 Christiana M. Neophytou1,2
Christiana M. Neophytou1,2 Chryso Pierides3Maria-Ioanna Christodoulou2Paul Costeas3,4Theodora-Christina Kyriakou2
Chryso Pierides3Maria-Ioanna Christodoulou2Paul Costeas3,4Theodora-Christina Kyriakou2 Panagiotis Papageorgis1,2*
Panagiotis Papageorgis1,2*- 1European University Research Centre, Nicosia, Cyprus
- 2Department of Life Sciences, School of Sciences, European University Cyprus, Nicosia, Cyprus
- 3The Center for the Study of Haematological Malignancies, Nicosia, Cyprus
- 4The Cyprus Cancer Research Institute, Nicosia, Cyprus
Myeloid cells include various cellular subtypes that are distinguished into mononuclear and polymorphonuclear cells, derived from either common myeloid progenitor cells (CMPs) or myeloid stem cells. They play pivotal roles in innate immunity since, following invasion by pathogens, myeloid cells are recruited and initiate phagocytosis and secretion of inflammatory cytokines into local tissues. Moreover, mounting evidence suggests that myeloid cells may also regulate cancer development by infiltrating the tumor to directly interact with cancer cells or by affecting the tumor microenvironment. Importantly, mononuclear phagocytes, including macrophages and dendritic cells (DCs), can have either a positive or negative impact on the efficacy of chemotherapy, radiotherapy as well as targeted anti-cancer therapies. Tumor-associated macrophages (TAMs), profusely found in the tumor stroma, can promote resistance to chemotherapeutic drugs, such as Taxol and Paclitaxel, whereas the suppression of TAMs can lead to an improved radiotherapy outcome. On the contrary, the presence of TAMs may be beneficial for targeted therapies as they can facilitate the accumulation of large quantities of nanoparticles carrying therapeutic compounds. Tumor infiltrating DCs, however, are generally thought to enhance cytotoxic therapies, including those using anthracyclines. This review focuses on the role of tumor-infiltrating and stroma myeloid cells in modulating tumor responses to various treatments. We herein report the impact of myeloid cells in a number of therapeutic approaches across a wide range of malignancies, as well as the efforts toward the elimination of myeloid cells or the exploitation of their presence for the enhancement of therapeutic efficacy against cancer.
Introduction
Immunity is the result of an intricate interaction between the innate and adaptive immune system. Innate immunity is the initial immunological response against an invading pathogen and has no immunological memory. On the other hand, adaptive immunity involves the development of immunological memory and enables the host to respond more efficiently to future exposure to the antigen. Following antigen processing, the degraded peptides associate with major histocompatibility complex (MHC) molecules within the interior of an antigen-presenting cell (APC) and are exposed on its surface. Myeloid cells, including DC and macrophages, are considered “professional” APCs. They present foreign antigens to helper T-cells on class II MHC molecules and can prime naïve T-cells. Other myeloid lineage types of cells, such as neutrophils, have no or very low expression of MHC-II and are inefficient at priming naïve T-cell responses (1). Nearly all nucleated cells can act as APCs by presenting antigens on class I MHC molecules to cytotoxic T-cells. Even though cancer cells are poor APCs, antigen presentation is involved in the body's defense against tumors.
Immune cells of various types and origins are integral components of the tumor microenvironment (TME) along with fibroblasts, endothelial cells, and extracellular components, such as collagen and hyaluronan. Cellular constituents from the lymphoid and myeloid lineage can elicit both immune suppressive and immune stimulatory functions and have an important role in regulating cancer progression and survival, as well as drug resistance (2–4). Tumors secrete factors which promote myelopoiesis and recruit circulating cells into the tumor mass, microenvironment or to secondary lymphoid organs, such as lymph nodes and spleen, and polarize their functionality to serve their survival and growth.
Hematopoietic stem cells (HSC) differentiate in the bone marrow into common myeloid progenitors (CMPs) which can give rise to DCs and to tumor-associated myeloid cells (TAMCs). TAMCs include at least four different myeloid cell populations: (a) Tumor-associated macrophages (TAMs) that exert crucial roles in regulating cancer-related inflammation, (b) monocytes that express the angiopoietin receptor Tie2 (tunica internal endothelial kinase 2) and have a pivotal function in tumor angiogenesis, (c) myeloid-derived suppressor cells (MDSCs) that can be further characterized as monocytic and granulocytic (m-MDSCs, g-MDSCs) depending on their morphology, phenotype and immune suppression functions and (d) tumor-associated neutrophils (TANs) that express pro-angiogenic factors and participate in tumor promotion (5).
Ideally, a competent immune system recognizes tumor-specific and embryonic antigens; however, cancer cells manage to escape immune surveillance by secreting immune escape variants, recruiting myeloid-derived cells and either maintaining them in an immunosuppressive phenotype or polarizing them into tumor-promoting cell types (6, 7). The mechanisms of recruitment of myeloid cells to the TME are often the targets of anti-cancer therapy. Emerging evidence indicates that TAMCs interfere with or facilitate most therapeutic approaches, including conventional chemotherapy, targeted approaches, radiotherapy and immunotherapy. TAMCs are found abundantly in the tumor stroma; high density of TAMCs has been significantly associated with poor prognosis in several cancer types including head and neck, breast, thyroid, liver, kidney, pancreatic, bladder, endometrial, ovarian, oral cancer, as well as Hodgkin lymphoma (8–10). Many studies have shown that TAMCs can induce chemoresistance against first-line chemotherapeutic drugs (Figure 1). In this review, we discuss the interplay between myeloid cells, mainly focusing on TAMs, MDSCs and DCs, and cancer therapy, the mechanisms of action by which they exert either positive or negative effects as well as provide insights related to current controversies in the field.
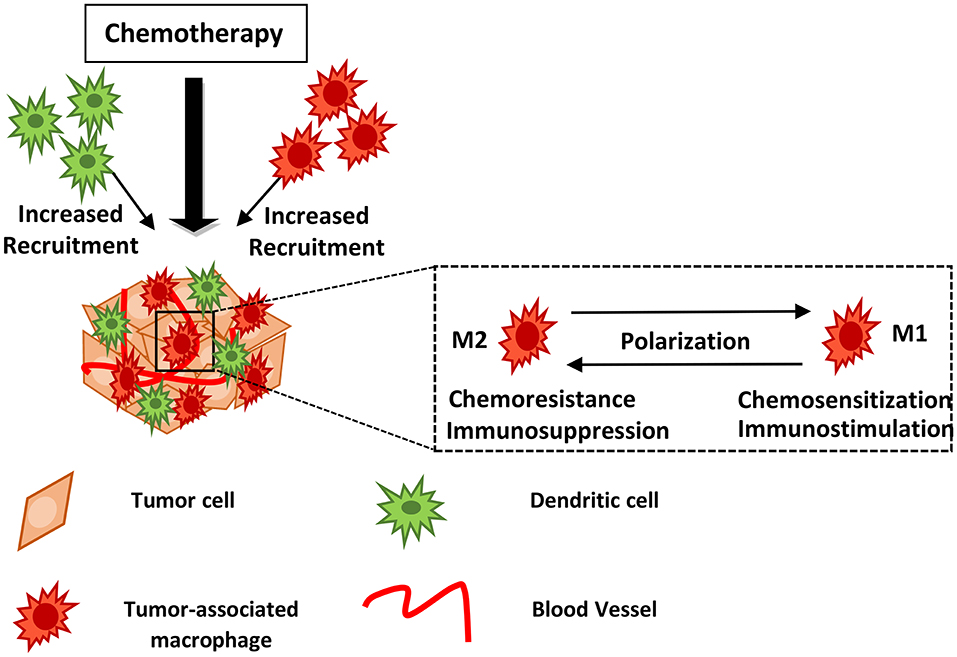
Figure 1. Myeloid cells may block or facilitate chemotherapy. Chemotherapy induces recruitment of innate immune cells including macrophages and dendritic cells into the treated tumor tissue. Drug treatment may lead to TAM polarization from the M2- to an M1-like phenotype, hindering tumor growth and metastasis. The mechanisms of action utilized by myeloid cells in supporting or blocking chemotherapy are described in the text and summarized in Table 1. DCs, Dendritic cells; TAMs, Tumor-associated macrophages.
Mechanisms of Recruitment and Functions of Myeloid Cell Populations in the Tumor Microenvironment
Cell populations of myeloid origin are critically important components of the TME as they play a central role in the regulation of anti-tumor immune responses. At the same time, inflammatory immune cells such as tumor-infiltrating lymphocytes (TILs), natural killer (NK) cells, NK T-cells, and B cells are also engaged within the TME, which further interact to affect the growth and function of cancer cells. While myeloid cells are required for enabling anti-tumor immunity, they can also have an immunosuppressive role in established tumors by promoting immune evasion, and facilitating primary tumor growth, progression and metastasis (11).
Colony-stimulating factor-1 (CSF-1) and colony-stimulating factor-2 (CSF-2), also known as macrophage colony-stimulating factor (M-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF), respectively, are secreted cytokines that regulate mature myeloid cell populations by affecting their activation, survival, mobilization and differentiation. They have also been implicated in the development of many diseases, including in tumor progression and metastasis (12). Cancer cells expressing high levels of M-CSF, recruit TAMs to the tumor site, via their receptor CSF-1R (13). The elevated expression of M-CSF in tumors, and consequently the presence of CSF-1R-positive macrophages, has been correlated with poor prognosis in patients with breast, bladder and ovarian cancer (9). M-CSF induces high expression of C-C motif chemokine ligand 2 (CCL2) by macrophages, a chemokine that acts as a chemoattractant driving them to the tumor but may also affect their polarization and survival (14, 15). Since M-CSF also mediates the polarization of macrophages to the tumor-promoting type (16), the targeting of the M-CSF/CSF-1R axis, represents an attractive therapeutic approach and has shown efficacy in cancer metastasis models and in several murine models of cancer (17–20).
A combination of cytokines, particularly granulocyte colony-stimulating factor (G-CSF) or GM-CSF, interleukin (IL)-6, and the transcriptional regulator CCAAT/enhancer-binding protein (C/EBP) are required for the differentiation of bone marrow progenitors into MDSCs (21, 22). Whilst solid indications demonstrate that MDSCs directly suppress cytotoxic leukocytes, conventional and plasmacytoid dendritic cells (pDC) can also have immunoregulatory effects in tumors (23). Consequently, a more comprehensive characterization of these subsets and a better understanding of their recruitment and expansion mechanisms are of paramount importance for the development of novel cancer therapeutic strategies as well as for the potential improvement of existing ones.
DCs are essential for the cross-priming of cytotoxic T lymphocytes against tumor-specific antigens; however tumor-residing DCs can cause cell anergy and tolerance by expressing low levels of costimulatory molecules and pro-inflammatory cytokines (24). TAMs that have a classic (M1) activation state are characterized by anti-tumor immunity, proinflammatory activity and the induction of T-cell responses (25, 26). The presence of M1-type macrophages in high numbers within the TME, has been associated with good prognosis in patients with non-small cell lung cancer (NSCLC), colorectal, hepatocellular, ovarian and gastric cancer (27). In malignant tumors, TAMs resemble M2-type macrophages, which undergo alternative (M2) activation. These cells have the ability to support tumor growth, inhibit immunity against the tumor, and promote tissue repair (28). These have been generally considered as a promising target for tumor therapy, with studies concentrating on the inhibition of macrophage recruitment, survival, and tumor-promoting activity in tumors, as well as, predominantly, on the shift of tumor-promoting M2 TAMs toward tumor-suppressive M1-type macrophages (29).
The importance of myeloid cells in facilitating the killing of tumor cells has been highlighted by many studies (30, 31). Myeloid cells can exert significant anti-tumor functions by activating NK and CD8+ T-cells. Cancer cells can be detected by NK cells through the expression of ligands for the receptor NKG2D (32). The binding of these ligands serves as a major signal of activation NK cells to stop aberrant cell proliferation and can be further enhanced through the function of myeloid cells. In fact, macrophages and DCs express Dectin-1, a receptor that recognizes N-glycan structures found on the surface of certain types of tumor cell. Activation of Dectin-1 induced a signaling pathway that directs the activity of NK cells against the tumor in a lung metastasis model of B16F1 melanoma cells (33). In addition, the expression of calreticulin on the surface of cancer cells can be recognized and processed by macrophages which then activate CD4+ and CD8+ T-cells. T-cells can then produce interferon gamma (IFN-γ) to induce cytolysis in cancer cells (34).
At the same time, tumor cells take advantage of the ability of myeloid cells to inhibit tumor-targeting immune responses and to mediate immunosuppressive effects. Tumor growth and progression is restrained to genetic or epigenetic alterations which, in turn, affect tumor development and invasion into the surrounding tissues. During this process, cancer cells reprogram infiltrating stromal cells to support an abnormally regulated inflammation that is hyporesponsive to the tumor (35). Cancer cells achieve this by producing immune effector molecules, such as tumor necrosis factor-α (TNFα) and interleukin-6 (IL-6), growth factors that regulate tumor proliferation and angiogenesis, such as transforming growth factor-β (TGF-β) and vascular endothelial growth factor (VEGF), and matrix metalloproteinases (MMPs) that degrade extracellular matrix proteins (31, 36). Abundance of tumor-infiltrating and circulating monocytes, MDSCs and neutrophils, is associated with advanced cancer progression, decreased disease-free and overall survival (37).
MDSCs can be subdivided into two major groups: monocytic MDSCs (m-MDSCs) and granulocytic MDSCs (g-MDSCs) that are morphologically similar to monocytes and granulocytes, respectively. In humans, m-MDSC have the same density fraction as monocytes. However, monocytes express the MHC class II cell surface receptor HLA-DR in high levels while m-MDSCs are characterized by low or no HLA-DR expression. Furthermore, m-MDSCs have a CD11b+HLA-DR−CD14+CD15− phenotype (38). The expansion of m-MDSCs is induced by a combination of soluble factors produced by tumor and/or surrounding cells such as stromal cells, T-cells or macrophages including VEGF, GM-CSF, M-CSF, IL-4, IL-6, IL-10, prostaglandin E2 (PGE2), MMP9, C-X-C motif chemokine ligand 5 (CXCL5), and CXCL12/ stromal-derived factor 1 alpha (SDF1-alpha) (39). Human g-MDSCs are phenotypically characterized as CD11b+HLA-DR−CD14−CD15+ (38).
In addition to their morphological and phenotypic differences, m-MDSCs and g-MDSCs also have different mechanisms by which they suppress immune function. TAMs and m-MDSCs have a shared mechanism for the expression of inducible nitric oxide synthase (iNOS) and arginase. Indoleamine 2,3-dioxygenase (IDO), inducible nitric oxide synthase (iNOS), arginase I and effector cytokine production have been proposed to be involved in suppression of T-cell proliferation and cytotoxicity (40). iNOS generates nitric oxide (NO) causing the inhibition of IL-2 receptor signaling, blocking T-cell activation and proliferation, thus leading to an immunosuppressive effect (41). TAMs that have a classic (M1) activation state (25), paradoxically express iNOS, whose immunosuppressive effect is, however, overawed by other proinflammatory and anti-tumor mediators. TAMs and MDSCs commonly employ another immunosuppressive mechanism involving the generation of reactive oxygen species (ROS) and reactive nitrogen species, such as peroxynitrite mediated by iNOS and arginase (42). Following T-cell receptor (TCR) modification of anti-tumor T-cells due to peroxynitrite, they become unable to bind to their equivalent MHC-peptide antigens presented by APCs in the tumors (43). Furthermore, m-MDSCs can indirectly suppress anti-tumor immunity, through the production of TGF-β and IL-10 cytokines, which inhibit anti-tumor TILs, generate regulatory T-cells (Tregs) in the tumor and induce DCs into a regulatory phenotype (44). Alternatively, immunosuppressive g-MDSCs share many of the immunosuppressive mechanisms of m-MDSCs, but they also produce ROS, which are able to alter the TCR of TILs through direct cell-to-cell contact (45, 46).
Understanding the interactions between the tumor and infiltrating cells will allow the prediction of tumor progression as well as the design of novel anticancer therapies which will target the tumor microenvironment. Working on the basis that cancer and myeloid cells use common pathways for immune system regulation, along with the fact that myeloid cells have the ability to network with different immune cell populations toward inducing an anti-tumor immune response, myeloid-based therapies have increasingly gained attention as possible adjuncts to improve efficacy of current therapies, including immune checkpoint inhibitors (ICIs), oncolytic viruses, dendritic cell vaccines, and traditional chemoradiation. Characterizing the myeloid compartment also allows for patient stratification on prognosis and response to immunotherapy based on the presence of myeloid-specific biomarkers in combination with tumor mutational burden, checkpoint expression, and T-cell receptor diversity (47).
TAMCs In Conventional Chemotherapy and Targeted Therapy
Negative Impact of TAMCs in Chemotherapy
The presence of TAMs in the TME provides cancer cells with cytokines, growth factors and proteases that mediate survival, chemoresistance and promote invasion. One example is cysteine cathepsin proteases that are produced by macrophages and have been shown to enhance pancreatic tumor growth and invasion (48). Paclitaxel, an anti-microtubule agent belonging to the Taxane family, is used for the treatment of ovarian, breast and non-small cell lung cancer. Following Paclitaxel treatment, the infiltration of macrophages in mammary tumors as well as cathepsin levels were increased. In co-culture experiments, macrophages protected cancer cells from Paclitaxel treatment by producing cathepsins B and S; this was reversed by cathepsin inhibition, suggesting that concurrent inhibition of TAMs along with chemotherapy may limit the development of resistance (49). The chemoresistance facilitated by macrophages was also observed against Etoposide and Doxorubicin (49).
As previously mentioned, CSF-1 and its receptor (CSF-1R) are also involved in the tumor-promoting functions of TAMs. The blockade of CSF-1 decreased macrophage infiltration and improved response of mammary and pancreatic carcinomas to chemotherapy (50). B-Raf is a serine/threonine protein kinase, that acts downstream of RAS and has an important role in the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway. The BRAF gene is commonly mutated in melanoma resulting in a constitutive function of the B-Raf protein (51). In a mouse model of melanoma, the concurrent treatment with CSF-1R inhibitor, PLX3397, and BRAF inhibitor, Vemurafenib, resulted in enhanced anti-tumor responses attributed to a significant reduction of tumor-infiltrating myeloid cells and an increase of tumor-infiltrating lymphocytes (52). Another specific CSF-1R inhibitor, GW2580, in combination with an anti-VEGFR-2 antibody, synergistically inhibited tumor angiogenesis in lung cancer and melanoma in vivo models; blocking CSF-R1 led to reduced tumor recruitment of TAMs and reverted a TAM-mediated compensatory antiangiogenic mechanism involving MMP-9 (53). The chemoresistance induced by tumor-infiltrating macrophages could also be mediated by the expression of IL-10; therapeutic blockade of IL-10 receptor (IL-10R) had similar effects to CSF-1 neutralization and enhanced tumor response to Paclitaxel and Carboplatin in the MMTV-PyMT transgenic model of luminal B-type mammary carcinoma (54).
In two human hepatocellular carcinoma xenograft mouse models (HCCLM3-R and SMMC7721) tumor growth, lung metastasis, and tumor angiogenesis were observed following treatment with Sorafenib, a multi-kinases inhibitor. Sorafenib caused a significant increase in macrophage peripheral recruitment and intratumoral infiltration accompanied with elevation of CSF-1R, CXCL12/SDF-1 alpha, and VEGF. SDF-1/CXCL12 has been correlated with cancer cell invasion by recruiting macrophages to the area surrounding the tumor (55). Targeting of macrophages using two specific drugs, Zoledronic acid (ZA) and Clodrolip, in combination with Sorafenib significantly hindered tumor progression, angiogenesis, and metastasis to the lungs compared with animals treated with Sorafenib alone (56). Serial low doses of Sorafenib augmented tumor inhibition and function of CD8+ T-cells by decreasing MDSCs and reversing the immunosuppressive microenvironment in an E.G7/OT-1 murine model (57).
The importance of TAM polarization is evident in patients treated with platinum-containing chemotherapy. Chemoresistance is associated with elevated levels of PGE2 and IL-6, two inflammatory mediators that are regulated by cyclooxygenase (COX), drive differentiation of monocytes to the M2 tumor-promoting phenotype. Treatment with Cisplatin or Carboplatin increased the potency of cervical and ovarian cell lines to induce M2 macrophages that produce IL-10. Tumor-produced IL-6 and PGE2 led to increased levels of activated Signal Transducer and Activator of Transcription 3 (STAT3) and decreased levels of activated STAT1 and STAT6, respectively. Blockade of canonical Nuclear Factor Kappa-light-chain-enhancer of activated B cells (NF-κB) reduced the production of PGE2 and/or IL-6 by the tumor cells and abrogated the effect of the chemotherapy. Blocking COX using the specific inhibitor indomethacin as well as inhibition of interleukin-6 receptor (IL-6R) with the clinical monoclonal antibody Tocilizumab, prevented M2 differentiation. These results propose that chemoresistance may be caused via an increase in the number of M2 macrophages and that concurrent therapy with COX inhibitors and/or anti-IL-6R antibodies might facilitate platinum-based chemotherapy in resistant tumors (58).
Studies have shown that B and T lymphocytes may exert pro-tumor effect by regulating the activity of myeloid cells, resulting in resistance to therapy and promoting metastasis in different malignancies, including epithelial hyperplasia, squamous carcinomas and prostate cancer (59–61). In the absence of a robust CD8+ CTL response, CD4+ T-effector lymphocytes enhance breast cancer metastasis to the lung by enhancing the activity of TAMCs (62). In a study using an aggressive transgenic mouse model of mammary adenocarcinoma development [MMTV–polyoma middle T (PyMT) mice (63)], combination of CSF1R-signaling antagonists that block infiltration of mammary tumors by CD68+ macrophages, in combination with Paclitaxel, improved survival, delayed primary tumor growth and reduced pulmonary metastasis. This study also showed that the presence of an enriched CD68high /CD4high /CD8low cell population, significantly correlates with reduced overall survival (OS) for patients with breast cancer (64).
Depletion of myeloid-lineage cells enhanced anti-cancer immunity associated with gemcitabine (GEM) treatment in mice with pancreatic ductal adenocarcinoma (PDAC) tumors (65). GEM is a nucleoside analog used as first-line treatment. Myeloid cells expressing the granulocytic marker (GR-1) were found in abundance in PDAC tumor tissues while CD4+ and CD8+ cells were present in small numbers. Following GEM treatment, myeloid cells in tumor tissues and in peripheral blood decreased while numbers of CD4+ or CD8+ cells increased suggesting that anti-cancer immunity was enhanced. In addition, concurrent treatment of mice with GEM and further depletion of myeloid cells using an anti-GR-1 antibody significantly prolonged survival (65). Inflammatory breast cancer (IBC) is often characterized by overexpression of epidermal growth factor receptors 1 and/or 2 (ErbB1, ErbB2) that activate downstream survival pathways phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) and MAPK (66). Lapatinib (a dual ErbB1/2 tyrosine kinase inhibitor) is for IBC patient treatment and functions by blocking ErbB1 and ErbB2 receptor phosphorylation and activation (67). Following combination treatment with Lapatinib and the anthracycline Doxorubicin in an MMTV-neu mice HER2-positive breast cancer model, CD8+ T-cells secreting IFN-γ contributed to the anti-tumor effects of these drugs. Increased effectiveness correlated with decreased content of immunosuppressive TAMs in the tumor bed induced by Doxorubicin (68).
In PDAC patients, TAMs may contribute to resistance to GEM by reducing GEM-induced apoptosis. In vitro co-culture of macrophages with cancer cells significantly reduced of Caspase-3 activation and apoptosis during GEM treatment. In in vivo PDAC models of mice, macrophages recruitment to the tumor using CSF1R-antagonist GW2580, enhanced the effect of GEM; the presence of TAMs in the tumor seems to convey resistance to GEM by inducing upregulation of the enzyme cytidine deaminase (CDA). CDA metabolizes GEM following its transfer into the cell. In PDAC cells, decreasing the expression of CDA inhibited the protective effect of TAMs against GEM (69).
TAMs have also been found to confer resistance to MAPK pathway inhibitors against melanoma. The mechanism of action involved expression of TNFα by TAMs and acted through the lineage transcription factor microphthalmia transcription factor (MITF). MITF plays a key role in melanocyte differentiation by transcriptional control of genes expressing enzymes involved in melanin synthesis; in addition, MITF has protumoral targets including B-cell lymphoma 2 (Bcl-2) and Hypoxia-Inducible Factor 1 (HIF-1) that convey survival signals (70). TNF binding to TNFR activates multiple signaling pathways, including MAPK and NF-κB and induces apoptosis and necroptosis pathways (71). Inhibition of TNFα signaling with IκB kinase inhibitors significantly improved the effectiveness of MAPK pathway inhibitors by targeting not only the melanoma cells but also the tumor microenvironment (72). Also, in melanoma cells macrophages conferred resistance to BRAF inhibitors in mouse and human tumor models, which was overcome by blocking the MAPK pathway or VEGF signaling. The presence of macrophages predicts early relapse following therapy in melanoma (73). Administration of the BRAF small molecule inhibitor PLX4720 had similar effects in a murine model of melanoma; PLX4720 reduced tumor growth by promoting the formation of a more immune stimulatory microenvironment correlated with a reduced accumulation of CD11b+/GR-1+ myeloid cells (74).
The polarization of macrophages into M1 or M2, has important implications for therapeutic strategies in human cancers (Figure 1). The M2 subtype is thought to support tumor growth. In a spontaneous mouse model of gastrointestinal stromal tumor (GIST) as well as upon analysis of freshly procured human GISTs, TAMs displayed an M1-like phenotype and function at baseline; however, treatment with Imatinib, that acts as a KIT oncoprotein inhibitor, induced TAMs to become M2-like, in both mice and humans. This process involved the interaction of TAMs with apoptotic tumor cells leading to the induction of C/EBP transcription factors and development of resistance to Imatinib (75). Re-programming of macrophages from an immune-inhibitory M2-like subtype toward an immune-stimulatory M1-like subtype, by targeting the VEGF receptor 2, enhanced anticancer efficacy in a CD8+ T-cell–dependent manner in murine breast cancer models, suggesting that combination of anti-angiogenic therapy with other types of drugs may facilitate anti-tumor effects by altering the phenotype of TAMs (76).
TAMCs Facilitate Chemotherapy
Tumor Associated Macrophages
Common chemotherapeutic drugs may act as alkylating agents. They function by adding an alkyl group to the guanine base of the DNA molecule, making the strands unable to uncoil and separate and causing breakage of the DNA strands and apoptosis. In response to alkylating agents, an immune response involving the activation of macrophages is initiated and this involves the high mobility group box 1 (HMGB1) protein. HMGB1 is an important chromatin protein that bends DNA, facilitates protein binding and helps regulate transcription (77). Activated macrophages and monocytes secrete HMGB1 which acts as a mediator of inflammation (78). In an athymic mouse tumor xenograft model where tumors were formed using immortalized murine embryonic fibroblasts (MEFs) overexpressing Bcl-xl, DNA alkylating therapy led to inhibition of protumor cytokines such as IL-4, IL-10, and IL-13, recruitment of innate immune cells including neutrophils, NK cells and macrophages into the treated tumor tissue and to complete tumor regression; loss of HMGB1 resulted in increased levels of protumor cytokines upon treatment and failure to activate innate immunity (79).
Contradictory to the chemoresistance developed by macrophages following exposure to Paclitaxel as described in the previous chapter, other studies suggest that the agent can promote anti-tumor immunity by polarizing M2 macrophages to the M1-like phenotype (80). As previously mentioned, macrophages that polarize as M1-type are considered pro-inflammatory and potentially mediate anti-tumor activities, whereas those that polarize as M2-type decrease inflammation and may promote cancer cell growth via angiogenesis and immunosuppression (81–83). Similarly, administration of Doxil nanomedicine combined with the TGFβ inhibitor Tranilast, increases immunostimulatory M1-type macrophage content over the M2-type in mouse models of triple-negative breast cancer (84). A possible explanation, which may also be applied to other observations, could be the improved blood vessel perfusion, oxygenation, and normalization of the TME. Moreover, macrophages polarize to the M1-type following exposure to IFN-γ and Lipopolysaccharide (LPS) (85). Paclitaxel induced TAMs toward an M1-like profile in mouse models of melanoma and breast tumors which was depended on the presence of Toll-Like receptor 4 (TLR4) on myeloid cells. Absence of TLR4 weakened the antitumor effect of Paclitaxel. This was confirmed using gene expression analysis of tumor samples from ovarian cancer patients that showed enrichment of genes correlated to the M1 macrophage activation profile following Paclitaxel treatment (80). A similar effect was observed using nanoparticles loaded with albumin-bound paclitaxel (nAb-PTX) (86). Using 3D-spheroid models of co-cultured breast cancer cells with macrophages as well as in vivo models, researchers showed increased drug accumulation within the macrophages and the tumor spheroids; this shifted the TME toward a pro-inflammatory, anti-tumorigenic state. In addition, the loaded nanoparticles (NPs) increased macrophage motility and delivery of the NPs toward the cancer cells promoting apoptosis and inhibiting proliferation. Importantly, the NPs loaded with PTX induced macrophage differentiation toward the anti-cancer M1 phenotype (86).
Following surgery, the OS of gastric carcinoma patients treated with 5-fluorouracil-based chemotherapy was positively correlated with increased numbers of CD68+ macrophages (87). In a cohort study involving 110 patients with PDAC, post-surgical adjuvant chemotherapy was shown to “re-educate” TAMs and help them elicit an anti-tumoral response. Cyclophosphamide (CTX) is a common chemotherapeutic agent, which acts as an alkylating agent, that is used for the treatment of several human malignancies (88). Enrichment of TAMs at the tumor–stroma interface positively correlated with responsiveness to CTX therapy in patients with PDAC, independently of the density of T-cells. A similar effect was observed in vitro, where in the presence of GEM, macrophages activated a cytotoxic gene expression program and switched to an anti-tumor phenotype (89). In patients with invasive ductal breast cancer receiving adjuvant multimodal chemotherapy, tumor infiltration by macrophages correlated with improved time-to-relapse (TTR) and OS (90). Similar observations were made in patients with colorectal carcinoma (CRC). In a study of 1,400 CRCs patients treated with adjuvant multimodal chemotherapy, the level of CD16+ macrophage infiltration correlated with that of CD3+ and CD8+ lymphocytes and with improved survival compared with patients with low infiltration (91). These results contradict with previous reports indicating that a low number of CD68+ macrophages infiltrates associates with improved patient survival (64); the role of TAMs in predicting patient response to treatment is therefore complex and remains to be further elucidated.
TAMs may also act as a slow-release reservoir for therapeutic nanoparticles (TNPs). TNPs are generally applied as a vehicle to deliver drugs specifically to the tumor site and increase their accumulation. TNPs comprised by a fluorescent platinum (IV) pro-drug and a polymer platform (PLGA-b-PEG) were shown to accumulate in TAMs. TAMs acted as a local drug depot and allowed for the slow release of the DNA-damaging drug to neighboring tumor cells. The depletion of TAMs led to a decrease of intratumoral TNP accumulation and treatment efficacy in a lung cancer animal model. The presence of TAMs can therefore affect the design and use of TNPs for tumor targeting (92).
Histidine-rich glycoprotein (HRG), is a host-produced immunomodulatory and antiangiogenic factor that regulates tumor vessel formation and inflammation. HRG is produced in the tumor stroma from plasma or platelets and has been reported to inhibit tumor growth and metastasis and to enhance chemotherapy in brain tumor models (93, 94). This effect is mediated through downregulation of placental growth factor (PlGF) followed by polarizing TAMs from the M2- to a tumor-inhibiting M1-like phenotype. It is likely that HRG/PIGF/M1-type TAMs enhance the antitumor immune response and facilitate vessel normalization, effects known to hinder tumor growth and metastasis and to facilitate chemotherapy (29).
Dendritic Cells
The therapeutic efficacy of anthracyclines, a group of conventional chemotherapeutic drugs that act by inhibiting topoisomerase II causing DNA damage, may depend on the presence of intratumoral dendritic cells. Specifically, mice bearing fibrosarcomas were treated with the anthracycline mitoxantrone (MTX). This caused cancer cell death which led to the release of Adenosine triphosphate (ATP), recruitment of myeloid cells and their differentiation locally into inflammatory DC-like cells. The presence of the DC-subset was responsible for the immune system-depended anti-tumor effect of anthracycline, by engulfing tumor antigens and presenting them to T-lymphocytes. Importantly, preventing tumor infiltration by myeloid cells, abolished the anti-tumor immune response following chemotherapy (95). The activation of autophagy in cancer cells is essential for increasing the recruitment of DC and improve the efficacy of chemotherapy. In mouse models of colorectal cancer and sarcomas, response to chemotherapy led to the release of ATP by autophagy-competent cancer cells, attracted dendritic cells and T lymphocytes into the tumor bed and restored chemotherapeutic responses (96). Exposure to chemotherapeutic agent Paclitaxel, does not significantly affect the viability of DCs in concentrations up to 100 μM (97). Furthermore, exposure of DCs to clinically relevant concentrations of Paclitaxel led to increased HLA class II expression which was similar to the expression observed when DCs are exposed to lipopolysaccharide (LPS). Paclitaxel also increased proliferation of allogeneic T-cells. This study suggests that Paclitaxel may induce immunostimulatory effects in certain concentrations and may find clinical applications in patients receiving DC vaccines (97).
DC-like cells are important in the anti-tumor immune response since they have enhanced abilities to activate CD8+ T-cells compared to TAMs. The effect of Oxaliplatin combined with Cyclophosphamide (Oxa-Cyt) treatment on tumors relied on TLR4 signaling; Oxa-Cyt treatment led to an increase of TLR4 selectively in DC cells within the tumor stroma and ultimately led to CD8+ T-cell anti-tumor immunity in lung adenocarcinoma mouse models (98). CTX has been shown to induce anti-cancer effects by stimulating immunomodulatory factors; in patients with hematologic malignancies, a single high-dose treatment with CTX induced an increase in the number of DCs (99). DC turnover in the spleen, liver, and tumor site as well as their expansion in the circulation, enhanced the beneficial anti-tumor effects of CTX in mice models. The expansion of DC (CD11c+CD11b+) induced by CTX was associated with proliferation of DCs in the bone marrow (BM) prior to their increase in the circulation in a melanoma mouse model (100). These newly recruited DCs secreted more IL-12 and less IL-10 compared with those from untreated animals and were able to induce anti-tumor T-cell responses in a colon cancer model (101).
In addition to facilitating chemotherapy, DCs may also contribute to specific cancer targeting induced by small molecule inhibitors. Overactivation of the Jak2/STAT3 signaling pathway induced by tumor-derived factors may be responsible for irregular DC differentiation and function in colon cancer (102). The use of a selective inhibitor of Jak2/STAT3, JSI-124, led to activation of the transcription factor NF-κB, promoted the differentiation of mature DCs and led to T-cell activation (103). JSI-124 has been previously shown to inhibit the growth of tumors with constitutively active STAT3 (104).
Finally, recent evidence suggest that dosing and scheduling of chemotherapy administration may also regulate anti-tumor immunity. In contrast to traditional chemotherapy protocols in which the anti-cancer agents are cyclically administered near the maximum tolerated dose (MTD) alternated with longer drug-free periods, metronomic chemotherapy protocols suggest a more frequent administration of doses as low as 1/10th of the MTD (105). Besides significantly reducing drug-mediated adverse effects, metronomic chemotherapy may enhance anti-tumor immune responses. More specifically, metronomic administration of CTX was found to increase infiltration of DCs, macrophages and NK cells in mouse models as well as end-stage patients of various cancer types (106, 107). In addition, metronomic chemotherapy regimens may also promote vascular normalization to enhance delivery of co-administered drugs, thus further improving the efficacy of anti-cancer treatments (108, 109). The mechanisms of action utilized by myeloid cells in supporting or hindering chemotherapy are summarized in Table 1.
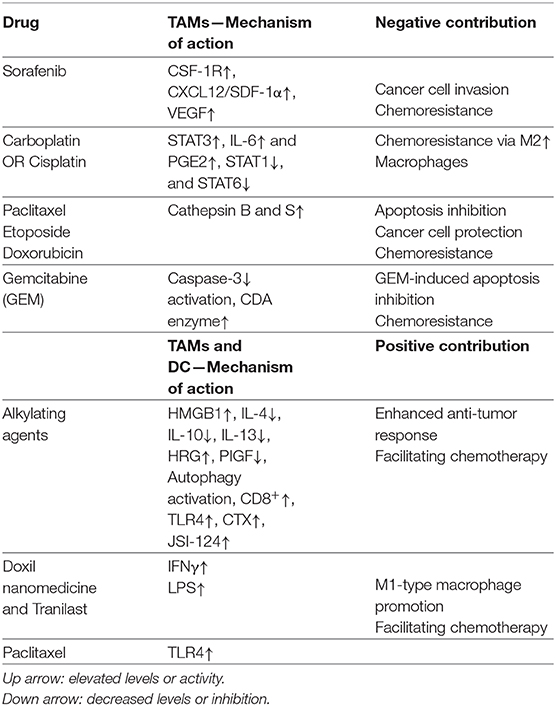
Table 1. Proteins implicated in TAM negative or positive contribution to chemotherapy.
TAMCs in Radiotherapy
Radiotherapy is an important and commonly used treatment approach in cancer; local radiotherapy allows for non-invasive, site-specific intervention. Even though the main mechanism of action is via tumor cell DNA damage, recent evidence suggests that irradiation activates tumor-specific immunity (Figure 2). The effects of radiotherapy include the induction of antigen release from dying tumor cells, the activation of APCs and the support of tumor-specific T-cell immigration and function (110–113). In a study using the RIP1-Tag5 (RT5), human melanoma xenografts mouse model, and human pancreatic cancer specimens derived from patients undergoing low-dose irradiation (LDI) of 0.5 Gy, researchers showed that neoadjuvant local LDI causes the CTL recruitment and activation in solid tumors. This was associated with the accumulation of iNOS-positive (iNOS+) macrophages and led to prolonged survival in xenotransplant mouse tumor models (114). Dendritic cells contribute to the immune response following high dose radiation. Local high-dose irradiation (10 Gy) leads to activation of tumor-associated DC that induce tumor-specific effector CD8+ T-cells (115).
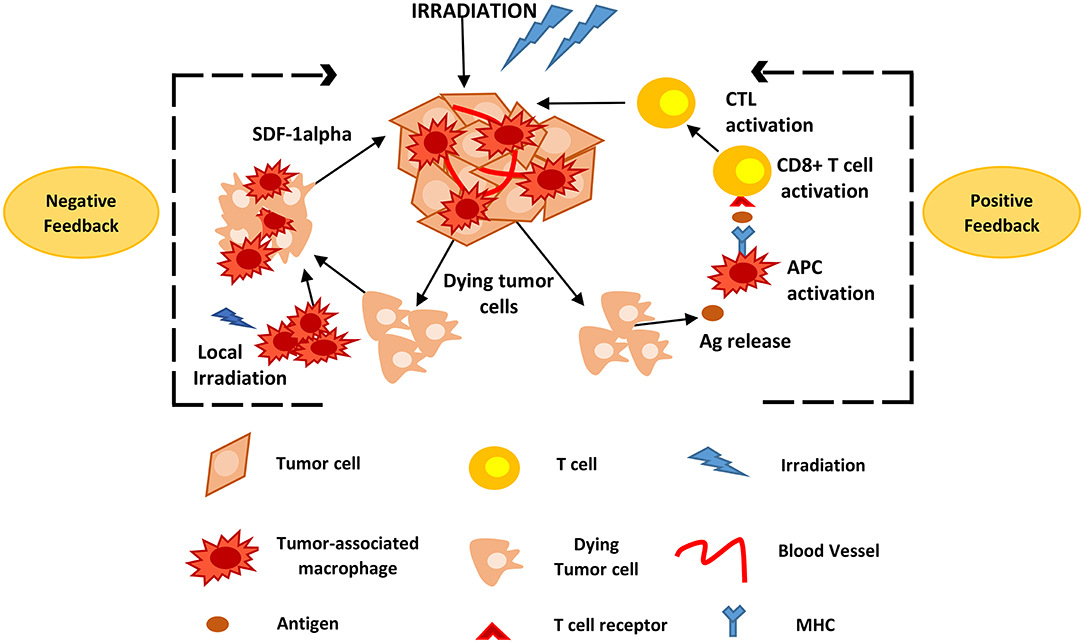
Figure 2. Positive and negative feedback loops of TAM activity during radiotherapy. Following high and low irradiation protocols, antigens are released from dying tumor cells and taken up by APCs, such as TAMs, that subsequently activate CD8+ T-cells. This causes CTL recruitment and activation that attack solid tumors. Local irradiation may also cause the accumulation of macrophages to the tumor site that promote tumor recurrence mainly via the expression of SDF-1alpha. Ag, Antigen; APCs, antigen presenting cells; CTL, cytotoxic T lymphocytes; TAMs, tumor associated macrophages; SDF-1alpha, stromal cell-derived factor-1 alpha.
Myeloid cells may also negatively affect radiotherapy. In a prostate cancer animal model, irradiation with a local daily dose of 3 Gy for 5 days led to a systemic increase of MDSCs in lymph nodes, lung, spleen, and peripheral blood and a 2-fold increase in CSF-1 in tumors. Blockade of CSFR1 by a selective inhibitor, decreased macrophage migration and in combination with radiotherapy repressed tumor growth more effectively than irradiation alone (116). In a similar approach, following radiotherapy of mammary tumor-bearing mice using localized gamma irradiation (5 Gy), the blockade of CSF-1 using a neutralizing monoclonal antibody (mAb) or a small molecule inhibitor against the CSF-1 receptor kinase (PLX3397), caused depletion of macrophages and significantly inhibited tumor growth. This was associated with increased numbers of CD8+ T-cells in tumors, and reduced the number of CD4+ T-cells, the main source of the Th2 cytokine IL4 which can lead to a pro-tumor advantage (117). Following local irradiation with 21 Gy in breast and lung carcinoma xenograft models, myeloid bone marrow-derived cells (BMDCs), primarily macrophages, rapidly accumulated in tumors. The levels of SDF-1alpha/CXCL12, a chemokine that promotes BMDCs retention in the tissue, were increased in the tumor, 2 days after local irradiation. Concurrent treatment with radiation and an inhibitor of SDF-1alpha receptor (AMD3100) significantly hindered tumor regrowth. These results suggest that macrophages promote tumor recurrence following radiation via increase in the expression of SDF-1alpha (118).
The contradicting reports concerning the role of myeloid cells on the efficacy of radiotherapy, may be attributed to the radiation dose and fractionation methodology. Both these parameters appear to affect the tumor microenvironment and the immune system. The conventional standard dose fractionation of 2 Gy per fraction is mostly used to achieve cell damage within the tumor (119). However, several pre-clinical studies suggest hypofractionated high doses of 6 or 8 Gy are more effective compared to a single high dose of radiation in inducing pro-immunogenic effects (120–122). In vitro studies have also shown that larger doses of radiation induce immunogenic cell death (ICD) (123). The effects of radiation of the TME should also be taken into account when trying to evoke an immune response. High single-fraction doses (8–16 Gy) induce increased permeability and apoptosis in endothelial cells (124). Even though a definitive radiotherapy regimen that effectively manipulates the TME and activates the immune system against the tumor has not been established, low, standard and high doses as well as different fractionation approaches have been found to be effective. It is also possible that optimized dosing and fractionation protocols may be suitable for different types and/or stages of cancer. The effects or radiation on the immune system and how it may promote tumor survival or destruction are detailed in a recent review (125).
Role of MDSCs, TAMs and DCs in Immunotherapy
The main challenge of tumor immunologists is to control the vicious cycle of inflammation-immunosuppression taking place within the TME. The main approaches followed include among others, targeting immune checkpoint molecules on myeloid cells, the inhibition of recruitment and survival of myeloid cells, while novel approaches of nanomedicine regulating MDSCs are also under investigation.
Targeting Immune-Checkpoint Molecules on MDSCs, TAMs, and DCs
Immune checkpoints are among the regulators of the immune system that defend self-tolerance. Various tumor cells utilize these regulators to evade immune responses (126). Inhibitory immune-checkpoint molecules, including the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1) and its ligand (PD-L1), T-cell immunoglobulin and mucin-domain containing molecule (TIM-3), ligands belonging to the B7 family, and others are promising targets for novel cancer immunotherapeutics. Antibodies against these inhibitory molecules are being tested in clinical trials, for their potential as mono- or part of combinatorial therapy against human neoplasias. Some of them have received approval by the Food and Drug Administration (F.D.A.) and entered the clinical routine practice for the therapeutic management of certain human tumors. These include (a) the human anti-CTLA-4 mAb ipilimumab for the treatment of metastatic melanoma, (b) the human anti-PD-1 mAbs nivolumab and pembrolizumab for the treatment of melanoma and unresectable/metastatic solid tumors, respectively, and (c) the anti-PD-L1 mAb atezolizumab for patients with metastatic non-small cell lung cancer (127). PD-1, PD-L1, TIM-3, and B7 molecules are expressed by subsets of TAMs and DCs, and consist therapeutic targets facilitating the inhibition of the function of these cells and the subsequent elimination of the tumor (Figure 3).
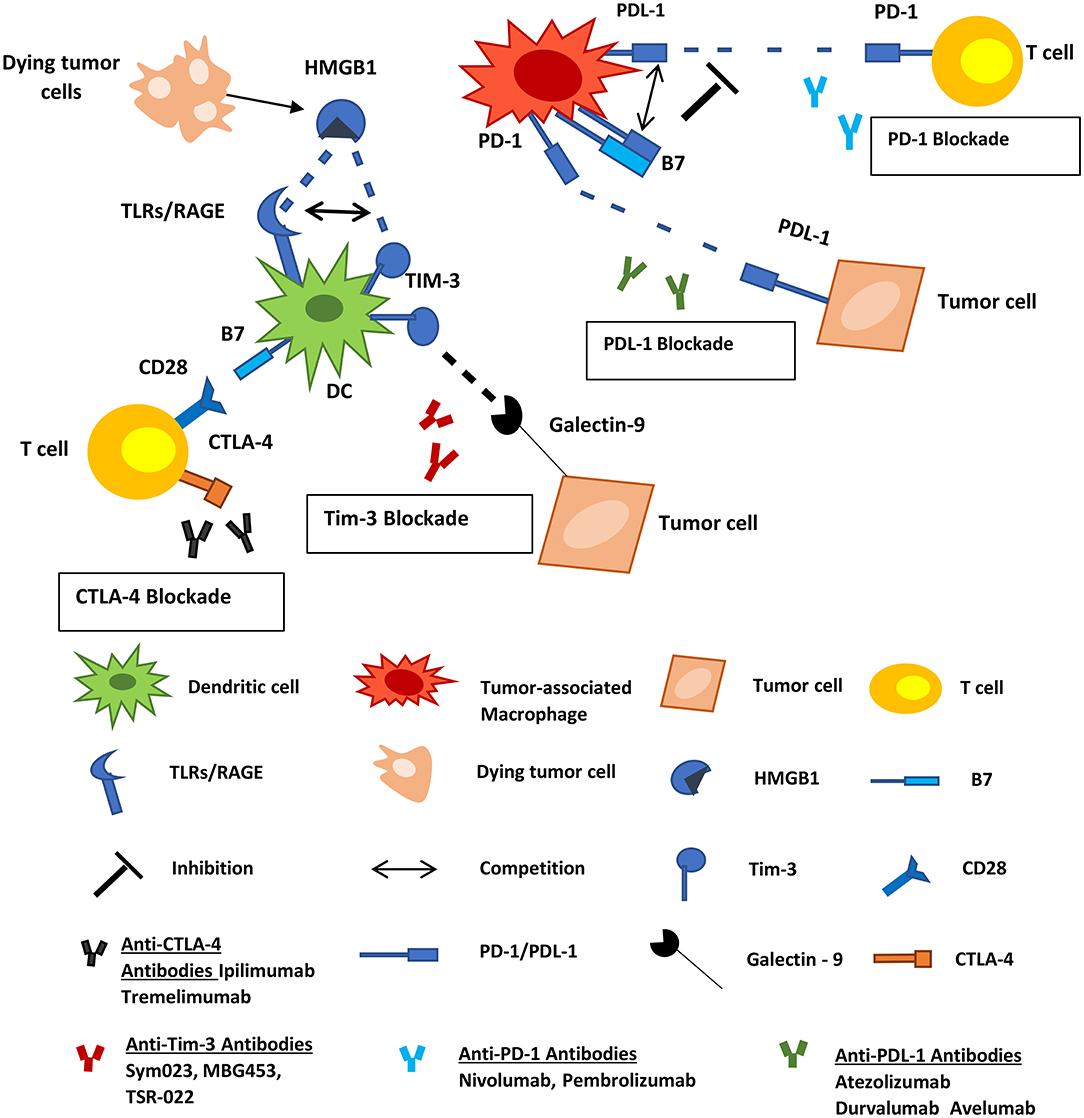
Figure 3. Inhibitory immune checkpoint molecules represent targets for cancer therapy. PD-1, PD-L1, TIM-3, and B7 molecules are expressed by subsets of myeloid-derived cells. PD-1 on macrophages interacts with PD-L1 on tumor cells and allows cancer progression by promoting escape from immune surveillance. TAMs also express PD-L1 and B7 molecules that can interact with the PD-1 on T-cells inhibiting the function of the latter. TIM-3 on infiltrating DCs binds to HMGB1 derived from dying tumor cells blocking anti-tumor immune responses. Tumor cells also express Galectin-9 which interacts with TIM-3 on DCs negatively regulating their function. Tumor Associated Macrophages; DCs, dendritic cells; HMGB1, high-mobility group protein 1; TLR, toll-like receptor; RAGE, receptor for advanced glycation end products.
As a response to hypoxia and specific cytokines, TAMs express elevated levels of CTLA-4 ligands and other immune-checkpoint inhibitors. CTLA-4 ligands such as B7 molecules are also highly expressed in DCs of the tumor microenvironment (128). This overexpression is associated with the downregulation of anti-tumor activities of T-cells, by inhibition of the co-stimulatory interaction with CD28, both in humans and in animal models (129–132). The use of anti-CTLA-4 antibodies would hamper this inhibition and promote T-cell co-stimulation via CD80/CD86–CD28 interaction. A subject of debate, however, is whether the anti-CTLA-4 antibodies act in favor of anti-tumor immune responses or not, in terms of targeting the CTLA-4 molecules expressed on regulatory T-cells (Tregs). CTLA-4 expression by Tregs comprise one their main cell contact-dependent mechanisms affecting antigen-presentation to T-cells. However, it seems that anti-CTLA-4 therapy favors the blockade of the inhibitory activity of CTLA-4 on both effector T-cells and Tregs (133). Indeed, ipilimumab administration in mice bearing melanoma tumors, amplified CD8+ T-cell activities against tumor cells and inhibited immunosuppressive functions of Tregs (126, 133, 134). The latter is achieved via Treg phagocytosis by TAMs expressing Fcγ receptors (133). In a murine model of head and neck squamous cell carcinoma (HNSCC), the blockade of CTLA-4 was correlated with reduced numbers of MDSCs and M2 macrophages, and enhancement of T-cell activation in both tumor microenvironment and macro-environment (132). In humans, it has been reported that patients with advanced melanoma receiving ipilimumab exhibited significantly decreased counts of MDSCs and significantly increased counts of CD8+ effector/memory T-cells in the circulation (135). In addition to that, in another recent study, the percentage of FcγRIIIA+ CD16+ peripheral blood monocytes was found to be higher in melanoma patients who respond to ipilimumab therapy compared to the non-responders (136). The above data underline the pivotal role of the CD8+ T/MDSC balance in the outcome of anti-tumor immune responses, and more importantly how this balance can be affected by treatment with anti-CTLA-4 antibodies. Additionally, a recent study supports that dual therapy with ipilimumab and the vitamin A derivative all-trans retinoic acid (ATRA), leads to a more pronounced reduction in circulating MDSCs compared to the anti-CTLA4 monotherapy (137). ATRA is the standard-of-care treatment for patients with acute promyelocytic leukemia (APL), where it induces terminal differentiation of immature myelocytic tumor cells leading to their death (138). In a similar way, ATRA promotes the differentiation of MDSCs, resulting in their decreased numbers and function (139). Based on these properties, combinatorial administration of ATRA and ipilimumab consists a promising enhanced weapon in the treatment against human cancers.
PD-1 is expressed by a subset of macrophages and DCs of the TME. This molecule may interact with PD-L1 on tumor cells leading to the negative regulation of TAMs and DCs. Expression of PD-1 by TAMs increases with the progression of tumor in mice and in advanced stages of the disease in humans, while it is negatively associated with their phagocytic activities against tumors cells (140, 141). In murine models of cancer, inhibition of the PD-1/PD-L1 interaction enhances macrophage phagocytosis, suppresses the growth of the tumor and prolongs the survival of animals (140). Another recent study showed that the results of anti-PD-L1 therapy in tumor-bearing mice was totally abolished in PD-L1-deficient animals, supporting the importance of blocking the PD-1/PD-L1 pathway as an anti-cancer therapeutic approach (142). Despite the promising evidence from pre-clinical studies, in human clinical trials the administration of nivolumab (anti-PD-L1) in patients with renal cell carcinoma, non-small cell lung cancer or metastatic melanoma with PD-L1-negative tumors was associated with reduced clinical response (NCT00730639; clinicaltrials.gov) (143, 144). Tumor-infiltrating DCs and TAMs also express PD-L1 and B7 molecules that can interact with the PD-1 on T-cells obstructing the function of the latter (145). Thus, it is essential that all the above mentioned evidence are carefully considered during the development of anti-cancer immunotherapeutic strategies to act in favor of anti-tumor immunity and not by promoting its inhibition. Future clinical studies are needed to evaluate the possible beneficial impact of this blockade in patients with specific types of tumors (141).
Certain myeloid cell subsets express TIM-3, which can bind to the phosphatidylserine (PS) revealed by apoptotic cells further contributing to their presentation to CD8+ T-cells (146). DCs express TIM-3 which can also bind to HMGB1 derived from dying cells (147). HMGB1 and nucleic acids from apoptotic cells stimulate anti-tumor immunity, and TIM-3 can block this stimulation by competition for binding to the HMGB1-nucleic acid complex (147). Tumor cells also express galectin-9 which interacts with TIM-3 on tumor-infiltrating DCs regulating their function. Indeed, it was recently shown that anti-Tim-3 blocking in combination with Paclitaxel administration amplified anti-tumor immune responses against breast cancer in vivo, and this inhibition was facilitated by the galectin-9-Tim-3 interaction, rather than the HMGB1-nucleic acid-Tim3 or PS-Tim3 interaction (148). This interplay between tumor cells and tumor-infiltrating DCs is probably pivotal for the development and perpetuation of other types of malignancies (149), since galectin-9 is highly expressed by several tumor cell types (150).
Nowadays, numerous phase II and III clinical trials are testing the potential of antibodies against CTLA-4 (ipilimumab, tremelimumab), or the PD-1/PDL-1 axis (nivolumab, pembrolizumab, atezolizumab, durvalumab, avelumab), and also the combinatorial use of anti-PD-1/anti-CTLA-4 antibodies (nivolumab plus ipilimumab) in a series of human malignancies. Moreover, novel antibodies, not yet FDA approved, targeting TIM-3 immune checkpoint (including INCAGN02390, Sym023, MBG453, TSR-022) are now being tested in phase I and II trials (127).
Inhibition of Recruitment and Survival of MDSCs
Cancer cells often express increased levels of the chemokine CCL2; also known as monocyte chemoattractant protein 1, MCP1), which recruits MDSCs (expressing the CCR2) in the site of tumor inflammation (151). Blocking the CCL2-CCR2 interaction could be another alternative to prevent the accumulation of these cells in the TME. Inhibition of this pathway has provided with promising results in murine models of pancreatic (50), hepatocellular (152) and prostate (153) cancer. Moreover, a phase II clinical trial using carlumab (anti-CCL2 mAb) in individuals with metastatic castration-resistant prostate cancer (NCT00992186; clinicaltrials.gov) supports that the withdrawal of anti-CCL2 treatment may lead to a rebound of CCL2 levels; in the participants of this study there was an upregulation of the CCL2 serum levels that exceeded those before treatment (154). Also, cessation of anti-CCL2 treatment was shown to accelerate metastasis in a murine model of breast cancer (155). Thus, therapeutic interventions against CCL2-CCR2 interaction need to be critically examined and future studies to evaluate their overall therapeutic efficiency.
Recent evidence supports that the interaction between CD200-CD200 receptor (CD200R) is essential for the control of immune responses in TME by regulating TAMCs. In humans, certain tumor cell types including melanoma cells (156), ovarian cancer cells (157), malignant B cells (158) and cells from some neuroendocrine neoplasms overexpress CD200. Within the TME, significantly high levels of CD200 can be also detected on endothelial cells, activated T and B cells, Tregs (159), as well as MDSCs, TAMs and DCs (160). The CD200 and CD200R molecules share structural similarities with the PD-PDL1 and CTLA4-B7 molecules and the CD200-CD200R axis is also considered as an immune-checkpoint regulator of tumor-related immune responses (161). The complicated network of interactions among these cell types can drive the outcome of immune responses in the TME and impact tumor progression.
The blockade of CD200-CD200R interactions is currently amongst the immunotherapeutic alternatives under investigation. Studies on hu-SCID (severe combined immunodeficiency) mice with established tumors (162–164) have shown that adoptively transferred peripheral blood mononuclear cells together with blockade of CD200 can lead to the rejection of the tumors. Nevertheless, there is a debate regarding the beneficial effects that the blockage of CD200-CD200R axis may have in cancer patients (141), since following treatment with chemotherapeutic agents such as doxorubicin, the recruitment of functional DCs in the TME claims the CD200-CD200R pathway (165). In humans, the anti-CD200 mAb Samalizumab has entered two phase I clinical trials: one on patients bearing solid tumors (NCT02987504) and the other on patients with B-cell chronic lymphocytic leukemia (B-CLL) or multiple myeloma (NCT00648739) (clinicaltrials.gov). Both studies were terminated, but the second one published results showing that administration of the drug was associated with reduced expression of CD200 on B-cells and CD4+ effector T-cells of B-CLL individuals, however inefficient in the three myeloma patients evaluated (166). What is more, it is expected that the use of CD200-depletion antibodies would have considerable side-effects, since CD200 is also expressed on normal cells. Therefore, the alternative way of targeting the CD200R, which is expressed by cancer but not by normal cells, would be more feasible for use in the clinical setting for the treatment of human malignancies. A study on mice with CD200-negative melanoma tumors showed that treatment with an agonistic anti-CD200R mAb inhibited the tumor formation and metastasis in the lungs of the animals, via inhibition of myeloid cell functions (167). A recent study on animals with colon cancer, suggests that co-treatment with anti-CD200R and a Toll-Like Receptor 7 (TLR-7) agonist promotes the anti-tumor effects of myeloid cells within the TME (168).
Future Perspectives: Nanomedicine Approaches to Deplete, Modulate or Recruit MDSCs or TAMs
Novel approaches for the enhancement of anti-cancer therapeutics also lie in the field of nanomedicine: Based on the enhanced permeability and retention (EPR) effect therapeutic strategies that use carrier materials of < 100 nm can enhance the uptake of chemotherapeutics specifically by tumor cells, thus lowering non-specific cytotoxicity (169, 170). In the field of cancer immunotherapy, the efforts focus on the use of nanoparticles to drive various immunoregulators to tumors and recruit myeloid-derived cells to the site of tumor inflammation (169).
MDSCs and TAMs can facilitate the use of nanoparticles in anti-cancer immunotherapeutics due to their phagocytic ability (171). It was shown that in tumor-bearing mice peripherally administered with nanoparticles, the monocytic and polymorphonuclear MDSCs were preferentially targeted uptaking 10-fold more of these carrier materials compared to the tumor cells (172). Other in vivo studies on mice with tumors or hematological malignancies have shown that intradermal administration of nanoparticles carrying the chemotherapeutic agents 6-thioguanine or a gemcitabine derivative were accumulated in macrophages and myeloid cells of the spleen and the tumors and finally led to the depletion of the MDSC compartments in these sites, promoting adoptive T-cell therapy (172–174).
Apart from depletion, nanoparticles have been used for the polarization of MDSCs to an anti-tumor immune phenotype, using stimulants of the innate immune system, such as TLR ligands (175, 176). A recent article describes how nanoparticles loaded with R848, a TLR7/8 agonist, can promote the polarization of TAMs toward an M1 phenotype, resulting in the control of the tumor growth and protection of the animals against tumor re-challenge (177). Interestingly, the co-administration of R848-nanoparticles with anti-PD1 therapy abolished the resistance of mice to anti-PD1 treatment and led to improved response rates (177). The approach of using nanoparticles carrying mimetics of “danger signals” to induce innate anti-tumor responses together with immune checkpoint inhibitors has already entered trials in the clinical setting: nanomaterials with a TLR9 agonist and the anti-PD-1 mAb pembrolizumab are now being tested in a phase Ib/II clinical study in patients with various metastatic solid tumors [NCT03684785; clinicaltrials.gov].
An alternative of reprogramming macrophages using small interfering RNAs (RNAi) or micro RNAs (miRNAs) that are loaded on nanoparticles was applied to mice with melanoma, colon carcinoma, non-small cell lung cancer and other tumors, leading to encouraging results (178–180). Recently, a research group used nanoparticles that have co-encapsulated both a chemoattract of MDSCs, the CCL2 chemokine, and an RNAi sequence interfering with Cebpb, critical for the immunosuppression phenotypes of these cells. The administration of capsules co-carrying these two protein- and RNA- factors, induced the attraction of MDSCs while reduced the differentiation of monocytes to macrophages in in vitro studies of primary MDSCs and in in vivo experiments on fibrosarcoma mice (181). Lastly, using ferumoxytol, an FDA-approved iron supplement composed of dextran-coated iron oxide nanoparticles, Zanganeh et al. (182) managed to inhibit tumor growth by inducing the pro-inflammatory M1 phenotype in the TME of early breast tumors, and liver metastases in mice with lung cancer.
Conclusions
It is unambiguously accepted that immune cells in the tumor stroma exert fundamental effects not only on cancer development and disease progression but also for treatment efficacy. In particular, myeloid cells residing in the tumor microenvironment, including MDSCs, TAMs, DCs and tumor-associated neutrophils (TANs), can either enhance tumor rejection or facilitate cancer progression based on their functional interplay with cancer cells. Moreover, while the variety of tumor treatment options gradually increase, including targeted therapies, nanotherapies and immunotherapies, reaching optimal levels of efficacy often appears to be hindered by the infiltration and complex interactions with myeloid cells. It is therefore critically important for future studies to further subcategorize immune cells of myeloid origin based on their pro- or anti-tumor properties. Moreover, it may be appropriate to tailor conventional treatment approaches, such as chemotherapy, nanotherapy and radiotherapy, in terms of dosing, fractionation and scheduling in order to achieve optimal conditions for activation of anti-tumor immune responses. Finally, identification and validation of exclusive cell surface marker panels for each subpopulation as well as better understanding the common pro-tumor traits of these cells will allow for better stratification of cancer patient prognosis and the development of more effective therapeutic interventions.
Author Contributions
CN wrote the paper and helped with illustrations. T-CK wrote part of the paper, prepared illustrations. CP wrote part of the paper. M-IC wrote part of the paper and helped with illustrations. PC wrote part of the paper. PP conceived the theme, wrote the paper, and prepared illustrations. All authors critically read and approved the manuscript.
Funding
This work was co-funded by the European Regional Development Fund and the Republic of Cyprus through the Research and Innovation Foundation (Project: POST-DOC/0916/0044).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ATP, adenosine triphosphate; APC, antigen-presenting cell; APL, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; BM, bone marrow; BMDCs, bone marrow-derived cells; B-CLL, B-Cell chronic lymphocytic leukemia; CCL-2, C-C motif chemokine ligand 2; CRC, colorectal carcinoma; CMPs, common myeloid progenitor cells; COX, cyclooxygenase; CDA, cytidine deaminase; CSF-1, colony-stimulating factor 1 receptor; CSF-1R, colony-stimulating factor-1; CSF-2, colony-stimulating factor-2; CXCL12, C-X-C motif chemokine ligand 12; CXCL5, C-X-C motif chemokine ligand 5; CTX, cyclophosphamide; CTL, cytotoxic T lymphocyte; DCs, dendritic cells; EPR, enhanced permeability and retention; ErbB1, epidermal growth factor receptor 1; ErbB2, epidermal growth factor receptor 2; ERK, extracellular signal-regulated kinase; FDA, food and drug administration; GIST, gastrointestinal stromal tumor; GEM, gemcitabine; GM-CSF, granulocyte macrophage colony-stimulating factor; g-MDSCs, granulocytic MDSCs; G-CSF, granulocyte colony-stimulating factor; HSC, hematopoietic stem cell; HNSCC, head and neck squamous cell carcinoma; HMGB1, high mobility group box 1; ICIs, immune checkpoint inhibitors; iNOS, inducible nitric oxide synthase; IDO, indoleamine 2,3-dioxygenase; IL-10, interleukin-10; IL-10R, interleukin-10 receptor; IL-2, interleukin-2; IL-4, interleukin-4; IL-6, interleukin-6; IBC, inflammatory breast cancer; IL-6R, interleukin-6 receptor; LDI, lipopolysaccharide (LPS)low-dose irradiation; MMP-9, matrix metalloproteinase 9; MTD, maximum tolerated dose; MAPK, mitogen-activated protein kinase; M-CSF, macrophage colony-S factor; MHC, major histocompatibility complex; MMPs, matrix metalloproteinases; MITF, microphthalmia transcription factor; MTX, mitoxantrone; MDSCs, myeloid-derived suppressor cells; MEFs, murine embryonic fibroblasts; NPs, nanoparticles; NO, nitric oxide; NK cells, natural killer (NK) cells; NSCLC, non-small cell lung cancer; OS, overall survival; PTX, paclitaxel; PDAC, pancreatic ductal adenocarcinoma; PS, phosphatidylserine; PI3K, phosphatidylinositol-3-kinase; PlGF, placental growth factor; pDC, plasmacytoid dendritic cells; PMNs, polymorphonuclear neutrophils; PGE2, prostaglandin E2; PD-1, programmed cell death protein 1; PD-L1, programmed cell death protein ligand 1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; Tregs, regulatory T-cells; STAT1, signal transducer and activator of transcription 1; STAT3, signal transducer and activator of transcription 3; STAT6, signal transducer and activator of transcription 6; SDF-1alpha, stromal-serived factor 1 alpha; TCR, T-cell receptor; TLR4, toll-like receptor 4; TNPs, therapeutic nanoparticles; TTR, time-to-relapse; C/EBP, transcriptional regulator CCAAT/enhancer-binding protein; TGF-β, transforming growth factor-β; TME, tumor microenvironment; TNFα, tumor necrosis factor-α; TAMs, tumor-associated macrophages; TAMCs, tumor-associated myeloid cells; TANs, tumor-associated neutrophils; TILs, tumor-infiltrating lymphocytes; VEGF, vascular endothelial growth factor; ZA, zoledronic acid.
References
1. Lin A, Lore K. Granulocytes: new members of the antigen-presenting cell family. Front Immunol. (2017) 8:1781. doi: 10.3389/fimmu.2017.01781
2. Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. (2015) 21:846–53. doi: 10.1038/nm.3915
3. Patel H, Nilendu P, Jahagirdar D, Pal JK, Sharma NK. Modulating secreted components of tumor microenvironment: a masterstroke in tumor therapeutics. Cancer Biol Ther. (2018) 19:3–12. doi: 10.1080/15384047.2017.1394538
4. Papageorgis P, Stylianopoulos T. Role of TGFbeta in regulation of the tumor microenvironment and drug delivery (review). Int J Oncol. (2015) 46:933–43. doi: 10.3892/ijo.2015.2816
5. Sica A, Porta C, Morlacchi S, Banfi S, Strauss L, Rimoldi M, et al. Origin and functions of tumor-associated myeloid cells (TAMCs). Cancer Microenviron. (2012) 5:133–49. doi: 10.1007/s12307-011-0091-6
6. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
7. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. (2015) 25:18–27. doi: 10.1016/j.semradonc.2014.07.008
8. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
9. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
10. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. (2010) 362:875–85. doi: 10.1056/NEJMoa0905680
11. Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. (2013) 43:2930–42. doi: 10.1002/eji.201343349
12. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. (2016) 100:481–9. doi: 10.1189/jlb.3RU0316-144R
13. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. (2001) 193:727–40. doi: 10.1084/jem.193.6.727
14. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. (2014) 192:3858–67. doi: 10.4049/jimmunol.1302821
15. Gschwandtner M, Derler R, Midwood KS. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol. (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
16. Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. (2008) 111:5457–66. doi: 10.1182/blood-2008-01-136895
17. Manthey CL, Johnson DL, Illig CR, Tuman RW, Zhou Z, Baker JF, et al. JNJ-28312141, a novel orally active colony-stimulating factor-1 receptor/FMS-related receptor tyrosine kinase-3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Mol Cancer Ther. (2009) 8:3151–61. doi: 10.1158/1535-7163.MCT-09-0255
18. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
19. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
20. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. (2013) 2:e26968. doi: 10.4161/onci.26968
21. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
22. Elpek KG, Cremasco V, Shen H, Harvey CJ, Wucherpfennig KW, Goldstein DR, et al. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol Res. (2014) 2:655–67. doi: 10.1158/2326-6066.CIR-13-0209
23. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
24. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. (2012) 125(Pt 23):5591–6. doi: 10.1242/jcs.116392
25. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
26. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014-22. doi: 10.1038/ni.2703
27. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
28. Xu M, Liu M, Du X, Li S, Li H, Li X, et al. Intratumoral delivery of IL-21 overcomes anti-Her2/Neu resistance through shifting tumor-associated macrophages from M2 to M1 phenotype. J Immunol. (2015) 194:4997–5006. doi: 10.4049/jimmunol.1402603
29. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. (2011) 19:31–44. doi: 10.1016/j.ccr.2010.11.009
30. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
31. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. (2008) 454:436–44. doi: 10.1038/nature07205
32. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and Its Ligands: “One for All, All for One”. Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
33. Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife. (2014) 3:e04177. doi: 10.7554/eLife.04177
34. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol. (2018) 9:1977. doi: 10.3389/fimmu.2018.01977
35. Srivastava MK, Andersson A, Zhu L, Harris-White M, Lee JM, Dubinett S, et al. Myeloid suppressor cells and immune modulation in lung cancer. Immunotherapy. (2012) 4:291–304. doi: 10.2217/imt.11.178
36. De Vlaeminck Y, Gonzalez-Rascon A, Goyvaerts C, Breckpot K. Cancer-associated myeloid regulatory cells. Front Immunol. (2016) 7:113. doi: 10.3389/fimmu.2016.00113
37. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
38. Elliott LA, Doherty GA, Sheahan K, Ryan EJ. Human tumor-infiltrating myeloid cells: phenotypic and functional diversity. Front Immunol. (2017) 8:86. doi: 10.3389/fimmu.2017.00086
39. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother. (2019) 68:687–97. doi: 10.1007/s00262-019-02302-2
40. Andersen MH. The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia. (2014) 28:1784–92. doi: 10.1038/leu.2014.108
41. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
42. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. (2003) 170:270–8. doi: 10.4049/jimmunol.170.1.270
43. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
44. Yang R, Cai Z, Zhang Y, Yutzy WHt, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. (2006) 66:6807–15. doi: 10.1158/0008-5472.CAN-05-3755
45. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
46. Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. (2010) 40:2969–75. doi: 10.1002/eji.201040895
47. Gnjatic S, Bronte V, Brunet LR, Butler MO, Disis ML, Galon J, et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J Immunother Cancer. (2017) 5:44. doi: 10.1186/s40425-017-0243-4
48. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. (2010) 24:241–55. doi: 10.1101/gad.1874010
49. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. (2011) 25:2465–79. doi: 10.1101/gad.180331.111
50. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. (2013) 73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731
51. Cheng L, Lopez-Beltran A, Massari F, MacLennan GT, Montironi R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward precision medicine. Mod Pathol. (2018) 31:24–38. doi: 10.1038/modpathol.2017.104
52. Mok S, Tsoi J, Koya RC, Hu-Lieskovan S, West BL, Bollag G, et al. Inhibition of colony stimulating factor-1 receptor improves antitumor efficacy of BRAF inhibition. BMC Cancer. (2015) 15:356. doi: 10.1186/s12885-015-1377-8
53. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. (2010) 115:1461–71. doi: 10.1182/blood-2009-08-237412
54. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
55. Odemis V, Moepps B, Gierschik P, Engele J. Interleukin-6 and cAMP induce stromal cell-derived factor-1 chemotaxis in astroglia by up-regulating CXCR4 cell surface expression. Implications for brain inflammation. J Biol Chem. (2002) 277:39801–8. doi: 10.1074/jbc.M200472200
56. Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. (2010) 16:3420–30. doi: 10.1158/1078-0432.CCR-09-2904
57. Chuang HY, Chang YF, Liu RS, Hwang JJ. Serial low doses of sorafenib enhance therapeutic efficacy of adoptive T cell therapy in a murine model by improving tumor microenvironment. PLoS ONE. (2014) 9:e109992. doi: 10.1371/journal.pone.0109992
58. Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. (2013) 73:2480–92. doi: 10.1158/0008-5472.CAN-12-3542
59. de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. (2005) 7:411–23. doi: 10.1016/j.ccr.2005.04.014
60. Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. (2010) 17:121–34. doi: 10.1016/j.ccr.2009.12.019
61. Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. (2010) 464:302–5. doi: 10.1038/nature08782
62. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. (2009) 16:91–102. doi: 10.1016/j.ccr.2009.06.018
63. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. (1992) 12:954–61. doi: 10.1128/MCB.12.3.954
64. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
65. Sakai Y, Miyazawa M, Komura T, Yamada T, Nasti A, Yoshida K, et al. Distinct chemotherapy-associated anti-cancer immunity by myeloid cells inhibition in murine pancreatic cancer models. Cancer Sci. (2019) 110:903–12. doi: 10.1111/cas.13944
66. Van den Eynden GG, Van der Auwera I, Van Laere S, Colpaert CG, van Dam P, Merajver S, et al. Validation of a tissue microarray to study differential protein expression in inflammatory and non-inflammatory breast cancer. Breast Cancer Res Treat. (2004) 85:13–22. doi: 10.1023/B:BREA.0000021028.33926.a8
67. Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, et al. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. (2002) 21:6255–63. doi: 10.1038/sj.onc.1205794
68. Hannesdottir L, Tymoszuk P, Parajuli N, Wasmer MH, Philipp S, Daschil N, et al. Lapatinib and doxorubicin enhance the Stat1-dependent antitumor immune response. Eur J Immunol. (2013) 43:2718–29. doi: 10.1002/eji.201242505
69. Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. (2014) 33:3812–9. doi: 10.1038/onc.2013.357
70. Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. (2010) 23:27–40. doi: 10.1111/j.1755-148X.2009.00653.x
71. Hayden MS, Ghosh S. Regulation of NF-κB by TNF family cytokines. Semin Immunol. (2014) 26:253–66. doi: 10.1016/j.smim.2014.05.004
72. Smith MP, Sanchez-Laorden B, O'Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. (2014) 4:1214–29. doi: 10.1158/2159-8290.CD-13-1007
73. Wang T, Xiao M, Ge Y, Krepler C, Belser E, Lopez-Coral A, et al. BRAF Inhibition stimulates melanoma-associated macrophages to drive tumor growth. Clin Cancer Res. (2015) 21:1652–64. doi: 10.1158/1078-0432.CCR-14-1554
74. Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, Kaech SM. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNγ. Cancer Res. (2014) 74:3205–17. doi: 10.1158/0008-5472.CAN-13-3461
75. Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. (2013) 210:2873–86. doi: 10.1084/jem.20130875
76. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
77. Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. (2005) 15:496–506. doi: 10.1016/j.gde.2005.08.007
78. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. (1999) 285:248–51. doi: 10.1126/science.285.5425.248
79. Guerriero JL, Ditsworth D, Catanzaro JM, Sabino G, Furie MB, Kew RR, et al. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol. (2011) 186:3517–26. doi: 10.4049/jimmunol.1003267
80. Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. (2018) 78:5891–900. doi: 10.1158/0008-5472.CAN-17-3480
81. Mills CD. M1 and M2 Macrophages: oracles of health and disease. Crit Rev Immunol. (2012) 32:463–88. doi: 10.1615/CritRevImmunol.v32.i6.10
82. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. (2008) 66:1–9. doi: 10.1016/j.critrevonc.2007.07.004
83. Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. (2004) 40:1660–7. doi: 10.1016/j.ejca.2004.03.016
84. Panagi M, Voutouri C, Mpekris F, Papageorgis P, Martin MR, Martin JD, et al. TGF-β inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics. (2020) 10:1910–22. doi: 10.7150/thno.36936
85. Huang X, Li Y, Fu M, Xin HB. Polarizing macrophages in vitro. Methods Mol Biol. (2018) 1784:119–26. doi: 10.1007/978-1-4939-7837-3_12
86. Leonard F, Curtis LT, Ware MJ, Nosrat T, Liu X, Yokoi K, et al. Macrophage polarization contributes to the anti-tumoral efficacy of mesoporous nanovectors loaded with albumin-bound paclitaxel. Front Immunol. (2017) 8:693. doi: 10.3389/fimmu.2017.00693
87. Wang B, Xu D, Yu X, Ding T, Rao H, Zhan Y, et al. Association of intra-tumoral infiltrating macrophages and regulatory T cells is an independent prognostic factor in gastric cancer after radical resection. Ann Surg Oncol. (2011) 18:2585–93. doi: 10.1245/s10434-011-1609-3
88. Bass KK, Mastrangelo MJ. Immunopotentiation with low-dose cyclophosphamide in the active specific immunotherapy of cancer. Cancer Immunol Immunother. (1998) 47:1–12. doi: 10.1007/s002620050498
89. Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. (2016) 65:1710–20. doi: 10.1136/gutjnl-2015-309193
90. Mohammed ZM, Going JJ, Edwards J, Elsberger B, McMillan DC. The relationship between lymphocyte subsets and clinico-pathological determinants of survival in patients with primary operable invasive ductal breast cancer. Br J Cancer. (2013) 109:1676–84. doi: 10.1038/bjc.2013.493
91. Sconocchia G, Zlobec I, Lugli A, Calabrese D, Iezzi G, Karamitopoulou E, et al. Tumor infiltration by FcgammaRIII (CD16)+ myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer. (2011) 128:2663–72. doi: 10.1002/ijc.25609
92. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. (2015) 6:8692. doi: 10.1038/ncomms9692
93. Klenotic PA, Huang P, Palomo J, Kaur B, Van Meir EG, Vogelbaum MA, et al. Histidine-rich glycoprotein modulates the anti-angiogenic effects of vasculostatin. Am J Pathol. (2010) 176:2039–50. doi: 10.2353/ajpath.2010.090782
94. Thulin A, Ringvall M, Dimberg A, Karehed K, Vaisanen T, Vaisanen MR, et al. Activated platelets provide a functional microenvironment for the antiangiogenic fragment of histidine-rich glycoprotein. Mol Cancer Res. (2009) 7:1792–802. doi: 10.1158/1541-7786.MCR-09-0094
95. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
96. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. (2011) 334:1573–7. doi: 10.1126/science.1208347
97. John J, Ismail M, Riley C, Askham J, Morgan R, Melcher A, et al. Differential effects of Paclitaxel on dendritic cell function. BMC Immunol. (2010) 11:14. doi: 10.1186/1471-2172-11-14
98. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. (2016) 44:343–54. doi: 10.1016/j.immuni.2015.11.024
99. Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, et al. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients' blood cells: implications for cancer chemoimmunotherapy. Clin Cancer Res. (2013) 19:4249–61. doi: 10.1158/1078-0432.CCR-12-3666
100. Salem ML, Al-Khami AA, El-Naggar SA, Diaz-Montero CM, Chen Y, Cole DJ. Cyclophosphamide induces dynamic alterations in the host microenvironments resulting in a Flt3 ligand-dependent expansion of dendritic cells. J Immunol. (2010) 184:1737–47. doi: 10.4049/jimmunol.0902309
101. Radojcic V, Bezak KB, Skarica M, Pletneva MA, Yoshimura K, Schulick RD, et al. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother. (2010) 59:137–48. doi: 10.1007/s00262-009-0734-3
102. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. (2004) 172:464–74. doi: 10.4049/jimmunol.172.1.464
103. Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol. (2005) 175:4338–46. doi: 10.4049/jimmunol.175.7.4338
104. Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. (2003) 63:1270–9.
105. Maiti R. Metronomic chemotherapy. J Pharmacol Pharmacother. (2014) 5:186–92. doi: 10.4103/0976-500X.136098
106. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. (2007) 56:641–8. doi: 10.1007/s00262-006-0225-8
107. Liu P, Jaffar J, Hellstrom I, Hellstrom KE. Administration of cyclophosphamide changes the immune profile of tumor-bearing mice. J Immunother. (2010) 33:53–9. doi: 10.1097/CJI.0b013e3181b56af4
108. Mpekris F, Baish JW, Stylianopoulos T, Jain RK. Role of vascular normalization in benefit from metronomic chemotherapy. Proc Natl Acad Sci USA. (2017) 114:1994–9. doi: 10.1073/pnas.1700340114
109. Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. (2012) 7:383–8. doi: 10.1038/nnano.2012.45
110. Antonia S, Mule JJ, Weber JS. Current developments of immunotherapy in the clinic. Curr Opin Immunol. (2004) 16:130–6. doi: 10.1016/j.coi.2004.01.012
111. Tesniere A, Apetoh L, Ghiringhelli F, Joza N, Panaretakis T, Kepp O, et al. Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol. (2008) 20:504–11. doi: 10.1016/j.coi.2008.05.007
112. Cao ZA, Daniel D, Hanahan D. Sub-lethal radiation enhances anti-tumor immunotherapy in a transgenic mouse model of pancreatic cancer. BMC Cancer. (2002) 2:11. doi: 10.1186/1471-2407-2-11
113. Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. (2002) 62:1462–70.
114. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
115. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. (2012) 189:558–66. doi: 10.4049/jimmunol.1200563
116. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. (2013) 73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981
117. Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2-Polarized CD4(+) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res. (2015) 3:518–25. doi: 10.1158/2326-6066.CIR-14-0232
118. Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. (2010) 70:5679–85. doi: 10.1158/0008-5472.CAN-09-4446
119. Demaria S, Formenti SC. Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front Oncol. (2012) 2:153. doi: 10.3389/fonc.2012.00153
120. Golden EB, Formenti SC. Is tumor (R)ejection by the immune system the “5th R” of radiobiology? Oncoimmunology. (2014) 3:e28133. doi: 10.4161/onci.28133
121. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
122. Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys. (2012) 83:1306–10. doi: 10.1016/j.ijrobp.2011.09.049
123. Filatenkov A, Baker J, Mueller AM, Kenkel J, Ahn GO, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. (2015) 21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824
124. Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. (2001) 293:293–7. doi: 10.1126/science.1060191
125. Carvalho HA, Villar RC. Radiotherapy and immune response: the systemic effects of a local treatment. Clinics. (2018) 73(Suppl. 1):e557s. doi: 10.6061/clinics/2018/e557s
126. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
127. Christodoulou MI, Zaravinos A. New clinical approaches and emerging evidence on immune-checkpoint inhibitors as anti-cancer therapeutics: CTLA-4 and PD-1 pathways and beyond. Crit Rev Immunol. (2019) 39:379–408. doi: 10.1615/CritRevImmunol.2020033340
128. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. doi: 10.3389/fonc.2018.00086
129. Anfray C, Ummarino A, Andon FT, Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells. (2019) 9:E46. doi: 10.3390/cells9010046
130. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
131. Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, et al. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. (1996) 5:285–93. doi: 10.1016/S1074-7613(00)80323-4
132. Yu GT, Bu LL, Zhao YY, Mao L, Deng WW, Wu TF, et al. CTLA4 blockade reduces immature myeloid cells in head and neck squamous cell carcinoma. Oncoimmunology. (2016) 5:e1151594. doi: 10.1080/2162402X.2016.1151594
133. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
134. Du Four S, Maenhout SK, Niclou SP, Thielemans K, Neyns B, Aerts JL. Combined VEGFR and CTLA-4 blockade increases the antigen-presenting function of intratumoral DCs and reduces the suppressive capacity of intratumoral MDSCs. Am J Cancer Res. (2016) 6:2514–31.
135. de Coana YP, Wolodarski M, Poschke I, Yoshimoto Y, Yang Y, Nystrom M, et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget. (2017) 8:21539–53. doi: 10.18632/oncotarget.15368
136. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
137. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol. (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
138. Degos L, Wang ZY. All trans retinoic acid in acute promyelocytic leukemia. Oncogene. (2001) 20:7140–5. doi: 10.1038/sj.onc.1204763
139. Fang J, Chen SJ, Tong JH, Wang ZG, Chen GQ, Chen Z. Treatment of acute promyelocytic leukemia with ATRA and As2O3: a model of molecular target-based cancer therapy. Cancer Biol Ther. (2002) 1:614–20. doi: 10.4161/cbt.308
140. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
141. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. (2020) 17:1–12. doi: 10.1038/s41423-019-0306-1
142. Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. (2018) 128:1708. doi: 10.1172/JCI120803
143. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
144. Gandini S, Massi D, Mandala M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: a systematic review and meta-analysis. Crit Rev Oncol Hematol. (2016) 100:88–98. doi: 10.1016/j.critrevonc.2016.02.001
145. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
146. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. (2009) 113:3821–30. doi: 10.1182/blood-2008-10-185884
147. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
148. de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell. (2018) 33:60–74.e6. doi: 10.1016/j.ccell.2017.11.019
149. Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. (2010) 7:708–17. doi: 10.1016/j.stem.2010.11.014
150. Heusschen R, Griffioen AW, Thijssen VL. Galectin-9 in tumor biology: a jack of multiple trades. Biochim Biophys Acta. (2013) 1836:177–85. doi: 10.1016/j.bbcan.2013.04.006
151. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
152. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
153. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. (2007) 67:9417–24. doi: 10.1158/0008-5472.CAN-07-1286
154. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. (2013) 31:760–8. doi: 10.1007/s10637-012-9869-8
155. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
156. Petermann KB, Rozenberg GI, Zedek D, Groben P, McKinnon K, Buehler C, et al. CD200 is induced by ERK and is a potential therapeutic target in melanoma. J Clin Invest. (2007) 117:3922–9. doi: 10.1172/JCI32163
157. Moreaux J, Veyrune JL, Reme T, De Vos J, Klein B. CD200: a putative therapeutic target in cancer. Biochem Biophys Res Commun. (2008) 366:117–22. doi: 10.1016/j.bbrc.2007.11.103
158. Moreaux J, Hose D, Reme T, Jourdan E, Hundemer M, Legouffe E, et al. CD200 is a new prognostic factor in multiple myeloma. Blood. (2006) 108:4194–7. doi: 10.1182/blood-2006-06-029355
159. Love JE, Thompson K, Kilgore MR, Westerhoff M, Murphy CE, Papanicolau-Sengos A, et al. CD200 expression in neuroendocrine neoplasms. Am J Clin Pathol. (2017) 148:236–42. doi: 10.1093/ajcp/aqx071
160. Wang L, Liu JQ, Talebian F, El-Omrani HY, Khattabi M, Yu L, et al. Tumor expression of CD200 inhibits IL-10 production by tumor-associated myeloid cells and prevents tumor immune evasion of CTL therapy. Eur J Immunol. (2010) 40:2569–79. doi: 10.1002/eji.201040472
161. Liu JQ, Hu A, Zhu J, Yu J, Talebian F, Bai XF. CD200-CD200R Pathway in the regulation of tumor immune microenvironment and immunotherapy. Adv Exp Med Biol. (2020) 1223:155–65. doi: 10.1007/978-3-030-35582-1_8
162. Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol. (2007) 178:5595–605. doi: 10.4049/jimmunol.178.9.5595
163. Kretz-Rommel A, Qin F, Dakappagari N, Cofiell R, Faas SJ, Bowdish KS. Blockade of CD200 in the presence or absence of antibody effector function: implications for anti-CD200 therapy. J Immunol. (2008) 180:699–705. doi: 10.4049/jimmunol.180.2.699
164. Kretz-Rommel A, Bowdish KS. Rationale for anti-CD200 immunotherapy in B-CLL and other hematologic malignancies: new concepts in blocking immune suppression. Expert Opin Biol Ther. (2008) 8:5–15. doi: 10.1517/14712598.8.1.5
165. Ma Y, Mattarollo SR, Adjemian S, Yang H, Aymeric L, Hannani D, et al. CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res. (2014) 74:436–45. doi: 10.1158/0008-5472.CAN-13-1265
166. Mahadevan D, Lanasa MC, Farber C, Pandey M, Whelden M, Faas SJ, et al. Phase I study of samalizumab in chronic lymphocytic leukemia and multiple myeloma: blockade of the immune checkpoint CD200. J Immunother Cancer. (2019) 7:227. doi: 10.1186/s40425-019-0710-1
167. Talebian F, Liu JQ, Liu Z, Khattabi M, He Y, Ganju R, et al. Melanoma cell expression of CD200 inhibits tumor formation and lung metastasis via inhibition of myeloid cell functions. PLoS ONE. (2012) 7:e31442. doi: 10.1371/journal.pone.0031442
168. Pilch Z, Tonecka K, Braniewska A, Sas Z, Skorzynski M, Boon L, et al. Antitumor activity of TLR7 is potentiated by CD200R antibody leading to changes in the tumor microenvironment. Cancer Immunol Res. (2018) 6:930–40. doi: 10.1158/2326-6066.CIR-17-0454
169. Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. (2020) 20:321–34. doi: 10.1038/s41577-019-0269-6
170. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. (2010) 7:653–64. doi: 10.1038/nrclinonc.2010.139
171. Torres Andon F, Alonso MJ. Nanomedicine and cancer immunotherapy - targeting immunosuppressive cells. J Drug Target. (2015) 23:656–71. doi: 10.3109/1061186X.2015.1073295
172. Kourtis IC, Hirosue S, de Titta A, Kontos S, Stegmann T, Hubbell JA, et al. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS ONE. (2013) 8:e61646. doi: 10.1371/journal.pone.0061646
173. Jeanbart L, Kourtis IC, van der Vlies AJ, Swartz MA, Hubbell JA. 6-Thioguanine-loaded polymeric micelles deplete myeloid-derived suppressor cells and enhance the efficacy of T cell immunotherapy in tumor-bearing mice. Cancer Immunol Immunother. (2015) 64:1033–46. doi: 10.1007/s00262-015-1702-8
174. Sasso MS, Lollo G, Pitorre M, Solito S, Pinton L, Valpione S, et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials. (2016) 96:47–62. doi: 10.1016/j.biomaterials.2016.04.010
175. Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, et al. Immunomodulatory spherical nucleic acids. Proc Natl Acad Sci USA. (2015) 112:3892–7. doi: 10.1073/pnas.1502850112
176. Guan C, Chernyak N, Dominguez D, Cole L, Zhang B, Mirkin CA. RNA-based immunostimulatory liposomal spherical nucleic acids as potent TLR7/8 modulators. Small. (2019) 15:e1903338. doi: 10.1002/smll.201903338
177. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. (2018) 2:578–88. doi: 10.1038/s41551-018-0236-8
178. Zilio S, Vella JL, De la Fuente AC, Daftarian PM, Weed DT, Kaifer A, et al. 4PD Functionalized dendrimers: a flexible tool for in vivo gene silencing of tumor-educated myeloid cells. J Immunol. (2017) 198:4166–77. doi: 10.4049/jimmunol.1600833
179. Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L, et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. (2017) 11:9536–49. doi: 10.1021/acsnano.7b05465
180. Parayath NN, Parikh A, Amiji MM. Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating MicroRNA-125b. Nano Lett. (2018) 18:3571–9. doi: 10.1021/acs.nanolett.8b00689
181. Ledo AM, Sasso MS, Bronte V, Marigo I, Boyd BJ, Garcia-Fuentes M, et al. Co-delivery of RNAi and chemokine by polyarginine nanocapsules enables the modulation of myeloid-derived suppressor cells. J Control Release. (2019) 295:60–73. doi: 10.1016/j.jconrel.2018.12.041
Keywords: myeloid cells, dendritic cells, macrophages, immunotherapy, Tumor-associated myeloid cells, nano-immunotherapy
Citation: Neophytou CM, Pierides C, Christodoulou M-I, Costeas P, Kyriakou T-C and Papageorgis P (2020) The Role of Tumor-Associated Myeloid Cells in Modulating Cancer Therapy. Front. Oncol. 10:899. doi: 10.3389/fonc.2020.00899
Received: 13 March 2020; Accepted: 07 May 2020;
Published: 09 June 2020.
Edited by:
George S. Karagiannis, Albert Einstein College of Medicine, United StatesReviewed by:
Fransisca Leonard, Houston Methodist Research Institute, United StatesLaetitia Aymeric, INSERM U1232 Centre de Recherche en Cancérologie et Immunologie Nantes Angers (CRCINA), France
Copyright © 2020 Neophytou, Pierides, Christodoulou, Costeas, Kyriakou and Papageorgis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis Papageorgis, cC5wYXBhZ2Vvcmdpc0BldWMuYWMuY3k=