 Shuhua Zheng
Shuhua Zheng Wensi Tao
Wensi Tao- 1College of Osteopathic Medicine, Nova Southeastern University, Fort Lauderdale, FL, United States
- 2Department of Radiation Oncology, University of Miami-Miller School of Medicine, Coral Gables, FL, United States
As a dynamic regulator for short-lived protein degradation and turnover, the ubiquitin-proteasome system (UPS) plays important roles in various biological processes, including response to cellular stress, regulation of cell cycle progression, and carcinogenesis. Over the past decade, research on targeting the cullin-RING (really interesting new gene) E3 ligases (CRLs) in the UPS has gained great momentum with the entry of late-phase clinical trials of its novel inhibitors MLN4924 (pevonedistat) and TAS4464. Several preclinical studies have demonstrated the efficacy of MLN4924 as a radiosensitizer, mainly due to its unique cytotoxic properties, including induction of DNA damage response, cell cycle checkpoints dysregulation, and inhibition of NF-κB and mTOR pathways. Recently, the PROteolysis TArgeting Chimeras (PROTACs) technology was developed to recruit the target proteins for CRL-mediated polyubiquitination, overcoming the resistance that develops inevitably with traditional targeted therapies. First-in-class cell-permeable PROTACs against critical radioresistance conferring proteins, including the epidermal growth factor receptor (EGFR), androgen receptor (AR) and estrogen receptor (ER), cyclin-dependent kinases (CDKs), MAP kinase kinase 1 (MEK1), and MEK2, have emerged in the past 5 years. In this review article, we will summarize the most important research findings of targeting CRLs for radiosensitization.
Introduction
Over 60% of cancer patients undergo radiotherapy (RT) during their course of illness, with an estimated 40% contribution toward curative cancer treatment (1, 2). While RT is an essential element for curative, adjuvant, and palliative treatment of a range of human malignancies, a key challenge in RT is to maximize radiation doses to the tumor mass while sparing the surrounding healthy tissue (1). To that end, various approaches combining RT with chemotherapies as radiosensitizers have been explored, which led to improvements in tumor response and higher overall survival (OS) rates (3). Despite a clear success, the favorable clinical outcome of chemoradiotherapy still comes at the sacrifice of increased toxicity in many clinical contexts, mainly due to the limited specificity of conventional chemotherapies (4). In the past two decades, several clinical trials have been conducted to test combining RT with targeted therapies against radioresistance conferring proteins such as epidermal growth factor receptor (EGFR), histone deacetylase (HDAC) and the B-rapidly accelerated fibrosarcoma (BRAF), aiming to develop combined-modality treatment regimens with fewer side effects (5–7). Clinical studies consistently suggested increased efficacy and improved survival rates of these new strategies, highlighting the clinical importance of using targeted agents as radiosensitizers (4). However, cancer cells will inevitably develop resistance toward these targeted therapies, leading to disease progression and relapse (8). Therefore, there is an urgent need to develop new strategies for radiosensitization.
In the past decade, targeting the activities of cullin-RING (really interesting new gene) E3 ligases (CRLs) in the ubiquitin-proteasome system (UPS) has gained considerable momentum for cancer treatment with the entry of several late-phase clinical trials of its first-in-class inhibitor MLN4924 (pevonedistat) (9). As early as the 1990s, the implication of targeting CRL for radiosensitization was suggested when its key component RING box protein 2 (Rbx2, a.k.a., SAG, ROC2) was identified as a redox inducible antioxidant protein (10). In recent years, studies showed that CRLs carry out the turnover of vital proteins involved in DNA damage response (DDR), as well as those in cell signaling pathways that are critical for radiosensitization (9, 11). Furthermore, in the past 5 years, the development of cell-permeable PROteolysis TArgeting Chimeras (PROTACs), which can selectively recruit radioresistance conferring proteins for CRL-mediated polyubiquitination, paved new methods in developing radiosensitizers that are less likely to develop chemoresistance (12). As such, it is crucial to systematically overview the mechanism of actions of CRL inhibitors for radiosensitization.
In this review article, we will summarize major strategies targeting CRLs and evaluate their potential as radiosensitizers based on the revised framework of the Steel hypothesis, originally described by George Steel in the 1970s (4). The revised hypothesis describes the scenario whereby combined-modality of targeted therapies and RT can improve the therapeutic outcomes by five mechanisms: (1) spatial cooperation, (2) temporal modulation, (3) biological cooperation, (4) cytotoxic enhancement, and (5) normal tissue protection (4).
A Glimpse of Traditional Radiosensitizers
Typical RT involves ionizing radiation (IR), which uses high-energy photon radiation, such as X-rays and gamma (γ) rays, and particle radiation, such as electron (e), carbon ion and proton (13, 14). The IR exerts cytotoxic effects via direct DNA damage, or indirectly via generation of free radicals, particularly reactive oxygen species (ROS) (15–17). Therefore, the radiosensitivity of cancer cells can be influenced by biological factors that regulate DNA damage repair, oxygen perfusion levels, and cell cycle stage (16). Traditional radiosensitizers target these underlying parameters for radiosensitization.
Platinum analogs, 5-fluorouracil (5-FU), and taxanes are the most common clinically used radiosensitizers. Platinum analogs, such as cisplatin and oxaliplatin, can bind to DNA and produce DNA-DNA crosslinking, which will lead to cell cycle arrest and exacerbating the radiation-induced DNA damage (18). Meanwhile, 5-FU, capecitabine (a 5-FU oral prodrug), and gemcitabine act as pseudo-substrates, incorporation of these nucleoside analogs can dysregulate cell cycle checkpoint in the S phase, disabling DNA damage repair machinery in cancer cells upon IR administration (19, 20). On the other hand, taxanes, such as paclitaxel and docetaxel, synchronize tumor cells at cell cycle G2-M phase and trigger chromosomal missegregation (21, 22). Meanwhile, tumors in the hypoxic microenvironment (low pO2) are more radioresistant than those well-oxygenated (13). At the presence of oxygen, RT-induced DNA damages will be “fixed” via the formation of peroxyl radicals in DNA that had been insulted by free radicals (23). The oxygen mimics such as nitroimidazole derivatives (i.e., pimonidazole and nimorazole), and hypoxia-specific toxins were investigated in clinical trials as radiosensitizers (24, 25). Wang et al. (16) provided a comprehensive review on the recent development of radiosensitizers based on these principles.
The therapeutic potential of radiosensitizer is largely determined by the enhanced efficacy and selectivity against cancer cells but not normal tissue. However, traditional radiosensitizers are also chemotherapeutic drugs, which can cause prominent side effects. For example, cisplatin can cause intolerable nausea, vomiting, hearing loss, and kidney damage (26). Targeted therapies, such as MLN4924 and PROTACs are highly selective and would have fewer side effects. In fact, clinical trials of MLN4924 showed that this compound is well-tolerated (27).
Targeting the Ubiquitin-Proteasome System (UPS)
Cellular protein levels are tightly controlled by both protein synthesis and degradation. The ubiquitin-proteasome system (UPS), first characterized in the mid-20th century, is a dynamically regulated multi-enzyme process that earmarks substrate proteins for proteasomal-mediated degradation via polyubiquitination (28). Targeted inhibition of the UPS via direct eradication of the proteasome activities using bortezomib, carfilzomib, or ixazomib has been proven clinically effective for treating multiple myeloma (MM) (29). Several clinical trials also investigated the UPS inhibitors for their potential as radiosensitizers in the treatment of metastatic melanoma (Phase I), head and neck cancer (Phase I), and glioblastoma multiforme (GBM; Phase II) (30–32). However, unexpected earlier tumor progression as a result of EGFR stabilization has been reported with the combined administration of bortezomib and conventional radiochemotherapy in head and neck cancer (32, 33). Such suboptimal response is conceivable as proteasome inhibition indiscriminately stabilizes the substrates, including EGFR and other oncogenic proteins, limiting the clinical applications in targeting proteasome as a radiosensitizing strategy (32). Instead of directly inhibiting the proteasome, recent studies have employed alternative strategies such as targeting the UPS via inhibition of the upstream ubiquitin (Ub) conjugation events or directly recruiting specific substrate protein for polyubiquitination using PROteolysis TArgeting Chimeras (PROTACs) (Figures 1, 2).
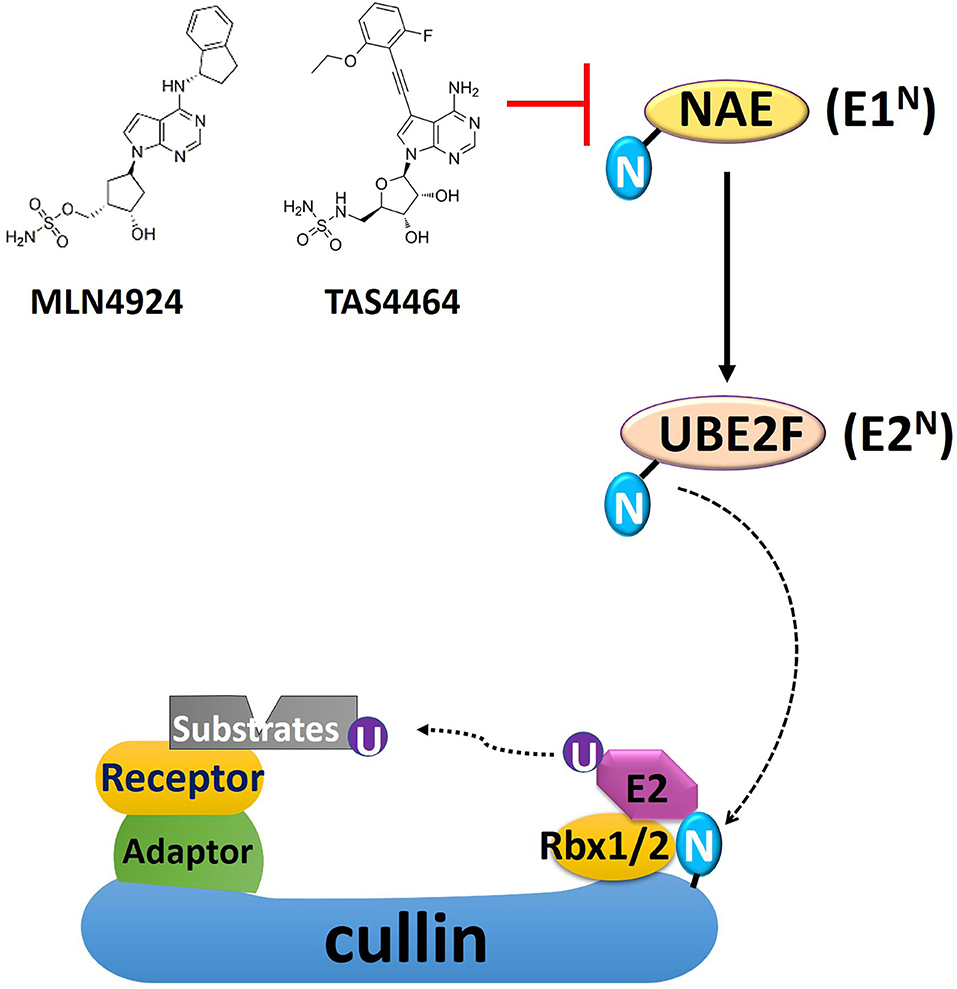
Figure 1. A schematic overview of cullin-RING E3 ligase (CRL) and NEDD8 conjugation. Conjugation of NEDD8 to the scaffold cullin protein in the CRL is carried out in three enzymatic steps involving NEDD8-activating enzyme (NAE, E1N), UBC12, and UBE2F (E2N). The substrate receptor protein, docked in the CRL complex by binding to the adaptor protein, recruits substrates for ubiquitin conjugation. MLN4924 and TAS4464 are specific NAE inhibitors, prohibiting NEDD8 conjugation and thus inhibit CRL activity. The 2-D structure of MLN4924 and TAS4464 was derived from Yu et al. (34). N, NEDD8; U, ubiquitin; Rbx, RING box protein.
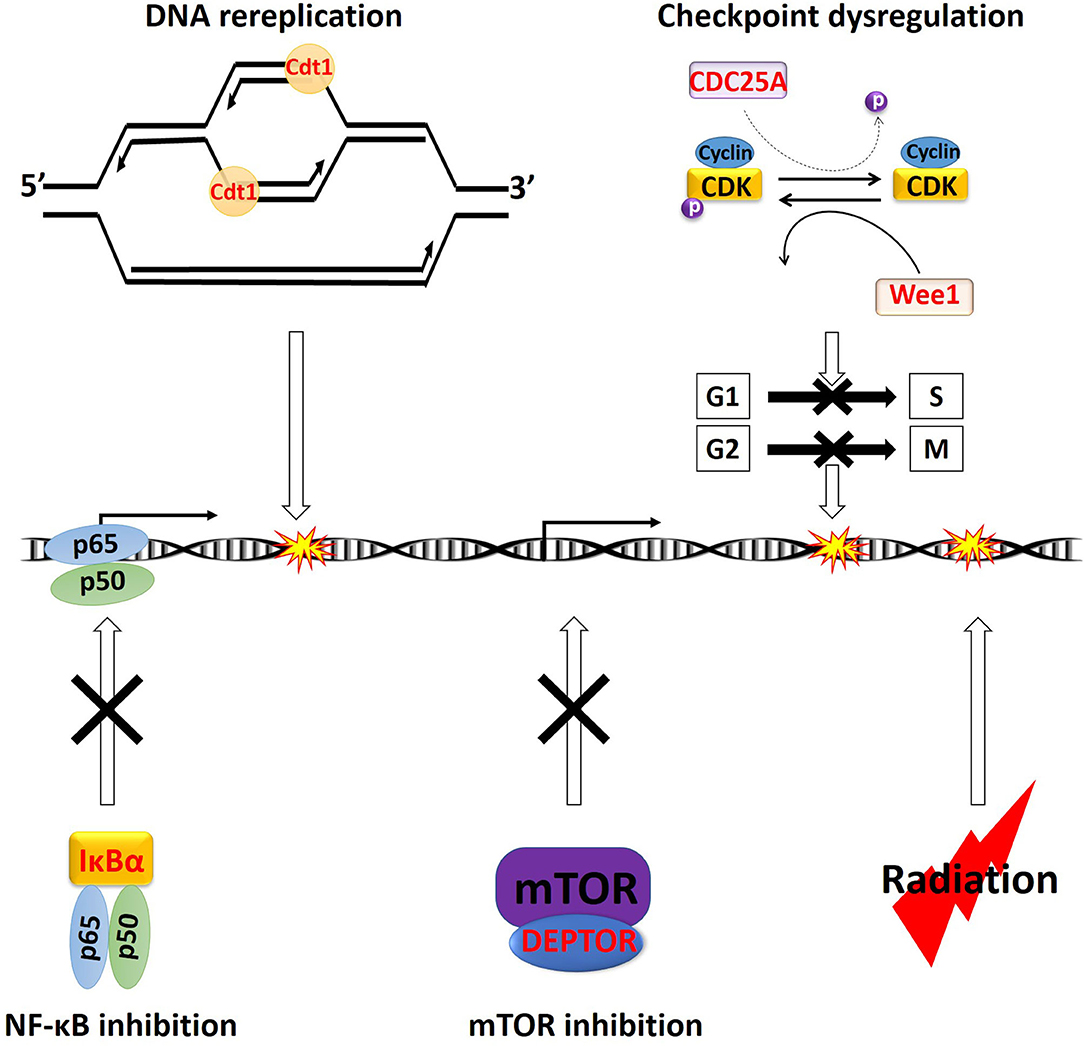
Figure 2. Overview of the major cytotoxic mechanism of NEDDylation inhibition. All the characters in “Red” color are CRL substrates. Accumulation of Cdt1 can trigger DNA rereplication, resulting in DNA damages. Stabilization of CDC25A can potentially cause dysregulation of both early (G1 phase to S phase) and late (G2 phase to M phase) cell cycle checkpoints (dash arrow). Meanwhile, the accumulation of Wee1, p21, and p27 will trigger cell cycle arrest at the G1 or G2 phase. Stabilization of IκBα will lead to sequestration NF-κB p65 and p50 heterodimer in the cytosol, leading to inhibition of its transcriptional activities. Stabilization of DEPTOR can directly inhibit mTOR. Ionizing radiation also triggers cell death via induction of DNA damages. p, phosphor group.
NEDDylation Inhibition
NEDD8 Conjugation Pathway
The cullin-RING E3 ligases (CRLs) are responsible for polyubiquitination of about 20% of cellular proteins degraded via the UPS, most of which are critically involved in cell cycle progression, DDR, and oncogenic signaling cascades (9). The CRL complex's core structure is formed with a scaffold protein cullin bound with the RING-finger containing proteins (Rbx1 or Rbx2) at the C-terminus of cullin (35). This core complex will be joined by the adaptor protein, which binds the cullin's N-terminus domain to form a complete CRL complex (Figure 1) (35). Fully activation of CRLs requires conjugation of an Ub-like protein called neural precursor cell-expressed developmentally downregulated 8 (NEDD8) to near the C-terminus of the cullin in the CRL complex (11). Conjugation of NEDD8 to cullins is carried out in three enzymatic steps involving NEDD8-activating enzyme (NAE; E1), UBC12 and UBE2F (E2s), and E3s (Figure 1). NAE adenylates NEDD8 on its C-terminal glycine, forming a NEDD8-NAE complex via a covalent thiol-ester bond, and then transfers NEDD8 to the E2s via another thiol-ester bond (36, 37). NEDD8 E3 ligases execute the final step in conjugating NEDD8 to cullins, forming an isopeptide bond with the ε-amino group of a substrate lysyl residue (38, 39). NEDD8 conjugation facilitates the CRL structural remodeling that will juxtapose the substrate toward the catalytically active ubiquitylation site of CRL (40–42). The substrate receptor protein, docked in the CRL complex by binding to the adaptor protein, recruits substrates for Ub conjugation once CRL is fully functional (43) (Figure 1). So far, seven different types of human cullin proteins (CUL1, 2, 3, 4A, 4B, 5, 7) have been identified, and new members of the receptor and adaptor proteins are emerging (44). A more detailed overview of NEDD8 conjugation in CRLs has been summarized by Petroski and Deshaies in their review paper (45).
MLN4924 is an adenosine sulfamate analog that inhibits NEDDylation via the formation of an MLN4924-NEDD8 adduct, blocking the downstream NEDD8 conjugation cascade within a few hours after administration (46) (Figure 1). TAS4464 is another NAE inhibitor recently developed with more potent inhibitory effects and prolonged duration of target-binding compared with MLN4924 (47, 48). NAE inhibitors MLN4924 and TAS4464 targeting the process of NEDD8 conjugation have shown particularly promising results in several clinical trials (phase I/II/III) for cancer treatment (46). NEDDylation inhibition appears to have a unique profile of sensitivity toward various types of malignancies. So far, the primary identified cytotoxic mechanisms of MLN4924 include induction of DNA rereplication, senescence, dysregulation of cell cycle checkpoints control, as well as inhibition of mTOR and NF-κB pathways (Figure 2) (46, 49–55). Substrates of CRLs, such as Wee1, checkpoint kinase 1 (CHK1), p21, and cell division cycle 25A (CDC25A), are key components of double-strand breaks (DSBs) repair proteins (56–60) (Figure 2). Mainly, the degradation of cell cycle proteins Cdt1, p21, and Set8 is mediated by CRL4Cdt2, in which Cdt2 plays as a substrate recognition protein (61–63). Genotoxic insults trigger binding of Cdt2 on to the DNA sliding clamp—proliferating cell nuclear antigen (PCNA)—loading CRL4Cdt2 on to DNA for the substrate degradation (61–63). Set8 stabilization leads to lysine 20 of histone H4 (H4K20) hypermethylation, triggering transcriptional downregulation of histone with the resultant chromatin decompaction and DNA damage activation, as depicted elegantly by Abbas et al. (61). Driven by the fact that the development of radioresistance is largely determined by factors such as DNA damage response DDR activation after ionizing radiation (IR), cell cycle checkpoints controls, and anti-apoptotic pathways dysregulation, it is essential to investigate the potential of using NEDDylation inhibitors as radiosensitizers (64).
NEDDylation Inhibitors as Radiosensitizers—DNA Damage Response
One of the major cytotoxic effects of MLN4924 is achieved through the stabilization of its substrate Cdt1, a so-called “DNA replication licensing factor,” which tightly regulates the cell cycle progression by facilitating the formation of the pre-replicative complexes (pre-RC) at the G1 phase of cell cycle (46, 65). To prevent relicensing, which will lead to multiple rounds of DNA replication initiation per cell cycle, Cdt1 is rapidly degraded by the CRL Skp1-cullin1-F-box protein (SCF) right after G1 phase (66) (Figure 2). MLN4924-mediated inhibition of SCF will lead to the accumulation of Cdt1, causing firing of several rounds of DNA replication initiations without cell division, as evidenced by the accumulation of cells with > 4N DNA in flow cytometry (46) (Figure 2). This process will lead to the collision of replication forks and the induction of overwhelming both single- and double-strand DNA damage (67).
The majority of IR-mediated cell killing is mediated by the massive induction of DNA DSBs (64). Radiosensitivity of tumor cells is largely decided by their ability to trigger the DDR, via activation of cell cycle checkpoints and DNA damage repair (64). MLN4924 functions as a radiosensitizer in several types of cancer by potentiating DNA damage and interfering with DDR activation. In the orthotopic xenograft mouse models of human pancreatic cancer and head and neck squamous cell carcinoma (HNSCC), MLN4924 overcame radioresistance via induction of DNA rereplication, leading to prominent induction of DSBs (68, 69). In pancreatic cancer cells, the maximal radiosensitizing effects of MLN4924 was achieved when MLN4924 was administered 24 h prior to receiving RT (69). MLN4924 pretreatment before RT administration will allow time for CRL substrates' accumulation. The radiosensitizing effect of MLN4924 was partially reversed in pancreatic cancer cells with Cdt1 knockdown (69). However, the exact involvement of Cdt1 stabilization in MLN4924-induced radiosensitization needs further investigation.
Expression levels of CRL components were significantly elevated in HNSCC cells compared with those in adjacent normal squamous mucosa of the oral cavity and nasopharynx (68). As a result, DNA rereplication was not observed in the cells of normal tissue (68). Besides HNSCC, hyperactivation of CRLs was also observed in GBM, breast cancer, and liver cancer (70). Therefore, the unique cytotoxic mechanism highlights the potential of NEDDylation inhibitors as radiosensitizers from the perspectives of “spatial cooperation,” “biological cooperation,” “normal tissue protection,” and “cytotoxic enhancement” based on the revised Steel framework.
MLN4924 as a Radiosensitizer—Cell Cycle Arrest
Due to the lethality of unrepaired DNA DSBs, developing new agents to prevent activation of cell cycle checkpoints in response to IR is critical to overcoming radioresistance (71). DDR is initiated by activation of ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and RAD3-related (ATR), which will locate the DNA damage and activate various downstream proteins (72). ATM is the major regulator of DDR following IR-induced DSBs, leading to phosphorylation of downstream CHK1 and CHK2 (72). Activated CHKs will then phosphorylate the isoforms of CDC25 phosphatases, triggering their polyubiquitination and degradation (73). Meanwhile, the dephosphorylation and activation of CDK2-cyclinE and CDK1-cyclinB depend on the phosphatase activities of CDC25 (73). As a result, with activation of DDR and subsequent CDC25 degradation, cell cycle arrest will occur at the end of G1 phase or the end of G2 phase to allow time for DNA repair (73) (Figure 2). Among the three isoforms of CDC25s (CDC25A, B, C), CDC25A regulates both early (G1 phase to S phase) and late (G2 phase to M phase) cell cycle checkpoints (Figure 2) (73). Rapid degradation of CDC25A is critical for activating cell cycle arrest upon IR-induced DNA damages (72). The ubiquitination of CDC25A is carried out by the CRL E3 ligase SCFbeta−TrCP, in which the beta-TrCP (β-transducin repeat-containing protein) facilitates the recruitment of the CDC25A for Ub conjugation (74). MLN4924-mediated inhibition of SCFbeta−TrCP will stabilize the CDC25A protein, causing cell cycle checkpoint dysregulation and potentially radiosensitization (Figure 2).
Accumulation of CRL substrates may also induce cell cycle arrest via checkpoint activation. The Wee1 kinase, which phosphorylates and keeps CDK1 in inactive form for activation of cell cycle checkpoints, is another major CRL substrate (Figure 2) (75). Meanwhile, degradation of members of the universal cyclin-dependent kinase inhibitors (CDKIs) family p21 (Cip1) and p27 (Kip1) is also mediated by CRLs (76). As such, several studies reported the activation of cell cycle checkpoints with MLN4924 treatment (77–79). In cell lines of hormone-refractory prostate cancer (HRPC), MLN4924 triggered cell cycle arrest at the G2 phase due to Wee1, p21, and p27 accumulation (79). In the colorectal cancer cell lines of HT-29 and HCT-116, and breast cancer cell lines of SK-BR-3 and MCF7, MLN4924 induced stabilization of p27 and p21, respectively, leading to cell cycle G2/M arrest (77, 78). MLN4924 and RT cotreatment induced a more significant accumulation of Wee1, p21, and p27 than either treatment modality alone, leading to prominent cell cycle arrest and unanimous sensitization all these types of cancer cells toward RT (77–79).
MLN4924 as a Radiosensitizer—Anti-apoptotic Pathways
Increased radioresistance of cancer cells is developed by the activation of several compensatory pro-survival cell signaling pathways, including phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathway, EGFR/mitogen-activated protein kinase (MAPK) pathway and NF-κB signaling pathway (80, 81). The classical theories of how radiation activates these anti-apoptotic pathways state that ionizing events in the cytosol and the mitochondria will generate large quantities of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that will inhibit protein phosphatase (PTPase) activities (81). Radiation can also promote membrane-associated receptor activation by lipid rafts aggregation, leading to activation of downstream pathways (82). Activation of the PI3K/AKT/mTOR, EGFR/MAPK, and NF-κB pathways can facilitate the development of radioresistance by promoting DNA damage repair, and transcriptional upregulation of a myriad of stress-responsive proteins (83, 84) (Figure 2). Therefore, it is critical to understand the impact of NEDDylation inhibition on these compensatory pro-survival pathways activated by RT.
Inhibition of NF-κB pathway is one of the major causes of MLN4924 induced cytotoxicity, as evidenced in the initial studies in acute myeloid leukemia (AML) (49). The inhibitor of nuclear factor kappa B (IκBα) binds to the NF-κB p65 and p50 complex and keeps the heterodimer in the cytosol as an inactive form (85). Activation of the pathway triggers rapid degradation of IκBα via the SCFβ−TrCP E3 ligase, releasing the p65 and p50 heterodimer for nuclear translocation and transcriptional upregulation of its target genes (86). Treatment of MLN4924 will inhibit the SCFβ−TrCP and prohibit RT-induced IκBα degradation, with resultant sequestration of p65 and p50 in the cytoplasm (49, 52, 87) (Figure 2). This mechanism is validated in studies showing that eradication of the RING-box protein Rbx2 in the SCFβ−TrCP complex triggered IκBα stabilization and NF-κB pathway inhibition, leading to re-sensitization of cancer cells toward RT (88) (Figure 2). Furthermore, the existing studies also suggested that the radiosensitizing effect of bortezomib is largely due to the inhibition of the NF-κB pathway (89). As a result, another major radiosensitizing mechanism of MLN4924 is achieved through the NF-κB pathway inhibition (Figure 2).
Several mTOR inhibitors, including everolimus and temsirolimus, are under early Phase (I/II) clinical trials as a radiosensitizer to treat several cancer types such as prostate cancer, GBM, and lung cancer (90–92). In human cancer cell lines of acute lymphoblastic leukemia (ALL), AML, cervical, breast, colon, GBM, and kidney, the activity of mTOR is downregulated by MLN4924 in an almost dose-dependent manner, as evidenced with dephosphorylation of mTOR downstream targets such as p70S6 kinase (51, 93–95). Intrinsic mTOR's upstream inhibitors, including the DEP domain containing MTOR interacting protein (DEPTOR) and the regulated in development and DNA damage responses 1 (REDD1), are substrates for SCFβTrCP and cullin4A-RING (CRL4A), respectively. These protein-drug interactions largely explain the unanimous response of mTOR inhibition toward NEDDylation inhibition (50, 51, 55). The significant inhibitory effect of MLN4924 on the PI3K/AKT/mTOR axis has implicated the NEDDylation inhibitors as potential therapeutic radiosensitizers (Figure 2).
In summary, NEDDylation inhibition can block key pro-survival pathways activated with RT via stabilization of their intrinsic upstream inhibitory proteins. The unique role of MLN4924 in blocking these compensatory pathways demonstrated its potential application as a radiosensitizer via “spatial cooperation,” “biological cooperation,” and “cytotoxic enhancement” (4).
PROteolysis Targeting Chimeras (PROTACs)
Neither bortezomib nor MLN4924 addresses specific proteins as they broadly inhibit the general machinery necessary for protein degradation. MLN4924 is not selective since all the CRL complexes in the cells are inhibited, blocking the activities of over 400 enzymes (70). The PROteolysis TArgeting Chimeras (PROTACs) technology was developed in recent years to overcome these limitations of targeting the protein degradation machinery (96). PROTACs are heterobifunctional molecules with two different ligands connected via a linker (Figure 3). One end of the PROTAC, i.e., the “warhead,” binds to the protein of interest (POI), and the other end binds to the receptor protein in the CRL complex, thereby promoting the physical interaction of the target protein with the E3 ligase for polyubiquitination (Figure 3) (97). Traditional targeted therapies using occupancy driven pharmacology only affect enzymatic function via competitive inhibition, which requires druggable active sites in those enzymes that are susceptible to mutations and protein overexpression (8). Whereas, polyubiquitinated POIs will be degraded by the proteasome with the eradication of both the enzymatic activities and the scaffold functions of target proteins (97). Furthermore, PROTACs-induced protein degradation is a catalytic process, as PROTACs will dissociate from the CRL complex after POI polyubiquitination and binds to a new target. This unique catalytic property of PROTACs can lead to efficient POI clearance at a very low dose5 (98) (Figure 3).
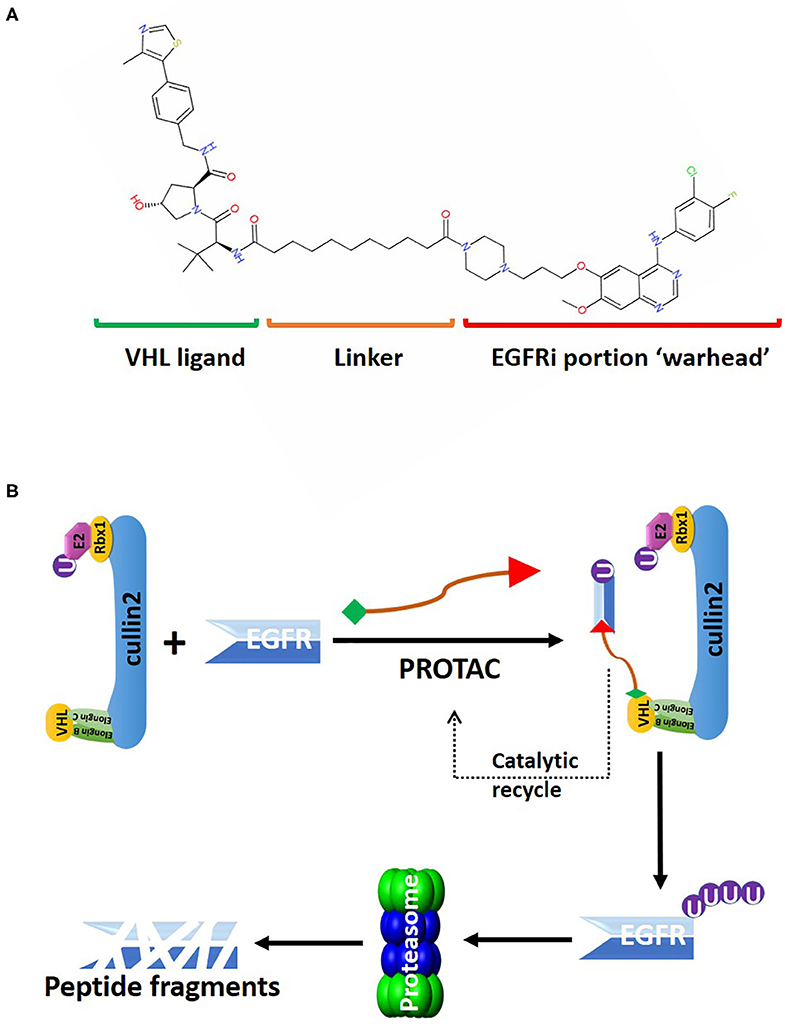
Figure 3. An overview of the PROTAC targeting EGFR for CRL2VHL-mediated polyubiquitination. (A) 2D structure of the PROTAC MS39 that targets EGFR. The “warhead” potion of the PROTAC is based on EGFR inhibitor (EGFRi) gefitinib. (B) MS39 can recruit EGFR for polyubiquitin conjugation by the CRL2VHL. MS39 mediated EGFR degradation is a catalytic process, as evidenced with dissociation of the PROTAC from the CRL2VHL complex after EGFR polyubiquitination.
Due to these unique characteristics, targeting CRL substrates related to the development of radioresistance with PROTACs could provide a new strategy to sensitize cancer cells toward RT (70) (Figure 4). Since 2015, over 30 small-molecule PROTACs have been reported, most of them utilize substrate receptor proteins von Hippel-Lindau (VHL) in the CRL2 complex (CRL2VHL), and cereblon (CRBN) in the cullin4-RING complex (CRL4CRBN) as the PROTAC binding sites in the E3 ligase (99) (Figure 4). PROTACs against radioresistance-related key substrates of CRL2 and CRL4 have been developed. These substrates include the EGFR, androgen (AR) and estrogen (ER) receptors, CDKs, MAP kinase kinase 1 (MEK1) and MEK2, anaplastic lymphoma kinase (ALK), Bruton tyrosine kinase (BTK), bromodomain and extra-terminal motif (BET) proteins, and bromodomain (BRD) proteins (Figure 4) (100–107). Since all these PROTACs are first-in-class protein degraders developed within the past 5 years, a comprehensive understanding of the underlying molecular mechanism of these novel compounds is essential for developing next-generation radiosensitizers.
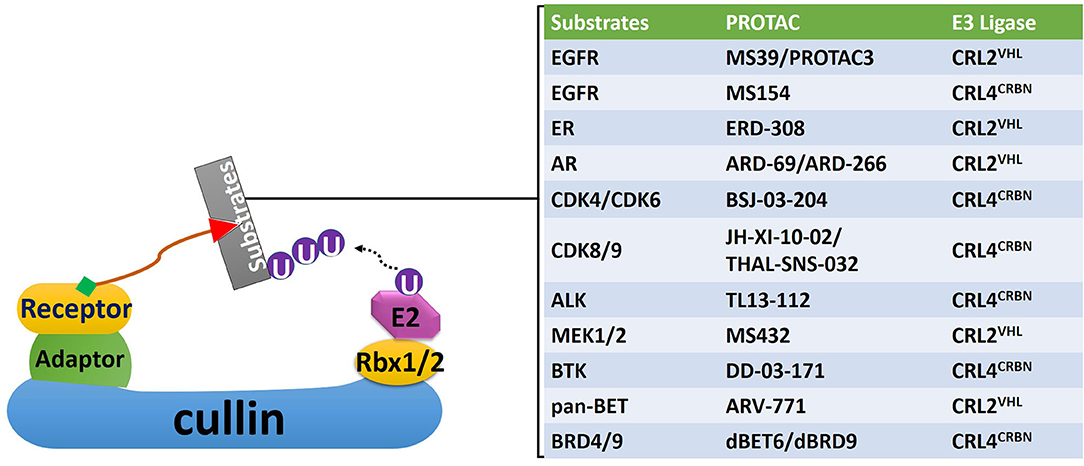
Figure 4. A non-exhaustive list of PROTACs targeting proteins related with the development of radioresistance. Corresponding CRLs that mediate polyubiquitination of these proteins were also identified. EGFR, epidermal growth factor receptor; ER, estrogen receptor; AR, androgen receptor; CDK, cyclin-dependent kinase; ALK, anaplastic lymphoma kinase; MEK, mitogen-activated protein kinase kinase; BTK, Bruton tyrosine kinase; BET, bromodomain and Extra-Terminal motif; BRD, bromodomain.
PROTACs as Radiosensitizers—Targeting Receptors
Targeting EGFR using traditional targeted therapies, such as monoclonal antibodies or tyrosine kinase inhibitors (TKIs), as radiosensitizers have gained moderate success in non-small-cell lung cancer (NSCLC), but failed in GBM and HNSCC to improve OS rates (108, 109). Genetic alterations, including amplification, rearrangement, altered splicing, and mutations, that regulate EGFR expression levels and protein activities in GBM and HNSCC, will eventually lead to the development of resistance toward EGFR inhibitors (108, 110). Lysosome-independent degradation of EGFR is mediated by CRL2VHL, consistent with the report in clinical trial showing stabilization of EGFR with bortezomib-mediated proteasome inhibition (32, 111). Recently, several PROTACs were developed to target EGFR for CRL2VHL-mediated degradation. PROTAC-based technology offers great flexibility in choosing the clinically relevant forms of EGFR proteins targeted for degradation by changing the “warhead” of the degrader (Figure 3). For example, the lapatinib-based PROTAC largely degraded the wildtype, and exon-20 insertion mutant forms of EGFR; the gefitinib-based PROTAC selectively degraded EGFR with exon-19 deletion, and L858R point mutation; afatinib-based PROTAC degraded double mutant (L858R/T790M) EGFR (112). All these PROTACs can efficiently eliminate EGFR at low-nanomolar concentrations, and exerted sustained inhibitory effects on cancer cell proliferation and downstream kinases signaling of EGFR (112).
RT-induced overexpression of hormonal receptors, including AR and ER, plays a vital role in mediating radioresistance in prostate and breast cancers, respectively (113, 114). In castration-resistant prostate cancer (CRPC) cells, the PROTAC ARD-61 can efficiently degrade AR and inhibited cancer cell proliferation with half-maximum inhibitory concentration (IC:50) values <500 nM, regardless of AR mutations, and expression status of AR splice variants, such as AR splice variant-7 (AR-V7) (102). Meanwhile, the viability of cells not expressing AR was not affected (102). Another AR degrader ARD-69 has DC50 values of <1 nM in prostate cancer cell lines LNCaP and VCaP (DC50: the concentration at which 50% of the target protein has been degraded) (115). The importance of these PROTACs as potential radiosensitizers for prostate cancer is emphasized by the study showing that targeted degradation of RT-increased AR with FDA-approved AR degradation enhancer, dimethylcurcumin (ASC-J9), significantly sensitized prostate cancer toward radiation in xenograft models, while conventional anti-androgen drugs, such as enzalutamide, has no radiosensitizing effects (114). Meanwhile, the ER degrader ERD-308 can induce over 95% of ER degradation at concentrations of <5 nM in ER+ breast cancer cell lines of MCF-7 and T47D (101). Given the synergistic effect of typical anti-estrogenic drugs with RT in the breast, and cervical cancers, these ER degraders can also be used as potential radiosensitizers alongside with RT (116). More importantly, these degraders can overcome the common resistant mechanism developed during anti-hormonal therapy.
PROTACs as Radiosensitizers—Targeting Oncogenic Kinases
It is well-known that many patients will eventually become drug resistant and develop disease relapse with prolonged treatment of protein kinase inhibitors (117). The mechanism of drug resistance is mainly attributed to the kinome rewiring effect, whereby reactivation of the oncogenic pathways restored via compensatory feedback activation of alternative kinases (117). Particularly, RT-induced activation of MEK1/2 is associated with radioresistance is found in several human malignancies (118, 119). Targeted inhibition of MEK1/2 using kinase inhibitors led to radiosensitization in several types of cancer, such as astrocytoma and pancreatic tumor (120, 121). Meanwhile, CDK inhibitors, especially those against CDK4 and CDK6, synergized with RT in killing GBM cells and prolonged survival in the orthotopic GBM model (122). However, resistance toward MEK1/2 and CDK inhibitors will inevitably occur with prolonged treatment (123).
Treatment with PROTACs will lead to catalytic degradation of specific kinases, offering sustained inhibition on their downstream targets (112). PROTACs for kinases critical for radioresistance, including CDKs, MEK1/2, ALK, and BTK, have been recently developed (103, 105–107). Given their unique pharmacological characteristics, these PROTACs carry great potential as radiosensitizers. For example, the PROTAC MS432 recruits MEK1/2 for CRL2VHL-mediated polyubiquitination (105). It can suppress extracellular signal-regulated kinase (ERK) phosphorylation and efficiently inhibit colorectal cancer cell proliferation with DC50 values of 31 nM and 17 nM for MEK1 and MEK2 in HT-29 cells, respectively (105). Meanwhile, the CDK6 degrader was recently developed by linking the FDA-approved CDK6 inhibitor palbociclib with a thalidomide derivative for targeted CDK6 polyubiquitination by CRL4CRBN (124). Given that the combination treatment of RT with CDK inhibitors palbociclib and ribociclib are well-tolerated in malignancies such as breast cancer and glioma (NCT 02607124), CDKs-targeting PROTACs are great pharmaceutical candidates for radiosensitizers (125).
Conclusion and Discussion
In summary, protein turnover is a dynamically regulated process influencing many important biological functions, including DDR, cell cycling, and signaling transductions. Pharmacological intervention of protein turnover offers a new therapeutic window for radiosensitization. Driven by their unique cytotoxic mechanisms, the novel strategies targeting the UPS with NEDDylation inhibitors and the PROTACs carry great potential as radiosensitizers to improve the efficacy of RT. The NEDDylation inhibitor MLN4924 exerts several cytotoxic functions, including DNA damage, cell cycle checkpoints dysregulation, and inhibition of NF-κB, and mTOR pathways (Figure 2). Preclinical studies had validated the efficacy of NEDDylation inhibitors as radiosensitizers. Meanwhile, recent progress in PROTAC technology has shown significant improvements in terms of the cellular permeability and substrate specificity. The PROTACs can selectively recruit key proteins related to radioresistance, such as EGFR, AR, ER, MEK1/2, and CDKs, for CRL-mediated polyubiquitin conjugation and subsequent degradation. Based on these strong basic and preclinical investigations hereby summarized, further clinical studies using NEDDylation inhibitors and PROTACs as radiosensitizers are warranted for the therapeutic gain of human malignancies.
Author Contributions
SZ wrote the original manuscript. WT edited and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by SCCC American Cancer Society (IRG) Pilot Project Grants (PI:WT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Barnett GC, West CM, Dunning AM, Elliott RM, Coles CE, Pharoah PD, et al. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer. (2009) 9:134–42. doi: 10.1038/nrc2587
2. Jagodinsky JC, Harari PM, Morris ZS. The promise of combining radiation therapy with immunotherapy. Int J Radiat Oncol Biol Phys. (2020). doi: 10.1016/j.ijrobp.2020.04.023. [Epub ahead of print].
3. Qin A, Lusk E, Daignault-Newton S, Schneider BJ. Chemotherapy and radiation versus chemotherapy alone for elderly patients with N3 stage IIIB NSCLC. Clin Lung Cancer. (2019) 20:e495–503. doi: 10.1016/j.cllc.2019.04.003
4. Morris ZS, Harari PM. Interaction of radiation therapy with molecular targeted agents. J Clin Oncol. (2014) 32:2886–93. doi: 10.1200/JCO.2014.55.1366
5. Anker CJ, Grossmann KF, Atkins MB, Suneja G, Tarhini AA, Kirkwood JM. Avoiding severe toxicity from combined BRAF inhibitor and radiation treatment: consensus guidelines from the eastern cooperative oncology group (ECOG). Int J Radiat Oncol Biol Phys. (2016) 95:632–46. doi: 10.1016/j.ijrobp.2016.01.038
6. Clement-Zhao A, Tanguy ML, Cottu P, De La Lande B, Bontemps P, Lemanski C, et al. Toxicity of locoregional radiotherapy in combination with bevacizumab in patients with non-metastatic breast cancer (TOLERAB): final long-term evaluation. PLoS ONE. (2019) 14:e0221816. doi: 10.1371/journal.pone.0221816
7. Barazzuol L, Jeynes JC, Merchant MJ, Wera AC, Barry MA, Kirkby KJ, et al. Radiosensitization of glioblastoma cells using a histone deacetylase inhibitor (SAHA) comparing carbon ions with X-rays. Int J Radiat Biol. (2015) 91:90–8. doi: 10.3109/09553002.2014.946111
8. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. (2018) 17:353–77. doi: 10.1038/nrd.2018.21
9. Zhao Y, Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Des. (2013) 19:3215–25. doi: 10.2174/13816128113199990300
10. Duan H, Wang Y, Aviram M, Swaroop M, Loo JA, Bian J, et al. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol. (1999) 19:3145–55. doi: 10.1128/MCB.19.4.3145
11. Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. (2018) 44:92–102. doi: 10.1016/j.cellsig.2018.01.009
12. Reynders M, Matsuura BS, Berouti M, Simoneschi D, Marzio A, Pagano M, et al. PHOTACs enable optical control of protein degradation. Sci Adv. (2020) 6:eaay5064. doi: 10.1126/sciadv.aay5064
13. Wardman P. Chemical radiosensitizers for use in radiotherapy. Clin Oncol (R Coll Radiol). (2007) 19:397–417. doi: 10.1016/j.clon.2007.03.010
14. Imai R, Kamada T, Araki N, Working Group for Bone and Soft Tissue Sarcomas. Carbon ion radiation therapy for unresectable sacral chordoma: an analysis of 188 cases. Int J Radiat Oncol Biol Phys. (2016) 95:322–7. doi: 10.1016/j.ijrobp.2016.02.012
15. Lin A, Maity A. Molecular pathways: a novel approach to targeting hypoxia and improving radiotherapy efficacy via reduction in oxygen demand. Clin Cancer Res. (2015) 21:1995–2000. doi: 10.1158/1078-0432.CCR-14-0858
16. Wang H, Mu X, He H, Zhang XD. Cancer radiosensitizers. Trends Pharmacol Sci. (2018) 39:24–48. doi: 10.1016/j.tips.2017.11.003
17. Buckley AM, Lynam-Lennon N, O'Neill H, O'Sullivan J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat Rev Gastroenterol Hepatol. (2020) 17:298–313. doi: 10.1038/s41575-019-0247-2
18. Wilson GD, Bentzen SM, Harari PM. Biologic basis for combining drugs with radiation. Semin Radiat Oncol. (2006) 16:2–9. doi: 10.1016/j.semradonc.2005.08.001
19. McGinn CJ, Lawrence TS. Recent advances in the use of radiosensitizing nucleosides. Semin Radiat Oncol. (2001) 11:270–80. doi: 10.1053/srao.2001.26002
20. Boeckman HJ, Trego KS, Turchi JJ. Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of nonhomologous end joining. Mol Cancer Res. (2005) 3:277–85. doi: 10.1158/1541-7786.MCR-04-0032
21. Orth M, Unger K, Schoetz U, Belka C, Lauber K. Taxane-mediated radiosensitization derives from chromosomal missegregation on tripolar mitotic spindles orchestrated by AURKA and TPX2. Oncogene. (2018) 37:52–62. doi: 10.1038/onc.2017.304
22. Hei TK, Piao CQ, Geard CR, Hall EJ. Taxol and ionizing radiation: interaction and mechanisms. Int J Radiat Oncol Biol Phys. (1994) 29:267–71. doi: 10.1016/0360-3016(94)90273-9
23. Grimes DR, Partridge M. A mechanistic investigation of the oxygen fixation hypothesis and oxygen enhancement ratio. Biomed Phys Eng Express. (2015) 1:045209. doi: 10.1088/2057-1976/1/4/045209
24. Metwally MA, Frederiksen KD, Overgaard J. Compliance and toxicity of the hypoxic radiosensitizer nimorazole in the treatment of patients with head and neck squamous cell carcinoma (HNSCC). Acta Oncol. (2014) 53:654–61. doi: 10.3109/0284186X.2013.864050
25. Dische S, Bennett MH, Orchard R, Stratford MR, Wardman P. The uptake of the radiosensitizing compound Ro 03-8799 (Pimonidazole) in human tumors. Int J Radiat Oncol Biol Phys. (1989) 16:1089–92. doi: 10.1016/0360-3016(89)90923-1
26. Astolfi L, Ghiselli S, Guaran V, Chicca M, Simoni E, Olivetto E, et al. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: a retrospective evaluation. Oncol Rep. (2013) 29:1285–92. doi: 10.3892/or.2013.2279
27. Shah JJ, Jakubowiak AJ, O'Connor OA, Orlowski RZ, Harvey RD, Smith MR, et al. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. (2016) 22:34–43. doi: 10.1158/1078-0432.CCR-15-1237
28. Meyer-Schwesinger C. The ubiquitin-proteasome system in kidney physiology and disease. Nat Rev Nephrol. (2019) 15:393–411. doi: 10.1038/s41581-019-0148-1
29. Dimopoulos MA, Goldschmidt H, Niesvizky R, Joshua D, Chng WJ, Oriol A, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. (2017) 18:1327–37. doi: 10.1016/S1470-2045(17)30578-8
30. Lao CD, Friedman J, Tsien CI, Normolle DP, Chapman C, Cao Y, et al. Concurrent whole brain radiotherapy and bortezomib for brain metastasis. Radiat Oncol. (2013) 8:204. doi: 10.1186/1748-717X-8-204
31. Kong XT, Nguyen NT, Choi YJ, Zhang G, Nguyen HN, Filka E, et al. Phase 2 study of bortezomib combined with temozolomide and regional radiation therapy for upfront treatment of patients with newly diagnosed glioblastoma multiforme: safety and efficacy assessment. Int J Radiat Oncol Biol Phys. (2018) 100:1195–203. doi: 10.1016/j.ijrobp.2018.01.001
32. Argiris A, Duffy AG, Kummar S, Simone NL, Arai Y, Kim SW, et al. Early tumor progression associated with enhanced EGFR signaling with bortezomib, cetuximab, and radiotherapy for head and neck cancer. Clin Cancer Res. (2011) 17:5755–64. doi: 10.1158/1078-0432.CCR-11-0861
33. Pugh TJ, Chen C, Rabinovitch R, Eckhardt SG, Rusthoven KE, Swing R, et al. Phase I trial of bortezomib and concurrent external beam radiation in patients with advanced solid malignancies. Int J Radiat Oncol Biol Phys. (2010) 78:521–6. doi: 10.1016/j.ijrobp.2009.07.1715
34. Yu Q, Jiang Y, Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. (2020) 10:746–65. doi: 10.1016/j.apsb.2019.09.005
35. Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. (2011) 11:347–56. doi: 10.2174/156800911794519734
36. Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL, et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. (2003) 12:1427–37. doi: 10.1016/S1097-2765(03)00452-0
37. Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. (2009) 33:483–95. doi: 10.1016/j.molcel.2009.01.011
38. Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. (2004) 118:83–97. doi: 10.1016/j.cell.2004.06.016
39. Kamura T, Conrad MN, Yan Q, Conaway RC, Conaway JW. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. (1999) 13:2928–33. doi: 10.1101/gad.13.22.2928
40. Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. (2008) 134:995–1006. doi: 10.1016/j.cell.2008.07.022
41. Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. (2008) 32:21–31. doi: 10.1016/j.molcel.2008.08.021
42. Baek K, Krist DT, Prabu JR, Hill S, Klugel M, Neumaier LM, et al. NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly. Nature. (2020) 578:461–6. doi: 10.1038/s41586-020-2000-y
43. Sailo BL, Banik K, Girisa S, Bordoloi D, Fan L, Halim CE, et al. FBXW7 in cancer: what has been unraveled thus far? Cancers. (2019) 11:246. doi: 10.3390/cancers11020246
44. Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. (2005) 123:1107–20. doi: 10.1016/j.cell.2005.09.033
45. Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. (2005) 6:9–20. doi: 10.1038/nrm1547
46. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. (2009) 458:732–6. doi: 10.1038/nature07884
47. Muraoka H, Yoshimura C, Kawabata R, Tsuji S, Hashimoto A, Ochiiwa H, et al. Activity of TAS4464, a novel NEDD8 activating enzyme E1 inhibitor, against multiple myeloma via inactivation of nuclear factor kappaB pathways. Cancer Sci. (2019) 110:3802–10. doi: 10.1111/cas.14209
48. Yoshimura C, Muraoka H, Ochiiwa H, Tsuji S, Hashimoto A, Kazuno H, et al. TAS4464, A highly potent and selective inhibitor of NEDD8-activating enzyme, suppresses neddylation and shows antitumor activity in diverse cancer models. Mol Cancer Ther. (2019) 18:1205–16. doi: 10.1158/1535-7163.MCT-18-0644
49. Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. (2010) 115:3796–800. doi: 10.1182/blood-2009-11-254862
50. Gu Y, Kaufman JL, Bernal L, Torre C, Matulis SM, Harvey RD, et al. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood. (2014) 123:3269–76. doi: 10.1182/blood-2013-08-521914
51. Zhao Y, Xiong X, Jia L, Sun Y. Targeting cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. (2012) 3:e386. doi: 10.1038/cddis.2012.125
52. Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. (2014) 20:1576–89. doi: 10.1158/1078-0432.CCR-13-0987
53. Jia L, Li H, Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. (2011) 13:561–9. doi: 10.1593/neo.11420
54. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. (2010) 70:10310–20. doi: 10.1158/0008-5472.CAN-10-2062
55. Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. (2011) 44:304–16. doi: 10.1016/j.molcel.2011.08.029
56. Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, et al. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. (2003) 17:3062–74. doi: 10.1101/gad.1157503
57. Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature. (2003) 423:885–9. doi: 10.1038/nature01747
58. Lovejoy CA, Lock K, Yenamandra A, Cortez D. DDB1 maintains genome integrity through regulation of Cdt1. Mol Cell Biol. (2006) 26:7977–90. doi: 10.1128/MCB.00819-06
59. Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. (2003) 114:599–610. doi: 10.1016/j.cell.2003.08.001
60. Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, et al. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell. (2009) 35:442–53. doi: 10.1016/j.molcel.2009.06.030
61. Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. (2010) 40:9–21. doi: 10.1016/j.molcel.2010.09.014
62. Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. (2006) 23:709–21. doi: 10.1016/j.molcel.2006.08.010
63. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. (2008) 22:2496–506. doi: 10.1101/gad.1676108
64. Morgan MA, Lawrence TS. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin Cancer Res. (2015) 21:2898–904. doi: 10.1158/1078-0432.CCR-13-3229
65. Nishitani H, Taraviras S, Lygerou Z, Nishimoto T. The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J Biol Chem. (2001) 276:44905–11. doi: 10.1074/jbc.M105406200
66. Li X, Zhao Q, Liao R, Sun P, Wu X. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J Biol Chem. (2003) 278:30854–8. doi: 10.1074/jbc.C300251200
67. Paiva C, Godbersen JC, Berger A, Brown JR, Danilov AV. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic leukemia B cells to alkylating agents. Cell Death Dis. (2015) 6:e1807. doi: 10.1038/cddis.2015.161
68. Vanderdys V, Allak A, Guessous F, Benamar M, Read PW, Jameson MJ, et al. The neddylation inhibitor pevonedistat (MLN4924) suppresses and radiosensitizes head and neck squamous carcinoma cells and tumors. Mol Cancer Ther. (2018) 17:368–80. doi: 10.1158/1535-7163.MCT-17-0083
69. Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. (2012) 72:282–93. doi: 10.1158/0008-5472.CAN-11-2866
70. Fouad S, Wells OS, Hill MA, D'Angiolella V. Cullin ring ubiquitin ligases (CRLs) in cancer: responses to ionizing radiation (IR) treatment. Front Physiol. (2019) 10:1144. doi: 10.3389/fphys.2019.01144
71. Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. (2016) 17:102. doi: 10.3390/ijms17010102
72. Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM signaling network in development and disease. Front Genet. (2013) 4:37. doi: 10.3389/fgene.2013.00037
73. Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. (2003) 4:671–7. doi: 10.1038/sj.embor.embor887
74. Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. (2003) 426:87–91. doi: 10.1038/nature02082
75. Jang SM, Redon CE, Aladjem MI. Chromatin-bound cullin-ring ligases: regulatory roles in DNA replication and potential targeting for cancer therapy. Front Mol Biosci. (2018) 5:19. doi: 10.3389/fmolb.2018.00019
76. Lu Z, Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle. (2010) 9:2342–52. doi: 10.4161/cc.9.12.11988
77. Wan J, Zhu J, Li G, Zhang Z. Radiosensitization of human colorectal cancer cells by MLN4924: an inhibitor of NEDD8-activating enzyme. Technol Cancer Res Treat. (2016) 15:527–34. doi: 10.1177/1533034615588197
78. Yang D, Tan M, Wang G, Sun Y. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS ONE. (2012) 7:e34079. doi: 10.1371/journal.pone.0034079
79. Wang X, Zhang W, Yan Z, Liang Y, Li L, Yu X, et al. Radiosensitization by the investigational NEDD8-activating enzyme inhibitor MLN4924 (pevonedistat) in hormone-resistant prostate cancer cells. Oncotarget. (2016) 7:38380–91. doi: 10.18632/oncotarget.9526
80. Hein AL, Ouellette MM, Yan Y. Radiation-induced signaling pathways that promote cancer cell survival (review). Int J Oncol. (2014) 45:1813–9. doi: 10.3892/ijo.2014.2614
81. Valerie K, Yacoub A, Hagan MP, Curiel DT, Fisher PB, Grant S, et al. Radiation-induced cell signaling: inside-out and outside-in. Mol Cancer Ther. (2007) 6:789–801. doi: 10.1158/1535-7163.MCT-06-0596
82. Zeng J, Zhang H, Tan Y, Sun C, Liang Y, Yu J, et al. Aggregation of lipid rafts activates c-met and c-Src in non-small cell lung cancer cells. BMC Cancer. (2018) 18:611. doi: 10.1186/s12885-018-4501-8
83. Karimian A, Mir SM, Parsian H, Refieyan S, Mirza-Aghazadeh-Attari M, Yousefi B, et al. Crosstalk between phosphoinositide 3-kinase/Akt signaling pathway with DNA damage response and oxidative stress in cancer. J Cell Biochem. (2019) 120:10248–72. doi: 10.1002/jcb.28309
84. Bai M, Ma X, Li X, Wang X, Mei Q, Li X, et al. The accomplices of NF-κB lead to radioresistance. Curr Protein Pept Sci. (2015) 16:279–94. doi: 10.2174/138920371604150429152328
85. Giridharan S, Srinivasan M. Mechanisms of NF-kappaB p65 and strategies for therapeutic manipulation. J Inflamm Res. (2018) 11:407–19. doi: 10.2147/JIR.S140188
86. Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. (1998) 396:590–4. doi: 10.1038/25159
87. Russell JS, Raju U, Gumin GJ, Lang FF, Wilson DR, Huet T, et al. Inhibition of radiation-induced nuclear factor-kappaB activation by an anti-Ras single-chain antibody fragment: lack of involvement in radiosensitization. Cancer Res. (2002) 62:2318–26.
88. Tan M, Zhu Y, Kovacev J, Zhao Y, Pan ZQ, Spitz DR, et al. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kappaB activation in mouse embryonic stem cells. Free Radic Biol Med. (2010) 49:976–83. doi: 10.1016/j.freeradbiomed.2010.05.030
89. Pordanjani SM, Hosseinimehr SJ. The role of NF-kB inhibitors in cell response to radiation. Curr Med Chem. (2016) 23:3951–63. doi: 10.2174/0929867323666160824162718
90. Narayan V, Vapiwala N, Mick R, Subramanian P, Christodouleas JP, Bekelman JE, et al. Phase 1 trial of everolimus and radiation therapy for salvage treatment of biochemical recurrence in prostate cancer patients following prostatectomy. Int J Radiat Oncol Biol Phys. (2017) 97:355–61. doi: 10.1016/j.ijrobp.2016.10.013
91. Chinnaiyan P, Won M, Wen PY, Rojiani AM, Werner-Wasik M, Shih HA, et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: results of NRG oncology RTOG 0913. Neuro Oncol. (2018) 20:666–73. doi: 10.1093/neuonc/nox209
92. Waqar SN, Robinson C, Bradley J, Goodgame B, Rooney M, Williams K, et al. A phase I study of temsirolimus and thoracic radiation in non–small-cell lung cancer. Clin Lung Cancer. (2014) 15:119–23. doi: 10.1016/j.cllc.2013.11.007
93. Guo N, Azadniv M, Coppage M, Nemer M, Mendler J, Becker M, et al. Effects of neddylation and mTOR inhibition in acute myelogenous leukemia. Transl Oncol. (2019) 12:602–13. doi: 10.1016/j.tranon.2019.01.001
94. Zheng S, Leclerc GM, Li B, Swords RT, Barredo JC. Inhibition of the NEDD8 conjugation pathway induces calcium-dependent compensatory activation of the pro-survival MEK/ERK pathway in acute lymphoblastic leukemia. Oncotarget. (2018) 9:5529–44. doi: 10.18632/oncotarget.23797
95. Leclerc GM, Zheng S, Leclerc GJ, DeSalvo J, Swords RT, Barredo JC. The NEDD8-activating enzyme inhibitor pevonedistat activates the eIF2α and mTOR pathways inducing UPR-mediated cell death in acute lymphoblastic leukemia. Leuk Res. (2016) 50:1–10. doi: 10.1016/j.leukres.2016.09.007
96. Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, et al. PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther. (2019) 4:64. doi: 10.1038/s41392-019-0101-6
97. Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, et al. Light-induced control of protein destruction by opto-PROTAC. Sci Adv. (2020) 6:eaay5154. doi: 10.1126/sciadv.aay5154
98. Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. (2019) 50:111–9. doi: 10.1016/j.cbpa.2019.02.022
99. Cully M. PROTAC moves in on lymphoma. Nat Rev Drug Discov. (2019) 18:584. doi: 10.1038/d41573-019-00113-9
100. Cheng M, Yu X, Lu K, Xie L, Wang L, Meng F, et al. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J Med Chem. (2020) 63:1216–32. doi: 10.1021/acs.jmedchem.9b01566
101. Hu J, Hu B, Wang M, Xu F, Miao B, Yang CY, et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J Med Chem. (2019) 62:1420–42. doi: 10.1021/acs.jmedchem.8b01572
102. Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. (2020) 22:111–9. doi: 10.1016/j.neo.2019.12.003
103. Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES, et al. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem Biol. (2019) 26:300–6.e9. doi: 10.1016/j.chembiol.2018.11.006
104. Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol. (2018) 14:405–12. doi: 10.1038/s41589-018-0010-y
105. Wei J, Hu J, Wang L, Xie L, Jin MS, Chen X, et al. Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J Med Chem. (2019) 62:10897–911. doi: 10.1021/acs.jmedchem.9b01528
106. Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. (2019) 133:952–61. doi: 10.1182/blood-2018-07-862953
107. Powell CE, Gao Y, Tan L, Donovan KA, Nowak RP, Loehr A, et al. Chemically induced degradation of anaplastic lymphoma kinase (ALK). J Med Chem. (2018) 61:4249–55. doi: 10.1021/acs.jmedchem.7b01655
108. Byeon HK, Ku M, Yang J. Beyond EGFR inhibition: multilateral combat strategies to stop the progression of head and neck cancer. Exp Mol Med. (2019) 51:1–14. doi: 10.1038/s12276-018-0202-2
109. Su Y, Cui J, Xu D, Wang M, Xu T, Tian H, et al. p16(INK4a) status and survival benefit of EGFR inhibitors in head and neck squamous cell cancer: a systematic review and meta-analysis. Crit Rev Oncol Hematol. (2018) 124:11–20. doi: 10.1016/j.critrevonc.2018.02.006
110. Eskilsson E, Rosland GV, Solecki G, Wang Q, Harter PN, Graziani G, et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. (2018) 20:743–52. doi: 10.1093/neuonc/nox191
111. Cai W, Yang H. The structure and regulation of cullin 2 based E3 ubiquitin ligases and their biological functions. Cell Div. (2016) 11:7. doi: 10.1186/s13008-016-0020-7
112. Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. (2018) 25:67–77.e3. doi: 10.1016/j.chembiol.2017.09.009
113. Rong C, Meinert E, Hess J. Estrogen receptor signaling in radiotherapy: from molecular mechanisms to clinical studies. Int J Mol Sci. (2018) 19:713. doi: 10.3390/ijms19030713
114. Chou FJ, Chen Y, Chen D, Niu Y, Li G, Keng P, et al. Preclinical study using androgen receptor (AR) degradation enhancer to increase radiotherapy efficacy via targeting radiation-increased AR to better suppress prostate cancer progression. EBioMedicine. (2019) 40:504–16. doi: 10.1016/j.ebiom.2018.12.050
115. Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J Med Chem. (2019) 62:941–64. doi: 10.1021/acs.jmedchem.8b01631
116. Segovia-Mendoza M, Jurado R, Mir R, Medina LA, Prado-Garcia H, Garcia-Lopez P. Antihormonal agents as a strategy to improve the effect of chemo-radiation in cervical cancer: in vitro and in vivo study. BMC Cancer. (2015) 15:21. doi: 10.1186/s12885-015-1016-4
117. Wilson LJ, Linley A, Hammond DE, Hood FE, Coulson JM, MacEwan DJ, et al. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. (2018) 78:15–29. doi: 10.1158/0008-5472.CAN-17-2291
118. Barker FG II, Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, et al. EGFR overexpression and radiation response in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. (2001) 51:410–8. doi: 10.1016/S0360-3016(01)01609-1
119. Kim DW, Choy H. Potential role for epidermal growth factor receptor inhibitors in combined-modality therapy for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. (2004) 59(Suppl. 2):11–20. doi: 10.1016/j.ijrobp.2003.11.042
120. Studebaker A, Bondra K, Seum S, Shen C, Phelps DA, Chronowski C, et al. Inhibition of MEK confers hypersensitivity to X-radiation in the context of BRAF mutation in a model of childhood astrocytoma. Pediatr Blood Cancer. (2015) 62:1768–74. doi: 10.1002/pbc.25579
121. Estrada-Bernal A, Chatterjee M, Haque SJ, Yang L, Morgan MA, Kotian S, et al. MEK inhibitor GSK1120212-mediated radiosensitization of pancreatic cancer cells involves inhibition of DNA double-strand break repair pathways. Cell Cycle. (2015) 14:3713–24. doi: 10.1080/15384101.2015.1104437
122. Whittaker S, Madani D, Joshi S, Chung SA, Johns T, Day B, et al. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. (2017) 3:17033. doi: 10.1038/cddiscovery.2017.33
123. Poulikakos PI, Solit DB. Resistance to MEK inhibitors: should we co-target upstream? Sci Signal. (2011) 4:pe16. doi: 10.1126/scisignal.2001948
124. Su S, Yang Z, Gao H, Yang H, Zhu S, An Z, et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem. (2019) 62:7575–82. doi: 10.1021/acs.jmedchem.9b00871
Keywords: NEDDylation, EGFR, PROTAC, cullin-RING E3 ligase, MLN4924
Citation: Zheng S and Tao W (2020) Targeting Cullin-RING E3 Ligases for Radiosensitization: From NEDDylation Inhibition to PROTACs. Front. Oncol. 10:1517. doi: 10.3389/fonc.2020.01517
Received: 17 June 2020; Accepted: 15 July 2020;
Published: 21 August 2020.
Edited by:
Zhe-Sheng Chen, St. John's University, United StatesReviewed by:
Tarek Abbas, University of Virginia, United StatesMikihiko Naito, National Institute of Health Sciences (NIHS), Japan
Copyright © 2020 Zheng and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuhua Zheng, c3oyMjdAbXluc3Uubm92YS5lZHU=; Wensi Tao, d3Rhb0BtZWQubWlhbWkuZWR1