 Alessia Varone
Alessia Varone Daniela Spano
Daniela Spano Daniela Corda
Daniela Corda- 1Institute of Biochemistry and Cell Biology, National Research Council, Naples, Italy
- 2Department of Biomedical Sciences, National Research Council, Rome, Italy
Shp1 is a cytosolic tyrosine phosphatase that regulates a broad range of cellular functions and targets, modulating the flow of information from the cell membrane to the nucleus. While initially studied in the hematopoietic system, research conducted over the past years has expanded our understanding of the biological role of Shp1 to other tissues, proposing it as a novel tumor suppressor gene functionally involved in different hallmarks of cancer. The main mechanism by which Shp1 curbs cancer development and progression is the ability to attenuate and/or terminate signaling pathways controlling cell proliferation, survival, migration, and invasion. Thus, alterations in Shp1 function or expression can contribute to several human diseases, particularly cancer. In cancer cells, Shp1 activity can indeed be affected by mutations or epigenetic silencing that cause failure of Shp1-mediated homeostatic maintenance. This review will discuss the current knowledge of the cellular functions controlled by Shp1 in non-hematopoietic tissues and solid tumors, the mechanisms that regulate Shp1 expression, the role of its mutation/expression status in cancer and its value as potential target for cancer treatment. In addition, we report information gathered from the public available data from The Cancer Genome Atlas (TCGA) database on Shp1 genomic alterations and correlation with survival in solid cancers patients.
What is Shp1?
Src homology region 2 (SH-2) domain-containing phosphatase 1 (Shp1) is a non-receptor tyrosine phosphatase encoded by the PTPN6 gene that is located on human chromosome 12p13 and contains two promoter regions (within exon 1 and 2), giving rise to two forms of Shp1 which differ in their N-terminal amino acid sequences but have a similar phosphatase activity. Promoter I is active in non-hematopoietic cells, while promoter II in hematopoietic-derived cells (1, 2); in some epithelial cancer cells both promoters may function and generate various Shp1-alternative transcripts (3). The two Shp1 isoforms show different subcellular localizations: form I is mainly located in the nucleus, while form II is in the cytoplasm (2), suggesting that they have different targets.
Shp1 is a 595 amino acid protein composed of two tandem N-terminal SH2 domains (N-SH2 and C-SH2), a classic catalytic protein tyrosine phosphatase (PTP) domain, and a C-terminal tail containing several phosphorylation sites (4–6). Its crystal was first resolved by Yang et al. (4) and revealed a structure in which the N-SH2 is bound to the catalytic site of the protein through charge-charge interaction (4). In this auto-inhibited inactive state the access of substrates to the active site is prevented, but binding of phosphotyrosine residues to the SH2 domains causes a conformational change that impairs the interaction between the N-SH2 and the catalytic domains. This opens the conformation to allow the access of substrate and is further stabilized by new interactions between SH2 domains and the catalytic domain (6). These molecular rearrangements determine a sophisticated regulatory mechanism controlled by substrate recruitment.
An additional mechanism of activation is mediated by the phosphorylation of amino acids within the C-terminal tail. So far, three phosphorylation sites have been found, two tyrosine (Tyr536 and Tyr564) and a serine (Ser591) residues. Tyr536 and Tyr564 become phosphorylated upon various stimuli (i.e., insulin stimulation or apoptosis inducers), giving rise to an increased Shp1 activity (7–9). The molecular mechanism is not clear, although it has been proposed that Tyr phosphorylations could lead to interaction with the N-SH2 domain, releasing the inhibitory effect of this domain on the PTPase activity (10). Shp1 activity can also be negatively regulated by protein kinase C (PKC) or mitogen-activated protein kinases (MAPKs) through phosphorylation at Ser591, whose mechanism of inhibition has not been well-characterized (11).
Shp1 and Cancer
Protein-tyrosine phosphorylation is a reversible post-translational modification, tightly regulated by both kinases and phosphatases. Any deviation in the phosphorylation/dephosphorylation balance can promote the intracellular accumulation of tyrosine-phosphorylated proteins, which cause an altered regulation of cellular processes including cell growth, migration, invasion, differentiation, survival, and cellular trafficking (12, 13). In this scenario, Shp1 acts as a classical tumor suppressor, mainly involved in the homeostatic maintenance of potentially all these processes. Shp1 function is indeed altered in both solid and hematological human cancers through somatic mutations or epigenetic mechanisms. Besides its well-documented role in the regulation of hematopoietic cell biology [widely discussed in recent reviews to which the reader is referred (14–16)], Shp1 has now been correlated to a number of signal transduction pathways relevant to cancer pathogenesis and progression.
Here we discuss the recent knowledge on Shp1 pathways relevant to cancer (Figure 1), its alteration in tumors and relationship with the clinic including some therapeutic approaches and known drug candidates that target this protein.
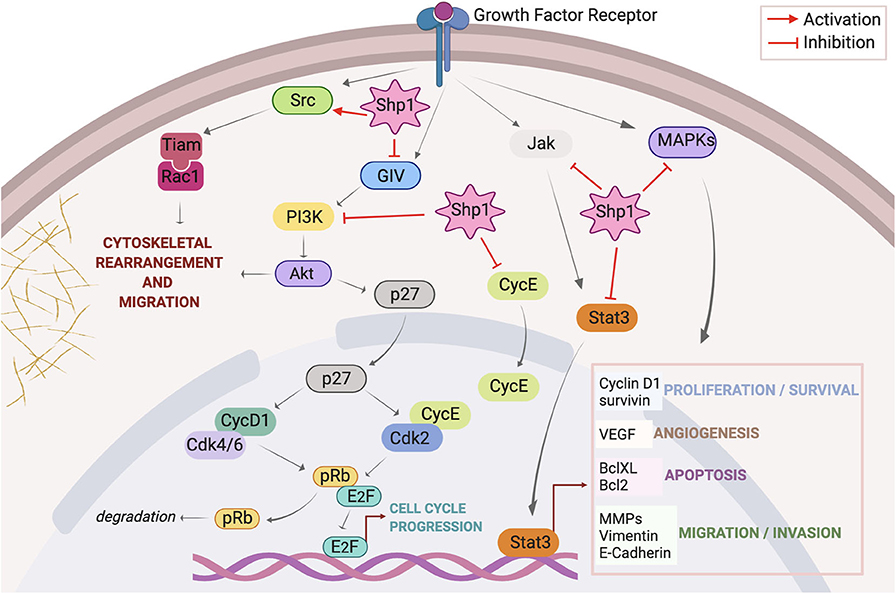
Figure 1. Overview of signaling pathways regulated by Shp1. Activation of growth factor receptors initiates signaling through Src, PI3K/Akt, JAK/STAT, and MAPKs axes. Once activated Src triggers a signal pathway that culminates with the assembly of Tiam/Rac1 complex at the plasma membrane and consequent formation of peripheral ruffles with stimulation of cell motility; Shp1 dephosphorylates Src on the inhibitory pTyr527 thus activating this cascade. Membrane receptor activation triggers the tyrosine phosphorylation of GIV that directly binds and activates PI3K; this in turn activates Akt inducing cytoskeletal rearrangements. Shp1 dephosphorylates GIV and inhibits Akt activation via the GIV-PI3K axis. Akt also phosphorylates p27(kip1) (p27) at Thr157, promoting its cytoplasmic localization and abolishing its inhibitory effect on cell cycle components like cyclin D1-Cdk 4/6 and cyclin E-Cdk 2 complexes; this results in increased pRb phosphorylation and dissociation from E2F, stimulation of transcriptional activity with consequent cell cycle progression. Shp1 blocks p27(kip1) nuclear localization through the regulation of PI3K/Akt activity. Shp1 also controls cell cycle progression regulating cyclin E localization. Following receptor stimulation, activated JAK phosphorylates STAT3, resulting in the translocation of activated STAT3 (p-STAT3) to the nucleus; Shp1 can directly dephosphorylate STAT3 or its upstream JAK thereby hampering STAT3-regulated cellular proliferation and survival, angiogenesis, apoptosis, migration, and invasion. Finally, Shp1 is involved in the attenuation of growth factors induced cell proliferation trough inhibition of MAPKs pathway.
Shp1 in Cell-Cycle Progression
Shp1 is generally considered as a negative regulator of cell proliferation, but current knowledge provides a more complex view of its role in cell signaling pathways, so that this enzyme can function both as a positive or negative mediator, depending on the target molecules and the specific cell type. Evidence suggests a role for Shp1 in the regulation of cell cycle, due to the control of mitogenic pathways activated by receptor tyrosine kinases or to its ability to modulate cell cycle components through direct interaction (17).
In endothelial cells Shp1 activation by tumor necrosis factor alpha (TNFα) inhibits proliferative responses to growth factors such as vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF), through attenuation of extracellular signal-regulated kinase (ERK) phosphorylation and MAPKs pathway. Here the overexpression of dominant-negative Shp1 prevents TNFα-induced inhibition of cell growth (18). In hepatic stellate cells Shp1 mediates the anti-proliferative signals downstream of platelet-derived growth factor (PDGF) receptor-β by regulating both proliferative and survival markers, such as the expression of cyclin D1 and the phosphorylation of Akt and ERK1/2 kinases (19). Importantly, this effect can be positively regulated by molecules that stimulate Shp1 expression, suggesting a potential therapeutic implication of Shp1 in liver fibrosis treatment (19). Along these lines, other studies report the involvement of Shp1 in the mechanisms of action of anti-proliferative stimuli, such as somatostatin. Somatostatin negatively regulates insulin signaling by controlling Shp1 recruitment to the insulin receptor. As for the PDGF receptor, the recruitment of Shp1 to the membrane induces receptor dephosphorylation and inactivation leading to the termination of insulin signaling and relative mitogenic response (20). Besides this mechanism, Shp1 can further regulate somatostatin signaling through the up-regulation of the cyclin-dependent kinase (Cdk) inhibitor p27(Kip1), inhibition of Cdk4 and Cdk2 activities with a consequent dephosphorylation of the Retinoblastoma protein and induction of cell-cycle arrest (21).
A mechanism alternative to the receptor regulation through which Shp1 controls cell cycle is the direct interaction with cell-cycle components. In intestinal epithelial cells, Shp1 expression and enzymatic activity induce cell cycle block and differentiation (22). Indeed, the overexpression of Shp1 in intestinal crypt cells decreases the expression of cyclin D1 and c-myc genes through the inhibition of E2F-dependent transcriptional activity, thus affecting the transition from G0/G1 to S phase (22). Later on, the same authors identified Cdk2 as a novel Shp1 interactor in epithelial cells (23). In proliferating intestinal cells Cdk2 interacts with Shp1 and promotes its proteasome-dependent truncation through direct Cdk2-dependent phosphorylation, similarly to the Cdk2-mediated degradation of other cell cycle regulators, such as p27(Kip1) (24) and cyclin E (25). Other groups also confirmed Cdk2-Shp1 interaction in different cell systems (26, 27). In a reciprocal manner Shp1 regulates Cdk2 localization and activity modulating cyclin E levels (28). This Cdk2 regulation has been proposed as a mechanism through which Shp1 regulates p27(Kip1), since Cdk2 phosphorylates p27(kip1) and promotes its degradation (28).
However, the involvement of p27(kip1) in the Shp1-mediated regulation of cell proliferation is controversial. In CHO cells, Shp1 mediates the anti-proliferative effect of somatostatin by the upregulation of p27(kip1) (21). Similarly, in microvascular endothelial cells Shp1 mediates the anti-proliferative activity of the tissue inhibitor metalloproteinase-2 (TIMP2) by increasing the de novo synthesis of p27(kip1) with the parallel inhibition of Cdk2 and Cdk4 activities (29).
Several studies also describe positive effects of Shp1 on mitogenic signaling. In HEK293 cells the overexpression of a catalytically-inactive mutant of Shp1 reduces cell growth and DNA synthesis through the suppression of mitogen-activated pathways (30). The same in ovarian cancer cells where the inhibition of Shp1 activity reduces tumor growth by increasing intracellular levels of the Cdk2/p27(kip1) and Cdk2/Shp1 complexes (26).
Therefore, the role of Shp1 in the regulation of cell cycle is still quite debated and further studies are needed to clarify its contribution to the control of cancer cell cycle. However, these apparent contradictions in Shp1 function might be due to the overexpression of dominant-negative mutants which could have non-specific effects. Nevertheless, we cannot rule out that the different Shp1 activity may lie in a differential interaction with tissue-specific proteins.
Shp1 in Cell Motility and Invasion
Shp1 has been recently correlated to a number of processes controlling epithelial to mesenchymal transition (EMT), cell migration and invasion. Studies conducted in several cancers define Shp1 as a negative regulator of these processes, particularly controlling the signal transducer and activator of transcription (STAT) pathway (31). Hyperactivation of oncogenic STAT3 has been observed in various malignant tumors where it controls cellular signaling regulating proliferation (e.g., cyclin D1), cell survival (e.g., Bcl-xl, Bcl2, survivin, Mcl-1 and c-Myc), but also migration and invasion [e.g., Rho, Rac, matrix metalloproteinase (MMP)-2, MMP-7, and MMP-9], as well as angiogenesis [e.g., VEGF; (32, 33)]. The major phosphorylation sites in STAT3 are Tyr705 and Ser727; Shp1 has been shown to directly dephosphorylate STAT3 on Tyr705 thus silencing the downstream pathway (34).
Protein levels of Shp1 indeed negatively correlate with p-STAT3Tyr705 and EMT markers in several hepatocellular carcinoma (HCC) cell lines (35). Cells not expressing Shp1 show higher levels of p-STAT3Tyr705 and vimentin, but a significantly lower or also loss of E-cadherin expression. As a result, these cells have a greater migratory and invasive capacity compared to other cell lines. Moreover, the Shp1 re-overexpression abolishes transforming growth factor (TGF)-β1-induced STAT3Tyr705 phosphorylation, EMT and invasion. The relationship between Shp1 and EMT was further confirmed in colorectal cancer (CRC) cells where Shp1 controls TGF-β1-induced EMT by suppressing p-STAT3Tyr705 (36). Several authors also reported how Shp1 negatively regulates the Janus kinase (JAK)-STAT pathway targeting p-STAT3Tyr705 in different systems, downregulating its target genes including those involved in invasion and metastasis (37–43).
Other studies demonstrate that Shp1 regulates migration and invasion through mechanisms other than the STAT3 pathway. Gα-interacting vesicle-associated protein (GIV) is a multidomain protein required by growth factors to enhance Akt activation and directly link Akt to the actin cytoskeleton, actin remodeling and cell migration (44). Shp1 antagonizes phospho-GIV by directly binding and dephosphorylating two phosphotyrosine residues within the C-terminal tail of GIV that serves as docking sites for p85α-regulatory subunit of phosphoinositide 3-kinase (PI3K). With this mechanism Shp1 may abrogate phospho-GIV-dependent enhancement of PI3K-Akt activity, resulting in the suppression of TGF-β1-induced EMT (44).
Our group has recently shown that Shp1 regulates actin remodeling and cell motility through yet another mechanism involving Src kinase (45). We identified Shp1 as the cellular receptor of the bioactive metabolite glycerophosphoinositol-4 phosphate (GroPIns4P) and as a novel component in the signaling pathway activated by EGF to control cell motility (45). In NIH3T3 cells, activation of the EGF receptor leads to activation of the cytosolic phospholipase A2 and results in the hydrolysis of phosphatidylinositol-4 phosphate and formation of GroPIns4P that binds to Shp1 and promotes its association with Src. This in turn activates Src by direct dephosphorylation of the inhibitory phosphotyrosine in position 530 and triggers a signaling cascade which ends with the formation of the Tiam/Rac complex at the cell membrane and the concomitant induction of plasma membrane ruffles leading to cell motility (45, 46).
Shp1 in Cell Death and Apoptosis
Along with the cellular processes described above, Shp1 regulates cell death and apoptosis. The antitumor-action of Shp1 is mainly due to its negative regulation of STAT3 oncogenic signaling, through direct regulation of JAK (47) and STAT3 (48). Indeed, besides being a master regulator of a plethora of cellular functions, STAT3 controls a series of target genes encoding for anti-apoptotic and proliferation-associated proteins (such as Bcl-xL, Bcl-2, cyclin D1, and Survivin) (33).
Numerous studies report how Shp1-mediated STAT3-downregulation represents a promising anti-cancer strategy to inhibit tumor growth and induce apoptosis in cancer cells. Indeed, a number of chemotherapeutic drugs (49–52) as well as Shp1-targeting natural compounds (40, 53, 54) induce Shp1-mediated dephosphorylation of p-STAT3Tyr705; this downregulates STAT3 transcriptional activity causing a block of tumor cell proliferation and induction of apoptosis. These compounds might have therapeutic relevance in cancer therapy through Shp1 activation and/or stabilization as discussed in the next sections.
Shp1 Alteration in Human Solid Cancers
Genomic alterations as well as epigenetic changes are critical for cancer onset, development and progression (55–58). To shed light on the Shp1 contribution to tumorigenesis we mined The Cancer Genome Atlas (TCGA) data for mutations and alterations of PTPN6 gene. PTPN6 genetic alterations, including mutations, fusion, amplification, deep deletion and multiple alterations, are found in several types of cancer. The total percentage of PTPN6 alterations in cancer amounts at 2.7% (with amplifications and mutations as the most frequent ones) whereas uterine carcinosarcoma (7.01%), testicular germ cell cancer (6.04%), ovarian cancer (5.82%), and melanoma (5.18%) show the highest frequency of PTPN6 genomic alterations.
As reported in Table 1, tumor-associated mutations may occur in the whole Shp1 sequence with most mutations (42.27%) in the phosphatase domain; this implies that each specific protein region may be pathologically relevant to cancer development. Most of these mutations (81.4%) are reported as missense, but to our knowledge there are no data regarding their effects on Shp1 function. The TCGA database reports just a few non-sense mutations that generate non-functional Shp1-truncated products. Finally, the major genomic alteration described in the PTPN6 gene is the loss of heterozygosity, mainly observed in HCC patients (59).
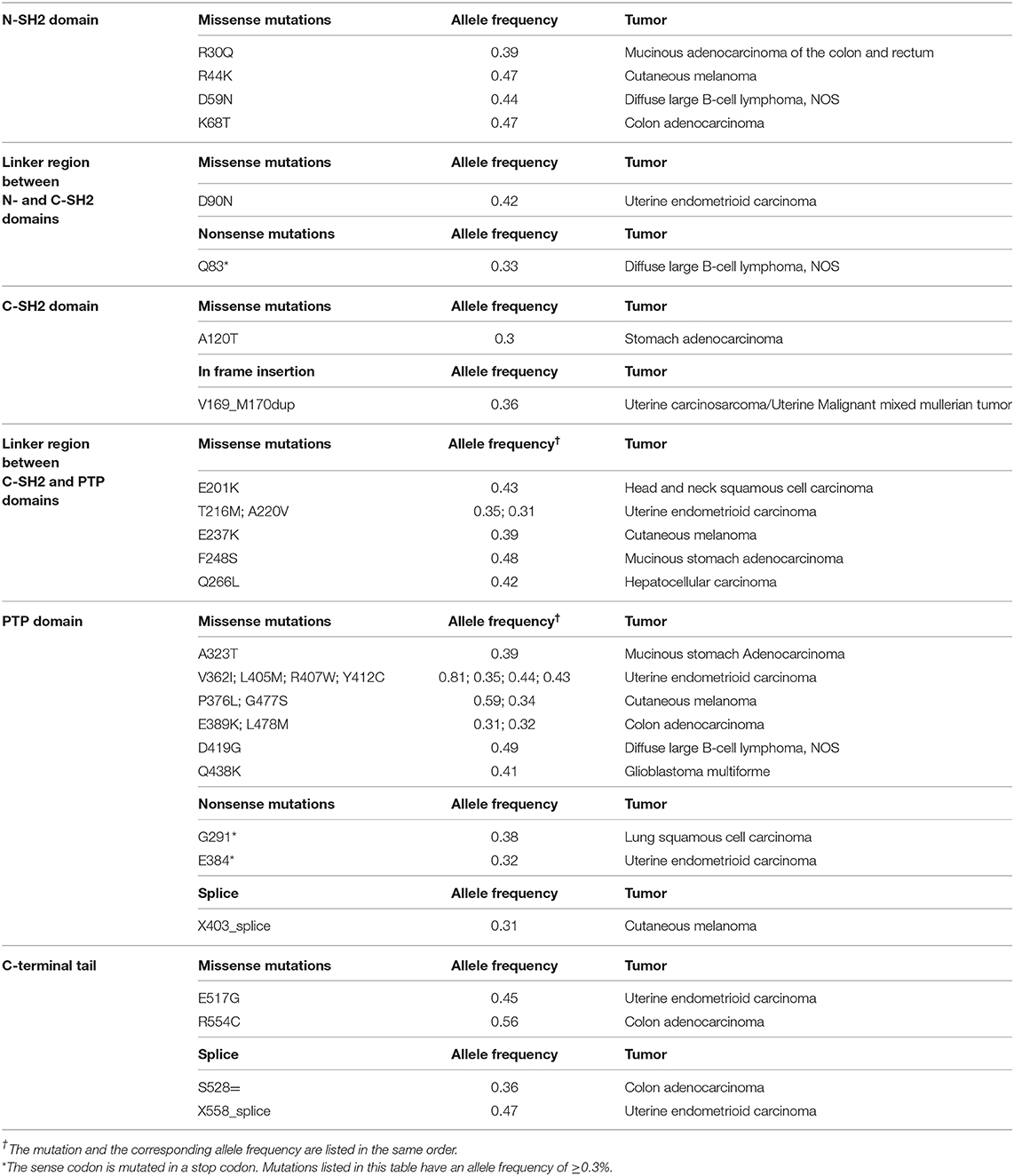
Table 1. PTPN6 gene mutations.
Epigenetic silencing of Shp1 expression is also observed in several cancer types, often due to the presence of hypermethylated CpG islands in the PTPN6 promoter. Although this alteration regards mainly hematological malignancies (60–64), it has been shown to occur also in a set of solid tumors, including esophageal squamous cell carcinoma (65), gastric adenocarcinoma (66), HCC (59), breast (67), and endometrial carcinoma (68). In hematopoietic cancers, besides the hypermethylation, enrichment of histone markers of silencing (e.g., trimethylation at lysine 9 of histone H3, trimethylation at lysine 27 of histone H3 and acetylation at lysine 9 of histone H3) is detected at the PTPN6 promoter (69), thus implying that also these histone modifications intervene in PTPN6 expression regulation. Although a similar mechanism has not yet been reported for solid tumors, we cannot exclude that it contributes, jointly to the hypermethylation, to the PTPN6 gene silencing.
Taken together, these alterations result in a diminished or abolished expression of Shp1 in most cancer cell lines and tissues examined; this deregulates the oncogenic pathways described above promoting malignant transformation.
Shp1 Expression and Clinical Correlation in Solid Cancers
According to our analysis of the TCGA database, cancer patients with PTPN6 genetic alteration(s) have a worse overall survival compared to those without PTPN6 alteration(s). To our knowledge, no data are available regarding the correlation between PTPN6 genetic alteration(s) and the progression-free survival in cancer patients. We therefore analyzed the TCGA data and found that the relationship between Shp1 expression and overall survival may be different in different solid cancers. Thus, higher Shp1 mRNA levels are associated to a worse overall survival outcome in kidney-renal clear-cell carcinoma and rectum adenocarcinoma patients, and to a better survival outcome in bladder carcinoma, breast cancer, cervical squamous cell carcinoma, kidney renal papillary cell carcinoma, lung adenocarcinoma, pancreatic ductal adenocarcinoma, sarcoma, stomach adenocarcinoma, and uterine corpus endometrial carcinoma patients. No correlation between Shp1 expression and overall survival was detected in other cancer types. Therefore, although PTPN6 is generally considered as a tumor suppressor gene, Shp1 could have different roles in tumorigenesis depending on the different biological background. This could explain the opposite correlations between Shp1 expression and overall survival observed in different type of cancers.
As mentioned, Shp1 expression changes in different types of cancer. In the hematological malignancies and in some solid tumors Shp1 expression is reduced or absent as a result of PTPN6 promoter hypermethylation (59–68). On the contrary, increased Shp1 levels are detected in a subset of high-grade breast tumors (70) and in ovarian cancers (71).
Several studies have been devoted to assess the prognostic and diagnostic value of Shp1 expression and promoter methylation, with the aim of identifying new biomarkers of cancer development. Shp1 decreased expression and PTPN6 hypermethylation are associated with tumor staging, pathological differentiation and poor survival in patients with esophageal squamous cell carcinoma (65). In endometrial carcinoma, PTPN6 hypermethylation is associated with age and tumor differentiation, while no correlation is found with muscular infiltration depth and lymphatic metastasis (68). In prostate cancer and high-grade glioma, PTPN6 promoter methylation and reduced expression of Shp1 correlate with increased malignancy and poor prognosis (72–75). Moreover, the PTPN6 high-methylation level is associated with early relapse of non-small cell lung cancer (NSCLC) (76). In addition, the PTPN6 high-methylation level of lymph nodes from CRC patients (77) and from stage I NSCLC patients (76) is associated with recurrence and/or poor prognosis.
Interestingly, a good correlation of PTPN6 methylation between cell-free circulating DNA from plasmas/sera and matched tumoral tissue is observed in glioma patients (75). Similarly, PTPN6 promoter methylation is significantly increased in plasma from NSCLC patients compared to healthy controls (78); its methylation level is also associated with the rate of survival in advanced NSCLC (78). These data suggest that PTPN6 methylation in plasma, in combination with clinical analysis, may be a promising biomarker for NSCLC diagnosis and prognosis.
Therapeutic Implications and Emergent Drugs
From all the above it appears that Shp1 represents an attractive target for drug development in cancer treatment. Inhibitors targeting the Shp1 phosphatase activity have been under development for some times, and some have now entered preclinical studies, including NSC-87877, sodium stibogluconate (SSG), tyrosine phosphatase inhibitor 1 (TPI-1), and suramine; however, only a few of them have been shown to be active in experimental tumor models (79). SSG has been through Phase I trials for both malignant melanoma (NCT00498979) and advanced malignancies (NCT00629200); the drug was administrated in combination with interferons followed or not by chemotherapy treatment. Unfortunately, no effect was seen against tumor development (80, 81), with the most common toxic side-effects being thrombocytopenia, elevated serum lipase, fatigue, fever, chills, anemia, hypokalemia, pancreatitis, and skin rash (observed in up to 68% of patients). At present, no Shp1 inhibitor is under Phase II trial.
Shp1 might represent an interesting target to modulate oncogenic STAT3 activities and thus inhibit tumor growth, promote apoptosis and prevent chemo- and radio-resistance even in tumors in which it is not mutated (82, 83). Guggulsterone [a phytosteroid extracted from the guggul plant (84)], plumbagin [a vitamin K3 analog derived from a medicinal plant (38, 85)], and morin [a flavonol extracted from mulberry figs and old fustic (86)] modulate STAT3 activities through induction of Shp1 expression; instead, the multi-targeted kinase inhibitors dovitinib (87), sorafenib (50), and its derivatives (88, 89) such as regorafenib (90, 91) and SC-60 (92) have antitumor activities by direct binding to, and enhancement of, Shp1 phosphatase activity.
Despite the pharmacological potential reported for these molecules in in-vitro and in-vivo studies performed on multiple cancer types (including leukemia, head and neck cancer, melanoma, and HCC), only sorafenib and regorafenib have been so far approved by the Food and Drug Administration for cancer treatment. Shp1 inhibitors instead need to be further developed to exploit the inhibition of the Shp1-affected pathways within cancer cells and/or enhance the efficacy of existing chemotherapeutic agents.
Shp1 and Cancer Immunotherapy
The repertoire of Shp1 functions is continuously expanding to include the modulation of the tumor microenvironment, thus suggesting new potential therapeutic implications. In recent years cancer immunotherapy, which exploits T-cells to arm the immune system against tumoral cells, has shown promising results based on a multitude of targeting strategies such as immunomodulatory monoclonal antibodies, adoptive cell transfers, high-dose immunostimulatory cytokines, and others (93). Among these, encouraging results have been achieved through the targeting of inhibitory crucial molecules of the antitumor T-cell response such as programmed-cell death 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (93).
As a downstream target of several receptors, Shp1 is also involved in T-cells signaling where it limits T-cell responsiveness either through direct dephosphorylation of the T-cell receptor (TCR)-ζ chain or dephosphorylation of downstream adaptor proteins including Lck, ZAP70, Vav, and PI3K (14, 94). Studies on CD8+ T-cells have demonstrated that the absence of Shp1 allows cells to form more stable and longer-lasting synapses with antigen presenting cells (APCs) (95), leading to reduced T-cells activation thresholds and to the stimulation and proliferation of low-affinity T-cells; this causes an increased number of tumor specific effector T-cells and a more efficient control of tumor growth (96, 97). Shp-1 depleted CD8+ T-cells result also to be more resistant to suppression by regulatory T-cells (Treg) (98), which is crucial for their survival into the tumor microenvironment. Similarly, also tumor-infiltrating lymphocytes (TILs) signaling has been reported to be regulated by Shp1, whose abrogation in TILs was found to restore TIL lytic function in vitro (99).
Shp1 has also been shown to inhibit Treg suppressor function (100), and thus its inhibition in these specific cell population may support a tolerant microenvironment, promoting tumor growth and progression. Collectively, these data suggest the need to modulate Shp1 activity in specific and individual immune cell populations rather than perform a global inhibition of its activity, as obtained by systemic administration of Shp1 inhibitors. This consideration could also explain the disappointing results of the Phase I clinical studies discussed above.
Moreover, these findings raise the question of how modulation of TCR activation and signaling through Shp1 inhibition might improve responsiveness to existing checkpoint blockade immunotherapy. Several studies have indeed proven that Shp1 can be recruited to the cytoplasmic tail of PD-1 by binding to its immunoreceptor tyrosine-based switch motifs (ITSM) thus dephosphorylating and inactivating proximal signaling molecules of TCR activation (101, 102). Similarly, Snooke et al. have recently demonstrated that Shp1 knockdown potentiates the antitumor activity of T-cells responding to low-affinity tumor antigen, particularly in combinatorial studies blocking both PD-1 and CTLA-4 (97). Shp1 has also been successfully targeted in preclinical models of adoptive T-cell immunotherapy (103, 104). Here the abrogation of Shp1 in tumor-specific effector T-cells, either by in-vitro transduction of Shp-1 shRNA-expressing-T-cells (104) or T-cells conjugated with Shp1 inhibitor-loaded nanoparticles (103), significantly enhances the effector function and tumor clearance in the therapy of disseminated leukemia and advanced prostate cancer, respectively.
Therefore, these studies suggest that the inhibition of Shp1 in effector T-cells could be a therapeutic strategy to be used alone or in combination with other immunomodulating strategies (e.g., PD-1 or CTLA-4 inhibition) and amenable to increase therapeutic activity of the tumor-specific T-cells.
Concluding Remarks
The above lines of evidence delineate Shp1 as a tumor suppressor gene contributing to tumorigenesis via the control of different cancer-related signaling and point at the potential of Shp1 as a target for therapy in a wide range of cancers. Synthetic lethality-based and rational combinatorial strategies are potential approaches to address the loss-of-function of Shp1 that occurs in several tumors and to restore the onco-suppressive activity of Shp1. Given the importance of genomic mechanisms regulating Shp1, epigenetic therapy through pharmacological modulation of histone modifications or promoter methylation could be particularly attractive. A few cancers are associated with elevated Shp1 expression; in this context Shp1 inhibition might represent an additional way to treat these tumors. Similarly, the role of Shp1 in tumor immunomodulation also inspires studies aimed at the identification and/or development of specific and safe Shp1 inhibitors.
The diverse Shp1 activities in different tumors demand for a detailed characterization of the Shp1-regulated pathway(s) altered in each specific case; similarly, the functional role of each Shp1 mutation needs to be functionally characterized in order to develop precise therapeutic approaches that will restore the Shp1 physiological action and hopefully provide improved cancer therapy.
Author Contributions
AV, DS, and DC analyzed and discussed the data reported and wrote the manuscript.
Funding
This work in the authors' laboratory was supported by the PRIN program from the Italian Ministry of University and Research (20177XJCHX), the SATIN POR Campania region project 2014-2020 and the Italian Association for Cancer Research (IG18776 to DC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. (2003) 306:1–12. doi: 10.1016/S0378-1119(03)00400-1
2. Tsui FW, Martin A, Wang J, Tsui HW. Investigations into the regulation and function of the SH2 domain-containing protein-tyrosine phosphatase, SHP-1. Immunol Res. (2006) 35:127–36. doi: 10.1385/IR:35:1:127
3. Evren S, Wan S, Ma XZ, Fahim S, Mody N, Sakac D, et al. Characterization of SHP-1 protein tyrosine phosphatase transcripts, protein isoforms and phosphatase activity in epithelial cancer cells. Genomics. (2013) 102:491–9. doi: 10.1016/j.ygeno.2013.10.001
4. Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ, et al. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem. (2003) 278:6516–20. doi: 10.1074/jbc.M210430200
5. Poole AW, Jones ML. A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. (2005) 17:1323–32. doi: 10.1016/j.cellsig.2005.05.016
6. Wang W, Liu L, Song X, Mo Y, Komma C, Bellamy HD, et al. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J Cell Biochem. (2011) 112:2062–71. doi: 10.1002/jcb.23125
7. Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol. (1994) 14:1824–34. doi: 10.1128/MCB.14.3.1824
8. Uchida T, Matozaki T, Noguchi T, Yamao T, Horita K, Suzuki T, et al. Insulin stimulates the phosphorylation of Tyr538 and the catalytic activity of PTP1C, a protein tyrosine phosphatase with Src homology-2 domains. J Biol Chem. (1994) 269:12220–8.
9. Yoshida K, Kharbanda S, Kufe D. Functional interaction between SHPTP1 and the Lyn tyrosine kinase in the apoptotic response to DNA damage. J Biol Chem. (1999) 274:34663–8. doi: 10.1074/jbc.274.49.34663
10. Zhang Z, Shen K, Lu W, Cole PA. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J Biol Chem. (2003) 278:4668–74. doi: 10.1074/jbc.M210028200
11. Liu Y, Kruhlak MJ, Hao JJ, Shaw S. Rapid T cell receptor-mediated SHP-1 S591 phosphorylation regulates SHP-1 cellular localization and phosphatase activity. J Leukoc Biol. (2007) 82:742–51. doi: 10.1189/jlb.1206736
12. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. (2001) 411:355–65. doi: 10.1038/35077225
13. Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol. (2001) 13:182–95. doi: 10.1016/S0955-0674(00)00196-4
14. Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. (2009) 228:342–59. doi: 10.1111/j.1600-065X.2008.00760.x
15. Abram CL, Lowell CA. Shp1 function in myeloid cells. J Leukoc Biol. (2017) 102:657–75. doi: 10.1189/jlb.2MR0317-105R
16. Garg M, Wahid M, Khan FD. Regulation of peripheral and central immunity: understanding the role of Src homology 2 domain-containing tyrosine phosphatases, SHP-1 and SHP-2. Immunobiology. (2020) 225:151847. doi: 10.1016/j.imbio.2019.09.006
17. Lopez-Ruiz P, Rodriguez-Ubreva J, Cariaga AE, Cortes MA, Colas B. SHP-1 in cell-cycle regulation. Anticancer Agents Med Chem. (2011) 11:89–98. doi: 10.2174/187152011794941154
18. Nakagami H, Cui TX, Iwai M, Shiuchi T, Takeda-Matsubara Y, Wu L, et al. Tumor necrosis factor-alpha inhibits growth factor-mediated cell proliferation through SHP-1 activation in endothelial cells. Arterioscl Thromb Vasc Biol. (2002) 22:238–42. doi: 10.1161/hq0202.104001
19. Tibaldi E, Zonta F, Bordin L, Magrin E, Gringeri E, Cillo U, et al. The tyrosine phosphatase SHP-1 inhibits proliferation of activated hepatic stellate cells by impairing PDGF receptor signaling. Biochimica et Biophysica Acta. (2014) 1843:288–98. doi: 10.1016/j.bbamcr.2013.10.010
20. Bousquet C, Delesque N, Lopez F, Saint-Laurent N, Esteve JP, Bedecs K, et al. sst2 somatostatin receptor mediates negative regulation of insulin receptor signaling through the tyrosine phosphatase SHP-1. J Biol Chem. (1998) 273:7099–106. doi: 10.1074/jbc.273.12.7099
21. Pages P, Benali N, Saint-Laurent N, Esteve JP, Schally AV, Tkaczuk J, et al. sst2 somatostatin receptor mediates cell cycle arrest and induction of p27(Kip1). Evidence for the role of SHP-1. J Biol Chem. (1999) 274:15186–93. doi: 10.1074/jbc.274.21.15186
22. Duchesne C, Charland S, Asselin C, Nahmias C, Rivard N. Negative regulation of beta-catenin signaling by tyrosine phosphatase SHP-1 in intestinal epithelial cells. J Biol Chem. (2003) 278:14274–83. doi: 10.1074/jbc.M300425200
23. Simoneau M, Boulanger J, Coulombe G, Renaud MA, Duchesne C, Rivard N. Activation of Cdk2 stimulates proteasome-dependent truncation of tyrosine phosphatase SHP-1 in human proliferating intestinal epithelial cells. J Biol Chem. (2008) 283:25544–56. doi: 10.1074/jbc.M804177200
24. Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. (1997) 11:1464–78. doi: 10.1101/gad.11.11.1464
25. Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. (2003) 12:381–92. doi: 10.1016/S1097-2765(03)00287-9
26. Caron D, Savard PE, Doillon CJ, Olivier M, Shink E, Lussier JG, et al. Protein tyrosine phosphatase inhibition induces anti-tumor activity: evidence of Cdk2/p27 kip1 and Cdk2/SHP-1 complex formation in human ovarian cancer cells. Cancer Lett. (2008) 262:265–75. doi: 10.1016/j.canlet.2007.12.012
27. Fiset A, Xu E, Bergeron S, Marette A, Pelletier G, Siminovitch KA, et al. Compartmentalized CDK2 is connected with SHP-1 and beta-catenin and regulates insulin internalization. Cell Signal. (2011) 23:911–9. doi: 10.1016/j.cellsig.2011.01.019
28. Rodriguez-Ubreva FJ, Cariaga-Martinez AE, Cortes MA, Romero-De Pablos M, Ropero S, Lopez-Ruiz P, et al. Knockdown of protein tyrosine phosphatase SHP-1 inhibits G1/S progression in prostate cancer cells through the regulation of components of the cell-cycle machinery. Oncogene. (2010) 29:345–55. doi: 10.1038/onc.2009.329
29. Seo DW, Li H, Qu CK, Oh J, Kim YS, Diaz T, et al. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J Biol Chem. (2006) 281:3711–21. doi: 10.1074/jbc.M509932200
30. Su L, Zhao Z, Bouchard P, Banville D, Fischer EH, Krebs EG, et al. Positive effect of overexpressed protein-tyrosine phosphatase PTP1C on mitogen-activated signaling in 293 cells. J Biol Chem. (1996) 271:10385–90. doi: 10.1074/jbc.271.17.10385
31. Huang TT, Su JC, Liu CY, Shiau CW, Chen KF. Alteration of SHP-1/p-STAT3 signaling: a potential target for anticancer therapy. Int J Mol Sci. (2017) 18:1234. doi: 10.3390/ijms18061234
32. Subramaniam A, Shanmugam MK, Perumal E, Li F, Nachiyappan A, Dai X, et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochimica et Biophysica Acta. (2013) 1835:46–60. doi: 10.1016/j.bbcan.2012.10.002
33. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et Biophysica Acta. (2014) 1845:136–54. doi: 10.1016/j.bbcan.2013.12.005
34. Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ, et al. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol. (2011) 55:1041–8. doi: 10.1016/j.jhep.2011.01.047
35. Fan LC, Shiau CW, Tai WT, Hung MH, Chu PY, Hsieh FS, et al. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene. (2015) 34:5252–63. doi: 10.1038/onc.2014.445
36. Fan LC, Teng HW, Shiau CW, Tai WT, Hung MH, Yang SH, et al. Regorafenib (Stivarga) pharmacologically targets epithelial-mesenchymal transition in colorectal cancer. Oncotarget. (2016) 7:64136–47. doi: 10.18632/oncotarget.11636
37. Wang J, Zhang L, Chen G, Zhang J, Li Z, Lu W, et al. Small molecule 1'-acetoxychavicol acetate suppresses breast tumor metastasis by regulating the SHP-1/STAT3/MMPs signaling pathway. Breast Cancer Res Treat. (2014) 148:279–89. doi: 10.1007/s10549-014-3165-6
38. Joo MK, Park JJ, Kim SH, Yoo HS, Lee BJ, Chun HJ, et al. Antitumorigenic effect of plumbagin by induction of SH2-containing protein tyrosine phosphatase 1 in human gastric cancer cells. Int J Oncol. (2015) 46:2380–8. doi: 10.3892/ijo.2015.2935
39. Zhang T, Li S, Li J, Yin F, Hua Y, Wang Z, et al. Natural product pectolinarigenin inhibits osteosarcoma growth and metastasis via SHP-1-mediated STAT3 signaling inhibition. Cell Death Dis. (2016) 7:e2421. doi: 10.1038/cddis.2016.305
40. Chen H, Zhu B, Zhao L, Liu Y, Zhao F, Feng J, et al. Allicin inhibits proliferation and invasion in vitro and in vivo via SHP-1-mediated STAT3 signaling in cholangiocarcinoma. Cell Physiol Biochem Int J Exp Cell Physiol Bioch Pharmacol. (2018) 47:641–53. doi: 10.1159/000490019
41. Kim SH, Yoo HS, Joo MK, Kim T, Park JJ, Lee BJ, et al. Arsenic trioxide attenuates STAT-3 activity and epithelial-mesenchymal transition through induction of SHP-1 in gastric cancer cells. BMC Cancer. (2018) 18:150. doi: 10.1186/s12885-018-4071-9
42. Koh JS, Joo MK, Park JJ, Yoo HS, Choi BI, Lee BJ, et al. Inhibition of STAT3 in gastric cancer: role of pantoprazole as SHP-1 inducer. Cell Biosci. (2018) 8:50. doi: 10.1186/s13578-018-0248-9
43. Li W, Du H, Zhou G, Song D. Euxanthone represses the proliferation, migration, and invasion of glioblastoma cells by modulating STAT3/SHP-1 signaling. Anatom Record. (2020). doi: 10.1002/ar.24363. [Epub ahead of print].
44. Mittal Y, Pavlova Y, Garcia-Marcos M, Ghosh P. Src homology domain 2-containing protein-tyrosine phosphatase-1 (SHP-1) binds and dephosphorylates G(alpha)-interacting, vesicle-associated protein (GIV)/Girdin and attenuates the GIV-phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway. J Biol Chem. (2011) 286:32404–15. doi: 10.1074/jbc.M111.275685
45. Varone A, Mariggio S, Patheja M, Maione V, Varriale A, Vessichelli M, et al. A signalling cascade involving receptor-activated phospholipase A2, glycerophosphoinositol 4-phosphate, Shp1 and Src in the activation of cell motility. Cell Commun Signal. (2019) 17:20. doi: 10.1186/s12964-019-0329-3
46. Filippi BM, Mariggio S, Pulvirenti T, Corda D. SRC-dependent signalling regulates actin ruffle formation induced by glycerophosphoinositol 4-phosphate. Biochimica et Biophysica Acta. (2008) 1783:2311–22. doi: 10.1016/j.bbamcr.2008.07.021
47. Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. (1996) 16:6985–92. doi: 10.1128/MCB.16.12.6985
48. Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ, et al. Corrigendum to signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol. (2017) 66:666–8. doi: 10.1016/j.jhep.2016.12.007
49. Chai H, Luo AZ, Weerasinghe P, Brown RE. Sorafenib downregulates ERK/Akt and STAT3 survival pathways and induces apoptosis in a human neuroblastoma cell line. Int J Clin Exp Pathol. (2010) 3:408–15.
50. Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, et al. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res. (2010) 16:5189–99. doi: 10.1158/1078-0432.CCR-09-3389
51. Huang S, Sinicrope FA. Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol Cancer Ther. (2010) 9:742–50. doi: 10.1158/1535-7163.MCT-09-1004
52. Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther. (2010) 9:953–62. doi: 10.1158/1535-7163.MCT-09-0947
53. Saraswati S, Alhaider A, Abdelgadir AM, Tanwer P, Korashy HM. Phloretin attenuates STAT-3 activity and overcomes sorafenib resistance targeting SHP-1-mediated inhibition of STAT3 and Akt/VEGFR2 pathway in hepatocellular carcinoma. Cell Commun Signal. (2019) 17:127. doi: 10.1186/s12964-019-0430-7
54. Zhang H, Tan YP, Zhao L, Wang L, Fu NJ, Zheng SP, et al. Anticancer activity of dietary xanthone alpha-mangostin against hepatocellular carcinoma by inhibition of STAT3 signaling via stabilization of SHP1. Cell Death Dis. (2020) 11:63. doi: 10.1038/s41419-020-2227-4
55. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. (2005) 2(Suppl. 1):S4–11. doi: 10.1038/ncponc0354
56. Baylin SB, Chen WY. Aberrant gene silencing in tumor progression: implications for control of cancer. Cold Spring Harb Symp Quant Biol. (2005) 70:427–33. doi: 10.1101/sqb.2005.70.010
57. Galm O, Herman JG, Baylin SB. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev. (2006) 20:1–13. doi: 10.1016/j.blre.2005.01.006
58. Garraway LA, Lander ES. Lessons from the cancer genome. Cell. (2013) 153:17–37. doi: 10.1016/j.cell.2013.03.002
59. Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. (2006) 130:1117–28. doi: 10.1053/j.gastro.2006.01.006
60. Takeuchi S, Matsushita M, Zimmermann M, Ikezoe T, Komatsu N, Seriu T, et al. Clinical significance of aberrant DNA methylation in childhood acute lymphoblastic leukemia. Leukem Res. (2011) 35:1345–9. doi: 10.1016/j.leukres.2011.04.015
61. Li Y, Yang L, Pan Y, Yang J, Shang Y, Luo J. Methylation and decreased expression of SHP-1 are related to disease progression in chronic myelogenous leukemia. Oncol Rep. (2014) 31:2438–46. doi: 10.3892/or.2014.3098
62. Kucuk C, Hu X, Jiang B, Klinkebiel D, Geng H, Gong Q, et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. (2015) 21:1699–711. doi: 10.1158/1078-0432.CCR-14-1216
63. Ding K, Chen X, Wang Y, Liu H, Song W, Li L, et al. Plasma DNA methylation of p16 and shp1 in patients with B cell non-Hodgkin lymphoma. Int J Clin Oncol. (2017) 22:585–92. doi: 10.1007/s10147-017-1100-7
64. Liu J, Wang Y, Sun X, Ji N, Sun S, Liu F, et al. Promoter methylation attenuates SHP1 expression and function in patients with primary central nervous system lymphoma. Oncol Rep. (2017) 37:887–94. doi: 10.3892/or.2016.5308
65. Liu L, Zhang S, Liu X, Liu J. Aberrant promoter 2 methylationmediated downregulation of protein tyrosine phosphatase, nonreceptor type 6, is associated with progression of esophageal squamous cell carcinoma. Mol Med Rep. (2019) 19:3273–82. doi: 10.3892/mmr.2019.9971
66. Joo MK, Park JJ, Yoo HS, Lee BJ, Chun HJ, Lee SW, et al. Epigenetic regulation and anti-tumorigenic effects of SH2-containing protein tyrosine phosphatase 1 (SHP1) in human gastric cancer cells. Tumour Biol. (2016) 37:4603–12. doi: 10.1007/s13277-015-4228-y
67. Hachana M, Trimeche M, Ziadi S, Amara K, Korbi S. Evidence for a role of the Simian Virus 40 in human breast carcinomas. Breast Cancer Res Treat. (2009) 113:43–58. doi: 10.1007/s10549-008-9901-z
68. Sheng Y, Wang H, Liu D, Zhang C, Deng Y, Yang F, et al. Methylation of tumor suppressor gene CDH13 and SHP1 promoters and their epigenetic regulation by the UHRF1/PRMT5 complex in endometrial carcinoma. Gynecol Oncol. (2016) 140:145–51. doi: 10.1016/j.ygyno.2015.11.017
69. Witzig TE, Hu G, Offer SM, Wellik LE, Han JJ, Stenson MJ, et al. Epigenetic mechanisms of protein tyrosine phosphatase 6 suppression in diffuse large B-cell lymphoma: implications for epigenetic therapy. Leukemia. (2014) 28:147–54. doi: 10.1038/leu.2013.251
70. Insabato L, Amelio I, Quarto M, Zannetti A, Tolino F, De Mauro G, et al. Elevated expression of the tyrosine phosphatase SHP-1 defines a subset of high-grade breast tumors. Oncology. (2009) 77:378–84. doi: 10.1159/000276765
71. Mok SC, Kwok TT, Berkowitz RS, Barrett AJ, Tsui FW. Overexpression of the protein tyrosine phosphatase, nonreceptor type 6 (PTPN6), in human epithelial ovarian cancer. Gynecol Oncol. (1995) 57:299–303. doi: 10.1006/gyno.1995.1146
72. Amara K, Trimeche M, Ziadi S, Laatiri A, Hachana M, Korbi S. Prognostic significance of aberrant promoter hypermethylation of CpG islands in patients with diffuse large B-cell lymphomas. Ann Oncol. (2008) 19:1774–86. doi: 10.1093/annonc/mdn374
73. Tassidis H, Brokken LJ, Jirstrom K, Ehrnstrom R, Ponten F, Ulmert D, et al. Immunohistochemical detection of tyrosine phosphatase SHP-1 predicts outcome after radical prostatectomy for localized prostate cancer. Int J Cancer. (2010) 126:2296–307. doi: 10.1002/ijc.24917
74. Zhang Y, Zhao D, Zhao H, Wu X, Zhao W, Wang Y, et al. Hypermethylation of SHP-1 promoter in patient with high-risk myelodysplastic syndrome and it predicts poor prognosis. Med Oncol. (2012) 29:2359–63. doi: 10.1007/s12032-012-0163-6
75. Zhang L, Wang M, Wang W, Mo J. Incidence and prognostic value of multiple gene promoter methylations in gliomas. J Neuro Oncol. (2014) 116:349–56. doi: 10.1007/s11060-013-1301-5
76. Vinayanuwattikun C, Chantranuwat P, Sriuranpong V, Mutirangura A. The role of SHP-1 promoter 2 hypermethylation detection of lymph node micrometastasis in resectable stage I non-small cell lung cancer as a prognostic marker of disease recurrence. Int J Clin Oncol. (2014) 19:586–92. doi: 10.1007/s10147-013-0605-y
77. Rattanatanyong P, Keelawat S, Kitkumthorn N, Mutirangura A. Epithelial-specific SHP1-P2 methylation - a novel universal tumor marker for detection of colorectal cancer lymph node metastasis. Asian Pacif J Cancer Prevent. (2016) 17:4117–23.
78. Vinayanuwattikun C, Sriuranpong V, Tanasanvimon S, Chantranuwat P, Mutirangura A. Epithelial-specific methylation marker: a potential plasma biomarker in advanced non-small cell lung cancer. J Thor Oncol. (2011) 6:1818–25. doi: 10.1097/JTO.0b013e318226b46f
79. Watson HA, Wehenkel S, Matthews J, Ager A. SHP-1: the next checkpoint target for cancer immunotherapy? Biochem Soc Trans. (2016) 44:356–62. doi: 10.1042/BST20150251
80. Yi T, Pathak MK, Lindner DJ, Ketterer ME, Farver C, Borden EC. Anticancer activity of sodium stibogluconate in synergy with IFNs. J Immunol. (2002) 169:5978–85. doi: 10.4049/jimmunol.169.10.5978
81. Naing A, Reuben JM, Camacho LH, Gao H, Lee BN, Cohen EN, et al. Phase I dose escalation study of sodium stibogluconate (SSG), a protein tyrosine phosphatase inhibitor, combined with interferon alpha for patients with solid tumors. J Cancer. (2011) 2:81–9. doi: 10.7150/jca.2.81
82. Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kunnumakkara AB, et al. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann NY Acad Sci. (2006) 1091:151–69. doi: 10.1196/annals.1378.063
83. Gupta SC, Kannappan R, Reuter S, Kim JH, Aggarwal BB. Chemosensitization of tumors by resveratrol. Ann NY Acad Sci. (2011) 1215:150–60. doi: 10.1111/j.1749-6632.2010.05852.x
84. Ahn KS, Sethi G, Sung B, Goel A, Ralhan R, Aggarwal BB. Guggulsterone, a farnesoid X receptor antagonist, inhibits constitutive and inducible STAT3 activation through induction of a protein tyrosine phosphatase SHP-1. Cancer Res. (2008) 68:4406–15. doi: 10.1158/0008-5472.CAN-07-6696
85. Sandur SK, Pandey MK, Sung B, Aggarwal BB. 5-hydroxy-2-methyl-1,4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: potential role in chemosensitization. Mol Cancer Res. (2010) 8:107–18. doi: 10.1158/1541-7786.MCR-09-0257
86. Gupta SC, Phromnoi K, Aggarwal BB. Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells through activation of protein tyrosine phosphatase SHP1. Biochem Pharmacol. (2013) 85:898–912. doi: 10.1016/j.bcp.2012.12.018
87. Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH, Lin MW, et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol Cancer Ther. (2012) 11:452–63. doi: 10.1158/1535-7163.MCT-11-0412
88. Chen KF, Tai WT, Hsu CY, Huang JW, Liu CY, Chen PJ, et al. Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP-1 phosphatase activity. Eur J Med Chem. (2012) 55:220–7. doi: 10.1016/j.ejmech.2012.07.023
89. Tai WT, Shiau CW, Chen PJ, Chu PY, Huang HP, Liu CY, et al. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology. (2014) 59:190–201. doi: 10.1002/hep.26640
90. Fan LC, Teng HW, Shiau CW, Lin H, Hung MH, Chen YL, et al. SHP-1 is a target of regorafenib in colorectal cancer. Oncotarget. (2014) 5:6243–51. doi: 10.18632/oncotarget.2191
91. Tai WT, Chu PY, Shiau CW, Chen YL, Li YS, Hung MH, et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin Cancer Res. (2014) 20:5768–76. doi: 10.1158/1078-0432.CCR-14-0725
92. Liu CY, Su JC, Huang TT, Chu PY, Huang CT, Wang WL, et al. Sorafenib analogue SC-60 induces apoptosis through the SHP-1/STAT3 pathway and enhances docetaxel cytotoxicity in triple-negative breast cancer cells. Mol Oncol. (2017) 11:266–79. doi: 10.1002/1878-0261.12033
93. Rothlin CV, Ghosh S. Lifting the innate immune barriers to antitumor immunity. J Immunother Cancer. (2020) 8:e000695. doi: 10.1136/jitc-2020-000695
94. Kilgore NE, Carter JD, Lorenz U, Evavold BD. Cutting edge: dependence of TCR antagonism on Src homology 2 domain-containing protein tyrosine phosphatase activity. J Immunol. (2003) 170:4891–5. doi: 10.4049/jimmunol.170.10.4891
95. Sathish JG, Dolton G, Leroy FG, Matthews RJ. Loss of Src homology region 2 domain-containing protein tyrosine phosphatase-1 increases CD8+ T cell-APC conjugate formation and is associated with enhanced in vivo CTL function. J Immunol. (2007) 178:330–7. doi: 10.4049/jimmunol.178.1.330
96. Sathish JG, Johnson KG, Leroy FG, Fuller KJ, Hallett MB, Brennan P, et al. Requirement for CD28 co-stimulation is lower in SHP-1-deficient T cells. Eur J Immunol. (2001) 31:3649–58. doi: 10.1002/1521-3765(20020816)8:16<3671::AID-CHEM3671>3.0.CO;2-9
97. Snook JP, Soedel AJ, Ekiz HA, O'connell RM, Williams MA. Inhibition of SHP-1 expands the repertoire of antitumor T cells available to respond to immune checkpoint blockade. Cancer Immunol Res. (2020) 8:506–17. doi: 10.1158/2326-6066.CIR-19-0690
98. Mercadante ER, Lorenz UM. T cells deficient in the tyrosine phosphatase SHP-1 resist suppression by regulatory T cells. J Immunol. (2017) 199:129–37. doi: 10.4049/jimmunol.1602171
99. Monu N, Frey AB. Suppression of proximal T cell receptor signaling and lytic function in CD8+ tumor-infiltrating T cells. Cancer Res. (2007) 67:11447–54. doi: 10.1158/0008-5472.CAN-07-1441
100. Iype T, Sankarshanan M, Mauldin IS, Mullins DW, Lorenz U. The protein tyrosine phosphatase SHP-1 modulates the suppressive activity of regulatory T cells. J Immunol. (2010) 185:6115–27. doi: 10.4049/jimmunol.1000622
101. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
102. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. (2015) 6:27359–77. doi: 10.18632/oncotarget.4751
103. Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. (2012) 33:5776–87. doi: 10.1016/j.biomaterials.2012.04.029
104. Stromnes IM, Fowler C, Casamina CC, Georgopolos CM, Mcafee MS, Schmitt TM, et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J Immunol. (2012) 189:1812–25. doi: 10.4049/jimmunol.1200552
Keywords: Shp1, cancer, signaling, tyrosine phosphatase, cancer therapy, TCGA
Citation: Varone A, Spano D and Corda D (2020) Shp1 in Solid Cancers and Their Therapy. Front. Oncol. 10:935. doi: 10.3389/fonc.2020.00935
Received: 04 April 2020; Accepted: 12 May 2020;
Published: 11 June 2020.
Edited by:
Gennaro Ilardi, Federico II University Hospital, ItalyReviewed by:
Simona Camorani, Gaetano Salvatore Institute for Endocrinology and Oncology, Italian National Research Council, ItalyBarbara Salvatore, Institute of Biostructure and Bioimaging (IBB), Italian National Research Council, Italy
Copyright © 2020 Varone, Spano and Corda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessia Varone, YWxlc3NpYS52YXJvbmVAaWJiYy5jbnIuaXQ=