 Franz Geisslinger
Franz Geisslinger Martin Müller
Martin Müller Angelika M. Vollmar
Angelika M. Vollmar Karin Bartel
Karin Bartel- Pharmaceutical Biology, Department Pharmacy, Ludwig-Maximilians-University of Munich, Munich, Germany
To date, cancer remains a worldwide leading cause of death, with a still rising incidence. This is essentially caused by the fact, that despite the abundance of therapeutic targets and treatment strategies, insufficient response and multidrug resistance frequently occur. Underlying mechanisms are multifaceted and extensively studied. In recent research, it became evident, that the lysosome is of importance in drug resistance phenotypes. While it has long been considered just as cellular waste bag, it is now widely known that lysosomes play an important role in important cellular signaling processes and are in the focus of cancer research. In that regard lysosomes are now considered as so-called “drug safe-houses” in which chemotherapeutics are trapped passively by diffusion or actively by lysosomal P-glycoprotein activity, which prevents them from reaching their intracellular targets. Furthermore, alterations in lysosome to nucleus signaling by the transcription factor EB (TFEB)—mTORC1 axis are implicated in development of chemoresistance. The identification of lysosomes as essential players in drug resistance has introduced novel strategies to overcome chemoresistance and led to innovate therapeutic approaches. This mini review gives an overview of the current state of research on the role of lysosomes in chemoresistance, summarizing underlying mechanisms and treatment strategies and critically discussing open questions and drawbacks.
Introduction
Cancer is still the second leading cause of death worldwide. In total, the annual number of new cancer diagnoses is 18 million with rising incidence and about 25–30% of all deaths worldwide are connected to cancer and cancer-related diseases (1, 2). Cancer cells possess several features distinct from healthy cells, described as the hallmarks of cancer, which enable them to survive and constantly proliferate (3).
Lysosomes are part of the endolysosomal system (ES), which has long simply been considered as the cell's recycling or waste compartment. Interestingly, the ES was discovered to be also important for sustaining several cancer hallmarks, including migration of invasive cancer cells and neoangiogenesis of endothelial cells (4–7). The ES consists of early, late and recycling endosomes as well as lysosomes, which are separated from the cytosol by a lipid bilayer (69). The ES biogenesis is predominantly regulated by TFEB and mTORC1 signaling and frequently altered upon oncogenic transformation. ES organelles are distinguished by their characteristic intraluminal composition, acidic pH and expression of surface proteins (8, 9). In particular, the lysosome contains various hydrolases, such as proteases and lipases, and displays a luminal pH of about 4.5–5. Today it is recognized as important regulator of nutrient homeostasis, apoptosis, autophagy and membrane trafficking, processes cancer cells critically depend on (7, 69). Consequently, targeting lysosomes gained interest in cancer therapy. Current research is focused on several of its membrane proteins, e.g., the V-ATPase and lysosomal cation channels, like TRPMLs and TPCs, for which excellent reviews are available (9–13).
A major drawback in cancer therapy is the phenomenon of multidrug resistance (MDR), in which tumor cells become unresponsive to treatment, despite the availability of a high variety of targets and related treatment strategies (14–16). The underlying mechanisms are very versatile. A prominent pathway, however, is linked to aberrant drug efflux mediated by the drug transporter P-glycoprotein (P-gp), an ATP-dependent efflux pump (17). Despite intensive research, all clinical trials evaluating P-gp inhibitors failed to date (18).
This review is divided into two parts. Firstly, we critically discuss literature connecting lysosomes and chemoresistance and secondly, we provide an overview of lysosome-based treatment strategies to overcome drug resistance.
Lysosomes and Chemoresistance
Notably, the lysosome recently emerged as promising target to overcome chemoresistance, as increasing evidence suggests that it is involved in P-gp trafficking, serves as drug safe house and links lysosomal biogenesis, induced by transcription factor TFEB, to chemoresistance phenotypes (19–21). Therefore, targeting lysosomal function might improve response to chemotherapy as explained in detail below.
Lysosomal Function Is Crucial for P-Glycoprotein Trafficking
P-gp is a membrane-bound protein, usually embedded in the plasma membrane, actively transporting cargo into the extracellular space. Recently, in resistant cancer cells, lysosomal overexpression of P-gp has been reported. Mechanistically, it is hypothesized that P-gp is incorporated into lysosomal membranes during trafficking and recycling events, rather than being redistributed after de-novo synthesis (19, 20, 22).
The ES is substantially involved in protein trafficking, a process that is dependent on proper function of ES membrane-integrated proteins and ion channels (5, 23, 24). Disturbance of ES membrane protein function in cancer cells is a promising treatment strategy. For instance, V-ATPase inhibition induces lysosomal alkalization and subsequently disrupts receptor trafficking (24–26). Additionally, inhibiting TPC2 and TRPML1 ion channels also impairs trafficking and induces apoptosis (5, 27). Extensively studied players of regulation of vesicular trafficking are the RabGTPases. This large family of small GTPases controls membrane identity, vesicle budding, uncoating, motility and fusion during vesicular trafficking (28–32). Rab GTPases show a distinct intracellular distribution pattern of different members to the respective organelle, thereby regulating directed vesicle trafficking. Lysosome associated Rab GTPases are e.g., Rab4, Rab5, and Rab7 (33). Although their impacts on P-gp trafficking are controversial and dependent on the cell type, Rab4 and Rab5 were shown to affect intracellular localization of P-gp (34). Nevertheless, Ferrandiz-Huertas et al. (35) were able to demonstrate that overexpression of Rab4 leads to a decrease in membranous P-gp abundance and subsequently to increased intracellular daunorubicin concentrations. Furthermore, Rab4 levels seem to correlate with resistance status in this study, as resistant cells have decreased Rab4 levels as compared to the parental cell line (35).
Based on these studies, evidence is given that modulating endolysosomal function impairs P-gp membrane trafficking and recycling. Hence, targeting the ES might be a reasonable approach to overcome multidrug resistance (Pathway A in Figure 1). However, most of the currently available studies remain to prove a therapeutic benefit and the effects are strongly cell type dependent.
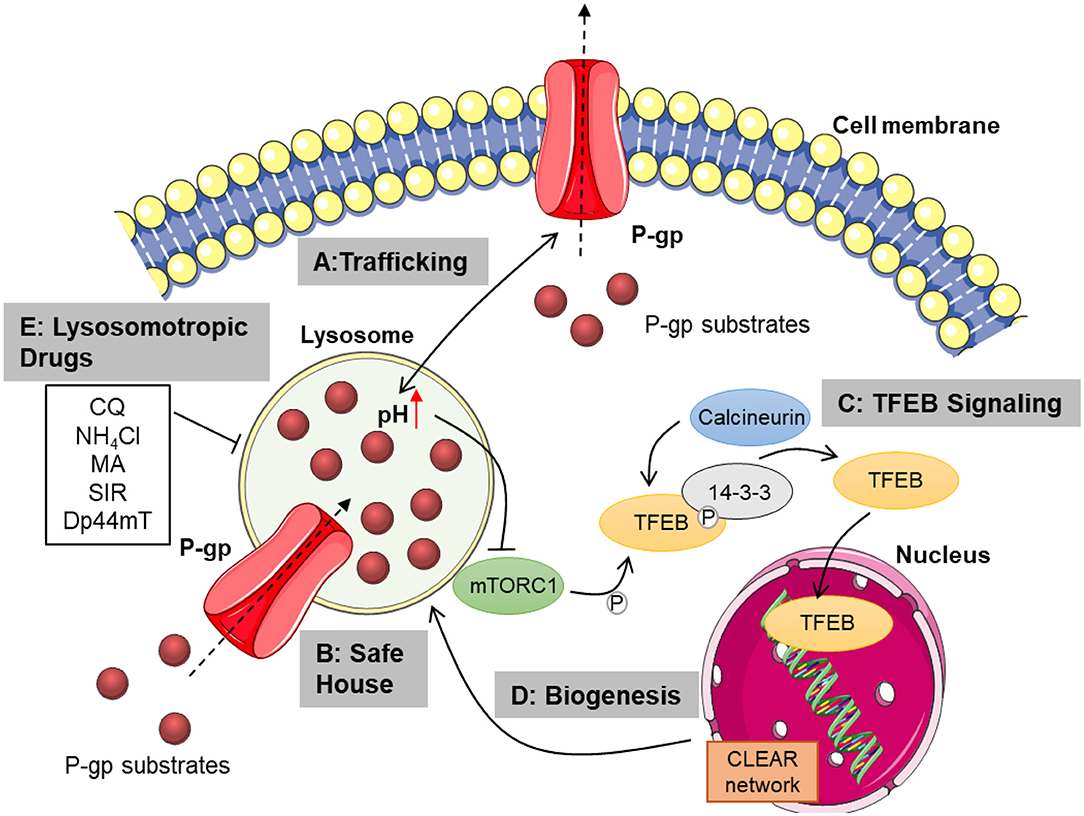
Figure 1. Overview of lysosomal mechanisms contributing to chemoresistance. Lysosomal function is pivotal for proper trafficking of P-glycoprotein to the cell membrane. Membranous P-gp transports cytostatics from the cytosol to the extracellular space (A). Lysosomal P-gp pumps its substrates into the lysosomal lumen, where they are sequestered in dependence of their physicochemical properties. Passive diffusion of hydrophobic weak bases also contributes to lysosomal drug sequestration (B). Subsequent disturbance of lysosomal function leads to TFEB activation mediated by mTORC1 inhibition and calcineurin activation. TFEB is then released from 14-3-3 and translocates to the nucleus, transcribing genes from the CLEAR network (C). This promotes lysosomal biogenesis, increasing lysosomal mass and thus sequestration capacity (D). Inhibiting lysosomal function by treatment with lysosomotropic or lysosome damaging agents as well as elevating lysosomal pH, may overcome chemoresistance mediated by lysosomal drug sequestration (E). CQ, chloroquine; NH4Cl, ammonium chloride; MA, methylamine; SIR, siramesine; Dp44mT, Di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone; P-gp, P-glycoprotein; mTORC1, mammalian target of rapamycin complex 1; TFEB, transcription factor EB; CLEAR, coordinated lysosomal expression and regulation; P, phosphate. This figure was created using images from Servier Medical Art Commons Attribution 3.0 Unported License. (http://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
The Lysosome as Drug Safe House
An important lysosomal mechanism contributing to chemoresistance is the so-called drug safe house effect. The acidity of lysosomes facilitates luminal accumulation of cytostatic weak bases and leads to their protonation, reducing their ability to pass the lysosomal membrane, causing lysosomal drug sequestration (36). Upon lysosomal trapping, cytostatics are prevented from reaching their intracellular targets, which are usually located in the nucleus or the cytosol and therefore fail to exert cytotoxicity (37) (Pathway B in Figure 1). Cytostatics enter the lysosome either by passive diffusion along the pH gradient or may be actively transported across the membrane by inward turned P-gp drug efflux pumps embedded in the lysosomal membrane. Lysosomal sequestration capacity therefore greatly depends on the physicochemical properties of the cytostatic and on the lysosomal features like pH and lysosomal volume (19, 37). Typically, weak bases can enter the lysosome via passive diffusion and are trapped upon protonation, yet also hydrophilicity of the protonated or deprotonated drug is an important factor as it strongly correlates with membrane permeability (19). In that regard, pKa values, which display acidity or basicity of a compound and logP/D values, which represent hydrophilicity of non-ionizable and ionizable molecules, respectively, can be used to estimate lysosomal sequestration of a drug (38, 39). For an active lysosomal trapping, the abundance of lysosomal P-gp activity is a determining factor. As discussed above, P-gp can be trafficked to the lysosomal membrane under stress conditions, which has been reported to tremendously enhance resistance to cytostatic P-gp substrates (20). In turn, stress factors like reactive oxygen species can be induced by cytostatics, further increasing lysosomal P-gp abundance and hence resistance.
However, the interplay between lysosomal properties, active or passive drug accumulation and effective sequestration are complex and cell line dependent. For instance, the weak base doxorubicin undergoes lysosomal trapping in UMUC-3 cells, but not in KB31 cells, most likely as result of different lysosomal properties. Interestingly, in the P-gp overexpressing KB31-subline KBV1, doxorubicin is effectively sequestered, an effect which is reversed by P-gp inhibition (19). Moreover, lysosomes of resistant cell lines often show a more acidic pH than their parental cell lines, probably further enhancing drug sequestration capability (40). Yet, the non-basic and lipophilic P-gp substrate paclitaxel is neither sequestered in KB31 nor KBV1 cells, owing to its ability to freely permeate the lysosomal membrane (19).
The fate of drugs sequestered in lysosomes remains to be clearly elucidated. It is suggested that they either stay trapped in lysosomes or are eliminated from the cell by drug-induced lysosomal exocytosis, preventing lysosomal damage. Supporting the hypothesis of lysosomal exocytosis, treatment with cytostatics leads to redistribution of basally perinuclear lysosomes to the plasma membrane. Furthermore, lysosomal content is increasingly released into the extracellular compartment, such as cathepsin D and V-ATPase (41, 71). Enhanced membrane fusion and subsequent exocytosis could be mediated by lysosomal calcium release via activation of lysosomal cation channels, such as TRPML1 (42). After lysosomal exocytosis, former trapped cytostatics are again abundant in the extracellular compartment, enabling repeated diffusion into the cell. Therefore, it is doubtful whether this mechanism contributes to chemoresistance, requiring further investigation. Nevertheless, the implication of lysosomes in chemoresistance is not restricted to its role as “drug safe house.” In fact, lysosomal abundance of cytostatics leads to changes in lysosomal properties, including pH and lysosomal volume, further influencing lysosomal signaling.
TFEB Signaling Regulating Drug Resistance
Lysosomal properties are mainly regulated by the transcription factor EB (TFEB), the master regulator of lysosomal biogenesis, which when activated transcribes genes belonging to the CLEAR (coordinated lysosomal expression and regulation) network. TFEB is usually retained inactive in the cytosol, by binding to the regulatory protein 14-3-3, which prevents nuclear translocation. The binding of TFEB to 14-3-3 depends on its phosphorylation status, which is regulated by the kinase mTORC1 and the phosphatase calcineurin. mTORC1 phosphorylates TFEB facilitating binding, while calcineurin dephosphorylates TFEB, leading to dissociation and subsequent nuclear translocation (43, 44).
By accumulating in the lysosomal lumen, cytostatic weak bases act like classic lysosomotropic compounds, raising lysosomal pH and increasing lysosomal volume. Lysosomotropism is the propensity of a typically basic substance to specifically accumulate in lysosomes and consequently alter lysosomal features and induce lysosomal membrane permeabilization (45). In particular, an increase of lysosomal volume after treatment with the cytostatic weak base sunitinib, but not with the non-basic cytostatics 5-fluorouracil and pemetrexed, positively correlates with drug resistance. In detail, treatment with cytostatic weak bases, such as trametinib, vincristine, sunitinib and doxorubicin leads to increased lysosomal volume in several cancer cell lines (39, 46, 71). Furthermore, a different study shows that LAMP1, a lysosomal marker, is upregulated in breast cancer cells upon development of resistance by continuous exposure to doxorubicin (47).
In line with an increase in lysosomal volume, TFEB is activated when exposed to cytostatic weak bases and upregulated upon development of chemoresistance, as shown for doxorubicin or mitoxantrone treatment. Further, mTORC1 activity is inhibited upon treatment with the cytostatic weak bases sunitinib and siramesine, while calcineurin activity is enhanced, leading to TFEB activation (21, 39) (Pathway C in Figure 1). Causal for mTORC1 inhibition might be a change in lysosomal properties, especially pH. If lysosomal pH is increased, for instance by inhibition of the V-ATPase, a proton pump maintaining lysosomal acidity, TFEB is activated and translocated to the nucleus (43). Weak base cytostatics accumulating in lysosomes rise lysosomal pH in a similar manner, as shown for doxorubicin in cardiomyocytes, for example (48). Consequently, TFEB and lysosomes could build a bidirectional axis with lysosomal drug accumulation activating TFEB and TFEB inducing lysosomal biogenesis, which increases lysosomal sequestration capacity and exerts a feedback loop (Pathway D in Figure 1). Additionally, this feedback loop might be enhanced by the influence of lysosomal calcium signaling. TFEB regulation is closely linked to lysosomal calcium signaling through the TRPML1 ion channel. Upon lysosomal stress, TRPML1 channels release calcium into the cytosol, creating a calcium enriched microenvironment in which the calcium-dependent phosphatase calcineurin is activated and dephosphorylates TFEB (44, 49). Increasing evidence suggests that TRPML1 channel function is important for cancer cells as genetic depletion leads to decreased cell survival and it has also been reported that TRPML1 and mTORC1 signaling are essential for aggressive cancer cells (50–52). Thus, TFEB and its regulators mTORC1 and calcineurin are considered as potential target for drug resistant tumors. However, which mechanism of those explained above contributes to the sensitization effect remains unknown and further studies addressing this topic are currently missing.
Furthermore, TFEB acts as transcription factor for several proteins essential for autophagy, a complex process promoting cell survival during stress conditions (53). Autophagy is a physiological “self-eating” recycling process which removes defective proteins or organelles, to maintain cellular homeostasis. In cancer cells, autophagy is a paradox which is on the one hand able to prevent tumor initiation by preventing cell damage, but on the other driving tumor progression by facilitating adaptations to stress conditions like nutrient deprivation and hypoxia (54). There is furthermore growing evidence that autophagy is a driving factor for chemoresistance. Autophagic signaling is also altered upon treatment with cytostatics, namely vincristine and doxorubicin, indicated by enhanced LC3-II and p62 levels, protein markers used for visualizing inhibited autophagy. Subsequent promotion of cell survival can be inhibited by knockdown of Atg5, an important regulator of autophagosome formation (46, 47, 55). Evidence further suggests a protective role for autophagy regulators ATG3, 5, 6, 7, and 12 in resistant cells, however, detailed mechanisms are not yet fully understood and hence require further studies (56–60). Interestingly, besides TFEB-regulated lysosomal alterations upon treatment with cytostatics, TFEB also contributes to cell survival by mechanisms independent of the aforementioned. For instance, TFEB influences DNA repair, leading to inhibition of apoptosis. Therefore, knockdown of TFEB prevents DNA repair and thus sensitizes MDA-MB-231 cells to doxorubicin treatment, promoting cell death induction (70). Brady et al. hypothesize that this TFEB-dependent DNA repair mechanism is mediated by p53-related signaling and is, besides TFEB, also activated by TFE3, a transcription factor closely related to TFEB (61). Therefore, lysosomal proteins not only favor resistance by drug sequestration being the “final destination” for cytostatics, but they also serve as signaling hub, activating cell survival pathways.
Lysosome-Based Treatment Strategies
Contribution of lysosomes to chemoresistance raised interest in lysosome-targeting strategies to sensitize tumor cells to chemotherapy. These strategies mainly focus on lysosomotropic compounds, which act by accumulating in the lysosomal lumen, thereby elevating lysosomal pH because of their weak base characteristics and usually inducing lysosomal membrane permeabilization (45). Lysosomotropic compounds commonly used in in vitro experiments are chloroquine, ammonium chloride, methylamine and siramesine (19, 62). By inhibiting lysosomal drug sequestration as consequence of pH elevation, lysosomotropic adjuvants cause intracellular redistribution of cytostatics from the lysosomal lumen to the cytosol and subsequently to their sites of action (Pathway E in Figure 1). This opens the possibility for combination therapies to overcome resistance. Indeed, combination of chloroquine, ammonium chloride or methylamine strikingly sensitizes resistant KBV1 cells to treatment with vincristine and doxorubicin. Additionally, combining vincristine with siramesine is superior to monotherapy in vitro and in vivo in breast cancer (19, 46).
As these data suggest a synergistic effect of cytostatics and inhibition of lysosomal function, also the V-ATPase might be considered as reasonable target due to its implication in sustaining lysosomal acidity. Evidence suggests that V-ATPase overexpression is associated with chemoresistance and V-ATPase knockdown sensitizes doxorubicin-resistant MCF-7 cells to doxorubicin and vincristine (63). Yet, studies addressing pharmacological V-ATPase inhibition regarding chemoresistance are scarce, requiring further research.
Another interesting approach focuses on the experimental metal chelator Di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT). By accumulating within lysosomes and forming redox-active copper complexes, Dp44mT potently induces lysosomal damage and subsequently lysosomal cell death (64), which is characterized by cathepsin D release from the lysosome into the cytosol and subsequent mitochondrial release of cytochrome c, initiating apoptosis (65). Therefore, Dp44mT toxicity is dependent on its lysosomal trapping. As Dp44mT is a weak base P-gp substrate, it accumulates stronger in P-gp expressing, resistant cells, enabling specific killing of resistant cells, while sensitive cells are less affected (66). Furthermore, Dp44mT could be used in combination with basic cytostatics due to its inhibitory effect on lysosomal drug sequestration. In this context, targeting acid sphingomyelinase, a lipase responsible for lysosomal hydrolysis of sphingomyelin, was proven to be another effective strategy to successfully treat multidrug resistant tumors. Siramesine and numerous approved basic amphiphilic drugs, including tricyclic antidepressants and antihistamines, can disrupt lysosomal membrane integrity by inhibiting acid sphingomyelinase and thereby achieve cancer-specific cytotoxicity (62). Thus, introducing lysosomal membrane permeabilization exemplarily emphasizes the potential of lysosome-targeted approaches to overcome chemoresistance.
Conclusion
Despite high efforts to improve chemotherapy, treatment failure and resistance mechanisms remain a major challenge. Exemplarily, for the efflux pump P-gp, which can limit efficacy of cytostatic drugs, no clinically approved treatment options are available to date. As summarized in this review, the contribution of lysosomes to drug resistance opened a new research field in the context of multidrug resistance to overcome chemoresistance. This implication is not restricted to direct lysosomal mechanisms, but also includes lysosome-associated signaling pathways. Evidence suggests that interfering with lysosomal function might be a promising approach enabling sensitization to chemotherapy, influencing several survival-promoting mechanisms, such as trafficking of efflux transporters, drug sequestration and pathways regulated by TFEB, including autophagy and DNA repair. Additionally, inhibition of lysosomal function could target P-glycoprotein-driven chemoresistance, giving hope for lysosome-targeted adjuvants in the future.
However, also lysosome-based strategies have their drawbacks. Targeting lysosomal function in general is not a cancer-specific approach, suggesting that it could lead to severe side effects, as lysosomal function is pivotal for virtually all types of cells, especially for immune cells (67, 68). Cancer-specific or cancer-enriched targets in this concern are still rare, but there is a high variety of lysosomal proteins, e.g., Rab GTPases, ion channels, lipases or the V-ATPase, which could serve as specific targets to disturb lysosomal function and thus drug resistance phenotypes of cancer cells more selectively. Therefore, further studies addressing the drawbacks and therapeutic potential of lysosome-based combination strategies are needed.
Author Contributions
FG and KB conception. FG wrote the first draft of the manuscript. KB and MM wrote sections of the manuscript. AV and KB critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the German Research Foundation (DFG VO 376/19-1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
3. Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
4. Favia A, Desideri M, Gambara G, D'alessio A, Ruas M, Esposito B, et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc Natl Acad Sci USA. (2014) 111:E4706–15. doi: 10.1073/pnas.1406029111
5. Nguyen ON, Grimm C, Schneider LS, Chao YK, Atzberger C, Bartel K, et al. Two-pore channel function is crucial for the migration of invasive cancer cells. Cancer Res. (2017) 77:1427–38. doi: 10.1158/0008-5472.CAN-16-0852
6. Pafumi I, Festa M, Papacci F, Lagostena L, Giunta C, Gutla V, et al. Naringenin impairs two-pore channel 2 activity and inhibits VEGF-induced angiogenesis. Sci Rep. (2017) 7:5121. doi: 10.1038/s41598-017-04974-1
7. Faris P, Shekha M, Montagna D, Guerra G, Moccia F. Endolysosomal Ca(2+) signalling and cancer hallmarks: two-pore channels on the move, TRPML1 lags behind! Cancers. (2018) 11:27. doi: 10.3390/cancers11010027
8. Schwake M, Schroder B, Saftig P. Lysosomal membrane proteins and their central role in physiology. Traffic. (2013) 14:739–48. doi: 10.1111/tra.12056
9. Grimm C, Bartel K, Vollmar AM, Biel M. Endolysosomal cation channels and cancer-a link with great potential. Pharmaceuticals. (2018) 11:4. doi: 10.3390/ph11010004
10. Kirkegaard T, Jaattela M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta. (2009) 1793:746–54. doi: 10.1016/j.bbamcr.2008.09.008
11. Stransky L, Cotter K, Forgac M. The function of V-ATPases in cancer. Physiol Rev. (2016) 96:1071–91. doi: 10.1152/physrev.00035.2015
12. Fennelly C, Amaravadi RK. Lysosomal biology in cancer. Methods Mol Biol. (2017) 1594:293–308. doi: 10.1007/978-1-4939-6934-0_19
13. Whitton B, Okamoto H, Packham G, Crabb SJ. Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. (2018) 7:3800–11. doi: 10.1002/cam4.1594
14. Chakraborty S, Rahman T. The difficulties in cancer treatment. Ecancermedicalscience. (2012) 6:ed16. doi: 10.3332/ecancer.2012.ed16
15. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. (2016) 38:1551–66. doi: 10.1016/j.clinthera.2016.03.026
16. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. (2019) 575:299–309. doi: 10.1038/s41586-019-1730-1
17. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. (2002) 2:48–58. doi: 10.1038/nrc706
18. Dash RP, Jayachandra Babu R, Srinivas NR. Therapeutic potential and utility of elacridar with respect to P-glycoprotein inhibition: an insight from the published in vitro, preclinical and clinical studies. Eur J Drug Metab Pharmacokinet. (2017) 42:915–33. doi: 10.1007/s13318-017-0411-4
19. Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem. (2013) 288:31761–71. doi: 10.1074/jbc.M113.514091
20. Al-Akra L, Bae DH, Sahni S, Huang MLH, Park KC, Lane DJR, et al. Tumor stressors induce two mechanisms of intracellular P-glycoprotein-mediated resistance that are overcome by lysosomal-targeted thiosemicarbazones. J Biol Chem. (2018) 293:3562–87. doi: 10.1074/jbc.M116.772699
21. Zhitomirsky B, Yunaev A, Kreiserman R, Kaplan A, Stark M, Assaraf YG. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. (2018) 9:1191. doi: 10.1038/s41419-018-1227-0
22. Noack A, Gericke B, Von Kockritz-Blickwede M, Menze A, Noack S, Gerhauser I, et al. Mechanism of drug extrusion by brain endothelial cells via lysosomal drug trapping and disposal by neutrophils. Proc Natl Acad Sci USA. (2018) 115:E9590–9. doi: 10.1073/pnas.1719642115
23. Grimm C, Holdt LM, Chen CC, Hassan S, Muller C, Jors S, et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat Commun. (2014) 5:4699. doi: 10.1038/ncomms5699
24. Schneider LS, Von Schwarzenberg K, Lehr T, Ulrich M, Kubisch-Dohmen R, Liebl J, et al. Vacuolar-ATPase inhibition blocks iron metabolism to mediate therapeutic effects in breast cancer. Cancer Res. (2015) 75:2863–74. doi: 10.1158/0008-5472.CAN-14-2097
25. Von Schwarzenberg K, Lajtos T, Simon L, Muller R, Vereb G, Vollmar AM. V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Mol Oncol. (2014) 8:9–19. doi: 10.1016/j.molonc.2013.08.011
26. Bartel K, Winzi M, Ulrich M, Koeberle A, Menche D, Werz O, et al. V-ATPase inhibition increases cancer cell stiffness and blocks membrane related Ras signaling - a new option for HCC therapy. Oncotarget. (2017) 8:9476–87. doi: 10.18632/oncotarget.14339
27. Colletti GA, Miedel MT, Quinn J, Andharia N, Weisz OA, Kiselyov K. Loss of lysosomal ion channel transient receptor potential channel mucolipin-1. (TRPML1) leads to cathepsin B-dependent apoptosis. J Biol Chem. (2012) 287:8082–91. doi: 10.1074/jbc.M111.285536
28. Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. (1998) 394:494–8. doi: 10.1038/28879
29. Nielsen E, Severin F, Backer JM, Hyman AA, Zerial M. Rab5 regulates motility of early endosomes on microtubules. Nat Cell Biol. (1999) 1:376–82. doi: 10.1038/14075
30. Carroll KS, Hanna J, Simon I, Krise J, Barbero P, Pfeffer SR. Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science. (2001) 292:1373–6. doi: 10.1126/science.1056791
31. Del Conte-Zerial P, Brusch L, Rink JC, Collinet C, Kalaidzidis Y, Zerial M, et al. Membrane identity and GTPase cascades regulated by toggle and cut-out switches. Mol Syst Biol. (2008) 4:206. doi: 10.1038/msb.2008.45
32. Semerdjieva S, Shortt B, Maxwell E, Singh S, Fonarev P, Hansen J, et al. Coordinated regulation of AP2 uncoating from clathrin-coated vesicles by rab5 and hRME-6. J Cell Biol. (2008) 183:499–511. doi: 10.1083/jcb.200806016
33. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. (2009) 10:513–25. doi: 10.1038/nrm2728
34. Fu D, Arias IM. Intracellular trafficking of P-glycoprotein. Int J Biochem Cell Biol. (2012) 44:461–4. doi: 10.1016/j.biocel.2011.12.009
35. Ferrandiz-Huertas C, Fernandez-Carvajal A, Ferrer-Montiel A. Rab4 interacts with the human P-glycoprotein and modulates its surface expression in multidrug resistant K562 cells. Int J Cancer. (2011) 128:192–205. doi: 10.1002/ijc.25310
36. Macintyre AC, Cutler DJ. The potential role of lysosomes in tissue distribution of weak bases. Biopharm Drug Dispos. (1988) 9:513–26. doi: 10.1002/bod.2510090602
37. Seebacher N, Lane DJR, Richardson DR, Jansson PJ. Turning the gun on cancer: utilizing lysosomal P-glycoprotein as a new strategy to overcome multi-drug resistance. Free Radic Biol Med. (2016) 96:432–45. doi: 10.1016/j.freeradbiomed.2016.04.201
38. Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, et al. Lysosomal sequestration. (trapping) of lipophilic amine. (cationic amphiphilic) drugs in immortalized human hepatocytes. (Fa2N-4 cells). Drug Metab Dispos. (2013) 41:897–905. doi: 10.1124/dmd.112.050054
39. Zhitomirsky B, Assaraf YG. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget. (2015) 6:1143–56. doi: 10.18632/oncotarget.2732
40. Schindler M, Grabski S, Hoff E, Simon SM. Defective pH regulation of acidic compartments in human breast cancer cells. (MCF-7) is normalized in adriamycin-resistant cells. (MCF-7adr). Biochemistry. (1996) 35:2811–7. doi: 10.1021/bi952234e
41. Zhitomirsky B, Assaraf YG. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget. (2017) 8:45117–32. doi: 10.18632/oncotarget.15155
42. Di Paola S, Medina DL. TRPML1-/TFEB-dependent regulation of lysosomal exocytosis. Methods Mol Biol. (2019) 1925:143–4. doi: 10.1007/978-1-4939-9018-4_12
43. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. (2012) 31:1095–108. doi: 10.1038/emboj.2012.32
44. Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. (2015) 17:288–99. doi: 10.1038/ncb3114
45. Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. (2013) 126:1905–12. doi: 10.1242/jcs.091181
46. Groth-Pedersen L, Ostenfeld MS, Hoyer-Hansen M, Nylandsted J, Jaattela M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. (2007) 67:2217–25. doi: 10.1158/0008-5472.CAN-06-3520
47. Guo B, Tam A, Santi SA, Parissenti AM. Role of autophagy and lysosomal drug sequestration in acquired resistance to doxorubicin in MCF-7 cells. BMC Cancer. (2016) 16:762. doi: 10.1186/s12885-016-2790-3
48. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. (2016) 133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443
49. Di Paola S, Medina DL. Ca(2+)-Dependent regulation of TFEB and lysosomal function. Methods Mol Biol. (2019) 1925:145–55. doi: 10.1007/978-1-4939-9018-4_13
50. Jung J, Cho KJ, Naji AK, Clemons KN, Wong CO, Villanueva M, et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. (2019) 20:e46685. doi: 10.15252/embr.201846685
51. Jung J, Venkatachalam K. TRPing the homeostatic alarm - Melanoma cells are selectively vulnerable to TRPML1 deletion. Cell Calcium. (2019) 84:102082. doi: 10.1016/j.ceca.2019.102082
52. Xu M, Almasi S, Yang Y, Yan C, Sterea AM, Rizvi Syeda AK, et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium. (2019) 79:80–8. doi: 10.1016/j.ceca.2019.02.010
53. Di Malta C, Cinque L, Settembre C. Transcriptional regulation of autophagy: mechanisms and diseases. Front Cell Dev Biol. (2019) 7:114. doi: 10.3389/fcell.2019.00114
54. Høyer-Hansen M, Jäättelä M. Autophagy: an emerging target for cancer therapy. Autophagy. (2008) 4:574–80. doi: 10.4161/auto.5921
55. Fang L-M, Li B, Guan J-J, Xu H-D, Shen G-H, Gao Q-G, et al. Transcription factor EB is involved in autophagy-mediated chemoresistance to doxorubicin in human cancer cells. Acta Pharmacol Sin. (2017) 38:1305–16. doi: 10.1038/aps.2017.25
56. An Y, Zhang Z, Shang Y, Jiang X, Dong J, Yu P, et al. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. (2015) 6:e1766. doi: 10.1038/cddis.2015.123
57. Xu R, Liu S, Chen H, Lao L. MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol Rep. (2016) 35:1757–63. doi: 10.3892/or.2015.4497
58. Hua L, Zhu G, Wei J. MicroRNA-1 overexpression increases chemosensitivity of non-small cell lung cancer cells by inhibiting autophagy related 3-mediated autophagy. Cell Biol Int. (2018) 42:1240–9. doi: 10.1002/cbin.10995
59. Cai Q, Wang S, Jin L, Weng M, Zhou D, Wang J, et al. Long non-coding RNA GBCDRlnc1 induces chemoresistance of gallbladder cancer cells by activating autophagy. Mol Cancer. (2019) 18:82. doi: 10.1186/s12943-019-1016-0
60. Wu J, Li W, Ning J, Yu W, Rao T, Cheng F. Long noncoding RNA UCA1 targets miR-582-5p and contributes to the progression and drug resistance of bladder cancer cells through ATG7-mediated autophagy inhibition. Onco Targets Ther. (2019) 12:495–508. doi: 10.2147/OTT.S183940
61. Brady OA, Jeong E, Martina JA, Pirooznia M, Tunc I, Puertollano R. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife. (2018) 7:e40856. doi: 10.7554/eLife.40856.048
62. Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. (2013) 24:379–93. doi: 10.1016/j.ccr.2013.08.003
63. You H, Jin J, Shu H, Yu B, De Milito A, Lozupone F, et al. Small interfering RNA targeting the subunit ATP6L of proton pump V-ATPase overcomes chemoresistance of breast cancer cells. Cancer Lett. (2009) 280:110–9. doi: 10.1016/j.canlet.2009.02.023
64. Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P, Richardson DR. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. (2011) 71:5871–80. doi: 10.1158/0008-5472.CAN-11-1218
65. Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. (2004) 104:1450–8. doi: 10.1182/blood-2004-03-0868
66. Seebacher NA, Richardson DR, Jansson PJ. A mechanism for overcoming P-glycoprotein-mediated drug resistance: novel combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. (2016) 7:e2510. doi: 10.1038/cddis.2016.381
67. Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. (2015) 77:57–80. doi: 10.1146/annurev-physiol-021014-071649
68. Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. (2019) 21:133–42. doi: 10.1038/s41556-018-0244-7
69. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. (2020) 21:101–18. doi: 10.1038/s41580-019-0185-4
70. Slade L, Biswas D, Ihionu F, El Hiani Y, Kienesberger PC, Pulinilkunnil T. A lysosome independent role for TFEB in activating DNA repair and inhibiting apoptosis in breast cancer cells. Biochem J. (2020) 477:137–60. doi: 10.1042/BCJ20190596
Keywords: chemoresistance, cytostatics, cancer, lysosome, TFEB
Citation: Geisslinger F, Müller M, Vollmar AM and Bartel K (2020) Targeting Lysosomes in Cancer as Promising Strategy to Overcome Chemoresistance—A Mini Review. Front. Oncol. 10:1156. doi: 10.3389/fonc.2020.01156
Received: 06 April 2020; Accepted: 08 June 2020;
Published: 09 July 2020.
Edited by:
Boris Zhivotovsky, Karolinska Institutet (KI), SwedenReviewed by:
Karin Öllinger, Linköping University, SwedenXianping Dong, Faculty of Medicine, Dalhousie University, Canada
Copyright © 2020 Geisslinger, Müller, Vollmar and Bartel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Bartel, a2FyaW4uYmFydGVsQGN1cC51bmktbXVlbmNoZW4uZGU=