 Giovanna Nicora
Giovanna Nicora Francesca Vitali
Francesca Vitali Arianna Dagliati
Arianna Dagliati Nophar Geifman
Nophar Geifman Riccardo Bellazzi1*
Riccardo Bellazzi1*- 1Department of Electrical, Computer and Biomedical Engineering, University of Pavia, Pavia, Italy
- 2Center for Innovation in Brain Science, University of Arizona, Tucson, AZ, United States
- 3Department of Neurology, College of Medicine, University of Arizona, Tucson, AZ, United States
- 4Center for Biomedical Informatics and Biostatistics, University of Arizona, Tucson, AZ, United States
- 5Centre for Health Informatics, The University of Manchester, Manchester, United Kingdom
- 6The Manchester Molecular Pathology Innovation Centre, The University of Manchester, Manchester, United Kingdom
In recent years, high-throughput sequencing technologies provide unprecedented opportunity to depict cancer samples at multiple molecular levels. The integration and analysis of these multi-omics datasets is a crucial and critical step to gain actionable knowledge in a precision medicine framework. This paper explores recent data-driven methodologies that have been developed and applied to respond major challenges of stratified medicine in oncology, including patients' phenotyping, biomarker discovery, and drug repurposing. We systematically retrieved peer-reviewed journals published from 2014 to 2019, select and thoroughly describe the tools presenting the most promising innovations regarding the integration of heterogeneous data, the machine learning methodologies that successfully tackled the complexity of multi-omics data, and the frameworks to deliver actionable results for clinical practice. The review is organized according to the applied methods: Deep learning, Network-based methods, Clustering, Features Extraction, and Transformation, Factorization. We provide an overview of the tools available in each methodological group and underline the relationship among the different categories. Our analysis revealed how multi-omics datasets could be exploited to drive precision oncology, but also current limitations in the development of multi-omics data integration.
Introduction
The integration and analysis of high-throughput molecular assays is a major focus for precision medicine in enabling the understanding of patient and disease specific variations. Integrated approaches allow for comprehensive views of genetic, biochemical, metabolic, proteomic, and epigenetic processes underlying a disease that, otherwise, could not be fully investigated by single-omics approaches. Computational multi-omics approaches are based on machine learning techniques and typically aim at classifying patients into cancer subtypes (1–5), designed for biomarker discovery and drug repurposing (6, 7).
While complexities underling cancer still hampers our understanding of how this disease arises and progresses (8), multi-omics approaches have been suggested as promising tools to dissect patient's dysfunctions in multiple biological systems that may be altered by cancer mechanisms (9).
Several efforts have been made to generate comprehensive multi-omics profiles of cancer patients. The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) provides detailed clinical, genomics, transcriptomics, and proteomics data on about 20,000 subjects and plans to generate additional data in the next years for a variety of cancer types. Analysis of datasets generated by multi-omics sequencing requires the development of computational approaches spanning from data integration (10), statistical methods, and artificial intelligence systems to gain actionable knowledge from data.
Here we present a descriptive overview on recent multi-omics approaches in oncology, which summarizes current state-of-art in multi-omics data analysis, relevant topics in terms of machine learning approaches, and aims of each survey, such as disease subtyping, or patient similarity. We provide an overview on each methodology group, while then focusing on publicly available tools.
Methods
Search Strategy
We retrieved publications by querying the Scopus database as: (cancer OR tumor OR tumor OR oncolog*)AND(multi-omic* OR multiomic*OR mixomic*)AND(“machine learning” OR “data fusion” OR “network analysis”).
Eligibility Criteria
Since other review covered previous years (10, 11) we included peer-reviewed journal articles published from 2014 to 2020 (last query 04-22-2020). If a study appears in multiple publications, only the latest version was included. We selected relevant studies by screening titles and abstracts, then analyzing full-texts. We excluded papers accordingly to the following criteria:
• Review articles;
• Studies focused on non-human subjects;
• Studies intended to validate and/or apply previously developed tools;
• Studies published in conference proceedings.
• Studies that integrate different measurement of the same type of omics (such as, only proteomics measurement).
Categories and Analyses
For each article, we extracted the publication year and the number of citations. We categorize the selected publications according to:
• Data inputs (i.e., types of omics);
• Research Aims:
1. Stratified Medicine for subgroup discovery: studies aimed at finding groups of patients that exhibit different therapeutic/prognostic outcomes;
2. Biomarker discovery: studies that detect -omics characteristics indicating a disease state;
3. Pathways analysis: studies aimed at discovering relation among -omics terms, such as genes or proteins in normal and cancer condition;
4. Drug repurposing/discovery: studies aimed at identifying new drugs to or existing effective drugs originally developed for other conditions;
• Methods and algorithms: Deep network, Networks-based methods (Bayesian and Heuristic Networks), Clustering, Features Extraction, Feature Transformation, Factorization.
We highlight successful approaches for each criterion and identify promising ones that are either nascent or unexplored as potential opportunities.
Results
We retrieved 270 papers. The Scopus query did not retrieve 24 relevant works that were added manually based on our previous knowledge. After a screening of papers' abstracts, 58 papers meeting our criteria were selected. Retrieved papers were organized into a matrix table (Table 1) and analyzed with respect to the aforementioned categories. As highlighted in Figure 1A, categories are not mutually exclusive, thus we show links between groups, which relate papers applying multiple methods. Figure 1B depicts all considered publications by year of publication and the Field-Weighted Citation Impact, a metric that allows comparison of papers accounting for year of publication and number citations. Studies are shown with different colors and shapes according to method used and the aim/output type.
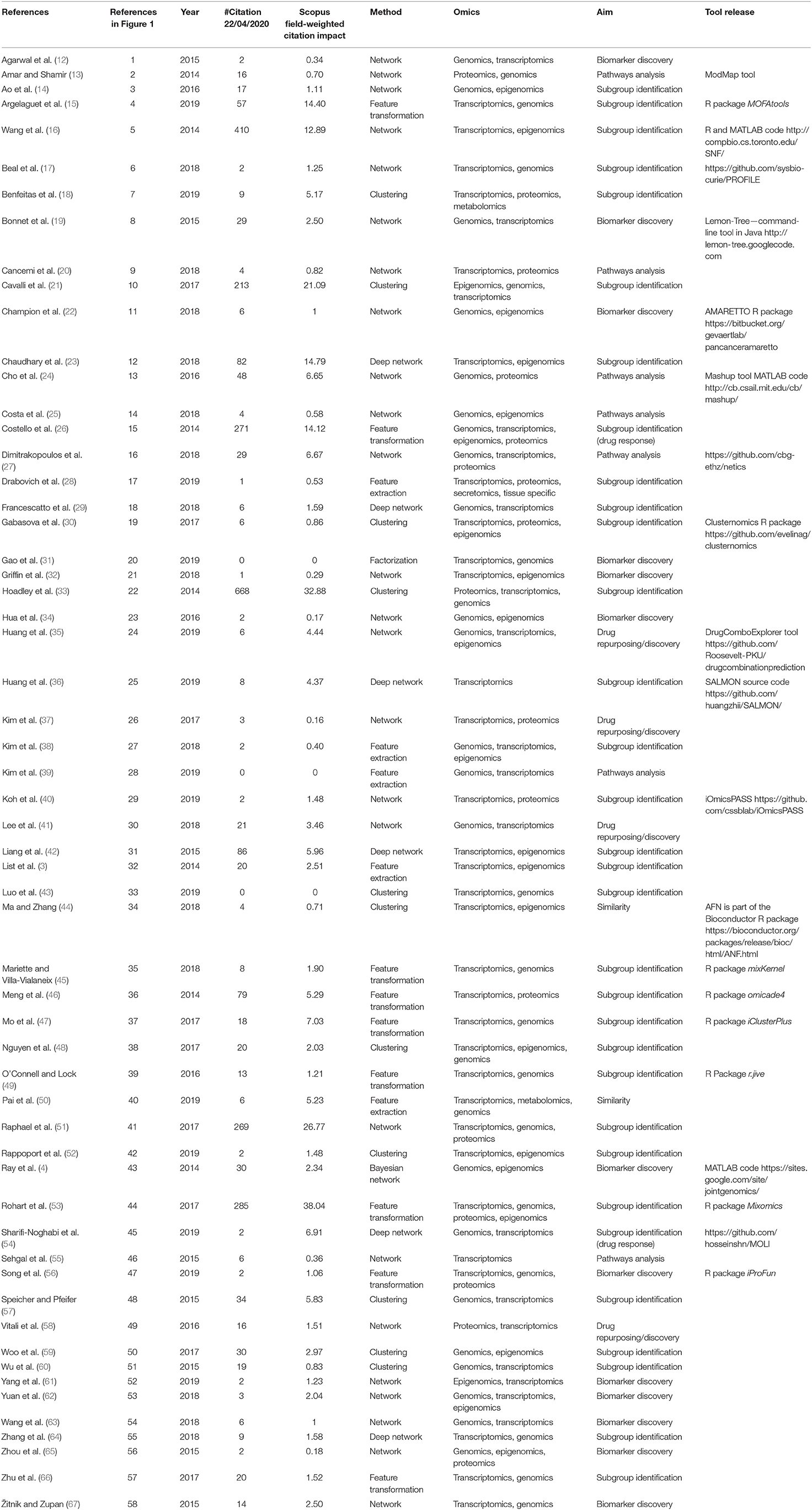
Table 1. Selected papers and categories.
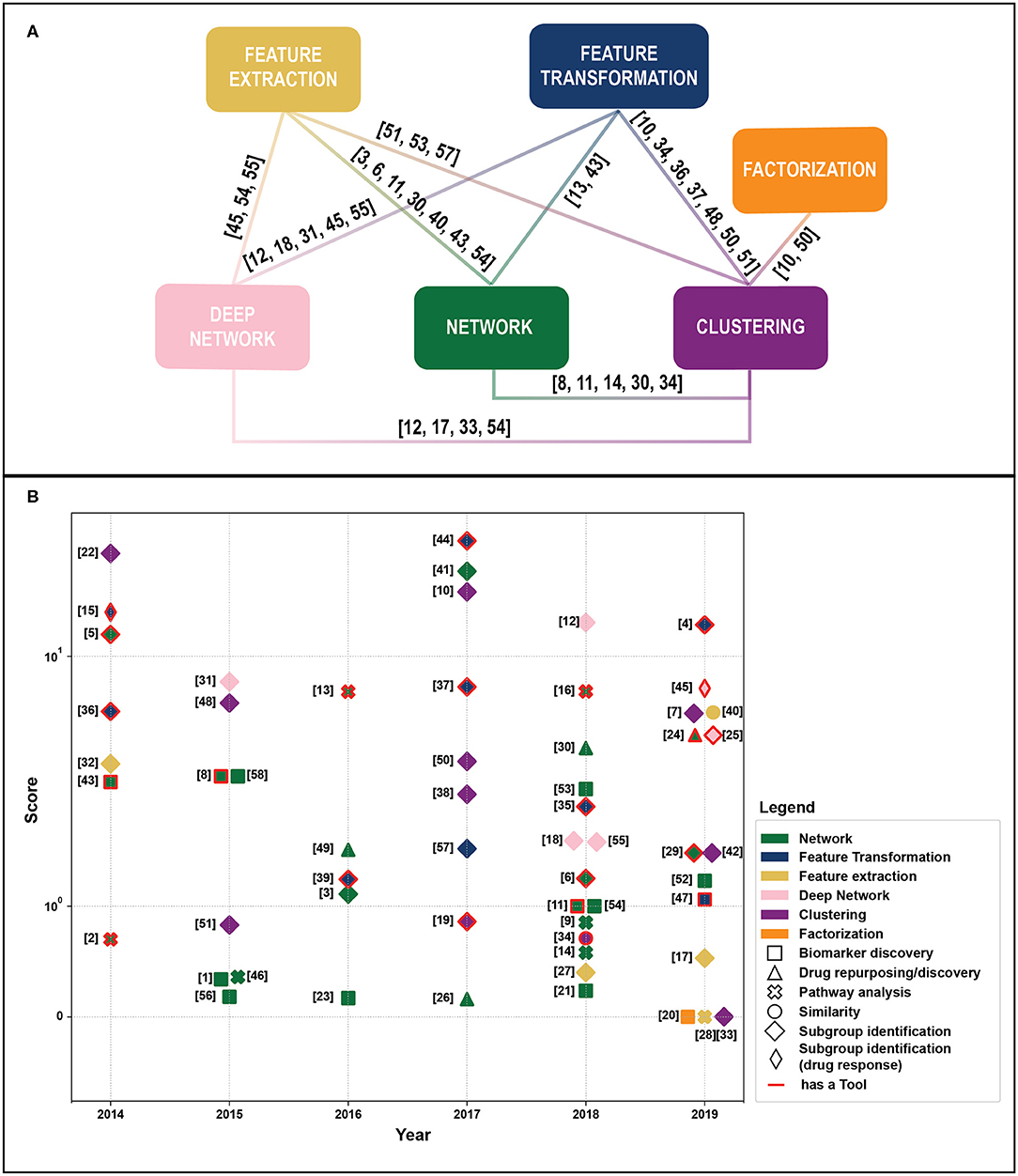
Figure 1. (A) Linkage between different methodological categories. References to papers (see Table 1). That could be categorized in different groups are reported near the link. (B) Publications by year of publication and Field-Weighted Citation Impact. Different colors indicate exploited methods, shapes aims, and outputs. Papers with red borders have source code or provide a tool. Papers in the “Subgroup identification” group and/or with free tool result to be the most cited across years. The reference numbers are reported in Table 1.
In the following sections, we describe the methodological categories that emerged from our literature review. For each methodological category, particular emphasis is placed on studies providing tools that can be exploited by other users, either with their own data or to reproduce their results.
Network-Based Methods
Network-based approaches were exploited to detect, reconstruct and study interactions among sub network modules (13, 19, 22, 25, 40); to assess functional correlation among multi-omics entities (12, 14, 20, 55, 61, 62); to integrate and fuse networks to create comprehensive view of a disease (16, 24, 32, 37, 41, 63, 65). A few work leverage Bayesian methods (4, 34) or Markov models (17, 67).
Some approaches integrate network analysis within frameworks that apply multiple algorithms (35, 51, 58). In (51) a multi-platform analysis exploited for profiling pancreatic adenocarcinoma, includes clustering and Similarity Network Fusion to integrate genomic, transcriptomic, and proteomic data from the different platforms. In (58) authors develop a framework for drug repurposing and multi-target therapies by constructing a protein network for the disease under study and fusing several data sources. In (27), a functional interaction network predicts variations in expressions caused by genomic alterations, and it is exploited to prioritize cancer genes. Few others interesting approaches (16, 19) have been discussed in (10).
iOmicsPass
iOmicsPASS (40) implements a network-based method for integrating multi-omics profiles over genome-scale biological networks. The tool provides analysis components to transform qualitative multi-omics data into scores for biological interaction, then it uses the resulting scores as input to select predictive sub-networks; finally, it selects predictive edges for phenotypic groups using a modified nearest shrunken centroid algorithm. Authors validate iOmicsPASS on Breast Invasive Ductal Carcinoma data, integrating mRNA expression, and protein abundance, with and without the normalization of the mRNA data by the DNA Copy Number Variation (CNV). When compared with the original nearest shrunken centroid classification algorithm, iOmicsPASS outperform the baseline method, indicating the importance of selecting predictive signature forms densely connected sub networks, thus limiting the search space of predictive features to known interactions.
AMARETTO
Amaretto (22) is an algorithm developed multiple omics profiles integration across different type of cancers. Authors illustrate how the algorithm identifies cancer driver genes based on multi-omics data fusion and detects subnetworks of modules across all cancers. The algorithm identifies potential cancer driver genes by investigating significant correlations between methylation, CNV and gene expression (GE) data. When the driver genes are identified it constructs a module network connecting them with the co-expressed target genes they control. This constricts a pan-cancer network that is able to identify novel pancancer driver genes.
DrugComboExplorer
DrugComboExplorer (35) identifies candidate drug combinations targeting cancer driver signaling networks by processing DNA sequencing, CNV, DNA methylation, and RNA-seq data from individual cancer patients using an integrated pipeline of algorithms. The pipeline is based on two components: the first one extracts dysregulated networks from transcriptome and methylation profiles of specific patients using bootstrapping-based simulated annealing and weighted co-expression network analysis. The second component generates a driver network signatures for each drug, evaluates synergistic effects of drug combinations on different driver signaling networks and ranks drug combinations according the synergistic effects. In (35) authors apply DrugComboExplorer on diffuse large B-cell-lymphoma and prostate cancer, demonstrating the ability of the tool to discover synergistic drug combinations and its higher prediction accuracy compared with existing computational approaches.
Deep Network
Deep Networks (DNs) are widely used to analyse omics-data (68). In a multi-omics scenario, clustering on DNs features showed different survival groups in neuroblastoma and liver cancer (23, 29, 64). In (42) authors integrated GE, methylation and miRNA in a restricted Boltzmann machine, where hidden layers represent different survival groups in breast cancer patients. Subnetworks are used in (54) to project different omics views in latent spaces that are further concatenated and fed into a final network to predict drug response.
SALMON
SALMON (Survival Analysis Learning with Multi-Omics Neural Networks) is a Deep Learning framework that integrates omics-data (mRNA and miRNA), clinical features and cancer biomarkers (36). Instead of feeding a neural network with mRNA and miRNA data, SALMON takes as input the eigengene matrices derived from co-expression analysis. Thus, it overcomes the high-dimensionality problem, reducing input features of about 99%. Authors assume that mRNA and miRNA data affect survival outcome independently, therefore the two corresponding eigengene matrices are connected to two different hidden layers whose output is linked to the final network with a Cox proportional hazards regression network. Results on breast cancer carcinoma patients showed improvements in survival prediction ability compared to single-omics.
Clustering
Multi-omics clustering approaches are exploited to detect regularities and patterns that reveal different cancer molecular subtypes (21, 33, 43, 48, 57, 60) and prognostic groups in hepatocellular carcinoma (59). In (18) consensus clustering is performed on transcriptomics, metabolomics, and proteomics data to stratify patients with hepatocellular carcinoma based on their redox response. Clustering applications are often preceded by feature selection and/or feature transformation of multi-omics data, such as factorization, low rank approximation, and neural network. An exhaustive review on multi-omics integrative clustering approaches can be found in (69).
Nemo
NEMO (NEighborhood based Multi-Omics clustering) is a similarity-based tool that computes inter-patient similarity matrices for each omics through a radial basis function kernel. Spectral clustering is performed on the resulting average similarity matrix (52). NEMO addresses the problem of partial datasets, where not all the omics are measured for all the patients, and the final average matrix is computed on the observed omics values, without performing imputation. NEMO clustering shows higher performance compared to the same approach with imputed data, while on TCGA cancer datasets it detects significant differences in survival for six out of 10 cancer types.
Clusternomics
The main assumption of multi-omics clustering approaches relies on the existence of a consistent clustering structure across heterogeneous datasets. Alternatively, in (30) authors introduced the context-dependent clustering Clusternomics. Each omics is seen as a context describing a particular aspect of the underlying biological process. The global clustering structure is inferred from the combination of Bayesian clustering assignments. Then, by separating cluster assignment on two levels, Clusternomics allows the number of clusters to vary on local or global structure. Its performances are evaluated on a simulated dataset, where it showed higher Adjuster Rank Index compared to other clustering techniques, but also on breast, lung and kidney cancer from TCGA repository, where it identified clinically meaningful clusters.
Affinity Network Fusion
Affinity Network Fusion (AFN) (44) is both a clustering and classification technique that applies graph clustering to a patient affinity matrix incorporating information from multiple views. For each omic, after feature selection and/or transformation, AFN computes patient pair-wise distances. kNN Graph Kernel applied to the distance metric creates a patient affinity matrix for each view. The final affinity matrix is the weighted sum of the computed affinity matrices. AFN approach showed improved clustering performance in detecting cancer subtypes on several TCGA datasets when compared to its application in single omics.
Feature Extraction
In multi-omics integration, variable selection to reduce the dimensionality of the omics dataset has a dominant role [(70), Figure 1A]. Recursive feature elimination was exploited to select subsets of expressed genes and methylation data to classify breast cancer disease subtypes with a Random Forest (3). Genes prioritization allowed prognosis prediction in different cancer types from epigenomics, transcriptomics, and genomics data (38), and biomarker discovery in prostate cancer (28). In (39) authors weight gene-gene interaction from transcriptomics and genomics data with a random walked-based method to select the most important interaction for survival prediction in breast cancer and neuroblastoma patients.
netDX
netDx is an algorithm that performs feature selection on Patient Similarity Networks (PSN) to classify patients in different prognostic groups (50). A PSN is built for each omics such that nodes represent patients and edges stand for the similarity of two nodes in the given view. Then netDx identifies which networks (i.e., which omics) strongly relate high- and low- risk patients through the GeneMANIA algorithm (71), which solves a regression problem to maximize the edges that connect query patients. Finally, each network is weighted according to its ability to relate patients of the same group and networks whose score exceeds a defined threshold are selected and combined in a single network by averaging their similarity scores. Authors benchmarked netDx against several machine-learning methods to predict survival outcomes on PanCancer TCGA multi-omics datasets, showing comparable results. On a breast cancer dataset, netDx selected features correspond to pathways known to be dysregulated in this type of cancer.
Feature Transformation
Feature transformation (FT) refers to algorithms that replace existing features with new features still function of the original ones. As shown in Figure 1B, the majority of FT techniques aims at identifying cancer subtypes, biomarkers, omics-signatures, and key features from multi-omics data. Zhu et al. (66) proposed a kernel machine-learning method for a pan-cancer prognostic assessment by integrating multi-omics data. This work is particularly interesting since it's the only FT method we reviewed that allows multi-omics profile integration individually and in combination with clinical factors. A Kernel-based approach, combined with non-linear regression and Bayesian inference, resulted to be the best performing algorithm in a drug sensitivity prediction challenge (26).
In the following, we will report selected FT approaches, although few other tools for subgroup discovery, such as iClusterBayes (47), Multi-Omics Factor Analysis (15), JIVE (49), and MCIA (46), are available.
MixOmics
One of the most recent and biggest efforts in this field resulted in an R package called mixOmics (53). MixOmics allows for multivariate analysis of omics data including data exploration, dimension reduction, and visualization. mixOmics can be applied in numerous of studies with different aims such as integration and biomarker identification from multi-omics studies. The package includes two different types of multi-omics integration. One aimed at integrating different type of omics data of the same biological samples, while the second focus on integrating independent data measured on the same predictors to increase sample size and statistical power (53). Both frameworks aim at extracting biologically relevant features, [i.e., molecular signatures, by applying FT techniques (53)]. In (53) authors presented the results on 150 samples of mRNA, miRNA and proteomics breast cancer data and showed its ability to correctly discriminate three types of breast cancers.
mixKernel
mixKernel (45) is a R package compatible with mixOmics, which allows integration of multiple datasets by representing each dataset through a kernel that provides pairwise information between samples. The single kernels are then combined into one meta-kernel in an unsupervised framework. These new meta-kernels can be used for exploratory analyses, such as clustering or more sophisticated analysis to get insights into the data integrated. The authors showed better performances of mixKernel applied to mRNA, miRNAs and methylation breast cancer data if compared with one kernel approach.
iProFun
iProFun (56) is a method aimed at elucidating proteogenomic functional consequences of CNV and methylation alterations. The authors integrated mRNA expression levels, global protein abundances, and phosphoprotein abundances of a certain cancer. The output consists in a list of genes whose CNVs and/or DNA methylations significantly influencing some or all of the data integrated. iProFun obtains summary statistics of data integrated based on a gene-level multiple linear regression. These statistics are then used to extract genes having a cascading effect of all cis-molecular traits of interests and genes whose functional regulations are unique at global protein levels. iProFun applied to ovarian cancer TCGA dataset showed its ability in extracting interesting genes that could be considered targets for future therapies.
Factorization
Traditional data mining methods are often inadequate to treat heterogeneous, sparse and noisy data such as multi-omics. Heavy pre-processing operations could modify, therefore loose, the inner structure of data coming from different sources. To discover latent characteristics hidden in huge amount of information, factorization techniques have been applied to highlight complex interactions among omics-data, hard to detect using standard approaches.
Gao et al. (31) developed an integrated Graph Regularized Non-negative Matrix Factorization model focused network construction by integrating gene expression data, CNV data, and methylation data. The authors used the factorization technique to decompose and fuse the multi-omics data. Then, by combining the results with network and mining analyses they showed how their method was able to find potential new cancer-related genes on two different TCGA datasets. Another method, based on factor analysis, aims at identifying latent factors in the multi-omics-data integrated in the model that can be used for subsequent analysis such as subgroup identification (15). Give its aim in extracting hidden features, we described this method in detail in the feature transformation section.
Discussion
Along with technological advances in high-throughput sequencing, which characterize multiple “omes” from biological samples, holistic systems for data integration and knowledge discovery with machine-learning algorithms are still under development. Precision oncology would greatly benefit from actionable knowledge gained from multi-omics assays. In this paper we provided an overview of recent works on this topic and highlight current achievements and limitations.
We reviewed relevant tools to perform analysis based on different combination of omics, and observed their growing numbers in recent years, indicating strong commitments to develop such tools. Several issues emerged, too. The majority of the proposed techniques were applied to TCGA dataset, and data integration was mainly focused on transcriptomics and genomics. Efforts should be devoted to make new data sources available to the research community (72), such as the UKBioBank (73) and DriverDBv3 (74), and to integrate other “omes,” such as metabolome, or patient-generated, and environmental data. Research in this field would greatly benefit from the development of databases specifically developed for containing and facilitating the analysis of multi-omics and clinical data, such as LinkedOmics (75). Another important improvement to increase usability and reproducibility would be to aim at developing methods that can be applied and generalized for all omics data type.
The complexity of multi-omics data analysis requires collaborative efforts among the clinical and machine-learning communities and the joint application of methodologies derived from heterogenous backgrounds. We noted that some promising methods, such as matrix-factorization have not been extensively exploited, while clustering and network-based approaches are the most extensively used, probably due to their flexibility and the possibility to be integrated in comprehensive frameworks that include feature extraction and transformation to deal with the curse of dimensionality. Deep learning methods, that are flexible and achieved outstanding results in other fields, are increasingly used, even though many works share the same “pipeline” (i.e., the exploitation of autoencoder hidden layers for clustering). Interestingly, the number open source tools have increased in the very last years (Figure 1B).
We are aware of some limitations of our review. An important aspect that has not been covered by this review is the quantitative comparison among tools (76), which could highlight possible overfitting (77) and issues that may prevent the actual translation of multi-omics approaches from bench to bedside. Although, by indicating works that provide a usable tool (Table 1), our review could be a starting point for a comprehensive quantitative comparison.
Author Contributions
RB conceived the study. GN, FV, and AD run the analyses and wrote the article. NG and RB revised the article. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Fondazione Regionale Ricerca Biomedica, Milan, Italy [FRRB project n. 2015-0042, Genomic profiling of rare hematologic malignancies, development of personalized medicine strategies, and their implementation into Rete Ematologica Lombarda (REL) clinical network] and by the NIHR Manchester BRC, MRC Molecular Pathology Node MMPathic (grant ref MR/N00583X/1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Simone Marini for his valuable help in the initial phases of the study.
References
1. Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. (2009) 25:2906–12. doi: 10.1093/bioinformatics/btp543
2. Lock EF, Hoadley KA, Marron JS, Nobel AB. Joint and individual variation explained (JIVE) for integrated analysis of multiple data types. Ann Appl Stat. (2013) 7:523–42. doi: 10.1214/12-AOAS597
3. List M, Hauschild A-C, Tan Q, Kruse TA, Mollenhauer J, Baumbach J, et al. Classification of breast cancer subtypes by combining gene expression and DNA methylation data. J Integr Bioinform. (2014) 11:236. doi: 10.2390/biecoll-jib-2014-236
4. Ray P, Zheng L, Lucas J, Carin L. Bayesian joint analysis of heterogeneous genomics data. Bioinformatics. (2014) 30:1370–6. doi: 10.1093/bioinformatics/btu064
5. Gligorijević V, Malod-Dognin N, PrŽulj N. Patient-specific data fusion for cancer stratification and personalised treatment. Pacific Symp Biocomput. (2016) 21:321–332. doi: 10.1142/9789814749411_0030
6. Gottlieb A, Stein GY, Ruppin E, Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol. (2011) 7:26. doi: 10.1038/msb.2011.26
7. Napolitano F, Zhao Y, Moreira VM, Tagliaferri R, Kere J, D'Amato M, et al. Drug repositioning: a machine-learning approach through data integration. J Cheminform. (2013) 5:30. doi: 10.1186/1758-2946-5-30
8. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
9. Knox SS. From “omics” to complex disease: a systems biology approach to gene-environment interactions in cancer. Cancer Cell Int. (2010) 10:11. doi: 10.1186/1475-2867-10-11
10. Huang S, Chaudhary K, Garmire LX. More is better: recent progress in multi-omics data integration methods. Front Genet. (2017) 8:84. doi: 10.3389/fgene.2017.00084
11. Li Y, Wu FX, Ngom A. A review on machine learning principles for multi-view biological data integration. Brief Bioinform. (2018) 19:325–40. doi: 10.1093/bib/bbw113
12. Agarwal M, Adhil M, Talukder AK. Multi-omics multi-scale big data analytics for cancer genomics. Lect Notes Comput Sci. (2015) 9498:228–43. doi: 10.1007/978-3-319-27057-9_16
13. Amar D, Shamir R. Constructing module maps for integrated analysis of heterogeneous biological networks. Nucleic Acids Res. (2014) 42:4208–19. doi: 10.1093/nar/gku102
14. Ao L, Song X, Li X, Tong M, Guo Y, Li J, et al. An individualized prognostic signature and multi-omics distinction for early stage hepatocellular carcinoma patients with surgical resection. Oncotarget. (2016) 7:24097–110. doi: 10.18632/oncotarget.8212
15. Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, et al. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. (2018) 14:e8124. doi: 10.15252/msb.20178124
16. Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. (2014) 11:333–7. doi: 10.1038/nmeth.2810
17. Beal J, Montagud A, Traynard P, Barillot E, Calzone L. Personalization of logical models with multi-omics data allows clinical stratification of patients. Front Physiol. (2019) 9:1965. doi: 10.3389/fphys.2018.01965
18. Benfeitas R, Bidkhori G, Mukhopadhyay B, Klevstig M, Arif M, Zhang C, et al. Characterization of heterogeneous redox responses in hepatocellular carcinoma patients using network analysis. EBioMedicine. (2019) 40:471–87. doi: 10.1016/j.ebiom.2018.12.057
19. Bonnet E, Calzone L, Michoel T. Integrative multi-omics module network inference with lemon-tree. PLoS Comput Biol. (2015) 11:3983. doi: 10.1371/journal.pcbi.1003983
20. Cancemi P, Buttacavoli M, Cara GD, Albanese NN, Bivona S, Pucci-Minafra I, et al. A multiomics analysis of S100 protein family in breast cancer. Oncotarget. (2018) 9:29064–81. doi: 10.18632/oncotarget.25561
21. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. (2017) 31:737–54.e6. doi: 10.1016/j.ccell.2017.05.005
22. Champion M, Brennan K, Croonenborghs T, Gentles AJ, Pochet N, Gevaert O. Module analysis captures pancancer genetically and epigenetically deregulated cancer driver genes for smoking and antiviral response. EBioMedicine. (2018) 27:156–66. doi: 10.1016/j.ebiom.2017.11.028
23. Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res. (2018) 24:1248–59. doi: 10.1158/1078-0432.CCR-17-0853
24. Cho H, Berger B, Peng J. Compact integration of multi-network topology for functional analysis of genes. Cell Syst. (2016) 3:540–8.e5. doi: 10.1016/j.cels.2016.10.017
25. Costa RL, Boroni M, Soares MA. Distinct co-expression networks using multi-omic data reveal novel interventional targets in HPV-positive and negative head-and-neck squamous cell cancer. Sci Rep. (2018) 8:5. doi: 10.1038/s41598-018-33498-5
26. Costello JC, Heiser LM, Georgii E, Gönen M, Menden MP, Wang NJ, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. (2014) 32:1202–12. doi: 10.1038/nbt.2877
27. Dimitrakopoulos C, Hindupur SK, Hafliger L, Behr J, Montazeri H, Hall MN, et al. Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics. (2018) 34:2441–8. doi: 10.1093/bioinformatics/bty148
28. Drabovich AP, Saraon P, Drabovich M, Karakosta TD, Dimitromanolakis A, Hyndman ME, et al. Multi-omics biomarker pipeline reveals elevated levels of protein-glutamine gamma-glutamyltransferase 4 in seminal plasma of prostate cancer patients. Mol Cell Proteomics. (2019) 18:1807–23. doi: 10.1074/mcp.RA119.001612
29. Francescatto M, Chierici M, Rezvan Dezfooli S, Zandonà A, Jurman G, Furlanello C, et al. Multi-omics integration for neuroblastoma clinical endpoint prediction. Biol Direct. (2018) 13:8. doi: 10.1186/s13062-018-0207-8
30. Gabasova E, Reid J, Wernisch L. Clusternomics: integrative context-dependent clustering for heterogeneous datasets. PLoS Comput Biol. (2017) 13:e1005781. doi: 10.1371/journal.pcbi.1005781
31. Gao Y-L, Hou M-X, Liu J-X, Kong X-Z. An integrated graph regularized non-negative matrix factorization model for gene co-expression network analysis. IEEE Access. (2019) 7:126594–602. doi: 10.1109/ACCESS.2019.2939405
32. Griffin PJ, Zhang Y, Johnson WE, Kolaczyk ED. Detection of multiple perturbations in multi-omics biological networks. Biometrics. (2018) 74:1351–61. doi: 10.1111/biom.12893
33. Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. (2014) 158:929–44. doi: 10.1016/j.cell.2014.06.049
34. Hua L, Zheng WY, Xia H, Zhou P. Detecting the potential cancer association or metastasis by multi-omics data analysis. Genet Mol Res. (2016) 15:e038987. doi: 10.4238/gmr.15038987
35. Huang L, Brunell D, Stephan C, Mancuso J, Yu X, He B, et al. Driver network as a biomarker: systematic integration and network modeling of multi-omics data to derive driver signaling pathways for drug combination prediction. Bioinformatics. (2019) 35:3709–17. doi: 10.1093/bioinformatics/btz109
36. Huang Z, Zhan X, Xiang S, Johnson TS, Helm B, Yu CY, et al. Salmon: survival analysis learning with multi-omics neural networks on breast cancer. Front Genet. (2019) 10:166. doi: 10.3389/fgene.2019.00166
37. Kim JY, Lee H, Woo J, Yue W, Kim K, Choi S, et al. Reconstruction of pathway modification induced by nicotinamide using multi-omic network analyses in triple negative breast cancer. Sci Rep. (2017) 7:7. doi: 10.1038/s41598-017-03322-7
38. Kim M, Oh I, Ahn J. An improved method for prediction of cancer prognosis by network learning. Genes. (2018) 9:1–11. doi: 10.3390/genes9100478
39. Kim SY, Jeong HH, Kim J, Moon JH, Sohn KA. Robust pathway-based multi-omics data integration using directed random walks for survival prediction in multiple cancer studies. Biol Direct. (2019) 14:8. doi: 10.1186/s13062-019-0239-8
40. Koh HWL, Fermin D, Vogel C, Choi KP, Ewing RM, Choi H, et al. iOmicsPASS: network-based integration of multiomics data for predictive subnetwork discovery. npj Syst Biol Appl. (2019) 5:22. doi: 10.1038/s41540-019-0099-y
41. Lee S-I, Celik S, Logsdon BA, Lundberg SM, Martins TJ, Oehler VG, et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun. (2018) 9:5. doi: 10.1038/s41467-017-02465-5
42. Liang M, Li Z, Chen T, Zeng J. Integrative data analysis of multi-platform cancer data with a multimodal deep learning approach. IEEE/ACM Trans Comput Biol Bioinforma. (2015) 12:928–37. doi: 10.1109/TCBB.2014.2377729
43. Luo Z, Wang W, Li F, Songyang Z, Feng X, Xin C, et al. Pan-cancer analysis identifies telomerase-associated signatures and cancer subtypes. Mol Cancer. (2019) 18:106. doi: 10.1186/s12943-019-1035-x
44. Ma T, Zhang A. Affinity network fusion and semi-supervised learning for cancer patient clustering. Methods. (2018) 145:16–24. doi: 10.1016/j.ymeth.2018.05.020
45. Mariette J, Villa-Vialaneix N. Unsupervised multiple kernel learning for heterogeneous data integration. Bioinformatics. (2018) 34:1009–15. doi: 10.1093/bioinformatics/btx682
46. Meng C, Kuster B, Culhane AC, Gholami AM. A multivariate approach to the integration of multi-omics datasets. BMC Bioinformatics. (2014) 15:162. doi: 10.1186/1471-2105-15-162
47. Mo Q, Shen R, Guo C, Vannucci M, Chan KS, Hilsenbeck SG. A fully Bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics. (2018) 19:71–86. doi: 10.1093/biostatistics/kxx017
48. Nguyen T, Tagett R, Diaz D, Draghici S. A novel approach for data integration and disease subtyping. Genome Res. (2017) 27:2025–39. doi: 10.1101/gr.215129.116
49. O'Connell MJ, Lock EF. R. JIVE for exploration of multi-source molecular data. Bioinformatics. (2016) 32:2877–9. doi: 10.1093/bioinformatics/btw324
50. Pai S, Hui S, Isserlin R, Shah MA, Kaka H, Bader GD, et al. netDx: interpretable patient classification using integrated patient similarity networks. Mol Syst Biol. (2019) 15:e8497. doi: 10.15252/msb.20188497
51. Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. (2017) 32:185–203.e13. doi: 10.1016/j.ccell.2017.07.007
52. Rappoport N, Shamir R, Schwartz R. NEMO: cancer subtyping by integration of partial multi-omic data. Bioinformatics. (2019) 35:3348–56. doi: 10.1093/bioinformatics/btz058
53. Rohart F, Gautier B, Singh A, Lê Cao KA. mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol. (2017) 13:e1005752. doi: 10.1371/journal.pcbi.1005752
54. Sharifi-Noghabi H, Zolotareva O, Collins CC, Ester M. MOLI: multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics. (2019) 35:i501–9. doi: 10.1093/bioinformatics/btz318
55. Sehgal V, Seviour EG, Moss TJ, Mills GB, Azencott R, Ram PT. Robust selection algorithm (RSA) for multi-omic biomarker discovery; integration with functional network analysis to identify miRNA regulated pathways in multiple cancers. PLoS ONE. (2015) 10:72. doi: 10.1371/journal.pone.0140072
56. Song X, Ji J, Gleason KJ, Yang F, Martignetti JA, Chen LS, et al. Insights into impact of DNA copy number alteration and methylation on the proteogenomic landscape of human ovarian cancer via a multi-omics integrative analysis. Mol Cell Proteomics. (2019) 18(8 Suppl.1):S52–65. doi: 10.1074/mcp.RA118.001220
57. Speicher NK, Pfeifer N. Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics. (2015) 31:i268–75. doi: 10.1093/bioinformatics/btv244
58. Vitali F, Cohen LD, Demartini A, Amato A, Eterno V, Zambelli A, et al. A network-based data integration approach to support drug repurposing and multi-Target therapies in triple negative breast cancer. PLoS ONE. (2016) 11:e0162407. doi: 10.1371/journal.pone.0162407
59. Woo HG, Choi J-H, Yoon S, Jee BA, Cho EJ, Lee J-H, et al. Integrative analysis of genomic and epigenomic regulation of the transcriptome in liver cancer. Nat Commun. (2017) 8:839. doi: 10.1038/s41467-017-00991-w
60. Wu D, Wang D, Zhang MQ, Gu J. Fast dimension reduction and integrative clustering of multi-omics data using lowrank approximation: application to cancer molecular classification. BMC Genomics. (2015) 16:1022. doi: 10.1186/s12864-015-2223-8
61. Yang Z, Liu B, Lin T, Zhang Y, Zhang L, Wang M, et al. Multiomics analysis on DNA methylation and the expression of both messenger RNA and microRNA in lung adenocarcinoma. J Cell Physiol. (2019) 234:7579–86. doi: 10.1002/jcp.27520
62. Yuan L, Guo LH, Yuan CA, Zhang Y, Han K, Nandi AK, et al. Integration of multi-omics data for gene regulatory network inference and application to breast cancer. IEEE/ACM Trans Comput Biol Bioinforma. (2019) 16:782–91. doi: 10.1109/TCBB.2018.2866836
63. Wang Z, Wei Y, Zhang R, Su L, Gogarten SM, Liu G, et al. Multi-omics analysis reveals a HIF network and hub gene EPAS1 associated with lung adenocarcinoma. EBioMedicine. (2018) 32:93–101. doi: 10.1016/j.ebiom.2018.05.024
64. Zhang L, Lv C, Jin Y, Cheng G, Fu Y, Yuan D, et al. Deep learning-based multi-omics data integration reveals two prognostic subtypes in high-risk neuroblastoma. Front Genet. (2018) 9:477. doi: 10.3389/fgene.2018.00477
65. Zhou Y, Liu Y, Li K, Zhang R, Qiu F, Zhao N, et al. ICan: an integrated co-alteration network to identify ovarian cancer-related genes. PLoS ONE. (2015) 10:e0116095. doi: 10.1371/journal.pone.0116095
66. Zhu B, Song N, Shen R, Arora A, Machiela MJ, Song L, et al. Integrating clinical and multiple omics data for prognostic assessment across human cancers. Sci Rep. (2017) 7:8. doi: 10.1038/s41598-017-17031-8
67. Žitnik M, Zupan B. Gene network inference by fusing data from diverse distributions. Bioinformatics. (2015) 31:i230–9. doi: 10.1093/bioinformatics/btv258
68. Tang B, Pan Z, Yin K, Khateeb A. Recent advances of deep learning in bioinformatics and computational biology. Front Genet. (2019) 10:214. doi: 10.3389/fgene.2019.00214
69. Wang D, Gu J. Integrative clustering methods of multi-omics data for molecule-based cancer classifications. Quant Biol. (2016) 4:58–67. doi: 10.1007/s40484-016-0063-4
70. Wu C, Zhou F, Ren J, Li X, Jiang Y, Ma S, et al. A selective review of multi-level omics data integration using variable selection. High-Throughput. (2019) 8:4. doi: 10.3390/ht8010004
71. Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. (2008) 9:S4. doi: 10.1186/gb-2008-9-s1-s4
72. Conesa A, Beck S. Making multi-omics data accessible to researchers. Sci Data. (2019) 6:251. doi: 10.1038/s41597-019-0258-4
73. Ollier W, Sprosen T, Peakman T. UK Biobank: from concept to reality. Pharmacogenomics. (2005) 6:639–46. doi: 10.2217/14622416.6.6.639
74. Liu SH, Shen PC, Chen CY, Hsu AN, Cho YC, Lai YL, et al. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res. (2020) 48:D863–70. doi: 10.1093/nar/gkz964
75. Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. (2018) 46:D956–63. doi: 10.1093/nar/gkx1090
76. Sathyanarayanan A, Gupta R, Thompson EW, Nyholt DR, Bauer DC, Nagaraj SH, et al. A comparative study of multi-omics integration tools for cancer driver gene identification and tumour subtyping. Brief Bioinform. (2019). doi: 10.1093/bib/bbz121. [Epub ahead of print].
Keywords: multi-omics, machine learning, tools, systematic review, oncology, cancer
Citation: Nicora G, Vitali F, Dagliati A, Geifman N and Bellazzi R (2020) Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 10:1030. doi: 10.3389/fonc.2020.01030
Received: 30 January 2020; Accepted: 26 May 2020;
Published: 30 June 2020.
Edited by:
Francesca Finotello, Innsbruck Medical University, AustriaReviewed by:
Federica Eduati, Eindhoven University of Technology, NetherlandsGiuseppe Jurman, Fondazione Bruno Kessler (FBK), Italy
Copyright © 2020 Nicora, Vitali, Dagliati, Geifman and Bellazzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccardo Bellazzi, cmljY2FyZG8uYmVsbGF6emlAdW5pcHYuaXQ=
†These authors have contributed equally to this work