO, both constitutively or after IFNγ exposure. This is further supported by a finding that shows direct correlation between higher Treg population in patients with AML and the blast IDO expression (87). Overall, the immunosuppressive effects of IDO make it an appealing therapeutic target, and efforts to block this pathway have led to the development of IDO inhibitors that are currently undergoing clinical testing in various types of malignancy including AML (clinicaltrials.gov identifier: 02835729) (88). In the context of CAR T cell therapy, several approaches that limit IDO expression and activity have been shown to improve CAR T cell efficacy in pre-clinical models. IDO1 gene is a putative target of miR-153, and overexpression of miR-153 was reported to suppress IDO expression and ultimately led to better CAR T cell efficacy in an in vivo model (89). Another study showed that the use of lymphodepleting drugs frequently used for pre-conditioning regimen, cyclophosphamide and fludarabine, downregulated IDO expression and augmented CD19-CAR T cell activity in vivo (90). Together, these observations support the combinatorial potential of IDO inhibition and CAR T cell therapy, and warrant further investigations including in myeloid malignancies.
Immunosuppressive Cellular Compartment
Cancer cells are able to recruit and/or promote the expansion of immunosuppressive cells including Tregs and myeloid-derived suppressor cells (MDSCs) (91). Increased Treg frequency has been reported in peripheral blood and bone marrow of patients with AML, and seems to correlate with poor prognosis (92, 93). AML cells can also produce large amounts of adenosine, as a result of adenosine triphosphate (ATP) or nicotinamide adenine dinucleotide (NAD+) breakdown mediated by ectonucleotideases that are expressed not only by the AML blasts, but also Tregs. The resulting adenosine accumulation leads to suppression of effector T cell function as well as augmentation of Treg activity (94). MDSCs are another immunosuppressive immune cell subset that are increased in patients with AML. In an in vitro co-culture assay, expansion of MDSCs was shown to be induced by both primary AML cells and AML cell lines in a contact-dependent manner. It was further identified that MUC-1 oncoprotein was responsible for the observed MDSC expansion (95). AML blasts themselves may possess many features of MDSCs, as discussed in a recent review (96). Notably, AML blasts, specifically the monocytic subtype, have been reported to directly suppress T cell anti-tumor responses by inducing large amounts of reactive oxygen species (ROS), subsequently triggering T cell apoptosis (97).
Several potential solutions could address the issue of abundant immunosuppressive cells in the context of CAR T cell therapy. Although not yet tested in AML, adenosine-mediated immunosuppression could be alleviated by combining CAR T cell therapy with antagonists targeting the adenosine receptor A2AR, as shown in a pre-clinical model for solid tumors (98). In addition, a number of clinical trials have been initiated in an attempt to reduce the abundance and recruitment of MDSCs in multiple malignancies by using small molecules such as liver-X nuclear hormone receptor (LXR) agonist and chemokine inhibitors (99). Given the T cell-inhibiting role of MDSCs, combining CAR T cells with small molecules that can deplete the MDSC population may result in synergistic effects. Indeed, at least in an in vivo model, MDSC depletion was shown to increase CAR T cell anti-tumor effects (100). In addition to impairing CAR T cell function, AML-induced immunosuppression is likely to also present a challenge for CAR T cells to persist in vivo. Though not yet tested specifically for AML, several strategies that have been shown to improve CAR T cell expansion and persistence include expression of IL-15 (101) or IL-18 transgene (102), addition of exogenous IL-7, IL-15, and/or IL-21 cytokines during CAR T cell ex vivo expansion (103, 104), and many others that are currently being developed to skew CAR T cell phenotype toward a stem cell/ central memory phenotype. Additionally, in an attempt to further improve CAR T cell efficacy, approaches that are able to boost CAR T cell activation, such as the use of immune agonists (100, 105) or co-stimulatory cytokines (106, 107) may lead to improved overall efficacy.
Upregulation of Inhibitory Ligands and Receptors
The roles of immune inhibitory ligands and their receptors have been extensively studied in a wide range of cancers. Decades of work invested in studying these immune checkpoints led to widespread clinical use of several monoclonal antibodies (mAbs) that block specific signaling pathways such as CTLA-4 and PD-1 (108, 109). The remarkable success of these mAbs have been highlighted by the FDA approval of α-PD-1, α-PD-L1, and α-CTLA-4 mAbs for a number of cancers, and more recently by the Nobel Prize in physiology and medicine to Tasuku Honjo and James Allison (110). Despite the overwhelming enthusiasm with which checkpoint inhibitors are being trialed in solid cancers, there are only limited data to date that support the role of a single inhibitory pathway in mediating AML immune-evasion. This discrepancy is thought to be due to, in part, the fact that AML has among the lowest mutational burdens in human cancer (111), thus presumably fewer neoantigens recognized by T cells.
In an autologous culture system, blockade of CTLA-4 was shown to augment the activity and expansion of AML-reactive T cells (112). To date, the most encouraging clinical data that supports the importance of CTLA-4 pathway in AML comes from a phase I trial exploring the use of ipilimumab in patients with hematological malignancies relapsing post allo-HSCT. In this study, the five patients achieving complete remission had AML. An interesting observation was that four of these patients had extramedullary disease (113).
The PD-1:PD-L1/PD-L2 axis has been investigated in AML, although it is unclear whether PD-L1 is consistently overexpressed on primary AML blasts (114, 115). PD-L1 may be upregulated upon exposure to inflammatory stimuli, such as toll-like receptor (TLR) ligands and IFNγ (116), and expression of PD-L1 and/or PD-L2 by AML blasts may be associated with poor prognosis (117). Expression of the PD-1 receptor on T cells has been described in patients with AML at relapse (117), and in an in vivo model, PD1−/− mice inoculated with AML had slower disease progression compared to PD-1-sufficient mice (118). As noted earlier, given the limited clinical efficacy of checkpoint inhibitors in AML as a single agent, researchers are now looking into combinatorial approaches with other anti-cancer modalities such as the hypomethylating agent, azacitidine, which is known to upregulate PD-1 and PD-L1 expression (clinicaltrials.gov identifier: 02775903, 03092674) (119). Other checkpoint pathways of interest in AML include T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT), T cell immunoglobulin domain and mucin domain 3 (TIM-3), and glucocorticoid induced tumor necrosis factor receptor related protein (GITR) (120–122).
CAR T cell combination with checkpoint inhibitors, either exogenously administered (123), or via genetic engineering of the CAR T cells themselves to synthesize the checkpoint blocking antibodies (124, 125), has been tested in pre-clinical models against several cancer types. These combination strategies have resulted in promising pre-clinical data, and more recently encouraging preliminary clinical data (126–128), which ultimately paved the way for their clinical translation. This has been highlighted by the numerous clinical trials currently ongoing to evaluate the safety and efficacy of the combined CAR T cell and ICB therapy, particularly for lymphomas and various solid tumors (129). As previously noted, the underwhelming effects of checkpoint inhibition in AML may be due to the low mutational burden nature of AML and thus the low frequency of AML-reactive T cells (111). We postulate that this therefore makes tumor-targeted CAR T cells an ideal partner for combination with checkpoint inhibitors in AML.
Conclusion and Future Directions
AML is an aggressive disease that, if not completely eliminated at first attempt, becomes resistant to further treatments. Robust therapies that eliminate all leukemia cells including putative leukemic stem cells are therefore needed to achieve deep and durable remissions. CAR T cell therapy has proven to be a potent immunotherapy weapon and is already being tested in the clinic for AML (and potentially other myeloid malignancies including MDS, MPN, and CML) (Table 1). However, there are a number of barriers that limit the full therapeutic potential of CAR T cells. First, there are no truly leukemia-specific cell-surface antigens that could be used as targets for CAR T cells. AML antigens are frequently shared by normal HSPCs or their progeny. Additionally, the manufacture of CAR T cells itself may also be a challenge in patients with active AML, potentially due to inhibition of T cell expansion by AML blasts, or prior exposure to T cell-damaging chemotherapy. Further, it is increasingly apparent that AML is a heterogeneous and complex disease capable of evading the immune system by means of various immunosuppressive mechanisms. Therefore, although CAR T cell therapy for AML is already in clinical trials, we are facing challenging yet surmountable obstacles to ensure the safe and effective use of CAR T cells for AML. As discussed in this review, there are many potential avenues to reduce toxicity to healthy tissues while maintaining, and even augmenting, the full therapeutic potential of CAR T cells. We strongly advocate for careful, rationally-designed clinical trials that are informed by a comprehensive and clear-eyed understanding of the AML—effector cell—microenvironment axis in order to advance toward a shared goal of successful AML immunotherapy.
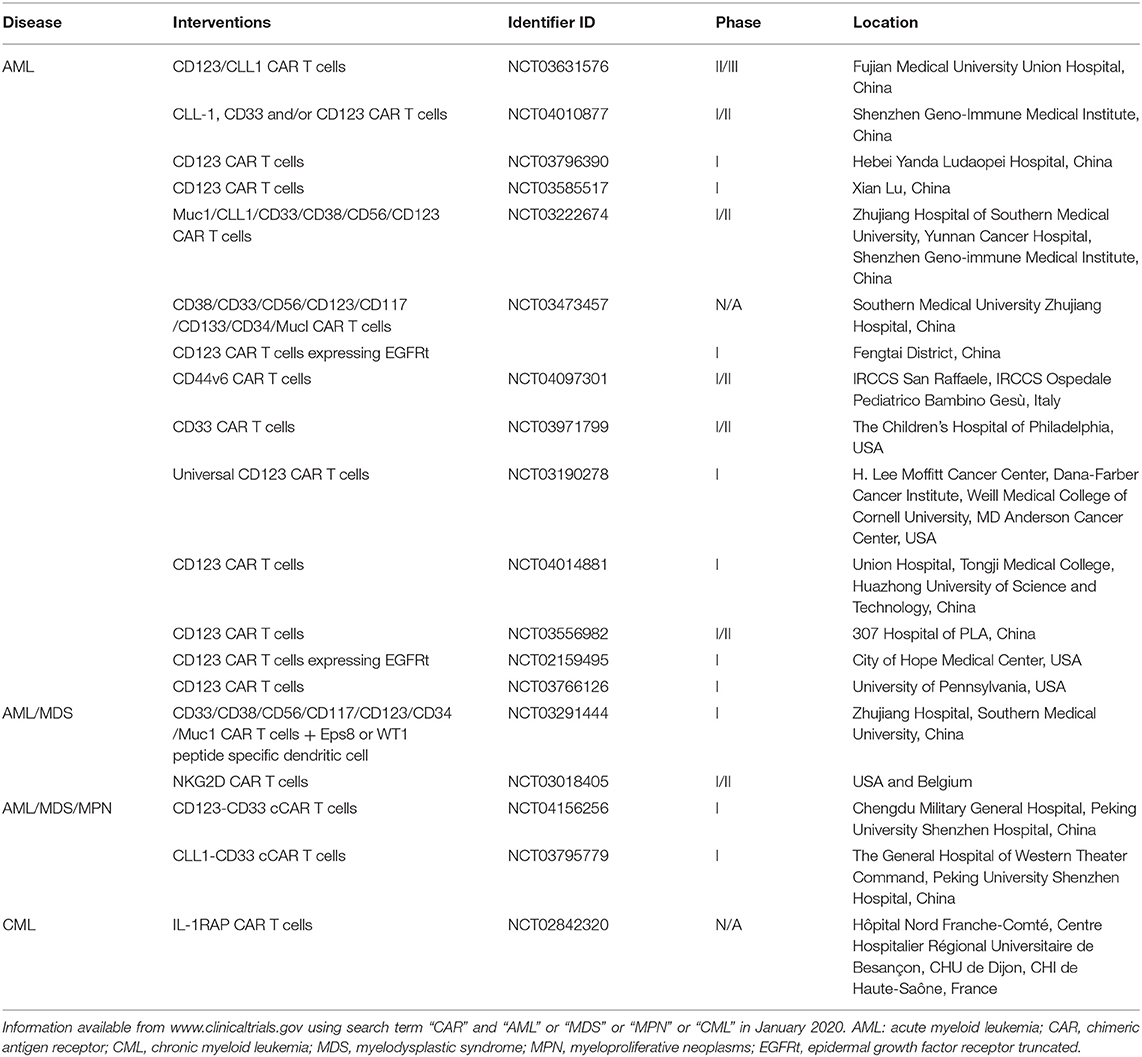
Table 1. CAR T cell trials in myeloid malignancies currently recruiting.
Author Contributions
SM and SG wrote the manuscript.
Funding
This work was supported by NIH CA 194256-05 (SG) and NIH 5P01CA214278-02 (SG).
Conflict of Interest
Research Funding: Novartis, Tmunity Therapeutics, Carisma Therapeutics. Stock and Ownership: Carisma Therapeutics. Consulting: Aileron, Fate, Sensei.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Murati A, Brecqueville M, Devillier R, Mozziconacci MJ, Gelsi-Boyer V, Birnbaum D. Myeloid malignancies: mutations, models and management. BMC Cancer. (2012) 12:304. doi: 10.1186/1471-2407-12-304
2. Dickinson AM, Norden J, Li S, Hromadnikova I, Schmid C, Schmetzer H, et al. Graft-versus-leukemia effect following hematopoietic stem cell transplantation for leukemia. Front Immunol. (2017) 8:496. doi: 10.3389/fimmu.2017.00496
3. Cibmtr. Data Resources. (2019). Available online at: https://www.cibmtr.org/Data/Resources/Pages/index.aspx.
4. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
5. Von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. (2016) 34:4381–9. doi: 10.1200/JCO.2016.67.3301
6. Hu B, Jacobs R, Ghosh N. Checkpoint inhibitors hodgkin lymphoma and non-hodgkin lymphoma. Curr Hematol Malig Rep. (2018) 13:543–54. doi: 10.1007/s11899-018-0484-4
7. Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood. (2018) 131:68–83. doi: 10.1182/blood-2017-07-740993
8. Hoseini SS, Cheung NK. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. (2017) 7:e522. doi: 10.1038/bcj.2017.2
9. Daver N, Boddu P, Garcia-Manero G, Yadav SS, Sharma P, Allison J, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia. (2018) 32:1094–105. doi: 10.1038/s41375-018-0070-8
10. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. (2014) 5:587–98. doi: 10.18632/oncotarget.1782
11. Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Blagitko-Dorfs N, et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget. (2016) 7:12840–56. doi: 10.18632/oncotarget.7326
12. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. (2013) 21:2122–9. doi: 10.1038/mt.2013.154
13. US National Library of Science. ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/results?term=CARandcond=AMLandSearch=Applyandrecrs=aandage_v=andgndr=andtype=andrslt=~[Online].
14. Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, Von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. (2014) 4:e218. doi: 10.1038/bcj.2014.39
15. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. (2015) 23:184–91. doi: 10.1038/mt.2014.164
16. Yi Luo LJC, Yongxian H, Lujia D, Guoqing W, He H. First-in-Man CD123-Specific Chimeric Antigen Receptor-Modified T Cells for the Treatment of Refractory Acute Myeloid Leukemia. Orlando, FL: ASH (2015).
17. Cummins K, Frey N, Nelson A, Schmidt A, Luger S, Hexner E, et al. Treating Relapsed/Refractory. (RR) AML With Biodegradable Anti-CD123 CAR Modified T Cells (2017) Atlanta, GA: Blood ASH.
18. Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. (2018) 132:901. doi: 10.1182/blood-2018-99-110579
19. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Kim M, Soderquist C, et al. Targeting CLEC12A with chimeric antigen receptor T cells can overcome the chemotherapy refractoriness of leukemia stem cells. Biol Blood Marrow Transplant. (2017) 23:S247–8. doi: 10.1016/j.bbmt.2016.12.413
20. Ma H, Padmanabhan IS, Parmar S, Gong Y. Targeting CLL-1 for acute myeloid leukemia therapy. J Hematol Oncol. (2019) 12:41. doi: 10.1186/s13045-019-0726-5
21. Bakker ABH, Van Den Oudenrijn S, Bakker AQ, Feller N, Van Meijer M, Bia JA, et al. C-type lectin-like molecule-1. Cancer Res. (2004) 64:8443. doi: 10.1158/0008-5472.CAN-04-1659
22. Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Am Soc Hematol. (2017) 130:811. doi: 10.1182/blood.V130.Suppl_1.811.811
23. Budde EL. Chimeric antigen receptor T-cell therapy for acute myeloid leukemia: targeting CD123. In: 1st European CAR T Cell Meeting (2019).
24. Cummins KD, Gill S. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin Hematol. (2019) 56:155–63. doi: 10.1053/j.seminhematol.2018.08.008
25. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. (2014) 123:2343–54. doi: 10.1182/blood-2013-09-529537
26. Kim MY, Yu K-R, Kenderian SS, Ruella M, Chen S, Shin T-H, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–53.e1419. doi: 10.1016/j.cell.2018.05.013
27. Petrov JC, Wada M, Pinz KG, Yan LE, Chen KH, Shuai X, et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. (2018) 32:1317–26. doi: 10.1038/s41375-018-0075-3
28. Bordignon C, Bonini C, Verzeletti S, Nobili N, Maggioni D, Traversari C, et al. Transfer of the HSV-tk gene into donor peripheral blood lymphocytes for in vivo modulation of donor anti-tumor immunity after allogeneic bone marrow transplantation. Hum Gene Ther. (1995) 6:813–9. doi: 10.1089/hum.1995.6.6-813
29. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:235. doi: 10.3389/fphar.2014.00235
30. Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, et al. Redesigning an FKBP–ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci USA. (1998) 95:10437–42. doi: 10.1073/pnas.95.18.10437
31. Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. (2005) 105:4247–54. doi: 10.1182/blood-2004-11-4564
32. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
33. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
34. Li H, Zhao Y. Increasing the safety and efficacy of chimeric antigen receptor T cell therapy. Protein Cell. (2017) 8:573–89. doi: 10.1007/s13238-017-0411-9
35. Ali R, Ramdial J, Algaze S, Beitinjaneh A. The role of anti-thymocyte globulin or alemtuzumab-based serotherapy in the prophylaxis and management of graft-versus-host disease. Biomedicines. (2017) 5:67. doi: 10.3390/biomedicines5040067
36. Zhao Z, Condomines M, Van der S, Sjoukje JC, Perna F, Kloss C, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
37. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
38. Finney OC, Brakke H, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. (2019) 129:2123–32. doi: 10.1172/JCI125423
39. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
40. Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomarker Res. (2014) 2:4. doi: 10.1186/2050-7771-2-4
41. Mardiana S, Lai J, House IG, Beavis PA, Darcy PK. Switching on the green light for chimeric antigen receptor T-cell therapy. Clin Transl Immunol. (2019) 8:e1046. doi: 10.1002/cti2.1046
42. Wirth TC, Kühnel F. Neoantigen targeting—dawn of a new era in cancer immunotherapy? Front Immunol. (2017) 8:1848. doi: 10.3389/fimmu.2017.01848
43. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
44. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
45. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
46. Mardis ER, Ding L, Dooling DJ, Larson DE, Mclellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. (2009) 361:1058–66. doi: 10.1056/NEJMoa0903840
47. Goswami M, Hourigan CS. Novel antigen targets for immunotherapy of acute myeloid leukemia. Curr Drug Targets. (2017) 18:296–303. doi: 10.2174/1389450116666150223120005
48. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. (2014) 512:324–7. doi: 10.1038/nature13387
49. Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Gotz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood. (2012) 120:1282–9. doi: 10.1182/blood-2011-11-394395
50. Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res. (2014) 20:1135–45. doi: 10.1158/1078-0432.CCR-13-0956
51. Adamia S, Bar-Natan M, Haibe-Kains B, Pilarski PM, Bach C, Pevzner S, et al. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia. (AML): new potential targets in AML. Blood. (2014) 123:2816–25. doi: 10.1182/blood-2013-02-481507
52. Casucci M, Nicolis Di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. (2013) 122:3461–72. doi: 10.1182/blood-2013-04-493361
53. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. (2017) 31:1788–97. doi: 10.1038/leu.2016.373
54. Chapuis AG, Egan DN, Bar M, Schmitt TM, Mcafee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
55. Orleans-Lindsay JK, Barber LD, Prentice HG, Lowdell MW. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function–implications for the adoptive immunotherapy of leukaemia. Clin Exp Immunol. (2001) 126:403–11. doi: 10.1046/j.1365-2249.2001.01692.x
56. Kenderian SS, June CH, Gill S. “Generating and Expanding Autologous Chimeric Antigen Receptor T Cells from Patients with Acute Myeloid Leukemia,” in Fortina P, Londin E, Park JY, Kricka LJ, editors. Acute Myeloid Leukemia: Methods and Protocols. New York, NY: Springer New York (2017). p. 267–76. doi: 10.1007/978-1-4939-7142-8_17
57. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
58. Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T cells: more than ease of access? Cells. (2018) 7:155. doi: 10.3390/cells7100155
59. Torikai H, Reik A, Liu P-Q, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. (2012) 119:5697–705. doi: 10.1182/blood-2012-01-405365
60. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. (2017) 20:3025–33. doi: 10.1016/j.celrep.2017.09.002
61. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. (2017) 9:2013. doi: 10.1126/scitranslmed.aaj2013
62. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. (2018) 11:132. doi: 10.1186/s13045-018-0677-2
63. Handgretinger R, Schilbach K. The potential role of γδ T cells after allogeneic HCT for leukemia. Blood. (2018) 131:1063–72. doi: 10.1182/blood-2017-08-752162
64. Rischer M, Pscherer S, Duwe S, Vormoor J, Jürgens H, Rossig C. Human γδ T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol. (2004) 126:583–92. doi: 10.1111/j.1365-2141.2004.05077.x
65. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human γ delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. (2018) 26:354–65. doi: 10.1016/j.ymthe.2017.12.001
66. Nussbaumer O, Koslowski M. The emerging role of γδ T cells in cancer immunotherapy. Immuno-Oncol Technol. (2019) 1:3–10. doi: 10.1016/j.iotech.2019.06.002
67. Wu D, Wu P, Qiu F, Wei Q, Huang J. Human γδT-cell subsets and their involvement in tumor immunity. Cell Mol Immunol. (2017) 14:245–53. doi: 10.1038/cmi.2016.55
68. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. (2014) 124:2824–33. doi: 10.1182/blood-2013-11-541235
69. Simon B, Wiesinger M, März J, Wistuba-Hamprecht K, Weide B, Schuler-Thurner B, et al. The generation of CAR-transfected natural killer t cells for the immunotherapy of melanoma. Int J Mol Sci. (2018) 19:2365. doi: 10.3390/ijms19082365
70. Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. JNCI. (2015) 108:djv375. doi: 10.1093/jnci/djv375
71. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
72. Kloess S, Kretschmer A, Stahl L, Fricke S, Koehl U. CAR-expressing natural killer cells for cancer retargeting. Transf Med Hemother. (2019) 46:4–13. doi: 10.1159/000495771
73. Curran EK, Godfrey J, Kline J. Mechanisms of immune tolerance in leukemia and lymphoma. Trends Immunol. (2017) 38:513–25. doi: 10.1016/j.it.2017.04.004
74. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MTL, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. (2009) 361:478–88. doi: 10.1056/NEJMoa0811036
75. Crucitti L, Crocchiolo R, Toffalori C, Mazzi B, Greco R, Signori A, et al. Incidence, risk factors and clinical outcome of leukemia relapses with loss of the mismatched HLA after partially incompatible hematopoietic stem cell transplantation. Leukemia. (2015) 29:1143–52. doi: 10.1038/leu.2014.314
76. Brouwer RE, Van Der Heiden P, Schreuder GM, Mulder A, Datema G, Anholts JD, et al. Loss or downregulation of HLA class I expression at the allelic level in acute leukemia is infrequent but functionally relevant, and can be restored by interferon. Hum Immunol. (2002) 63:200–10. doi: 10.1016/S0198-8859(01)00381-0
77. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. (2018) 379:2330–41. doi: 10.1056/NEJMoa1808777
78. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. (2018) 24:1504–6. doi: 10.1038/s41591-018-0146-z
79. Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, et al. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol. (2001) 167:6021–30. doi: 10.4049/jimmunol.167.10.6021
80. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, Mcewen-Smith RM, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. (2013) 122:749–58. doi: 10.1182/blood-2013-01-480129
81. Papadopoulos KP, Tsai FY-C, Bauer TM, Muigai L, Liang Y, Bennett MK, et al. CX-1158-101: a first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients. (pts) with solid tumors. J Clin Oncol. (2017) 35:3005. doi: 10.1200/JCO.2017.35.15_suppl.3005
82. Vacchelli E, Aranda F, Eggermont A, Sautes-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. (2014) 3:e957994. doi: 10.4161/21624011.2014.957994
83. Sharma MD, Baban B, Chandler P, Hou D-Y, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. (2007) 117:2570–82. doi: 10.1172/JCI31911
84. Baban B., Chandler P. R., Sharma M. D., Pihkala J., Koni P. A., Munn D. H., et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol.(2009) 183:2475–83. doi: 10.4049/jimmunol.0900986
85. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
86. Mabuchi R, Hara T, Matsumoto T, Shibata Y, Nakamura N, Nakamura H, et al. High serum concentration of L-kynurenine predicts unfavorable outcomes in patients with acute myeloid leukemia. Leuk Lymphoma. (2016) 57:92–8. doi: 10.3109/10428194.2015.1041388
87. Mansour I, Zayed RA, Said F, Latif LA. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology. (2016) 21:447–53. doi: 10.1080/10245332.2015.1106814
88. Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. (2016) 7:22928–38. doi: 10.18632/oncotarget.8216
89. Huang Q, Xia J, Wang L, Wang X, Ma X, Deng Q, et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol. (2018) 11:58. doi: 10.1186/s13045-018-0600-x
90. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase. (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
91. Vasievich EA, Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharmaceut. (2011) 8:635–41. doi: 10.1021/mp1004228
92. Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. (2009) 15:3325–32. doi: 10.1158/1078-0432.CCR-08-3010
93. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer. (2011) 129:1373–81. doi: 10.1002/ijc.25791
94. Dulphy N, Henry G, Hemon P, Khaznadar Z, Dombret H, Boissel N, et al. Contribution of CD39 to the immunosuppressive microenvironment of acute myeloid leukaemia at diagnosis. Br J Haematol. (2014) 165:722–5. doi: 10.1111/bjh.12774
95. Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. (2017) 129:1791–801. doi: 10.1182/blood-2016-07-730614
96. Barrett AJ. Acute myeloid leukaemia and the immune system: implications for immunotherapy. Br J Haematol. (2020) 188:147–58. doi: 10.1111/bjh.16310
97. Aurelius J, Thoren FB, Akhiani AA, Brune M, Palmqvist L, Hansson M, et al. Monocytic AML cells inactivate antileukemic lymphocytes: role of NADPH oxidase/gp91(phox) expression and the PARP-1/PAR pathway of apoptosis. Blood. (2012) 119:5832–7. doi: 10.1182/blood-2011-11-391722
98. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
99. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
100. Di S, Zhou M, Pan Z, Sun R, Chen M, Jiang H, et al. Combined adjuvant of poly I:C improves antitumor effects of CAR-T cells. Front Oncol. (2019) 9:241–241. doi: 10.3389/fonc.2019.00241
101. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci USA. (2016) 113:E7788–97. doi: 10.1073/pnas.1610544113
102. Avanzi MPG, Van Leeuwen D, Li X, Cheung K, Park H. IL-18 secreting CAR T cells enhance cell persistence, induce prolonged B cell aplasia and eradicate CD19+ tumor cells without need for prior conditioning. Blood. (2016) 128:816. doi: 10.1182/blood.V128.22.816.816
103. Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. (2011) 71:3516–27. doi: 10.1158/0008-5472.CAN-10-3843
104. Xu Y, Zhang M, Ramos C, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
105. Mardiana S, John LB, Henderson MA, Slaney CY, Von Scheidt B, Giuffrida L, et al. A multifunctional role for adjuvant anti-4-1BB therapy in augmenting antitumor response by chimeric antigen receptor T cells. Cancer Res. (2017) 77:1296–309. doi: 10.1158/0008-5472.CAN-16-1831
106. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
107. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. (2018) 36:346–51. doi: 10.1038/nbt.4086
108. Wolchok JD, Hoos A, Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. (2009) 15:7412. doi: 10.1158/1078-0432.CCR-09-1624
109. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. (2015) 348:124. doi: 10.1126/science.aaa1348
110. Mardiana S, Solomon BJ, Darcy PK, Beavis PA. Supercharging adoptive T cell therapy to overcome solid tumor–induced immunosuppression. Sci Transl Med. (2019) 11:eaaw2293. doi: 10.1126/scitranslmed.aaw2293
111. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. (2017) 9:34. doi: 10.1186/s13073-017-0424-2
112. Zhong RK, Loken M, Lane TA, Ball ED. CTLA-4 blockade by a human MAb enhances the capacity of AML-derived DC to induce T-cell responses against AML cells in an autologous culture system. Cytotherapy. (2006) 8:3–12. doi: 10.1080/14653240500499507
113. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. (2016) 375:143–53. doi: 10.1056/NEJMoa1601202
114. Salih HR, Wintterle S, Krusch M, Kroner A, Huang YH, Chen L, et al. The role of leukemia-derived B7-H1. (PD-L1) in tumor-T-cell interactions in humans. Exp Hematol. (2006) 34:888–94. doi: 10.1016/j.exphem.2006.03.006
115. Schnorfeil FM, Lichtenegger FS, Emmerig K, Schlueter M, Neitz JS, Draenert R, et al. T cells are functionally not impaired in AML: increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J Hematol Oncol. (2015) 8:93–93. doi: 10.1186/s13045-015-0189-2
116. Berthon C, Driss V, Liu J, Kuranda K, Leleu X, Jouy N, et al. In acute myeloid leukemia, B7-H1. (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-γ and can be reversed using MEK inhibitors. Cancer Immunol Immunother. (2010) 59:1839–49. doi: 10.1007/s00262-010-0909-y
117. Chen X, Liu S, Wang L, Zhang W-G, Ji Y, Ma X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol Therapy. (2008) 7:622–7. doi: 10.4161/cbt.7.5.5689
118. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. (2009) 114:1545–52. doi: 10.1182/blood-2009-03-206672
119. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. (2019) 9:370. doi: 10.1158/2159-8290.CD-18-0774
120. Baessler T, Krusch M, Schmiedel BJ, Kloss M, Baltz KM, Wacker A, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein ligand subverts immunosurveillance of acute myeloid leukemia in humans. Cancer Res. (2009) 69:1037–45. doi: 10.1158/0008-5472.CAN-08-2650
121. Li C, Chen X, Yu X, Zhu Y, Ma C, Xia R, et al. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int J Clin Exp Pathol. (2014) 7:6880–8.
122. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell Immunoglobulin and ITIM Domain. (TIGIT) Associates with CD8+ T cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res. (2016) 22:3057–66. doi: 10.1158/1078-0432.CCR-15-2626
123. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
124. Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor–modified T cells engineered to secrete checkpoint inhibitors. Clinical Cancer Res. (2017) 23:6982. doi: 10.1158/1078-0432.CCR-17-0867
125. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, Van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
126. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor. (CAR)-modified T cells: refueling the CAR. Blood. (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
127. Maude SL, Hucks GE, Seif AE, Talekar MK, Teachey DT, Baniewicz D, et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor. (CAR) T cells in relapsed acute lymphoblastic leukemia. (ALL). J Clin Oncol. (2017) 35:103. doi: 10.1200/JCO.2017.35.15_suppl.103
128. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 367:eaba7365. doi: 10.1126/science.aba7365
Keywords: chimeric antigen receptor, acute myeloid leukemia, engineered T cells, adoptive therapy, immunotherapy
Citation: Mardiana S and Gill S (2020) CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 10:697. doi: 10.3389/fonc.2020.00697
Received: 15 February 2020; Accepted: 14 April 2020;
Published: 06 May 2020.
Edited by:
Marta Coscia, University of Turin, ItalyReviewed by:
Antonio Curti, University of Bologna, ItalyRanganatha R. Somasagara, North Carolina Central University, United States
Copyright © 2020 Mardiana and Gill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saar Gill, c2Fhci5naWxsQHBlbm5tZWRpY2luZS51cGVubi5lZHU=