 Longyong Xu
Longyong Xu Weijie Zhang
Weijie Zhang Xiang H.-F. Zhang
Xiang H.-F. Zhang Xi Chen
Xi Chen- 1Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, United States
- 2Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX, United States
- 3Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX, United States
Metastases—the spreading of cancer cells from primary tumors to distant organs, including bone—is often incurable and is the major cause of morbidity in cancer patients. Understanding how cancer cells acquire the ability to colonize to bone and become overt metastases is critical to identify new therapeutic targets and develop new therapies against bone metastases. Recent reports indicate that the endoplasmic reticulum (ER) stress and, as its consequence, the unfolded protein response (UPR) is activated during metastatic dissemination. However, their roles in this process remain largely unknown. In this review, we discuss the recent progress on evaluating the tumorigenic, immunoregulatory and metastatic effects of ER stress and the UPR on bone metastases. We explore new opportunities to translate this knowledge into potential therapeutic strategies for patients with bone metastases.
Introduction
Bone is a frequent site of cancer metastases, and skeletal metastasis is much more common than the primary bone cancers (1). Metastatic spread of primary tumor cells to bone tissues comprises the following multiple-step cascade: (1) local invasion at the primary site; (2) intravasation; (3) survival in circulation; (4) arrest at distant organ sites; (5) extravasation to enter the parenchymal tissues of distant organs; (6) survival in the new microenvironment; and (7) proliferation to form macroscopic, clinically detectable secondary tumors, which is the step that eventually leads to morbidity (Figure 1) (2–4). Considerable research efforts have demonstrated that both intrinsic traits of cancer cells (the seeds) and the unique bone microenvironmental factors (the soil) contribute to the development of bone metastases (1, 3, 5–7). These efforts have led to approved treatment on bone metastases, exemplified by the introduction of bisphosphonates and denosumab (8–10). Meanwhile, several clinical trials are on-going based on the knowledge from these efforts. Further studies aim to understanding the molecular basis for each step of bone metastasis will be instrumental to manage the bone metastasis.
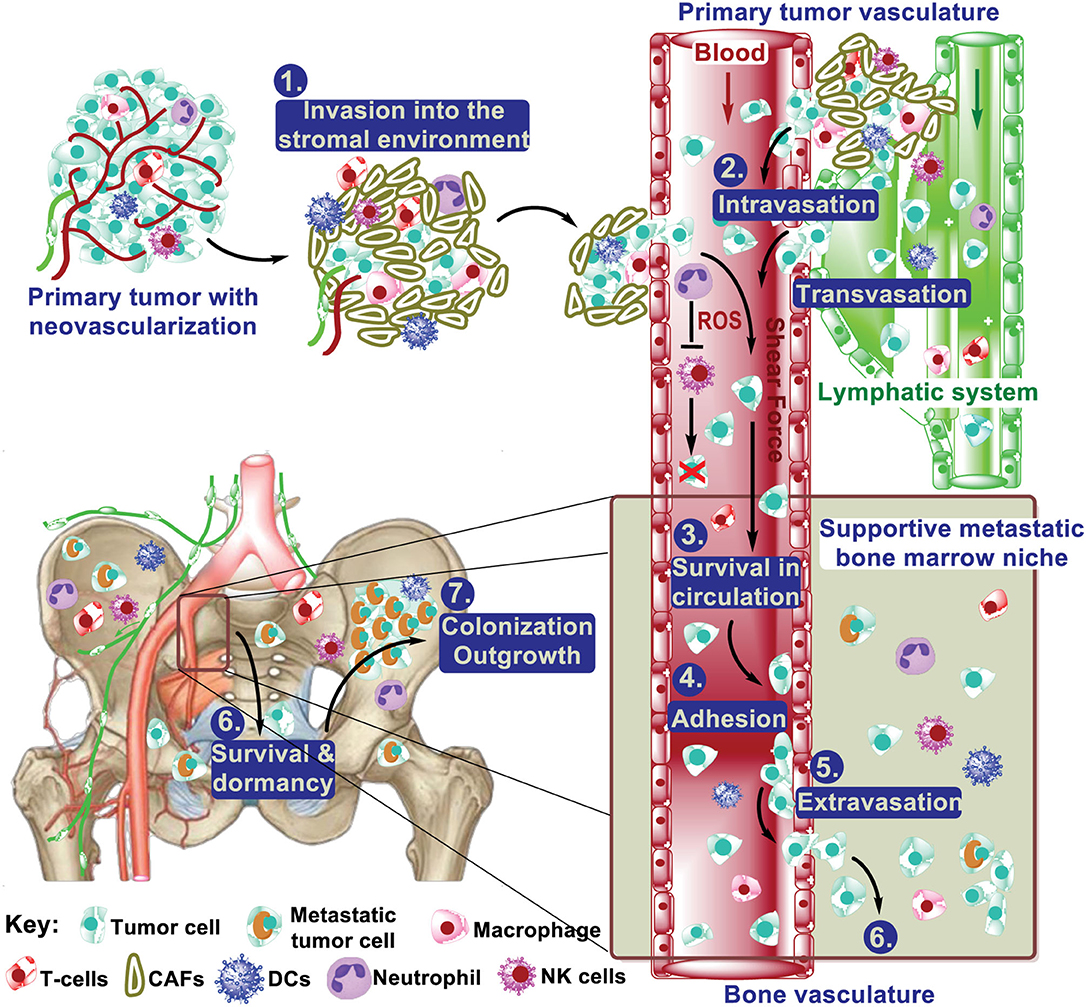
Figure 1. The multistep process from primary tumor to bone metastasis. During metastatic progression, tumor cells leave their primary site via local invasion into the surrounding tumor-associated stroma, followed by tumor cell intravasation into the blood and lymph vasculature. Once in the vasculature, tumor cells interact with neutrophils and NK cells, which regulate their survival in the circulation. These steps above are mostly common in metastasis to different distant organs. The extravasation of cancer cells from the blood vasculature into the bone marrow can occur very early. After extravasation, colonizing cancer cells must develop survival mechanisms to adapt to the local microenvironment and various treatment. This includes dormancy, interacting with (pre)metastatic niche cells (e.g., osteoblasts and osteoclasts), and resistance to immunity. Of those cells that survived, some will be reactivated after years, even decades, to form macrometastasis.
Tumor cells endure intrinsic (oncogenic) and extrinsic environmental stresses during metastatic dissemination (11). These stresses can either increase the protein synthesis, overwhelming the protein folding capacity of the endoplasmic reticulum (ER) or directly disrupt ER protein folding. This leads to the accumulation of unfolded and misfolded proteins (known as ER stress) (12, 13). An adaptive mechanism, termed the unfolded protein response (UPR), is consequently initiated by transmembrane sensors on the ER upon detection of ER stress to restore ER homeostasis (14). Multiple functions of the UPR in the development of primary tumors have been extensively studied, and targeting the UPR has been shown to be an effective therapeutic strategy in multiple cancers (15–28). However, its role in metastases remain far less documented. In this review, we will discuss the mechanisms of the UPR in tumor progression and its potential implications in bone metastases.
Bone Metastases
The relative incidence, median survival, and effect on bone homeostasis (osteolytic, osteoblastic, or mixed) of bone metastases vary greatly among different cancer types (Table 1) (1, 29, 30). Bone metastases are associated with multiple skeletal complications, including bone pain, impaired mobility, pathologic fractures, nerve compression, bone marrow aplasia, and hypercalcemia (31). The clinical detection of metastases may be a late event of disease progression; although, the dissemination to bone may occur early. Disseminated tumor cells (DTCs) can be detected in the bone marrow of patients and in mouse models even without the invasive diseases (32–38). However, because many more DTCs than macrometastases are present in the bone marrow in both patients and mouse models, it remains unclear whether these early DTCs ultimately cause metastatic outgrowth or if they are simply bystanders during the metastatic process (5, 9). Nevertheless, it is clear that the presence of DTCs in bone marrow is associated with poor prognosis and predicts eventual metastases to the bone as well as other organs (39–43). The microenvironment in primary tumors contribute to the selection of secondary tumors in bone. For example, cancer-associated fibroblasts select Src-hyperactivated bone metastatic seeds in triple-negative breast cancers (TNBC) (44). Meanwhile, premetastatic niches in the bone microenvironment are actively formed by secreted factors and/or exosomes from the primary tumor prior to DTC seeding (5, 45, 46). Upon arrival at the bone, DTCs that survive the hostile environment interact with the bone resident cells, forming the metastatic niche that determines the fate of DTCs (dormant, reactivated, or drug resistant) (5, 7, 9, 47).

Table 1. Incidence of bone metastases in cancer (1).
ER Stress and the UPR
The ER is the major organelle in eukaryotic cells responsible for intracellular Ca2+ homeostasis, lipid biosynthesis, and the folding of membrane and secreted proteins (14, 48–52). Protein folding in the ER is precisely regulated and highly sensitive to alterations in the protein load, mutations that affect the folding process, and the ER folding environment (e.g., redox state, nutrient status, and Ca2+ levels) (49, 53). The accumulation of unfolded or misfolded proteins in the ER causes ER stress, which can be detected and resolved by the UPR (48, 51, 54–57). There are three major UPR signaling branches named after their transmembrane sensors: (1) inositol-requiring enzyme 1α (IRE1α, encoded by ERN1), (2) PKR-like ER kinase (PERK, encoded by EIF2AK3), and (3) activating transcription factor 6α (ATF6α, encoded by ATF6) (58) (Figure 2). All of these three sensors are activated upon the dissociation of the binding immunoglobin protein (BiP, encode by HSPA5) (59) or by the direct binding of unfolded proteins (60, 61) under ER stress.
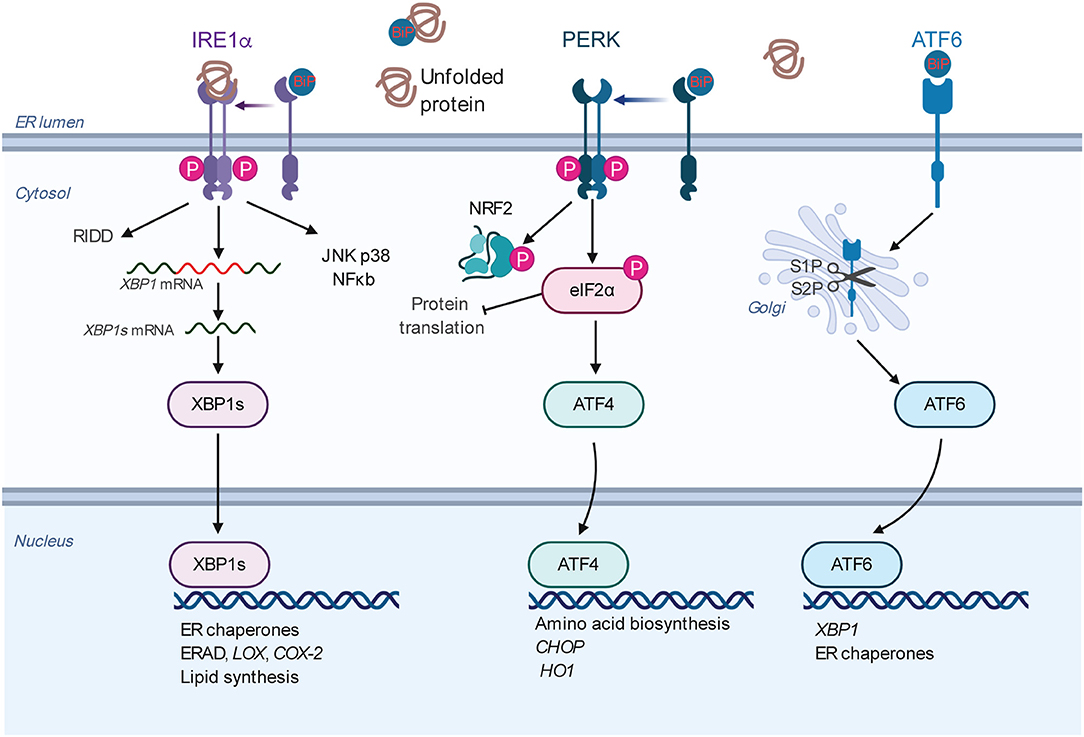
Figure 2. Overview of the mammalian UPR. The three ER resident sensors (IRE1α, PERK, and ATF6) transduce information about the protein folding status of the ER to the cytosol and nucleus to restore the protein folding capacity. Under normal conditions, the sensors are inactivated by binding to the chaperone BiP. Under ER stress conditions, the sensors are activated by BiP dissociation and/or direct misfolded protein binding. Each pathway uses a different mechanism for signal transduction upon activation. IRE1α dimerizes, autophosphorylates, and triggers its RNase activity. This leads to the splicing of the XBP1 mRNA to produce an active transcription factor, spliced XBP1 (XBP1s). XBP1s induces the transcription of the genes encoding protein chaperones, ERAD, and phospholipid synthesis. The RNase activity of IRE1α also degrades certain mRNAs through RIDD. Activated IRE1α can activate the JNK, p38, ERK, and NF-kB pathways, thus playing an XBP1-independent role to modulate diverse cellular responses. Upon activation, PERK phosphorylates eIF2α, leading to global translational attenuation while selectively mediating translation of ATF4. In turn, ATF4 induces the expression of genes involved in amino acid metabolism, proapoptotic factor DDIT3/CHOP, and antioxidant responses (HO1). PERK also phosphorylates and stabilizes NRF2, a transcription factor involved in redox metabolism. ATF6 is transported to the Golgi apparatus under ER stress, where it is processed by S1P and S2P, releasing its cytosolic domain fragment as a transcription factor. ATF6 activates genes encoding protein chaperones, ERAD components, and XBP1. Abbreviations: ATF, activating transcription factor; BiP, binding immunoglobulin protein; DDIT3, DNA damage inducible transcript 3; eIF2α, eukaryotic translation initiation factor 2 subunit 1; ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; HO1, heme oxygenase 1; IRE1α, inositol-requiring enzyme 1α; JNK, c-Jun N-terminal kinase; NF-kB, nuclear factor kappa light-chain enhancer of activated B cells; NRF2, NF-E2-related factor 2; PERK, PKR-like ER kinase; RIDD, regulated IRE1α dependent decay of mRNA; S1P and S2P, site 1 and site 2 proteases; UPR, unfolded protein response.
IRE1α is the most evolutionarily conserved branch of the UPR (62, 63). It is a bifunctional transmembrane kinase/endoribonuclease that dimerizes, and autophosphorylates upon luminal activation and then specifically cleaves 26 nucleotides from cytoplasmic X-box binding protein 1 (XBP1) mRNA (Hac1 in yeast) (54, 55, 64–66). This is the first step of a cytoplasmic splicing event that creates an active form of the transcription factor XBP1s which, among its various functions activates multiple ER quality control genes to enhance the protein folding capacity of the ER to reduce the misfolded proteins there. Meanwhile, activated IRE1α also degrades certain ER-localized cytoplasmic mRNAs in a process known as regulated IRE1-dependent decay (RIDD) to reduce the number of proteins entering the ER (67, 68). By interacting with different adaptor and modulator proteins, IER1α can also activate the JNK, ERK, p38, and NF-kB pathways (69–71).
PERK is a serine/threonine kinase, and its best characterized substrate is eIF2α (72). PERK-dependent phosphorylation of eIF2α reduces the protein load into the ER by inhibiting the 5′ cap-dependent translation, while selectively increasing the translation of ATF4. ATF4 subsequently activates multiple genes involved in the regulation of autophagy, amino acid metabolism, and antioxidant responses (73–75).
Under ER stress, ATF6 is translocated to the Golgi apparatus, where it is cleaved by site 1 protease (S1P) and S2P, releasing the cytoplasmic transcription factor fragment (76). ATF6 activates genes that are involved in protein folding in the ER. Collectively, the consequences of UPR activation—pro-survival or pro-apoptotic—depend on the duration and intensity of the stress stimuli (14, 15, 20, 51, 53, 56).
The UPR in Premetastatic Niche Formation
Survival and outgrowth of tumor cells in distant organs depend on their interaction with the microenvironment of the distal site (5–7). Several fundamental discoveries have revealed that cancer cells can remotely reprogram the microenvironments in distant organs to facilitate the later colonization, survival, and growth in a process termed prometastatic niche (PMN) formation (45, 46, 77). In the context of bone metastases, lysyl oxidase (LOX) is secreted by estrogen receptor–negative (ER−) breast tumors and mediates PMN formation in the bone (45). Hypoxia signature is correlated with increased risk of bone metastases, particularly in ER− breast tumors. Cox et al. found LOX is highly expressed in bone-tropic MDA-MB-231 subline 1833-BoT cells and is associated with bone tropism in ER− breast tumors. LOX secreted by the hypoxic primary tumor leads to the formation of premetastatic osteolytic lesions and promotes bone metastatic burden in a 4T1-BALB/c mouse model. The expression of LOX is induced by hypoxia inducible factor (HIF) under hypoxic conditions (45). Meanwhile, the UPR is known to be induced by hypoxia (15). Further study demonstrated that XBP1s interacts with HIF1α and is required for the upregulation of HIF1α-mediated hypoxia response pathway genes in TNBC tumors (18). This study implies that XBP1s may directly regulate the expression of LOX under hypoxic conditions. Indeed, XBP1 activates LOX expression in lung adenocarcinoma cells to promote the cell growth (78). Thus, the increased secretion of LOX in hypoxic tumors may be due to the activation of the UPR. It is compelling to note that blocking the IRE1a-XBP1 pathway may simultaneously inhibit the growth of both the primary TNBC tumors and bone metastases. Additional work will be required to test the universality of UPR involvement in the PMN formation during bone metastasis.
Role of the UPR on the Survival of Circulating Tumor Cells
In the vasculature, circulating tumor cells (CTCs) encounter various stresses, including the loss of extracellular matrix (ECM) detachment, oxidative stress, innate immune response, and physical shear force (3, 79, 80). Normally, cells undergo apoptosis when they lose contact with their ECM or neighboring cells. This specific type of apoptosis, termed anoikis, prevents adherent-independent cell growth, attachment to an inappropriate matrix, and thus colonization of distant organs (79, 80). Multiple studies suggest that the PERK-eIF2α branch of the UPR inhibits anoikis and is required for tumors to invade and metastasize (81–83). The PERK-eIF2α pathway was shown to activate in suspension-cultured MCF10A cells and sustains MCF10A cell survival (81). Cells that undergo epithelial-to-mesenchymal transition (EMT) are highly secretory, and the PERK axis of the UPR was found to be selectively activated (82). In addition, the inhibition of PERK compromised the ability of EMT cells to form tumorspheres and migrate in transwell assays (82). Human melanoma cells experience higher levels of oxidative stress in the circulation and distant tissues than in primary tumors (84). To manage such oxidative stress, metastasizing melanomas undergo reversible metabolic adaptations, including the synthesis of antioxidants, to survive and eventually metastasize to distant sites. A previous study (83) showed that the PERK downstream transcription factor ATF4 and NRF2, which is stabilized by PERK (85), activate the expression of major antioxidant enzyme heme oxygenase 1 (HO-1), and therefore protect the detached cells from oxidative stress.
CTCs are also vulnerable to immune attacks by innate immune cells, notably NK cells (86, 87). In contrast to NK cells, neutrophils seem to protect CTCs and favor the metastatic spreading (4, 88). The functions of the IRE1α-XBP1 branch during the CTC stage are complicated (89–92). On the one hand, XBP1s promotes NK cell proliferation and positively regulates cytolytic activity of NK cells (89, 91, 92). On the other hand, XBP1 stimulates the expression of lectin-type oxidized LDL receptor 1 (LOX-1) in human neutrophils and transforms them into immunosuppressive cells, possibly promoting CTC survival (90). Overall, the PERK pathway could promote CTC survival by inhibiting anoikis and oxidative stress. Further in vivo studies are necessary to evaluate the overall effect of the IRE1α-XBP1 branch on the survival of CTCs in vivo.
Role of the UPR on Colonization and Dormancy
CTCs surviving in the circulation arrive at the bone marrow vasculatures and extravasate into bone marrow parenchyma. It is still unclear whether this process is completed by passive entry due to the discontinuous endothelium of bone marrow sinusoids or if any other pathway actively involved (6, 7, 93). Compared with other organs, the bone is unique for its mineral content, enriched vasculatures, low oxygen level, high local Ca2+ concentration, and acidosis (94). As a result, the newly arrived tumor cells are challenged in many aspects (5, 6). Meanwhile, DTCs in bone remain dormant state in a variable period, which is critical for their survival, adaptation, escaping systemic treatments, and final outgrowth (6, 9, 94).
The hostile microenvironment (e.g., hypoxia) in the bone may disrupt ER protein folding; therefore, UPR pathways are expected to be upregulated in these DTCs. Indeed, UPR target genes are upregulated in dormant cancer cells from patients and mouse models (95–99). In the bone marrow of breast cancer patients, both GRP78/BiP and GRP94 are selectively highly expressed by bone marrow (BM) DTCs (98). Interestingly, UPR target genes are also overexpressed in cells derived from bone marrow DTCs compared with those from primary tumors (98, 100). These studies suggest that UPR upregulation is a stable trait for BM DTCs.
This trait may arise from the selection of pre-existing UPR positive subpopulation by the hostile microenvironment (and treatment, see discussion below) from the heterogenous cancer cell population, adaptation of the survived cancer cells to the microenvironment, or both. Nevertheless, these UPR genes are thought to confer a survival advantage to DTCs within the bone microenvironment because cell lines derived from BM DTCs are more resistant to glucose and oxygen deprivation in vitro. Studies in the head and neck cancer cell line HEp3 indicated that p38 plays a critical role in the induction and maintenance of tumor dormancy (95–97, 101). Interestingly, p38 activates all three branches of UPR in the dormant HEp3 cells, which contributes to the survival of cancer cells under glucose deprivation or chemotherapeutic treatments. Meanwhile, the PERK pathway inhibits the translation of cyclin D1/D3 and CDK4 in these cells, thereby arresting the cells in the G0-G1 phase. These studies support a causal role for the UPR in the establishment of dormancy (95–97). The upregulation of UPR genes are also found in dormant pancreatic ductal adenocarcinoma DTCs from lives of patient samples and mouse model (99). Collectively, these data indicate that UPR activation may be a common strategy utilized by cancer cells to enter dormancy and promote their survival. Further studies would be worthwhile to follow up on these impressive results and answer the following questions: (1) what triggers and/or maintains UPR signaling in dormant cancer cells in which overall protein synthesis is attenuated (101); (2) can UPR activation contribute to the dormant state of bone marrow DTCs in vivo, and if so, how; (3) what determines the pro-survival or pro-apoptotic effects of UPR activation in these cells; and (4) can the inhibition of the UPR promote DTCs death or sensitize them to therapies targeting proliferating cells.
Role of the UPR on the Reactivation and Outgrowth of DTCs
Our current knowledge about the reactivation process of dormant DTCs, particularly in bone, is limited (4–6, 102). The autonomous traits of tumor cells alone cannot explain the asynchronized relapse of metastases after a long latency. Alternatively, local stimulation of the microenvironments may awaken dormant tumor cells. In bone, osteoclasts are key players in the microenvironmental support of osteolytic breast cancer cell growth and bone destruction. The upregulation of vascular cell adhesion molecule 1 (VCAM-1) in tumor cells promotes the transition from indolent micrometastasis to overt metastasis in breast cancer (103). DTCs with high VCAM-1 recruit integrin α4β+ osteoclast progenitors and induce local osteoclast activity. Therapeutically targeting the VCAM-1–integrin a4 interaction effectively inhibits the progression of bone metastasis and preserves bone structure in mouse models (103). Osteoblasts are another cell type found in the remodeling bone environment. It has been suggested that cancer cells interact with osteogenic cells through E-cadherin/N-cadherin and gap junctions and such interaction promotes early-stage bone colonization and outgrowth (104, 105). In multiple myeloma, XBP1 is required for the expression of VCAM-1, IL6, and RANKL and promotes osteolytic outgrowth (106). Given that XBP1 is one of the top transcription factors enriched in bone metastases compared with primary tumors and metastases in other organs (105), additional research is necessary to determine whether XBP1 regulates VCAM-1 in bone metastasis and promotes outgrowth.
Similarly to metastases in other organs, the immune system is absolutely critical in regulating the outgrowth of bone metastasis (94). In clinical practice, donor-derived cancer develops on rare occasions in immune-suppressed recipients who have received organs from cancer survivors (disease free for more than 10 years) or donors without diagnosable cancer at the time of transplantation (107–109). These observations suggest that the competent immune system may hold disseminated tumor cells in an asymptomatic state. Indeed, a higher ratio of CD56+ CD8+ T cells and memory CD4+ T cells were found in DTC-present bone marrow samples than in DTC-free samples in breast cancer patients (110). In a mouse model of spontaneous bone metastasis, the restoration of interferon regulatory factor 7 (Irf7) suppresses bone metastases through interferon signaling, whereas the deficiency of T and NK cell responses accelerates breast cancer bone metastases (111). A possible interpretation of these results is that cancer cell proliferation is balanced by immune-mediated cancer cell death (4, 112). The bone marrow is occupied by diverse immune cells including neutrophils (113–115). With age, hematopoietic stem cells gradually lose their self-renewal and regeneration capacity and are biased to differentiate into myeloid lineage including monocytes (giving rise to macrophages and dendritic cells), granulocytes (giving rise to basophils, neutrophils, and eosinophils), and megakaryocytes (116–118). This leads to an aged-related decline of the immune response (referred to as immunosenescence) and chronic, sterile, low-grade inflammation (named “inflamm-aging”) in older adults (119, 120). Inflammation is linked to the relapse of breast cancer (121). Sustained experimental inflammation and the accompanying formation of neutrophil extracellular traps in the lungs was found to convert dormant breast cancer cells to aggressive lung metastases in mice. Mechanistically, the neutrophil extracellular traps associated protease neutrophil elastase and matrix metalloproteinase 9 sequentially cleaves the ECM component laminin, leading to laminin remodeling. The remodeled laminin activates α3β1-FAK signaling in dormant cancer cells to induce their reactivation (122). As discussed above, XBP1 promotes neutrophils into immunosuppressive cells. In addition, the inhibition of the IRE1α RNase activity downregulates the expression and secretion of CXCL1 in multiple breast cancer cell lines (23), indicating the possibility that IRE1α promotes neutrophil recruitment by activating CXCL1. Furthermore, the IRE1a-XBP1 pathway is required for neutrophil extracellular trap formation during infection (123) Overall, the IRE1a-XBP1 pathway is known to promote neutrophil recruitment and function. Nevertheless, additional investigation is necessary to test whether neutrophils contribute to DTC reactivation in bone and whether/how the IRE1α-XBP1 pathway is involved in this process.
Dendritic cells (DCs) are responsible for the presentation of tumor antigens to T cells and initiation of the antitumor response (124). Activated T cells, especially cytotoxic CD8+ T cells and CD4+ T helper 1 cells, attack and destroy the target tumor cells (125). However, these processes are often inhibited by tumor cells via multiple strategies including at least by silencing the antigen presentation (hiding major histocompatibility complex I (MHCI) or making dendritic cells (DCs) dysfunctional), T-cell dysfunction, and the establishment of an immunosuppressive tumor microenvironment by myeloid-derived suppressor cells (MDSCs) (124–126). In contrast to the essential role of the IRE1α-XBP1 pathway in the physiology of antigen presentation cells under homeostatic conditions (127, 128), a study by the Laurie Glimcher's laboratory uncovered XBP1s as a critical driver of tumor-associated dendritic cell (tDC) dysfunction in the ovarian cancer microenvironment (129). IRE1α activation of XBP1s, stimulated by lipid peroxidation byproducts in tDCs, leads to abnormal lipid accumulation and subsequent inhibition of the antigen-presenting capacity of tDCs. Accordingly, DC-specific XBP1 inhibition restores their immunostimulatory capacity and extends survival in tumor-bearing mice. In addition, targeting the IRE1α-XBP1 pathway benefits T-cell function directly in the ovarian cancer microenvironment by increasing mitochondrial respiration activity (130) and attenuating cholesterol-induced CD8+ T-cell exhaustion (131). The PERK downstream target Chop (encode by Ddit3) is highly expressed in tumor-associated MDSCs, and the depletion of Chop compromises the function of MDSCs and delays tumor growth (132). Therefore, inhibition of the IRE1α and PERK pathways could boost the immune response in multiple tumors.
Taken together, these recent findings suggest that ER stress is induced in tumor cells and infiltrated immune cells in the tumor microenvironment. Thus, it would be interesting to test whether the abovementioned functions of the UPR are specific to the tumor microenvironment studied or can be generalized to other cancer types and different metastatic organs including bone.
Therapeutic Resistance and Metastatic-Related Morbidity
Metastatic cancer often represents a terminal illness and is the main cause of cancer death (133). Current treatments for metastatic lesions are essentially similar to those for the corresponding primary tumors, including chemotherapy, targeted therapy, hormone therapy, radiation therapy, and immunotherapy (4, 134). Unfortunately, therapeutic resistance often occurs (4) due to many mechanisms, including tumor dormancy, stem-like properties, EMT, and immune suppression as discussed above. In a mouse model of spontaneous lung metastases from mammary tumors, IRE1α expression was induced upon cyclophosphamide-mediated chemotherapy (135). This result is further supported by a study that reported that IRE1α RNase activity is induced upon paclitaxel treatment in TNBC cells (23). Importantly, the inhibition of IRE1α RNase activity increases paclitaxel-mediated tumor suppression and delays tumor relapse posttherapy (23). This is consistent with our recent finding that the inhibition of IRE1α RNase activity substantially enhances the efficacy of docetaxel-based chemotherapy in treating MYC-overexpressing primary tumors and lung metastases (26). In summary, hijacking and upregulation of the IRE1α-XBP1 pathway is one strategy tumor cells use to develop chemoresistance, yet further investigation is required on the detailed mechanisms about how this pathway is activated and how it leads to resistance.
Bone pain is one of the most frequent symptoms of bone metastases, impairing both life quality and expectancy (136). One of the extensively studied molecules that leads to bone pain is cyclooxygenase-2 (COX-2), which is the key enzyme in prostaglandin biosynthesis (136, 137). Prostaglandins bind to prostanoid receptors on sensory terminals, resulting in bone pain (136). Inhibition of COX-2 attenuates bone pain, tumor growth, and bone destruction in a mouse model (138). The two latter phenotypes can be explained by the fact that prostaglandins can also directly promote cancer cell proliferation and induce immunosuppression (137). Recently, the IRE1α-XBP1 pathway was identified as an important regulator in prostaglandin biosynthesis and pain management (139). In myeloid cells (including macrophages and monocytes), XBP1 directly activates the expression of COX-2 and mPGES-1. Genetically or pharmacologically inhibition of the IRE1α-XBP1 pathway diminished pain-related behaviors in mouse models. Given the established functions of COX2/PGES-1 in pain and immunosuppression, this finding not only revealed a new therapeutic approach for attenuating pain behavior but also indicated an alternative explanation how the IRE1α-XBP1 arm promotes immunosuppression.
Conclusions and Future Directions
In the past decade, great strides have been made in bone metastasis research to enhance our understanding of this disease in both patients and experimental models. However, some key questions still remain unanswered. What triggers DTC dormancy and reawakening? How do DTCs evade immune cell surveillance? And ultimately, can we cure bone metastases? To address these questions, both conceptual and technological advances must be made. Improved models need to be developed that faithfully mimic the natural history of bone metastases in patients. The advancement in single-cell RNA sequencing has broadened our knowledge about the heterogeneity of cancer and bone marrow niche cells (114, 115, 140). This technique alone or together with the metastatic-niche labeling strategy (141) will shed new light on the biology of bone metastases and may identify new therapeutic targets.
The activation of the UPR has been demonstrated to endow cancer cells with tumorigenic, metastatic, and drug-resistant capacities and provide tumors with an immunosuppressive microenvironment. Given the convincing underlying mechanisms discovered and the exciting therapeutic results so far, it would be very promising to translate our current knowledge on the functions of the UPR in primary tumors to the study of bone metastases (Figure 3). Further studies are required to characterize the functions of the UPR in different steps of bone metastasis and in different cancer models. What are the driver events that induce/inhibit the UPR during bone metastasis? How does the UPR interplay with other signaling during this process? Can these stress responses in cancer cells be transmitted to niche cells to promote bone metastasis (142, 143)? Importantly, due to the immunosuppressive function of the UPR and the availability of multitarget drugs, it is conceivable to combine these inhibitors to various forms of cancer immunotherapy strategies to control bone metastases.
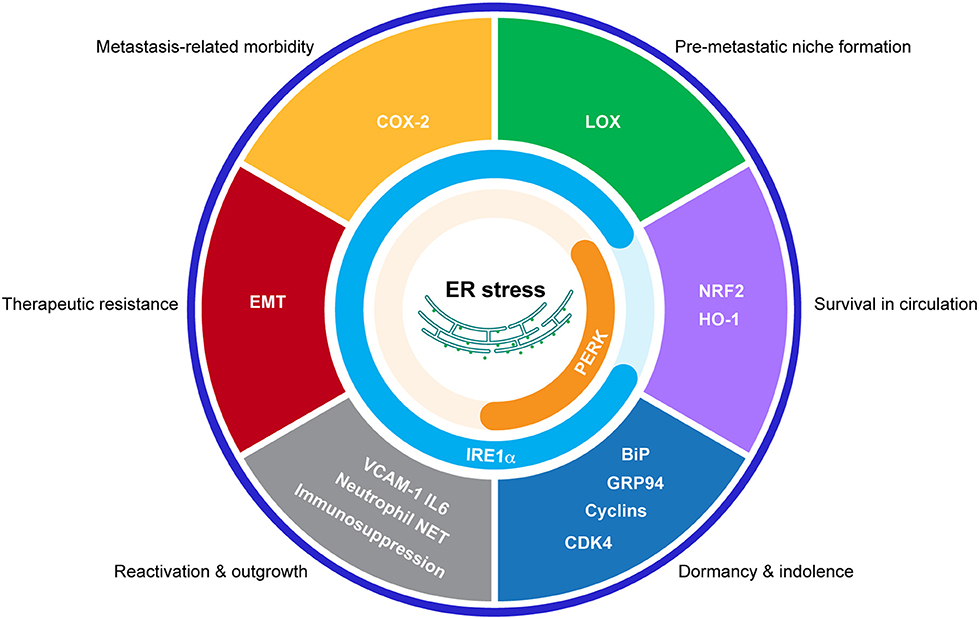
Figure 3. Potential connections between the UPR and bone metastasis. Schematic representation of the proposed effects of the UPR in bone metastasis by regulating indicated processes.
Author Contributions
LX and XC: wrote the manuscript. WZ and XZ: contributed to the bone metastasis part. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (R37CA228304 and R01HL146642 to XC; R01CA183898 to XZ), DOD/CDMRP (W81XWH1910524 to XC; W81XWH-16-1-0073 to XZ), American Cancer Society (RSG-18-181-01-TBE to XC), Cancer Prevention and Research Institute of Texas (RR150009 CPRIT scholar in Cancer Research award to XC), and Susan G. Komen for the Cure (CCR16380871 to XC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone metastases: an overview. Oncol Rev. (2017) 11:321. doi: 10.4081/oncol.2017.321
2. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
3. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. (2016) 529:298–306. doi: 10.1038/nature17038
4. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
5. Croucher PI, Mcdonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer. (2016) 16:373–86. doi: 10.1038/nrc.2016.44
6. Esposito M, Guise T, Kang Y. The biology of bone metastasis. Cold Spring Harb Perspect Med. (2018) 8:a031252. doi: 10.1101/cshperspect.a031252
7. Zhang W, Bado I, Wang H, Lo HC, Zhang XH. Bone metastasis: find your niche and fit in. Trends Cancer. (2019) 5:95–110. doi: 10.1016/j.trecan.2018.12.004
8. Esposito M, Kang Y. Targeting tumor-stromal interactions in bone metastasis. Pharmacol Ther. (2014) 141:222–33. doi: 10.1016/j.pharmthera.2013.10.006
9. Goddard ET, Bozic I, Riddell SR, Ghajar CM. Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol. (2018) 20:1240–9. doi: 10.1038/s41556-018-0214-0
10. D'oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: pathogenesis and therapeutic options: up-date on bone metastasis management. J Bone Oncol. (2019) 15:004–4. doi: 10.1016/j.jbo.2018.10.004
11. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
12. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. (2014) 14:581–97. doi: 10.1038/nrc3800
13. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. (2017) 168:692–706. doi: 10.1016/j.cell.2016.12.004
14. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
15. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. (2004) 4:966–77. doi: 10.1038/nrc1505
16. Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee A-H, Yoshida H, et al. XBP1 Is Essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. (2004) 64:5943–7. doi: 10.1158/0008-5472.CAN-04-1606
17. Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress–mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. (2012) 122:4621–34. doi: 10.1172/JCI62973
18. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. (2014) 508:103–7. doi: 10.1038/nature13119
19. Tang C-HA, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, et al. Inhibition of ER stress–associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. (2014) 124:2585–98. doi: 10.1172/JCI73448
20. Urra H, Dufey E, Avril T, Chevet E, Hetz C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer. (2016) 2:252–62. doi: 10.1016/j.trecan.2016.03.007
21. Ojha R, Amaravadi RK. Targeting the unfolded protein response in cancer. Pharmacol Res. (2017) 120:258–66. doi: 10.1016/j.phrs.2017.04.003
22. Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer. (2017) 3:643–58. doi: 10.1016/j.trecan.2017.07.002
23. Logue SE, Mcgrath EP, Cleary P, Greene S, Mnich K, Almanza A, et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat Commun. (2018) 9:3267. doi: 10.1038/s41467-018-05763-8
24. Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci Transl Med. (2018) 10:eaar2036. doi: 10.1126/scitranslmed.aar2036
25. Xie H, Tang C-HA, Song JH, Mancuso A, Del Valle JR, Cao J, et al. IRE1α RNase–dependent lipid homeostasis promotes survival in Myc-transformed cancers. J Clin Invest. (2018) 128:1300–16. doi: 10.1172/JCI95864
26. Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J Clin Invest. (2018) 128:1283–99. doi: 10.1172/JCI95873
27. Song M, Cubillos-Ruiz JR. Endoplasmic reticulum stress responses in intratumoral immune cells: implications for cancer immunotherapy. Trends Immunol. (2019) 40:128–41. doi: 10.1016/j.it.2018.12.001
28. Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol. (2019) 21:889–99. doi: 10.1038/s41556-019-0347-9
29. Arvelo F, Sojo F, Cotte C. Cancer and the metastatic substrate. Ecancermedicalscience. (2016) 10:701. doi: 10.3332/ecancer.2016.701
30. Svensson E, Christiansen CF, Ulrichsen SP, Rørth MR, Sørensen HT. Survival after bone metastasis by primary cancer type: a Danish population-based cohort study. BMJ Open. (2017) 7:e016022. doi: 10.1136/bmjopen-2017-016022
31. Weidle UH, Birzele F, Kollmorgen G, Rüger R. Molecular mechanisms of bone metastasis. Cancer Genomics Proteomics. (2016) 13:1–12. doi: 10.21873/cgp.20004
32. Melchior SW, Corey E, Ellis WJ, Ross AA, Layton TJ, Oswin MM, et al. Early tumor cell dissemination in patients with clinically localized carcinoma of the prostate. Clin Cancer Res. (1997) 3:249–56.
33. Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. (2008) 13:58–68. doi: 10.1016/j.ccr.2007.12.003
34. Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res. (2009) 15:677–83. doi: 10.1158/1078-0432.CCR-08-1754
35. Sanger N, Effenberger KE, Riethdorf S, Van Haasteren V, Gauwerky J, Wiegratz I, et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int J Cancer. (2011) 129:2522–6. doi: 10.1002/ijc.25895
36. Gruber IV, Hartkopf AD, Hahn M, Taran FA, Staebler A, Wallwiener D, et al. Relationship between hematogenous tumor cell dissemination and cellular immunity in DCIS patients. Anticancer Res. (2016) 36:2345–51.
37. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature. (2016) 540:588–92. doi: 10.1038/nature20609
38. Hosseini H, Obradovic MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. (2016) 540:552–8. doi: 10.1038/nature20785
39. Weckermann D, Müller P, Wawroschek F, Harzmann R, Riethmüller G, Schlimok G. Disseminated cytokeratin positive tumor cells in the bone marrow of patients with prostate cancer: detection and prognostic value. J Urol. (2001) 166:699–704. doi: 10.1016/S0022-5347(05)66046-6
40. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Eng J Med. (2005) 353:793–802. doi: 10.1056/NEJMoa050434
41. Janni W, Vogl FD, Wiedswang G, Synnestvedt M, Fehm T, Jückstock J, et al. Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse—a european pooled analysis. Clin Cancer Res. (2011) 17:2967–76. doi: 10.1158/1078-0432.CCR-10-2515
42. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. (2015) 520:353–7. doi: 10.1038/nature14347
43. Hoadley KA, Siegel MB, Kanchi KL, Miller CA, Ding L, Zhao W, et al. Tumor evolution in two patients with basal-like breast cancer: a retrospective genomics study of multiple metastases. PLoS Med. (2016) 13:e1002174. doi: 10.1371/journal.pmed.1002174
44. Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. (2013) 154:1060–73. doi: 10.1016/j.cell.2013.07.036
45. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. (2015) 522:106–10. doi: 10.1038/nature14492
46. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
47. Celià-Terrassa T, Kang Y. Metastatic niche functions and therapeutic opportunities. Nat Cell Biol. (2018) 20:868–77. doi: 10.1038/s41556-018-0145-9
48. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. (2007) 18:716–31. doi: 10.1016/j.semcdb.2007.09.003
49. Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. (2013) 5:a013201. doi: 10.1101/cshperspect.a013201
50. Jan CH, Williams CC, Weissman JS. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science. (2014) 346:1257521. doi: 10.1126/science.1257521
51. Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. (2015) 33:107–38. doi: 10.1146/annurev-immunol-032414-112116
52. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. (2016) 73:79–94. doi: 10.1007/s00018-015-2052-6
53. Braakman I, Bulleid NJ. Protein Folding and Modification in the Mammalian Endoplasmic Reticulum. Annu Rev Biochem. (2011) 80:71–99. doi: 10.1146/annurev-biochem-062209-093836
54. Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. (1996) 87:391–404. doi: 10.1016/S0092-8674(00)81360-4
55. Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T. Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells. (1996) 1:803–17. doi: 10.1046/j.1365-2443.1996.d01-274.x
56. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
57. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic reticulum stress signalling – from basic mechanisms to clinical applications. FEBS J. (2019) 286:241–78. doi: 10.1111/febs.14608
58. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
59. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. (2000) 2:326–32. doi: 10.1038/35014014
60. Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. (2011) 333:1891–4. doi: 10.1126/science.1209126
61. Karagöz GE, Acosta-Alvear D, Nguyen HT, Lee CP, Chu F, Walter P. An unfolded protein-induced conformational switch activates mammalian IRE1. Elife. (2017) 6:e30700. doi: 10.7554/eLife.30700.034
62. Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. (1993) 73:1197–206. doi: 10.1016/0092-8674(93)90648-A
63. Morl K, Ma W, Gething M-J, Sambrook J. A transmembrane protein with a cdc2+CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. (1993) 74:743–56. doi: 10.1016/0092-8674(93)90521-Q
64. Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. (1996) 15:3028–39. doi: 10.1002/j.1460-2075.1996.tb00666.x
65. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. (2001) 107:881–91. doi: 10.1016/S0092-8674(01)00611-0
66. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. (2002) 415:92–6. doi: 10.1038/415092a
67. Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. (2006) 313:104–7. doi: 10.1126/science.1129631
68. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. (2009) 186:323–31. doi: 10.1083/jcb.200903014
69. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. (2000) 287:664–6. doi: 10.1126/science.287.5453.664
70. Nguyên DT, Kebache S, Fazel A, Wong HN, Jenna S, Emadali A, et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol Biol Cell. (2004) 15:4248–60. doi: 10.1091/mbc.e03-11-0851
71. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol Cell Biol. (2006) 26:3071–84. doi: 10.1128/MCB.26.8.3071-3084.2006
72. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. (1999) 397:271–4. doi: 10.1038/16729
73. Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. (2004) 167:27–33. doi: 10.1083/jcb.200408003
74. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. (2004) 101:11269–74. doi: 10.1073/pnas.0400541101
75. Liu Z, Lv Y, Zhao N, Guan G, Wang J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. (2015) 6:e1822. doi: 10.1038/cddis.2015.183
76. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. (1999) 10:3787–99. doi: 10.1091/mbc.10.11.3787
77. Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. (2015) 527:329–35. doi: 10.1038/nature15756
78. Yang Y, Cheng BJ, Jian H, Chen ZW, Zhao Y, Yu YF, et al. XBP1-LOX Axis is critical in ER stress-induced growth of lung adenocarcinoma in 3D culture. Am J Transl Res. (2017) 9:700–7.
79. Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. (2008) 272:177–85. doi: 10.1016/j.canlet.2008.05.029
80. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
81. Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. (2011) 31:3616–29. doi: 10.1128/MCB.05164-11
82. Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JHL, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK–eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. (2014) 4:702–15. doi: 10.1158/2159-8290.CD-13-0945
83. Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest. (2015) 125:2592–608. doi: 10.1172/JCI78031
84. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. (2015) 527:186–91. doi: 10.1038/nature15726
85. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. (2003) 23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003
86. Hanna N, Fidler IJ. Role of natural killer cells in the destruction of circulating tumor Emboli23. J Natl Cancer Inst. (1980) 65:801–9. doi: 10.1093/jnci/65.4.801
87. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
88. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. (2019) 566:553–7. doi: 10.1038/s41586-019-0915-y
89. Mujaj S, Gandhi M, Vari F, Nourse J. Modulation of the unfolded protein response via XBP1 splicing: a novel mechanism that regulates natural killer cell effector function. (172.10). J Immunol. (2012) 188:172.110.
90. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
91. Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol. (2019) 20:865–78. doi: 10.1038/s41590-019-0388-z
92. Wang Y, Zhang Y, Yi P, Dong W, Nalin AP, Zhang J, et al. The IL-15–AKT–XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat Immunol. (2019) 20:10–7. doi: 10.1038/s41590-018-0265-1
93. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. (2015) 526:126–30. doi: 10.1038/nature15250
94. Johnson RW, Suva LJ. Hallmarks of bone metastasis. Calcif Tissue Int. (2018) 102:141–51. doi: 10.1007/s00223-017-0362-4
95. Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase–like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. (2006) 66:1702–11. doi: 10.1158/0008-5472.CAN-05-3092
96. Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. (2008) 68:3260–8. doi: 10.1158/0008-5472.CAN-07-6215
97. Schewe DM, Aguirre-Ghiso JA. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. (2008) 105:10519–24. doi: 10.1073/pnas.0800939105
98. Bartkowiak K, Effenberger KE, Harder S, Andreas A, Buck F, Peter-Katalinic J, et al. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J Proteome Res. (2010) 9:3158–68. doi: 10.1021/pr100039d
99. Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. (2018) 360:eaao4908. doi: 10.1126/science.aao4908
100. Bartkowiak K, Kwiatkowski M, Buck F, Gorges TM, Nilse L, Assmann V, et al. Disseminated tumor cells persist in the bone marrow of breast cancer patients through sustained activation of the unfolded protein response. Cancer Res. (2015) 75:5367–77. doi: 10.1158/0008-5472.CAN-14-3728
101. Ranganathan AC, Adam AP, Zhang L, Aguirre-Ghiso JA. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: an adaptive advantage for metastatic cells? Cancer Biol Ther. (2006) 5:729–35. doi: 10.4161/cbt.5.7.2968
102. Giancotti F. Mechanisms governing metastatic dormancy and reactivation. Cell. (2013) 155:750–64. doi: 10.1016/j.cell.2013.10.029
103. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging a4b1-positive osteoclast progenitors. Cancer Cell. (2011) 20:701–14. doi: 10.1016/j.ccr.2011.11.002
104. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. (2015) 27:193–210. doi: 10.1016/j.ccell.2014.11.017
105. Wang H, Tian L, Liu J, Goldstein A, Bado I, Zhang W, et al. The osteogenic niche is a calcium reservoir of bone micrometastases and confers unexpected therapeutic vulnerability. Cancer Cell. (2018) 34:823–39 e827. doi: 10.1016/j.ccell.2018.10.002
106. Xu G, Liu K, Anderson J, Patrene K, Lentzsch S, Roodman GD, et al. Expression of XBP1s in bone marrow stromal cells is critical for myeloma cell growth and osteoclast formation. Blood. (2012) 119:4205–14. doi: 10.1182/blood-2011-05-353300
107. Buell JF, Beebe TM, Trofe J, Gross TG, Alloway RR, Hanaway MJ, et al. Donor transmitted malignancies. Ann Transplant. (2004) 9:53–6.
108. Strauss DC, Thomas JM. Transmission of donor melanoma by organ transplantation. Lancet Oncol. (2010) 11:790–6. doi: 10.1016/S1470-2045(10)70024-3
109. Xiao D, Craig JC, Chapman JR, Dominguez-Gil B, Tong A, Wong G. Donor cancer transmission in kidney transplantation: a systematic review. Am J Transplant. (2013) 13:2645–52. doi: 10.1111/ajt.12430
110. Feuerer M, Rocha M, Bai L, Umansky V, Solomayer EF, Bastert G, et al. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int J Cancer. (2001) 92:96–105. doi: 10.1002/1097-0215(200102)9999:9999<::AID-IJC1152>3.0.CO;2-Q
111. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. (2012) 18:1224–31. doi: 10.1038/nm.2830
113. Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, et al. Bone marrow and the control of immunity. Cell Mol Immunol. (2012) 9:11–9. doi: 10.1038/cmi.2011.47
114. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. (2019) 177:1915–32 e1916. doi: 10.1016/j.cell.2019.04.040
115. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, et al. The bone marrow microenvironment at single-cell resolution. Nature. (2019) 569:222–8. doi: 10.1038/s41586-019-1104-8
116. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. (2008) 132:631–44. doi: 10.1016/j.cell.2008.01.025
117. Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. (2012) 10:120–36. doi: 10.1016/j.stem.2012.01.006
118. Geiger H, De Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. (2013) 13:376–89. doi: 10.1038/nri3433
119. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
120. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. (2018) 8:1960. doi: 10.3389/fimmu.2017.01960
121. Cole SW. Chronic inflammation and breast cancer recurrence. J Clin Oncol. (2009) 27:3418–9. doi: 10.1200/JCO.2009.21.9782
122. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
123. Abuaita BH, Sule GJ, Schultz TL, Gao F, Knight JS, O'riordan MX. The IRE1α stress signaling axis is a key regulator of neutrophil antimicrobial effector function. bioRxiv. (2019) 743336. doi: 10.1101/743336
124. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
125. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. (2016) 16:599–611. doi: 10.1038/nri.2016.80
126. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
127. Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. (2007) 204:2267–75. doi: 10.1084/jem.20070525
128. Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat Immunol. (2014) 15:248–57. doi: 10.1038/ni.2808
129. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 161:(2015) 1527–38. doi: 10.1016/j.cell.2015.05.025
130. Song M, Sandoval TA, Chae C-S, Chopra S, Tan C, Rutkowski MR, et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. (2018) 562:423–8. doi: 10.1038/s41586-018-0597-x
131. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–156.e145. doi: 10.1016/j.cmet.2019.04.002
132. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor Cells in tumors. Immunity. (2014) 41:389–401. doi: 10.1016/j.immuni.2014.08.015
133. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. (2006) 6:449–58. doi: 10.1038/nrc1886
135. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. (2015) 527:472–76. doi: 10.1038/nature15748
136. Aielli F, Ponzetti M, Rucci N. Bone metastasis pain, from the bench to the bedside. Int J Mol Sci. (2019) 20:280. doi: 10.3390/ijms20020280
137. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. (2010) 10:181–93. doi: 10.1038/nrc2809
138. Sabino MAC, Ghilardi JR, Jongen JLM, Keyser CP, Luger NM, Mach DB, et al. (2002). Simultaneous reduction in cancer pain, bone destruction, and tumor growth by selective inhibition of cyclooxygenase-2. Cancer Res. 62:7343–9.
139. Chopra S, Giovanelli P, Alvarado-Vazquez PA, Alonso S, Song M, Sandoval TA, et al. IRE1α-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science. (2019) 365:eaau6499. doi: 10.1126/science.aau6499
140. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. (2018) 20:1349–60. doi: 10.1038/s41556-018-0236-7
141. Ombrato L, Nolan E, Kurelac I, Mavousian A, Bridgeman VL, Heinze I, et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature. (2019) 572:603–8. doi: 10.1038/s41586-019-1487-6
142. Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. (2011) 108:6561–6. doi: 10.1073/pnas.1008942108
Keywords: bone metastases, seed and soil, metastatic niche, ER stress, unfolded protein response, immunotherapy
Citation: Xu L, Zhang W, Zhang XH-F and Chen X (2020) Endoplasmic Reticulum Stress in Bone Metastases. Front. Oncol. 10:1100. doi: 10.3389/fonc.2020.01100
Received: 10 March 2020; Accepted: 02 June 2020;
Published: 24 July 2020.
Edited by:
Maria Teresa Valenti, University of Verona, ItalyReviewed by:
Safikur Rahman, Babasaheb Bhimrao Ambedkar Bihar University, IndiaHanumantha Rao Madala, Harvard Medical School, United States
Copyright © 2020 Xu, Zhang, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Chen, WGkuQ2hlbkBiY20uZWR1