 Coralie Reger de Moura1,2
Coralie Reger de Moura1,2 Marco Prunotto3Anjum Sohail4Maxime Battistella2,5Fanelie Jouenne1,2
Marco Prunotto3Anjum Sohail4Maxime Battistella2,5Fanelie Jouenne1,2 Daniel Marbach6Celeste Lebbé2,7
Daniel Marbach6Celeste Lebbé2,7 Rafael Fridman4
Rafael Fridman4 Samia Mourah1,2*
Samia Mourah1,2*- 1Laboratory of Pharmacogenomics, Hôpital Saint-Louis, AP-HP, Paris, France
- 2INSERM, UMR_S976, Université de Paris, Paris, France
- 3School of Pharmaceutical Sciences, University of Geneva, Geneva, Switzerland
- 4Department of Pathology, School of Medicine, Karmanos Cancer Institute, Wayne State University, Detroit, MI, United States
- 5Department of Pathology, Hôpital Saint-Louis, AP-HP, Paris, France
- 6Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Basel, Switzerland
- 7Department of Dermatology, Hôpital Saint-Louis, AP-HP, Paris, France
Melanoma is a highly malignant skin cancer with high propensity to metastasize and develop drug resistance, making it a difficult cancer to treat. Current therapies targeting BRAF (V600) mutations are initially effective, but eventually tumors overcome drug sensitivity and reoccur. This process is accomplished in part by reactivating alternate signaling networks that reinstate melanoma proliferative and survival capacity, mostly through reprogramming of receptor tyrosine kinase (RTK) signaling. Evidence indicates that the discoidin domain receptors (DDRs), a set of RTKs that signal in response to collagen, are part of the kinome network that confer drug resistance. We previously reported that DDR1 is expressed in melanomas, where it can promote tumor malignancy in mouse models of melanoma, and thus, DDR1 could be a promising target to overcome drug resistance. In this review, we summarize the current knowledge on DDRs in melanoma and their implication for therapy, with emphasis in resistance to MAPK inhibitors.
DDR1, a Worse Prognostic Biomarker and an Emerging Target
Among the receptor families known to mediate the interaction of cells with collagen, the discoidin domain receptors (DDRs) constitute a major class. The DDRs are RTKs which undergo activation upon binding to collagens. There are two members in the DDR family, DDR1 and DDR2, with DDR1 comprising 5 isoforms, two of which are inactive or truncated receptors. There is only one DDR2 isoform. Structurally, full-length DDRs are multidomain type I transmembrane glycoproteins, comprising an extracellular discoidin domain, a transmembrane region, and an intracellular segment that includes a kinase domain [for structural details of DDRs see (1, 2)]. The reason for diversity in DDR1 isoforms is still unknown, but their structural differences may be necessary to activate distinct signaling pathways. The ability of DDRs to recognize collagens as ligands places these receptors in a unique category among the RTK family because the collagen family is comprised of 28 distinct members with different structural organizations, biomechanical properties, and tissue distributions (3). To recognize and respond to the various members of the collagen superfamily under various conditions and in different tissue locations, DDRs become versatile kinases, able to interact with distinct collagen types and initiate the downstream pathway in response to alterations on collagen properties in diverse physiological and pathological conditions. DDRs undergo receptor autophosphorylation in response to both fibril- and network-forming collagens. For instance, DDR1 and DDR2 are activated by several fibrillar collagens, albeit with different efficiencies. However, both receptors are efficiently activated by the ubiquitous fibrillar collagen type I (2, 4–6). In contrast, DDRs differentially respond to the network-forming collagen IV and X, with DDR1 being activated by collagen IV while DDR2 by collagen X (6). The ability of DDRs to recognize distinct collagen types has significant implications in conditions in which cells traffic through different tissue compartments. In cancer, for instance, premalignant and fully malignant carcinoma cells can express DDR1. Thus, as cells progress from normalcy to malignancy and acquire the ability to invade basement membranes (BM) and the underlying stromal matrix, the expression of DDR1 may modulate cancer cell behavior in response to both collagen IV and collagen I, possibly by initiating ligand-specific signaling pathways. On the other hand, DDR2, which is not usually expressed in epithelial cells, has been shown to be induced during the process of epithelial-to-mesenchymal transition (EMT), a molecular and cellular program that has been associated with enhanced invasive capacity (7, 8). Thus, DDR2 together with DDR1 may contribute to the activation of signaling pathways associated with interactions of carcinoma cells with both network-forming and fibrillar collagens, as they traffic through various matrix compartments. Although DDRs are implicated in normal organ development and function (2), there is multiple evidence showing that DDRs are critical players in cancer progression, regulating multiple aspects of malignancy including cell proliferation, migration, invasion, and drug resistance (9, 10). These effects of DDRs on malignant cell behavior appear to be mediated mostly via collagen-dependent receptor phosphorylation; however, evidence has shown that DDRs can also elicit pro-malignant activities in a kinase-independent manner (11, 12). In this regard, these studies highlight the importance DDR–collagen interaction through the discoidin domain, independent of kinase activity, in mediating the functions of DDRs in cells. However, more data are needed to establish a clear distinction between collagen-independent and -dependent effects of DDRs in cancer progression. While there is consensus on the pro-malignant effects of DDR2 in cancer, this is not the case for DDR1. Indeed, evidence suggests that DDR1 can elicit either tumor-promoting or -suppressing effects on cancer in a context-dependent manner [reviewed in (13)], possibly due to the fact that DDR1 plays a role in the maintenance of normal epithelial integrity by regulating cell–BM and cell–cell interactions (14–17). On the other hand, many studies have shown that overexpression of DDR1 in several cancer types correlates with disease progression (18–20). However, it is important to note that expression analyses in tissue samples are limited because a pro-malignant role for any gene cannot be asserted without functional studies. Regardless, the emerging picture for DDR1’s role in cancer progression is complex, likely involving tumor-suppressive and/or promotive effects. In this review, we will focus on DDR1 and melanoma, and its potential role in promoting malignant features and as a potential target to overcome drug resistance.
Melanoma-Targeted Therapies and Resistance
Over the past few years, numerous therapies have emerged in the management of advanced melanoma, which have profoundly transformed the therapeutic landscape and prognosis of this disease. Drug development has been driven by the unveiling of the molecular characteristics of melanomas, which provided new insights into the signaling networks that are operative in this disease (21). The mitogen-activated protein kinase (MAPK) pathway was found to be dysregulated in a significant proportion of melanomas. This dysregulation is mostly caused by the fact that the majority of melanomas harbor a mutation on the serine–threonine kinase BRAF (V600), which is part of the MAPK signaling pathway. Overall, over 90% of BRAFV600-mutated melanomas harbor a BRAFV600E mutation, 6% a BRAFV600R mutation, and 4% a BRAFV600E2, BRAFV600D, or a BRAFV600K mutation (22), and therefore mutated BRAF kinase became an attractive therapeutic target (23). As a result, several inhibitors (vemurafenib, dabrafenib, and encorafenib) were developed, which improved survival of melanoma patients when compared to conventional chemotherapy (24–26). Almost at the same time, inhibitors of MEK, a downstream signaling kinase of the MAPK pathway, were developed. These compounds (trametinib, cobimetinib, and binimetinib) also exhibited significant activity in BRAFV600-mutant melanoma (27, 28). Clinical trials evaluating the combination of BRAF inhibitors (BRAFi) with MEK inhibitors (MEKi) demonstrated significant clinical efficacy, as indicated by improved overall survival (OS) and progression-free survival (PFS). These promising results led to the approval of dabrafenib/trametinib and vemurafenib/cobimetinib combinations for patients with advanced, metastatic BRAFV600-mutant melanoma (29–31).
Despite these advances in melanoma treatment, acquired resistance to MAPK-targeted therapy is almost inescapable (32), and, as expected, resistance to BRAFi and MEKi was also found in melanoma patients. The mechanisms of resistance to MAPK inhibition are multiple and complex. In many cases, resistance is caused by reactivation of the MAPK pathway (RAS mutation, MEK and/or BRAF amplification, differential splicing leading to truncated variants of BRAF, activation of MAPKK), activation of the PI3K pathway through genetic alterations of PTEN, overexpression and activation of PDGF, IGF1, or c-Met receptors, or development of a pro-oncogenic tumor microenvironment (33–36). Another mechanism of resistance involves the action of ERBB3, a member of the EGF family of receptors, which is known to be over-expressed in human melanoma. Evidence has shown that BRAFi and MEKi therapeutic effects on BRAFV600-treated tumors are decreased by enhanced ERBB3 signaling, suggesting that ERBB3 is implicated in adaptive resistance to BRAF and MEK inhibitors. These observation suggested that a combination of ERBB2/EGFR inhibitor, which block NRG1/ERBB3 signaling, with BRAF and MEK inhibitors could overcome resistance (37, 38). It is worth mentioning that, eventually, these multiple and distinct mechanisms of resistance to MAPK inhibitors result in ERK reactivation, demonstrating the extent to which melanoma cells are addicted to ERK signaling for proliferation and/or survival (36). Recently, characterization of tumor cell and stromal/immune transcriptomic alterations in MAPKi-treated melanomas provided insight into the responses elicited by these inhibitors, even at early stages of treatment (39). Song et al. showed that an immune-phenotypic transition due to MAPK-targeted therapies could involve a loss of T-cell inflammation leading to an anti-PD1 resistance in melanoma, even at early stages of treatment. These studies suggested that several adaptive responses in both the tumor (intrinsic) and the immune system (extrinsic) are operative, which could offer new therapeutic opportunities to overcome resistance. The studies of Yan et al. also demonstrated that BRAF and MEK inhibitor-treated patients, showing complete responses, have preexisting tumor immunity transcriptomic signatures that are higher than those expressed in patients with progressive disease, suggesting that enriched immune infiltration improves response to BRAFi and MEKi combination (40). These observations highlight the crucial need for a better understanding of treatment effects on both the tumor and its microenvironment but also for more effective therapies aimed at overcoming or preventing drug resistance in melanoma patients. Several alternative strategies, including paradox breaker RAF inhibitors and ERK inhibitors, are currently under investigation in the BRAFi + MEKi resistance setting (41, 42). In this context, the validation of new and promising targets is the cornerstone of this challenge. Because DDRs are crucial regulators of tumor cell behavior in response to their immediate microenvironment and in light of our recent data on DDR1 in melanoma (43), we propose that DDR1 targeting in melanomas resistant to MAPK inhibitors is worth exploring.
DDR1 in Melanoma
Melanomas are derived from melanocytes, the melanin-producing cells in the epidermis. Melanocytes are located in the basal layer of the epidermis, making contact with the BM (44). Previous evidence demonstrated a role for DDR1, a major collagen IV receptor, in mediating the interaction of melanocytes with the BM. Adhesion of melanocytes to collagen IV, induced by overexpression of the matricellular protein CCN3, was mediated by upregulation of DDR1 protein expression, and silencing of DDR1 mRNA reduced CCN3-induced adhesion to collagen IV (45). However, whether this adhesive effect of DDR1 was mediated via its kinase activity was not determined. Regardless, CCN3-mediated DDR1 upregulation was proposed to play a major role in the adhesion of melanocytes to the BM and in the maintenance of skin homeostasis (45). While these in vitro studies suggested a role for DDR1 in melanocytes in normal skin, our recent immunohistochemical analyses in human skin sections demonstrated that DDR1 immunoreactivity was only detected in normal keratinocytes, albeit at relatively low levels of expression (43). Moreover, we found no detectable DDR1 expression in benign naevi in all cell types. Analyses of skin samples harboring melanoma showed a strong expression of DDR1 in the melanoma cells, which was positively correlated with invasive depth and patient survival. Our functional in vitro studies also showed a key role for DDR1 in melanoma cell proliferation, migration, invasion, and survival (43). Importantly, a pan-DDR inhibitor, DDR1-IN-1 (46), with higher selectivity toward DDR1 than to DDR2, decreased tumor growth in BRAF-mutated human melanoma xenograft models (43). Because melanoma and stromal cells also express DDR2, these preclinical studies with DDR1-IN-1 suggest that DDR1, and possibly DDR2, constitutes a potentially new target in melanoma (43). Based on these results, we posit that DDRs are promising therapeutic targets in BRAF-mutated melanomas.
To further examine the association between DDR1 and melanoma, we analyzed a curated set of seven non-redundant cutaneous melanoma cohorts from the cBioPortal site (47, 48). Out of a total of 667 patients, 114 (10.6%) were identified as harboring genetic alterations in DDR1. However, although the mutational burden in melanoma is higher compared to other types of cancers, no difference in survival was observed in patients harboring mutated DDR1. Analyses of TCGA database samples for DDR1 expression vs. BRAF mutational status showed DDR1 to be slightly upregulated in BRAF-mutated cancers (effect size 0.16, p = 0.00031; differential expression analysis of mutated vs. wild-type cases using a linear model) and with a similar tendency, but not statistically significant, between WT and mutated BRAF melanoma samples (effect size 0.27, p = 0.061, Figure 1A). However, we found that DDR1 and BRAF are co-expressed in the majority of skin melanoma samples (80%) regardless of BRAF mutational status (Figure 1B). Analysis of the same database for DDR1 expression vs. NRAS mutational status showed DDR1 to be slightly downregulated in all NRAS-mutated cancers (effect size −0.26, p = 0.00053) and with a similar tendency (effect size −0.29, p = 0.065, Figure 1C). As with BRAF, DDR1 and NRAS are co-expressed in almost all samples, regardless of NRAS mutational status (Figure 1D). Although not statistically significant, the analysis of the different skin melanoma subtypes showed that DDR1 expression is always high for BRAF and NF1 mutants, while there are few outliers with low DDR1 expression for RAS mutants and triple wild-type samples (Figure 1E).
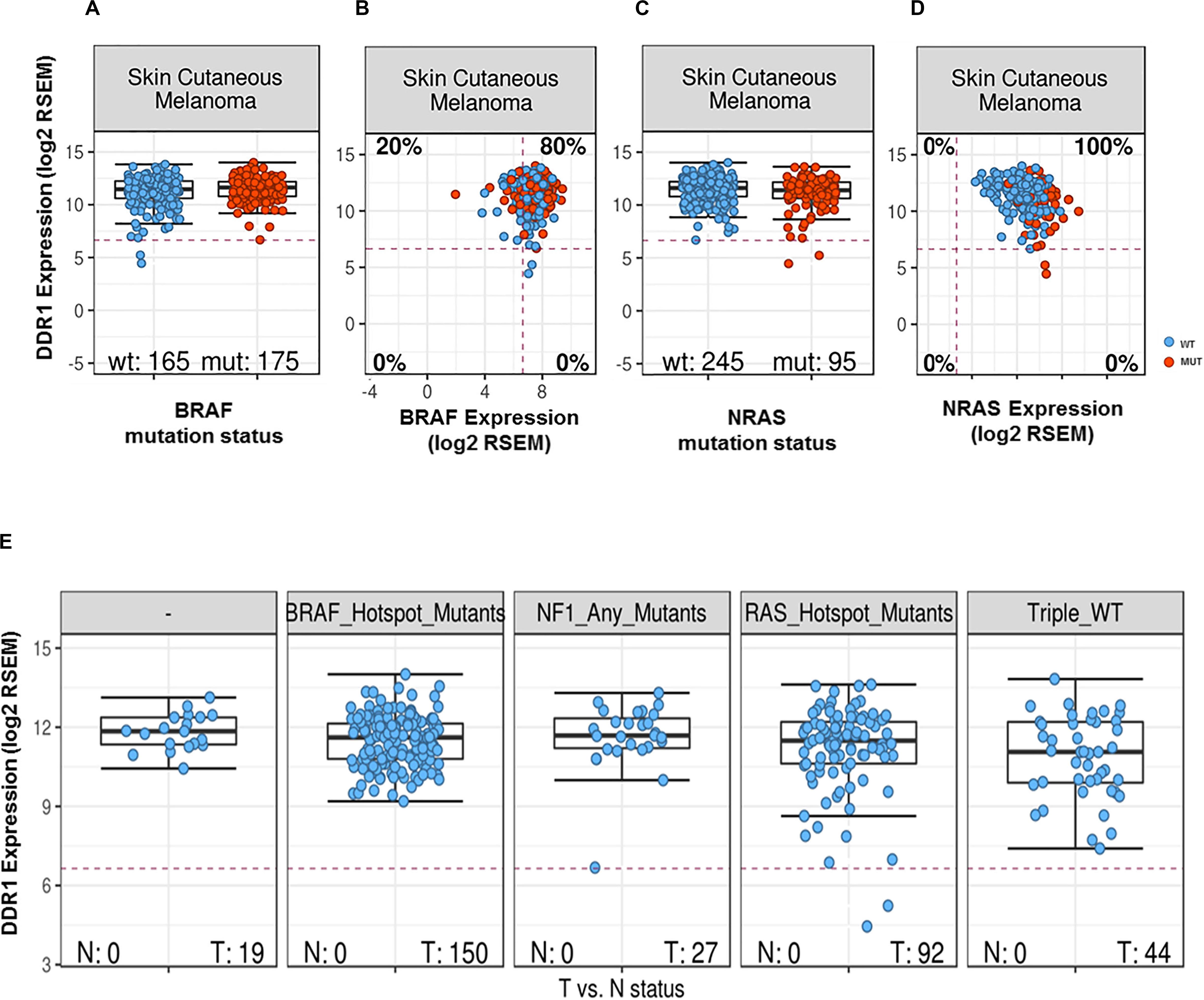
Figure 1. DDR1 expression in melanoma samples as a function of expression and mutational status of BRAF (A,B) or NRAS (C,D). Transcript abundances were quantified using RSEM (50) on a log2 scale. The red dashed lines indicate the typical cutoff for expressed genes [RSEM = log2(100)]: samples below the red lines are assumed to have no or very low expression. Classification of melanoma subtypes into mutant BRAF, mutant RAS, mutant NF1, and triple-WT (wild-type) was obtained from TCGA (51). (A) DDR1 expression for BRAF wild-type (left, blue) and mutated cases (right, red). (B) DDR1 versus BRAF expression for WT (blue) and BRAF-mutated cases (red). 80% of the samples show co-expression of DDR1 and BRAF (upper-right quadrant). (C) DDR1 expression for NRAS wild-type (left, blue) and mutated cases (right, red). (D) DDR1 versus NRAS expression for wild-type (blue) and cases with NRAS mutations (red). (E) DDR1 expression in melanoma molecular subtypes (BRAF, NRAS, NF1 mutated, and triple WT). There is no significant difference in the median expression of DDR1 (p-value > 0.05, Wilcoxon rank-sum test), but we note some outliers with low DDR1 expression in the NF1 mutant, RAS mutant, and triple WT cases (three left-most plots), while no outliers with low expression are observed for the BRAF mutant subtype. N, normal; T, tumor.
Conclusion
The emerging evidence suggests that DDRs are part of the signaling networks that contribute to melanoma progression. However, more studies are warranted to dissect the molecular mechanisms by which DDR-initiated signaling influences melanoma cell behavior. Melanomas are also characterized by a stroma rich in collagen (49), which constitutes a barrier for invading tumor cells but may also actively promote disease progression through DDR signaling. We posit that a DDR/collagen axis may contribute to the resistant phenotype of BRAF-mutated melanomas and therefore a rationale target to restore therapeutic efficacy.
Author Contributions
CR, MP, AS, RF, and SM contributed to development, writing, and final review of the article. MP and DM contributed to the database analysis. MB, FJ, DM, and CL contributed to the final review of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Institut National de la Santé et de la Recherche Medicale (INSERM), the Société Française de Dermatologie, SFD, and the Université de Paris.
Conflict of Interest
DM is employed by Roche, MP is an employee of Galapagos Ltd., at 80% employment rate, this work was performed during his 20% academic time at the University of Geneva. MB declares a consulting role for Histalim, BMS, and Innate Pharma. CL declares honoraria from Roche, advisory roles at Roche, GSK, Novartis, BMS, MSD, and Amgen and travel accommodation provided by Roche. SM declares a consulting role at Roche, Janssen, Biocartis and Novartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fu H-L, Valiathan RR, Arkwright R, Sohail A, Mihai C, Kumarasiri M, et al. Discoidin domain receptors: unique receptor tyrosine kinases in collagen-mediated signaling. J Biol Chem. (2013) 288:7430–7. doi: 10.1074/jbc.R112.444158
2. Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. (2014) 310:39–87. doi: 10.1016/B978-0-12-800180-6.00002-5
3. Gordon MK, Hahn RA. Collagens. Cell Tissue Res. (2010) 339:247–57. doi: 10.1007/s00441-009-0844-4
4. Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE, et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. (1997) 1:25–34. doi: 10.1016/S1097-2765(00)80004-0
5. Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. (1997) 1:13–23.
6. Leitinger B, Kwan APL. The discoidin domain receptor DDR2 is a receptor for type X collagen. Matrix Biol. (2006) 25:355–64. doi: 10.1016/j.matbio.2006.05.006
7. Walsh LA, Nawshad A, Medici D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol. (2011) 30:243–7. doi: 10.1016/j.matbio.2011.03.007
8. Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. (2013) 15:677–87. doi: 10.1038/ncb2743
9. Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. (2012) 31:295–321. doi: 10.1007/s10555-012-9346-z
10. Henriet E, Sala M, Abou Hammoud A, Tuariihionoa A, Di Martino J, Ros M, et al. Multitasking discoidin domain receptors are involved in several and specific hallmarks of cancer. Cell Adhes Migr. (2018) 12:363–77. doi: 10.1080/19336918.2018.1465156
11. Gao H, Chakraborty G, Zhang Z, Akalay I, Gadiya M, Gao Y, et al. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell. (2016) 166:47–62. doi: 10.1016/j.cell.2016.06.009
12. Grither WR, Longmore GD. Inhibition of tumor–microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc Natl Acad Sci USA. (2018) 115:E7786–94. doi: 10.1073/pnas.1805020115
13. Yeh YC, Lin HH, Tang MJ. Dichotomy of the function of DDR1 in cells and disease progression. Biochim Biophys Acta Mol Cell Res. (2019) 1866:118473. doi: 10.1016/j.bbamcr.2019.04.003
14. Søgaard PP, Ito N, Sato N, Fujita Y, Matter K, Itoh Y. Epithelial polarization in 3D matrix requires DDR1 signaling to regulate actomyosin contractility. Life Sci Alliance. (2019) 2:e201800276. doi: 10.26508/lsa.201800276
15. Vogel WF, Aszodi A, Alves F, Pawson T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol Cell Biol. (2001) 21:2906–17. doi: 10.1128/mcb.21.8.2906-2917.2001
16. Wang CZ, Hsu YM, Tang MJ. Function of discoidin domain receptor I in HGF-induced branching tubulogenesis of MDCK cells in collagen gel. J Cell Physiol. (2005) 203:295–304. doi: 10.1002/jcp.20227
17. Yeh Y-C, Wu C-C, Wang Y-K, Tang M-J. DDR1 triggers epithelial cell differentiation by promoting cell adhesion through stabilization of E-cadherin. Mol Biol Cell. (2011) 22:940–53. doi: 10.1091/mbc.E10-08-0678
18. Rammal H, Saby C, Magnien K, Van-Gulick L, Garnotel R, Buache E, et al. Discoidin domain receptors: potential actors and targets in cancer. Front Pharmacol. (2016) 7:55. doi: 10.3389/fphar.2016.00055
19. Jing H, Song J, Zheng J. Discoidin domain receptor 1: new star in cancer-targeted therapy and its complex role in breast carcinoma. Oncol Lett. (2018) 15:3403–8. doi: 10.3892/ol.2018.7795
20. Gadiya M, Chakraborty G. Signaling by discoidin domain receptor 1 in cancer metastasis. Cell Adhes Migr. (2018) 12:315–23. doi: 10.1080/19336918.2018.1520556
21. Flaherty KT, Hodi FS, Bastian BC. Mutation-driven drug development in melanoma. Curr Opin Oncol. (2010) 22:178–83. doi: 10.1097/cco.0b013e32833888ee
22. Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M, et al. The role of BRAF V600 mutation in melanoma. J Transl Med. (2012) 10:85. doi: 10.1186/1479-5876-10-85
23. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54.
24. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. (2012) 366:707–14. doi: 10.1056/NEJMoa1112302
25. McArthur GA, Chapman PB, Robert C, Larkin J. Safety and efficacy of vemurafenib in BRAF V600E and BRAF V600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study HHS public access. Lancet Oncol. (2014) 15:323–32. doi: 10.1016/S1470-2045(14)70012-9
26. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. (2012) 380:358–65.
27. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. (2012) 367:107–14.
28. Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. (2018) 19:603–15. doi: 10.1016/S1470-2045(18)30142-6
29. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. (2015) 372:30–9. doi: 10.1056/NEJMoa1412690
30. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. (2015) 386:444–51.
31. Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. (2016) 17:1248–60. doi: 10.1016/S1470-2045(16)30122-X
32. Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. (2015) 51:2792–9. doi: 10.1016/j.ejca.2015.08.022
33. Dummer R, Flaherty KT. Resistance patterns with tyrosine kinase inhibitors in melanoma: new insights. Curr Opin Oncol. (2012) 24:150–4. doi: 10.1097/CCO.0b013e32834fca92
34. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. (2012) 487:500–4. doi: 10.1038/nature11183
35. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. (2012) 487:505–9. doi: 10.1038/nature11249
36. Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. (2013) 32:1207–15. doi: 10.1038/onc.2012.160
37. Ruggiero CF, Malpicci D, Fattore L, Madonna G, Vanella V, Mallardo D, et al. ErbB3 phosphorylation as central event in adaptive resistance to targeted therapy in metastatic melanoma: early detection in CTCs during therapy and insights into regulation by autocrine neuregulin. Cancers (Basel). (2019) 11:1425. doi: 10.3390/cancers11101425
38. Abel EV, Basile KJ, Kugel CH, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. (2013) 123:2155–68. doi: 10.1172/JCI65780
39. Song C, Piva M, Sun L, Hong A, Moriceau G, Kong X, et al. Recurrent tumor cell–intrinsic and –extrinsic alterations during mapki-induced melanoma regression and early adaptation. Cancer Discov. (2017) 7:1248–65. doi: 10.1158/2159-8290.CD-17-0401
40. Yan Y, Wongchenko MJ, Robert C, Larkin J, Ascierto PA, Dréno B, et al. Genomic features of exceptional response in vemurafenib ± cobimetinib-treated patients with BRAF V600-mutated metastatic melanoma. Author Manuscr Publ OnlineFirst. Clin Cancer Res. (2019) 25:3239–46. doi: 10.1158/1078-0432.CCR-18-0720
41. Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. (2015) 27:85–96. doi: 10.1016/j.ccell.2014.11.006
42. Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, et al. An approach to suppress the evolution of resistance in BRAFV600E-mutant cancer. Nat Med. (2017) 23:929–37. doi: 10.1038/nm.4369
43. Reger de Moura C, Battistella M, Sohail A, Caudron A, Feugeas JP, Podgorniak MP, et al. Discoidin domain receptors: a promising target in melanoma. Pigment Cell Melanoma Res. (2019) 32:697–707. doi: 10.1111/pcmr.12809
44. Haass NK, Smalley KSM, Herlyn M. The role of altered cell-cell communication in melanoma progression. J Mol Histol. (2004) 35:309–18. doi: 10.1023/B:HIJO.0000032362.35354.bb
45. Fukunaga-Kalabis M, Martinez G, Liu Z-J, Kalabis J, Mrass P, Weninger W, et al. CCN3 controls 3D spatial localization of melanocytes in the human skin through DDR1. J Cell Biol. (2006) 175:563–9. doi: 10.1083/jcb.200602132
46. Kim H-G, Tan L, Weisberg EL, Liu F, Canning P, Choi HG, et al. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem Biol. (2013) 8:2145–50. doi: 10.1021/cb400430t
47. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
48. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) 6:11. doi: 10.1126/scisignal.2004088
49. Hutchenreuther J, Leask A. Why target the tumor stroma in melanoma? J Cell Commun Signal. (2018) 12:113–8. doi: 10.1007/s12079-017-0419-1
50. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. (2011) 12:323. doi: 10.1186/1471-2105-12-323
Keywords: DDR1, melanoma, drug resistance, MAPK inhibitors, therapeutic target
Citation: Reger de Moura C, Prunotto M, Sohail A, Battistella M, Jouenne F, Marbach D, Lebbé C, Fridman R and Mourah S (2020) Discoidin Domain Receptors in Melanoma: Potential Therapeutic Targets to Overcome MAPK Inhibitor Resistance. Front. Oncol. 10:1748. doi: 10.3389/fonc.2020.01748
Received: 05 March 2020; Accepted: 04 August 2020;
Published: 11 September 2020.
Edited by:
Petranel T. Ferrao, South Australian Health and Medical Research Institute (SAHMRI), AustraliaReviewed by:
Luigi Fattore, Sapienza University of Rome, ItalyAlvaro Galli, Pisa Research Area, Italian National Research Council, Italy
Copyright © 2020 Reger de Moura, Prunotto, Sohail, Battistella, Jouenne, Marbach, Lebbé, Fridman and Mourah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samia Mourah, c2FtaWEubW91cmFoQGFwaHAuZnI=