 Can Küçük
Can Küçük Li Wei
Li Wei Hua You
Hua You- 1Affiliated Cancer Hospital & Institute of Guangzhou Medical University, Guangzhou, China
- 2Izmir Biomedicine and Genome Center (IBG), Izmir, Turkey
- 3Department of Medical Biology, Dokuz Eylül University, İzmir, Turkey
- 4Department of Pathology, City of Hope Medical Center, Duarte, CA, United States
Indolent T-cell lymphoproliferative disease of the gastrointestinal tract (indolent GI T-LPD) is a benign neoplasm of CD4+ or CD8+ T cells that form primary tumors in the GI tract. Indolent GI T-LPD has recently been provisionally recognized as a distinct entity by the 2016 revision of the WHO classification of lymphoid neoplasms. Appropriate diagnosis of these cases is challenging as they may be misdiagnosed as T cell lymphoma that has an aggressive clinical course. Consequently, aggressive therapeutic approaches were usually chosen to treat these cases with no obvious benefit for most of the patients and potential side effects. Moreover, inflammatory diseases of the GI tract with similar symptoms may lead to misdiagnosis that leads to delays in administration of proper therapeutics against these cases. Therefore, it is of utmost importance to identify prognostic genetic biomarkers at the time of diagnosis for optimal medical care of these patients. TCR clonality analyses may not be useful for distinguishing these benign neoplasms from aggressive gastrointestinal T cell lymphomas; however, molecular genetic tests may prove useful as recurrent STAT3-JAK2 fusions, which may have diagnostic, prognostic or therapeutic value, have recently been identified. However, there is still lack of comprehensive information on the genetic and epigenetic factors associated with pathogenesis of indolent GI T-LPD. In this mini-review, we focus on the so far reported literature on indolent GI T-LPD cases, and discuss future directions for better differential diagnosis, risk stratification, and therapeutic target discovery with a special focus on the genetic and epigenetic alterations.
Background
Indolent T cell lymphoproliferative disease of the gastrointestinal tract (indolent GI T-LPD) is a benign lymphoid neoplasm provisionally recognized by the World Health Organization (WHO) classification of lymphoid neoplasms in 2016 as a distinct disease entity (1). These neoplasms mostly occur in the GI tract owing to clonal T cell proliferations. They may drive from CD8+ T cells and less often from CD4+ T cells (1). The infiltrating lymphocytes are mature small lymphocytes that are negative for CD56 expression; implying that they do not originate from NK cells. In general, these tumors show a non-invasive pattern and indolent clinical behavior. The proliferative index detected with Ki67 staining was very low (~5%) in indolent GI T-LPD cases (2). GI T-LPD is more common in males compared to females; and it is mainly observed in adults with a wide range of age (3).
Recognition of indolent GI T-LPD as a distinct neoplasm has important therapeutic implications especially for the type of therapeutic approach chosen after diagnosis. Importantly, misdiagnosis is currently one of the biggest challenge for these benign cases as they may be diagnosed as aggressive T cell lymphomas or inflammatory diseases, which may lead to improper management of these patients due to waste of time and wrong therapy applied (2). Our current knowledge on this neoplasm is still at the very beginning, and more research needs to be performed to have a deeper understanding of the genetic or epigenetic aberrations associated with the pathogenesis of indolent GI T-LPD cases.
In the following sections, we discuss (1) indolent GI T-LPD cases reported in the literature including unusual observations; (2) genetic alterations identified to overcome indolent GI T-LPD misdiagnosis problem; (3) prognosis and therapy of these cases; (4) future directions to shed light onto the possible research areas that may generate findings potentially useful in the clinical practice.
Case Reports
Nagaishi et al. observed an indolent GI T-LPD case with persistent diarrhea and villous atrophy in small intestine as the endoscopic finding (4). This case was treated with mogamulizumab due to surface expression of CCR4, which achieved durable remission in symptoms for more than 2 years. Interestingly, indolent GI T-LPD cases may unusually express CD20 which may lead to misdiagnosis as B-cell lymphomas thereby leading to unnecessary aggressive therapy (5). Guo et al. has recently reported an interesting GI T-LPD case with co-occurrence of diffuse large B cell lymphoma (6). This patient is a 46-year-old Chinese male initially diagnosed as GI T-LPD based on histological examination and T-cell receptor-gamma (TCRγ) clonal gene rearrangement assessment. This benign neoplasm may develop as a result of therapy against inflammatory diseases. Edison et al. reported an indolent GI T-LPD case developed in a patient with resistant Crohn's colitis as a result of tumor necrosis factor alpha (TNFα) therapy (7). Indolent GI T-LPD cases may rarely be associated with EBV infections. Wang et al. has recently reported a very rare case of adult-onset, immunocompetent EBV+ GI T-LPD (8). This case was a middle-aged woman with chronic diarrhea and severe intestinal bleeding. Importantly, she died 7 months post-diagnosis, which suggests that some GI T-LPD cases may show clinically aggressive behavior. The poor prognosis of this EBV positive case may be related to EBV-encoded oncogenic miRNAs as elevated levels of circulating miR-BART2-5p was shown to be associated with disease progression for EBV+ natural killer/T cell lymphoma (9). Given the lack of response of this GI T-LPD case to a variety of therapeutics, high expression of PD-L1 could be responsible for this patient's poor prognosis as EBV-encoded LMP1 upregulated PD-L1, and elevated PD-L1 resulted in adverse clinical course in NKTCL cases (10). EBV-induced epigenomic aberrations were reported in EBV+ malignancies (11). If these epigenomic changes are also present in EBV+ GI T-LPD cases, it will be worthwhile to address whether they affect patient survival or not. Although these observations imply that EBV infection is very likely responsible for poor prognosis of EBV+ GI T-LPD cases, we can not totally exclude the role of other potential pathogenic events. The characteristics of important cases reported are shown in Table 1.
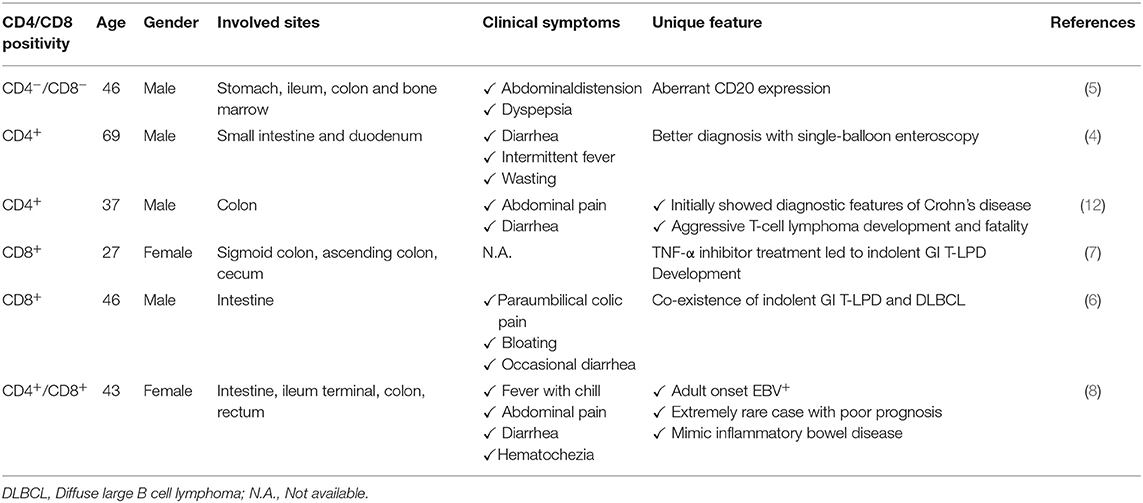
Table 1. Important case reports of indolent T-cell lymphoproliferative disease of the gastrointestinal tract.
Discovered Genetic Alterations to Avoid Indolent GI T-LPD Misdiagnosis
Indolent GI T-LPD cases may be misdiagnosed as enteropathy associated T cell lymphoma (EATL) or monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL), which are rare but aggressive lymphomas of the GI tract (2). The presenting symptoms of these aggressive gastrointestinal T cell lymphomas (e.g., abdominal pain, diarrhea) may be quite similar to those of indolent GI T-LPD cases (Table 2). Recent studies revealed recurrent activating mutations of STAT5B (18) and loss-of-function mutations of SETD2 (19) in MEITL cases, which can be useful for distinguishing MEITL cases from indolent GI T-LPD cases. By screening activating mutations located in the SH2 (Src Homology 2) domain of STAT3, it may be possible differentiate indolent GI T-LPD cases from EATL cases as the former did not have STAT3 mutations (2); whereas EATL cases harbor STAT3 mutations (13). Importantly, this neoplasm needs to be also differentiated from NK cell enteropathy, which is a recently described NK cell neoplasm appearing in gastrointestinal sites (20). Given that 30% frequency of JAK3 K563_C565del mutations were detected in NK cell enteropathy cases, these mutations may potentially be used to differentiate a subset of NK cell enteropathy cases from indolent GI T-LPD cases (17).
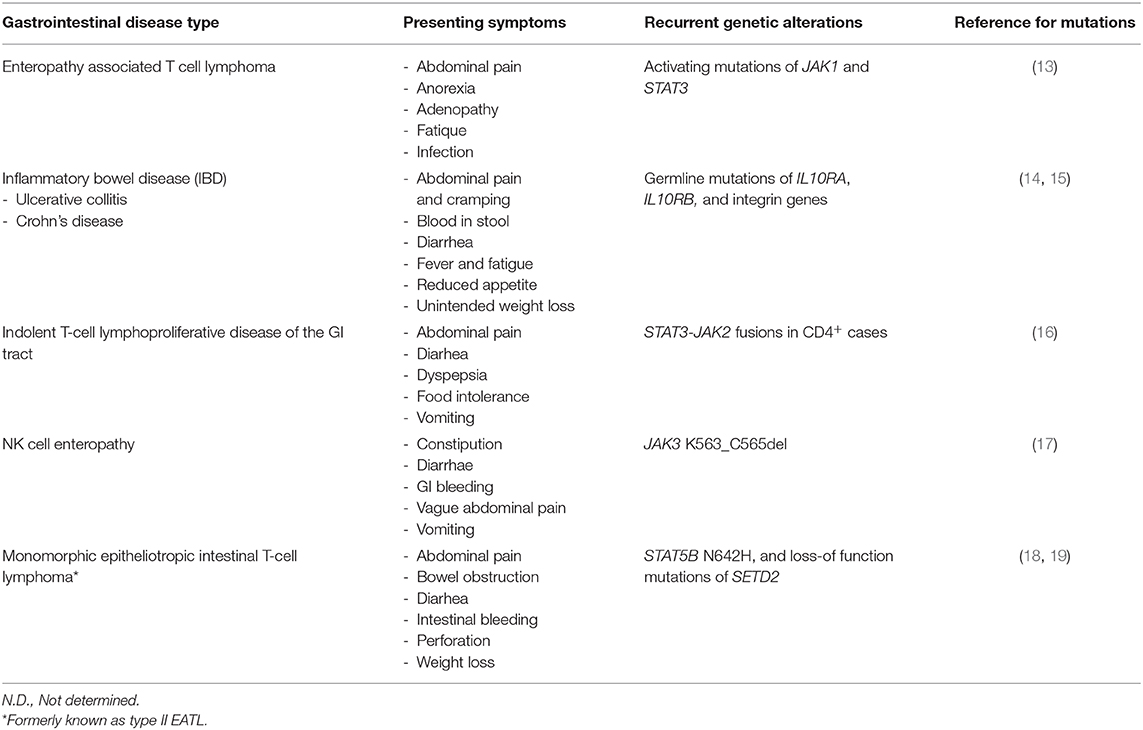
Table 2. Symptoms and recurrent genetic alterations in indolent GI T-LPD and similar gastrointestinal diseases.
The symptoms of inflammatory bowel disease (IBD) can be highly similar to those of indolent GI T-LPD cases, which in turn may lead to misdiagnosis of the latter ones (Table 2). Inflammatory bowel disease (IBD) is a heterogeneous disease that can further be subclassified as ulcerative collitis and Crohn's disease (21). There are few reports available investigating the genetic aberrations responsible for pathogenesis of IBD. Glocker et al. performed a pedigree-based analyses, and identified three distinct homozygous mutations in IL10RA and IL10RB genes, which form the IL10 receptor complex (14). These germline mutations prevented IL10 receptor signaling; thereby, disrupting a negative feedback regulation in peripheral blood mononuclear cells. Furthermore, a genome-wide association study (GWAS) revealed several new susceptibility loci, three of which include integrin genes (15). Given the difficulty of obtaining sufficiently large biopsy material from gastrointestinal sites, genetic screen for these inherited mutations in peripheral blood cells may be useful in routine clinical practice for distinguishing IBD and indolent GI T-LPD cases.
There are very few studies available, which focused on genetic aberrations that are associated with pathogenesis of indolent GI T-LPD cases. It is of utmost significance to identify genetic aberrations in this benign neoplasm to better diagnose, predict prognosis, and identify therapeutic targets. To address whether activating mutations of STAT3 previously reported in lymphomas derived from gamma delta T or NK cells (18) are present also in indolent GI T-LPD cases, Perry et al. screened six mutational hotspots in the SH2 domain of STAT3 in five indolent GI T-LPD case, which did not reveal any activating STAT3 mutation (2). Using complementary methodological approaches (i.e. FISH, RNA-Seq, Sanger sequencing), Sharma et al. has recently identified STAT3-JAK2 fusion genes in four of five CD4+ GI T-LPD cases but not in CD8+ or CD4+/CD8+ cases evaluated (16). If this finding is supported by future studies, STAT3-JAK2 fusion genes may be an important diagnostic biomarker which can be used in genetic tests for routine clinical applications. The indolent GI T-LPD cases with STAT3-JAK2 fusion genes did not show STAT3 activation but showed STAT5 activity based on immunohistochemistry. STAT3 Y705 residue phosphorylation is an indicator of STAT3 activation (18). It was unusual not to observe STAT3 activation in the CD4+ GI T-LPD cases with the STAT3-JAK2 fusion protein as this fusion gene retains the Y705 residue. Importantly, a recent study by Hu et al. ectopically expressed STAT3-JAK2 in different cell types, and showed that STAT3 is marginally phosphorylated, and have transcriptional activity albeit to a lesser extent than that of STAT5. Of note, STAT3-JAK2 fusion protein was observed to bind to and phosphorylate STAT5 much efficiently, consistent with the observation that STAT5 target genes were enriched in STAT3-JAK2 transduced T cell breast lymphoma 3 (TLBR3) cells but not STAT3 (22). The observed discrepancy related to STAT3 activation status may be due to relatively lower sensitivity of immunohistochemistry compared to that of western blot, high level of expression of phosphatases that dephosphorylate STAT3, or the intracellular amount of available STAT3 protein in indolent GI T-LPD cells. From clinical point of view, genetic detection of the STAT3-JAK2 using FISH coupled with STAT5 IHC may be the most suitable option during differential diagnosis, and decision for appropriate therapy for CD4+ T-LPDs. The clinical symptoms and recurrent genetic alterations in indolent GI T-LPD and similar gastrointestinal diseases are shown in Table 2.
Prognosis and Therapy
GI T-LPD usually presents as an indolent neoplasm with no progression to aggressive T cell lymphomas. These cases are often misdiagnosed as T cell lymphoma with little or no response to chemotherapy (23). As an example, an indolent GI T-LPD case with CD4−/CD8+ phenotype was initially misdiagnosed as MEITL (24). This case showed active but stable disease for 5 years post-administration of different chemotherapeutics. The fact that gastro-intestinal tract is the most common primary tumor site for extra-nodal lymphomas further underscores the importance of correct diagnosis of these benign neoplasms (3). Importantly, rare cases of indolent GI T-LPD that convert to aggressive lymphomas have been reported in the literature. Perry et al. has recently reported a 37 year-old male of an indolent GI T-LPD case who transitioned to peripheral T cell lymphoma 3 years after initial diagnosis (12). In general, genetic or epigenetic prognostic biomarkers that predict transition to peripheral T cell lymphoma in indolent GI T-LPD cases are unknown. It is possible that STAT3-JAK2 fusion gene promotes transition to aggressive T cell lymphoma as one of 5 CD4+ GI T-LPD case with the fusion protein was reported to develop T-cell lymphoma (16). However, more comprehensive studies need to be performed to address whether STAT3-JAK2 can be used as a prognostic biomarker for indolent GI T-LPD cases.
Radiotherapy may be a more effective option for treatment of indolent GI-T-LPD cases compared with chemotherapy. Recently, an indolent GI T-LPD case that have tumors localized in stomach has been treated successfully using involved field radiotherapy (IFRT) (25). This observation suggests that radiotherapy may be a good choice for treatment of cases with localized disease presenting in gastric sites. Nevertheless, it should be noted that longer follow-up is needed for this radiotherapy-treated case as it was followed-up only for 1 year post-treatment.
Edison et al. reported an unusual case of indolent GI T-LPD with resistant Chron's disease, which developed after tumor necrosis factor-α (TNF-α) inhibitor therapy (7). Interestingly, stopping TNF-α inhibitor therapy led to regression of the tumor, which suggests a causal relationship between this therapy and GI T-LPD. Furthermore, this observation suggests the need for avoiding TNF-α inhibitor treatment of patients with resistant Chron's disease to prevent development of indolent GI T-LPD.
Characterization of genetic lesions observed in indolent GI T-LPD as oncogenic drivers have potential to reveal novel therapeutic targets. Hu et al.'s study provided invaluable information on potential oncogenic role of STAT3-JAK2 in GI T-LPD pathogenesis. In that study, they showed that ectopic STAT3-JAK2 promotes growth of CD4+ T cells, and results in cytokine independent growth of Ba/F3, an IL3-dependent pro-B cell line cell. Of note, the main oncogenic function of this fusion protein was through activation of STAT5. Using an in vivo xenograft model, authors showed that STAT3-JAK2 promotes tumor growth, and this growth can be inhibited using JAK inhibitors. Altogether these observations imply that STAT3-JAK2 fusion protein is not important only as a diagnostic biomarker but also as a potential therapeutic target in CD4+ indolent GI T-LPD cases having this genetic lesion.
Future Directions
Our current knowledge on genetic and epigenetic alterations associated with indolent GI T-LPD is at the very beginning. There are several research areas to be focused on to improve differential diagnosis, predict prognosis, or identify therapeutic targets for better management of these cases (Figure 1). Most importantly, whole exome or genome sequencing of indolent GI T-LPD tumors have not been performed yet in the presence of patient-matched samples so the somatic mutation profiles of these tumors are still unknown. Obtaining sufficient tissue material from gastrointestinal sites of indolent T-LPD cases may be challenging for genetic testing. Hence, it would be interesting to see whether cancer-associated somatic mutations may be detected by targeted ultra-deep sequencing of plasma cell-free circulating DNA fragments, a method that proved highly useful for lymphoma types such as DLBCL (26) and classical Hodgkin's lymphoma (27). It would also be interesting to investigate whether there is genetic predisposition to indolent GI T-LPD by performing a genome-wide association study (GWAS) on a large cohort of patients to identify SNPs associated with indolent GI T-LPD, or perform pedigree-based genetic analyses to identify inherited mutations that contributed to development of these neoplasms. Identification of genetic predisposition factors that distinguish indolent T-LPD form other similar diseases may offer unique opportunities for improved diagnosis or prognosis through targeted genetic testing with a next-generation device or PCR-Sanger in routine clinic (28).
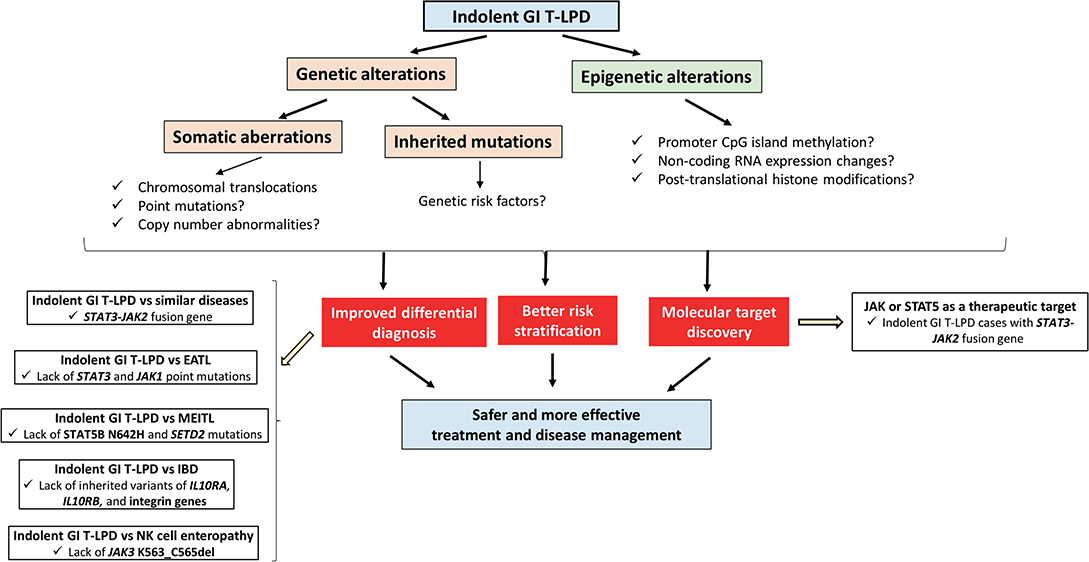
Figure 1. The schematic diagram showing the available and potential future genetic or epigenetic aberrations to be identified for better diagnosis, prognosis, and therapy of indolent GI T-LPD cases.
Epigenetic alterations such as promoter methylation-mediated tumor suppressor gene silencing (29), aberrant histone methylations (30), miRNA expression changes (31) can be used as biomarkers and/or therapeutic targets to improve diagnosis, risk stratification, and therapeutic efficacy of indolent GI T-LPD cases. To the best of our knowledge, no study has been reported investigating the epigenetic characteristics of indolent GI T-LPD cases. Given that epigenetic aberrations play key roles in regulating different “hallmarks” of cancer, it will be interesting to study the role of major epigenetic events in development of this neoplasm.
Conclusions
To sum up, there are many unknowns regarding the genetic and epigenetic aberrations associated with the development and pathogenesis of indolent GI T-LPD. Future investigations will reveal the genomic and epigenomic landscape that potentially can distinguish this neoplasm from other similar diseases to achieve accurate diagnosis; thereby leading to more effective therapeutic approaches with better disease management. Importantly, the presence of these disease-related aberrations will support distinguished recognition of indolent GI T-LPD as a distinct disease category by the medical authorities.
Author Contributions
CK, LW, and HY critically evaluated the literature related to the topic, identified references, and wrote the manuscript. HY financially supported the study. All authors contributed to the article and approved the submitted version.
Funding
The research activities in the CK research group were supported by the American Society of Hematology (ASH), The Scientific and Technological Research Council of Turkey (TÜBITAK), and the Young Scientists Award Program of the Turkish Academy of Sciences (TÜBA GEBIP 2017). The research activities in the HY laboratory were supported by Guangzhou Medical University. Moreover, HY was supported by the National Natural Science Foundation of China (81911530169, 81903088, 81670180, 81711540047, and 81850410547). Chongqing Science and Technology Bureau (cstc2017jxj1130005). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
GI T-LPD, T-cell lymphoproliferative disease of the gastrointestinal tract; WHO, World Health Organization; TCRγ, T-cell receptor-gamma; TNFα, tumor necrosis factor alpha; EATL, enteropathy associated T cell lymphoma; MEITL, monomorphic epitheliotropic intestinal T-cell lymphoma; SH2, Src Homology 2; IBD, inflammatory bowel disease; GWAS, genome-wide association study.
References
1. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. (2016) 127:2375–375 doi: 10.1182/blood-2016-01-643569
2. Perry AM, Warnke RA, Hu Q, Gaulard P, Copie-Bergman C, Alkan S, et al. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood. (2013) 122:3599–5992 doi: 10.1182/blood-2013-07-512830
3. C. van Vliet, Spagnolo DV. T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: review and update. Pathology. (2020) 52:128y141. doi: 10.1016/j.pathol.2019.10.001
4. Nagaishi T, Yamada D, Suzuki K, Fukuyo R, Saito E, Fukuda M, et al. Indolent T cell lymphoproliferative disorder with villous atrophy in small intestine diagnosed by single-balloon enteroscopy. Clin J Gastroenterol. (2019) 12:434–40. doi: 10.1007/s12328-019-00971-1
5. Wang X, Ng CS, Chen C, Yu G, Yin W. An unusual case report of indolent T-cell lymphoproliferative disorder with aberrant CD20 expression involving the gastrointestinal tract and bone marrow. Diagn Pathol. (2018) 13:82. doi: 10.1186/s13000-018-0762-4
6. Guo L, Wen Z, Su X, Xiao S, Wang Y. Indolent T-cell lymphoproliferative disease with synchronous diffuse large B-cell lymphoma: a case report. Medicine. (2019) 98:e15323. doi: 10.1097/MD.0000000000015323
7. Edison N, Belhanes-Peled H, Eitan Y, Guthmann Y, Yeremenko Y, Raffeld M, et al. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract after treatment with adalimumab in resistant Crohn's colitis. Hum Pathol. (2016) 57:45s. i doi: 10.1016/j.humpath.2016.06.021
8. Wang Y, Li Y, Meng X, Duan X, Wang M, Chen W, et al. Epstein-barr virus-associated T-cell lymphoproliferative disorder presenting as chronic diarrhea and intestinal bleeding: a case report. Front Immunol. (2018) 9:2583. doi: 10.3389/fimmu.2018.02583
9. Komabayashi Y, Kishibe K, Nagato T, Ueda S, Takahara M, Harabuchi Y. Circulating Epstein-Barr virus-encoded micro-RNAs as potential biomarkers for nasal natural killer/T-cell lymphoma. Hematol Oncol. (2017) 35:655063. doi: 10.1002/hon.2360
10. Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol. (2016) 9:109. doi: 10.1186/s13045-016-0341-7
11. Kucuk C, Wang J, Xiang Y, You H. Epigenetic aberrations in natural killer/T-cell lymphoma: diagnostic, prognostic and therapeutic implications. Ther Adv Med Oncol. (2020) 12:1758835919900856. doi: 10.1177/1758835919900856
12. Perry AM, Bailey NG, Bonnett M, Jaffe ES, Chan WC. Disease progression in a patient with indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Int J Surg Pathol. (2019) 27:102 7. doi: 10.1177/1066896918785985
13. Chander U, Leeman-Neill RJ, Bhagat G. Pathogenesis of enteropathy-associated T cell lymphoma. Curr Hemat Malign Rep. (2018) 13:308c17. doi: 10.1007/s11899-018-0459-5
14. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. (2009) 361:2033–033 doi: 10.1056/NEJMoa0907206
15. K.M., de Lange, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Gen. (2017) 49:256w261. doi: 10.1038/ng.3760
16. Sharma Oishi N, Boddicker RL, Hu G, Benson HK, Ketterling RP, et al. Recurrent STAT3-JAK2 fusions in indolent T-cell lymphoproliferative disorder of the gastrointestinal tract. Blood. (2018) 131:2262–6. doi: 10.1182/blood-2018-01-830968
17. Xiao W, Gupta GK, Yao J, Jang YJ, Xi L, Baik J, et al. Recurrent somatic JAK3 mutations in NK-cell enteropathy. Blood. (2019) 134:986–91. doi: 10.1182/blood.2019001443
18. Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. (2015) 6:6025. doi: 10.1038/ncomms7025
19. Roberti Dobay MP, Bisig B, Vallois D, Boechat C, Lanitis E, et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat Commun. (2016) 7:12602. doi: 10.1038/ncomms12602
20. Mansoor Pittaluga S, Beck PL, Wilson WH, Ferry JA, Jaffe ES. NK-cell enteropathy: a benign NK-cell lymphoproliferative disease mimicking intestinal lymphoma: clinicopathologic features and follow-up in a unique case series. Blood. (2011) 117:1447–447 doi: 10.1182/blood-2010-08-302737
21. Feakins RM. Ulcerative colitis or Crohn's disease? Pitfalls and problems. Histopathology. (2014) 64:317–35. doi: 10.1111/his.12263
22. Hu G, Phillips JL, Dasari S, Jacobs HK, Luchtel RA, Oishi N, et al. Targetability of STAT3-JAK2 fusions: implications for T-cell lymphoproliferative disorders of the gastrointestinal tract. Leukemia. (2020) 34:1467–71. doi: 10.1038/s41375-019-0678-3
23. Margolskee E, Jobanputra V, Lewis SK, Alobeid B, Green PH, Bhagat G. Indolent small intestinal CD4+ T-cell lymphoma is a distinct entity with unique biologic and clinical features. PLoS ONE. (2013) 8:e68343. doi: 10.1371/journal.pone.0068343
24. Kohri M, Tsukasaki K, Akuzawa Y, Tanae K, Takahashi N, Saeki T, et al. Peripheral T-cell lymphoma with gastrointestinal involvement and indolent T-lymphoproliferative disorders of the gastrointestinal tract. Leuk Res. (2020) 91:106336. doi: 10.1016/j.leukres.2020.106336
25. Takahashi N, Tsukasaki K, Kohri M, Akuzawa Y, Saeki T, Okamura D, et al. Indolent T-cell lymphoproliferative disorder of the stomach successfully treated by radiotherapy. J Clin Exp Hematop. (2020) 60:7ppto doi: 10.3960/jslrt.19022
26. Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood. (2017) 129:1947–57. doi: 10.1182/blood-2016-05-719641
27. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. (2018) 131:2413–25. doi: 10.1182/blood-2017-11-812073
28. Kucuk C. Genetic susceptibility to natural killer T-cell lymphoma. Lancet Oncol. (2019) 21:196–7. doi: 10.1016/S1470-2045(19)30820-4
29. Kucuk C, Hu X, Jiang B, Klinkebiel D, Geng H, Gong Q, et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. (2015) 21:1699–699: doi: 10.1158/1078-0432.CCR-14-1216
30. Jiang Y, Dominguez PM, Melnick AM. The many layers of epigenetic dysfunction in B-cell lymphomas. Curr Opin Hematol. (2016) 23:377mpho doi: 10.1097/MOH.0000000000000249
Keywords: Indolent T cell lymphoproliferative disease of the GI tract, misdiagnosis, prognosis, targeted therapy, T cell lymphoma, NK cell enteropathy, inflammatory bowel disease, genetic aberrations
Citation: Küçük C, Wei L and You H (2020) Indolent T-Cell Lymphoproliferative Disease of the GI Tract: Insights for Better Diagnosis, Prognosis, and Appropriate Therapy. Front. Oncol. 10:1276. doi: 10.3389/fonc.2020.01276
Received: 06 April 2020; Accepted: 19 June 2020;
Published: 07 August 2020.
Edited by:
Hong-Wu Xin, Yangtze University, ChinaReviewed by:
Fang Liu, Guangzhou University of Chinese Medicine, ChinaOzgur Kutuk, Başkent University, Turkey
Copyright © 2020 Küçük, Wei and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua You, eW91aHVhMzA3QDE2My5jb20=; Li Wei, d2VpbGlrd0BhbGl5dW4uY29t