 Tao Jiang
Tao Jiang Guangji Zhang
Guangji Zhang Zhaohuan Lou
Zhaohuan Lou- 1School of Basic Medical Sciences, Zhejiang Chinese Medical University, Hangzhou, China
- 2College of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, China
Metabolic changes are a major feature of tumors, including various metabolic forms, such as energy, lipid, and amino acid metabolism. Sterol regulatory element binding proteins (SREBPs) are important modules in regulating lipid metabolism and play an essential role in metabolic diseases. In the previous decades, the regulatory range of SREBPs has been markedly expanded. It was found that SREBPs also played a critical role in tumor development. SREBPs are involved in energy supply, lipid supply, immune environment and inflammatory environment shaping in tumor cells, and as a protective umbrella to support the malignant proliferation of tumor cells. Natural medicine and traditional Chinese medicine, as an important part of drug therapy, demonstrates the multifaceted effects of SREBPs regulation. This review summarizes the core processes in the involvement of SREBPs in tumors and provides a comprehensive understanding of the pathways through which natural drugs target the SREBP pathway and regulate tumor progression.
Introduction
Tumors are a major health challenge for humanity, with high annual mortality rates. In the continuous efforts to discover new treatments for tumors, metabolic changes have been receiving considerable attention. Metabolic changes involve numerous forms, including energy, sugar, lipids, and amino acids. Differing from that of normal cells, tumor metabolism is unique. As early as 1920, a study conducted by Warburg found that tumor cells obtained energy in a glycolysis anoxic environment (1). This led many scholars to focus on the direction of energy metabolism change and use it as a key point for tumor treatment (2). The massive proliferation of tumors requires a variety of substances, including energy, lipids, etc. To cope with these additional needs, tumor cells undergo extensive metabolic reprogramming (3–6). As a component of cellular basal metabolism, lipid metabolism plays a very important role in the metabolic programming of tumor cells (7). Lipids, including phospholipids and cholesterol, serve as a major component of the cell membrane and an additional energy supply to the cells (8–10). In addition, they play important biological regulatory roles as signaling molecules, specific receptors, and transcription factors (11–13). Growing evidence shows that tumor cells are involved in a large number of changes in lipid metabolism, which has become one of the important features of tumors (3, 7, 14). This also renders the key molecules of lipid metabolism and targeting the regulation of lipid metabolism a promising strategy for the treatment of tumors (15, 16).
For most normal cells, the growth rate is strictly regulated by the body, and lipids derived from de novo synthesis and dietary intake of hepatocytes can meet their growth needs. However, for proliferating malignant tumor cells, this is not sufficient; hence, the process of extra lipid production is activated in tumor cells (17). The production of fat involves multiple successive biological steps that can be controlled by numerous regulatory factors and different key enzymes. These enzymes and factors have exhibited a strong association with tumors. For example, high levels of fatty acid synthase (FASN) expression have been associated with invasive tumor phenotypes (18, 19), while both acetyl-CoA carboxylase and FASN have been shown to be highly expressed in malignant tumors and are also indicators of poor prognosis (7, 20, 21). These enzymes are regulated by a variety of complex mechanisms, and a large number of studies have shown that SREBPs are important molecules regulating these key enzymes and leading to lipid metabolism disorders (22–27). Many natural drugs can regulate SREBPs and various biological processes involved in different pathways. This review summarizes the key processes involved in targeting SREBPs through natural drugs for the treatment of tumors.
Overview of SREBPs
Sterol regulatory element-binding proteins (SREBPs) belong to a small family of membrane-bound proteins, and are basic helix-loop-helix leucine zipper transcription factors. There are three subtypes, namely SREBP-1a, SREBP-1c, and SREBP-2. Of those, SREBP-1a and SREBP-1c are encoded by the same gene, whereas SREBP-2 is encoded by a different gene (28). SREBP-1 is mainly regulated by caloric restriction (29, 30), while SREBP-2 is stimulated by thyroid hormone and itself (31, 32). SREBP-2 also preferentially participates in gene transcription in cholesterol biosynthesis (26, 33). Under physiological conditions, activation of SREBPs is tightly regulated by a negative feedback loop triggered by sterols in the endoplasmic reticulum (ER) (28, 34). The classical activation is mediated mainly by insulin-induced gene (INSIG) and SREBP cleavage-activating protein (SCAP) (Figure 1). Specifically, the SREBPs precursor protein transforms to a complex with another ER localized protein, termed SCAP. This complex interacts with the INSIG1 and INSIG2 proteins (35, 36). When the levels of cellular cholesterol are high, INSIGs become stable and allow the SCAP-SREBP complex to preserve in the ER (28, 37). When the levels of sterols in the ER decrease below the threshold, INSIGs are ubiquitylated and rapidly degraded (38) that can trigger the isolate of SCAP-SREBP complex from the ER (39). The isolated complex cannot be transported directly to the Golgi and needs specialized transport vesicles generated by coatomer complex II (COPII). In this process, the levels of sterol will lead to conformation changes in SCAP to determine whether SCAP can combine with COPII (40). These factors together lead to the transport of the SCAP-SREBP complex from the ER to Golgi. In the Golgi, SREBPs will be consecutively cleaved by two membrane-bound proteases site-1 protease (S1P) (41) and site-2 protease (S2P) (42). Then, cleaved SREBPs release the transcriptionally active NH2-terminal domains, that can enter into the nucleus and induce target gene expression including the SREBPs transcription factor itself (25, 43), consequently causing a series of downstream changes.
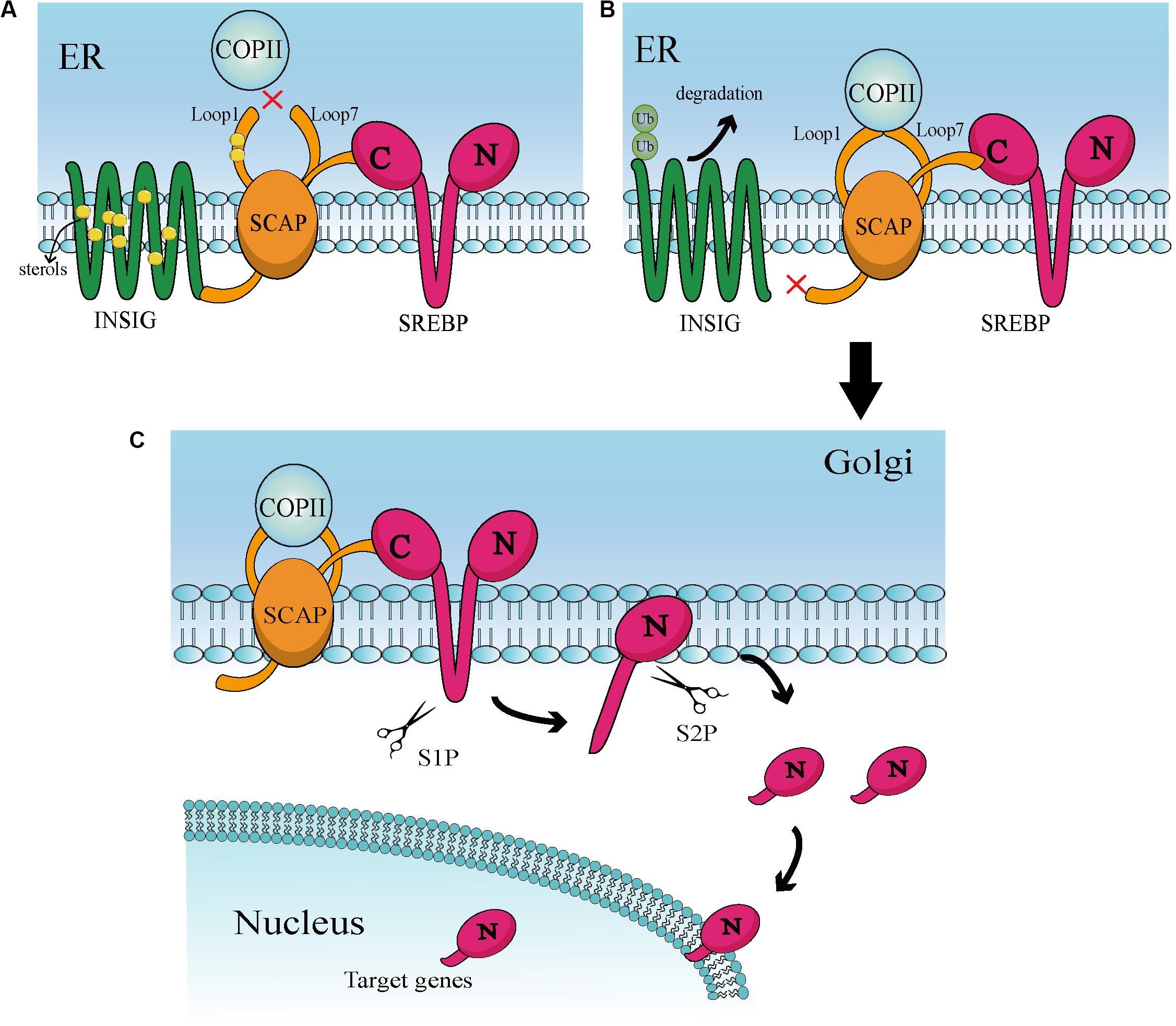
Figure 1. (A) At high sterol levels, the sterol binds to Loop 1 and induces conformational changes in SCAP, thereby increasing its affinity for INSIG. Then, binding of SCAP and INSIG triggers a second conformational changes in SACP, leading to the dissociation of Loop1 and Loop7. This conformation prevents COPII from binding to SCAP. In addition, sterol is combined with INSIG to keep the stability of INSIG. These interactions help the INSIG/SCAP/SREBP complex to be preserved on the ER membrane. (B) In a sterol-deprived environment, the reduction of sterol leads to ubiquitination and rapid degradation of INSIG. This gives the SCAP/SREBP complex an opportunity to escape ER. Deprivation of sterols also triggers conformational changes that enable Loop1 to bind to Loop7, which allows SCAP to bind to COPII. Then, COPII transports the complex to the Golgi apparatus via vesicles. (C) When in the Golgi, SREBP will be continuously cleaved by S1P and S2P proteases, ultimately releasing the N-terminal active domain. This active part enters the nucleus and binds to numerous target genes of SREBP, leading to downstream effects.
In addition to the strict restriction of transport and activation, SREBP is transcriptionally regulated through several mechanisms. As mentioned above, mature NH2-terminal domains of SREBPs enhances expression of itself (31). Liver X receptor (LXR) is one of the key molecules mediating SREBP mRNA transcription (44). Studies have shown that LXR plays an important role in tumors and is associated with SREBPs (45, 46). The same effect was observed for insulin via the mTOR pathway mechanism (47). Moreover, several miRNAs including miRNA-29, miRNA-185, and miRNA-342, are also responsible for the transcription of SREBP (48, 49).
Sterol regulatory element-binding proteins are involved in numerous biological processes, such as ER stress, inflammation, autophagy, and apoptosis. This also causes SREBPs to trigger a variety of diseases, including obesity, dyslipidemia, diabetes, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, chronic kidney disease, neurodegenerative diseases, and tumors (50). In the previous decades, the definitive function of SREBPs has been extended to many tumor-critical biological processes, highlighting the importance of lipids in cell and system homeostasis (19, 22). In an epidemiological study, long-term intake of cholesterol was also associated with an increased risk of gastrointestinal cancer (51). This suggests that uptake of exogenous lipids also exerts an effect on tumor progression. Multiple pathways mediated by SREBPs are associated with lipid metabolism and play a role in tumor progression. Although the sterol feedback regulation mechanism of SREBPs has been extensively studied, the mechanism through which SREBPs induce lipid metabolism and other specific regulatory functions in tumors remains unclear (52).
Drug Family Targeting SREBPs
In view of the special and powerful regulation of SREBPs in tumors, drugs targeting SREBPs have been developed. However, direct inhibition of SREBPs is a difficult task, as transcription factors are difficult to target for drugs (2). The current strategy is to inhibit the transport of SREBPs from the ER to Golgi and inhibit cleavage enzymes to block the release of the active domain. Besides this, inhibiting SREBPs transcription through key signaling pathways and restoring cholesterol sensitivity by inhibiting cholesterol droplets formation are also common interventions (53). Table 1 shows some of the common inhibitors of SCAP/SREBPs. Most of them are still in the preclinical stage, and a few of them have been studied clinically.
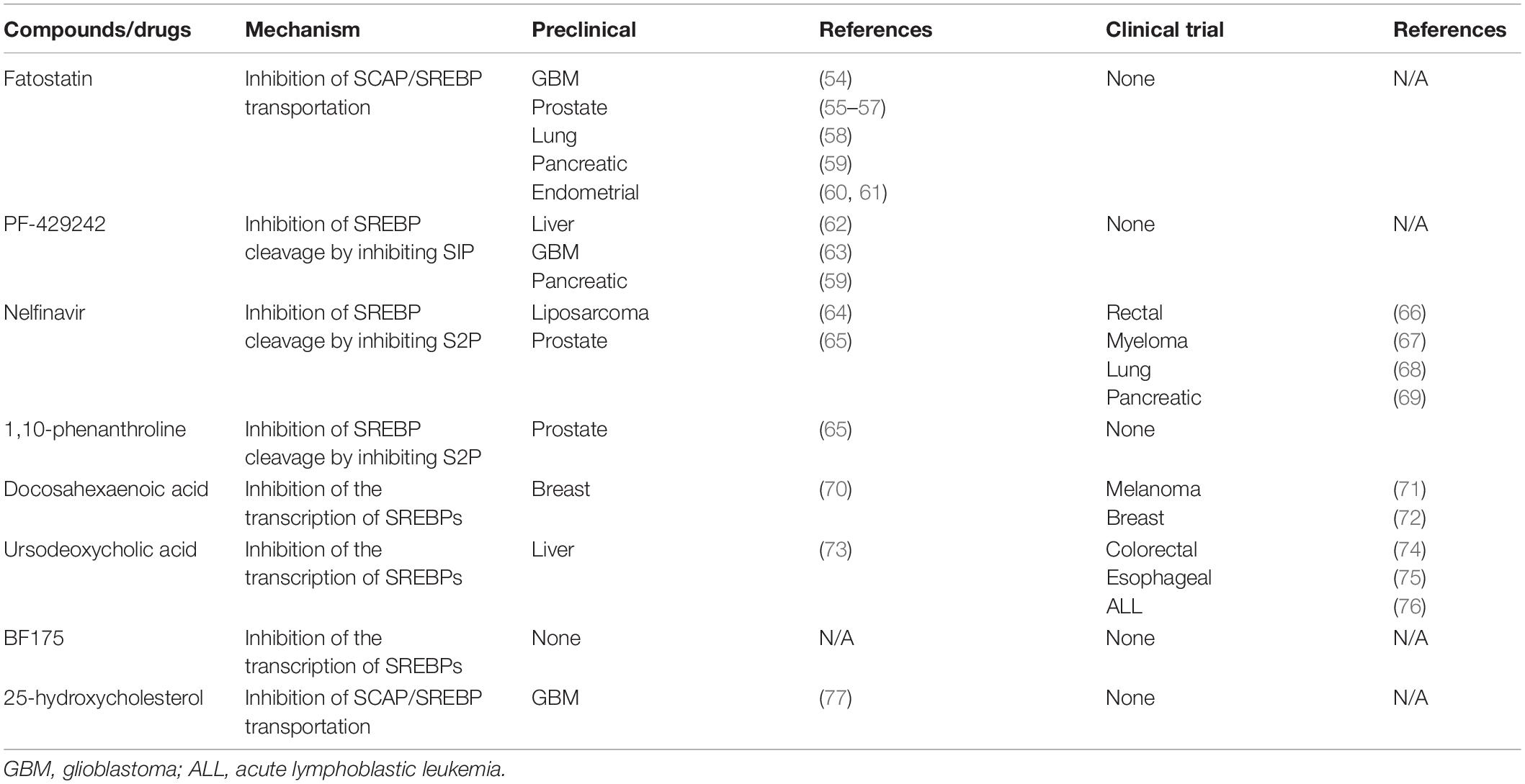
Table 1. The classical inhibitors targeting SCAP/SREBPs in cancer cells.
Although there have been many classical inhibitors, their application is still limited. SREBP inhibitors, including fatostatin, BF175, and 25-hydroxycholesterol cholesterol are only observed in a pre-clinical study. In part, these classical SREBP inhibitors such as 25-hydroxycholesterol, also activate LXR while inhibiting SREBPs, which will reactivate the SREBP-1c gene as a feedback loop. This can lead to enhanced expression of free fatty acid synthesis genes and increased plasma triacylglycerol content, thereby greatly limiting drug performance (78). Some drugs have entered clinical trials, but the analysis of SREBP is not sufficient. For example, nelfinavir is widely used in cancer research, but its main research course is to inhibit the Akt signaling pathway (79). Therefore, we need to find some emerging SREBPs inhibitors to avoid these side effects. Natural drugs have garnered increasing attention given their lower cost and minor side effects. Many of them are used in metabolic diseases and have shown the ability to inhibit SREBPs. Table 2 shows some of the natural drugs involved in the regulation of SREBPs, as well as a brief description of their mechanism of action. These compounds can target SACP/SREBPs, inhibit the detachment of SREBPs from the ER, inhibit the transport of SREBPs, and regulate the key molecular mechanisms involved in SREBPs. Although there are only few studies on tumors, they can serve as a guide for drug research and can be candidates for tumor treatment.
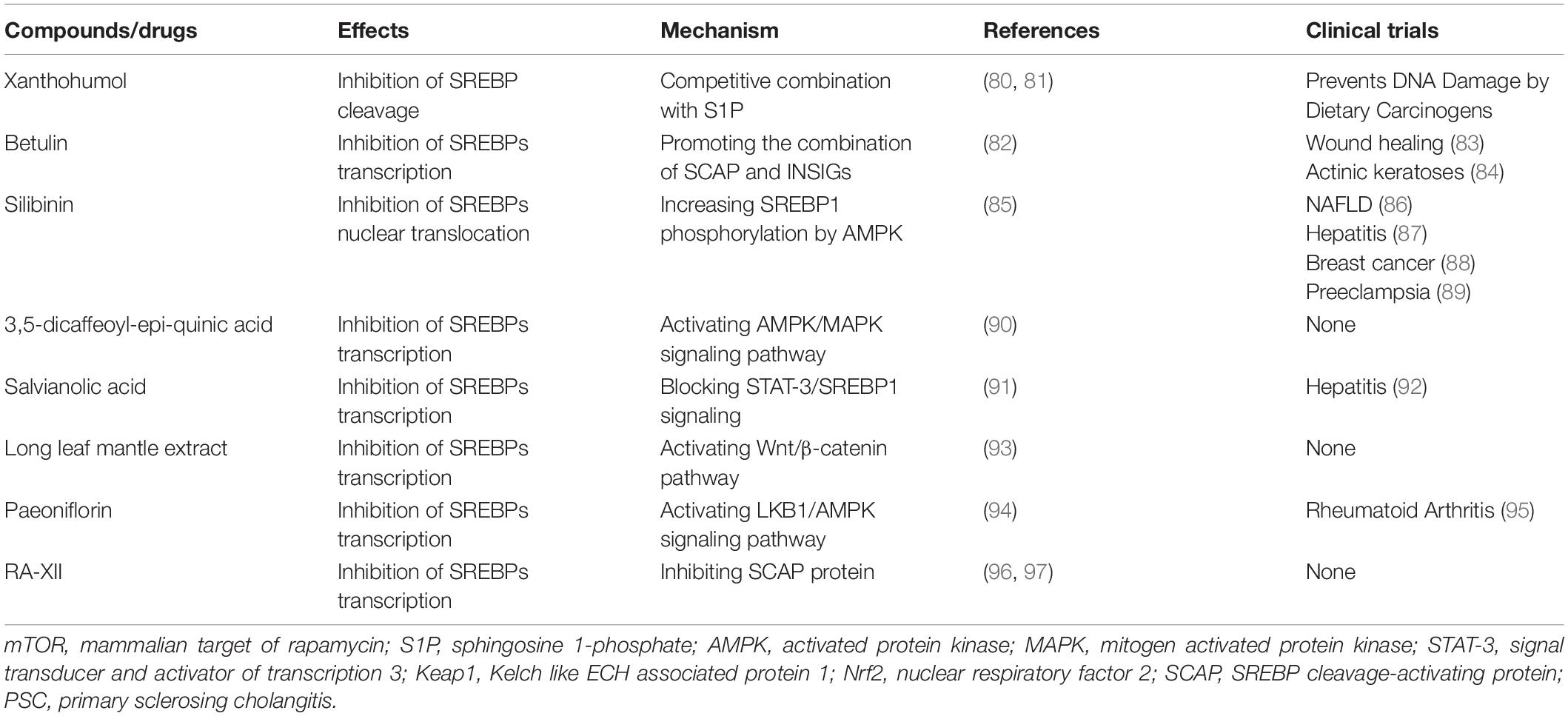
Table 2. Natural drugs targeting SREBPs and their mechanism of action.
The mechanism of these drugs is multi-targeted and involves a variety of common signaling pathways. Their impact is also multifaceted, involving a variety of biological processes, such as cellular lipid supply, energy supply, glucose supply, cell protection, and microenvironment modeling. In the following sections, we provide a more detailed description of the biological functions involved in the regulation of SREBPs by natural medicines.
Regulation of Tumor Cell Energy Supply
Cells require energy to maintain their vitality, growth, and normal physiological functions (98). ATP is the currency of cell energy supply (99). In normal cells, ATP is produced by glycolysis in the cytoplasm, oxidative phosphorylation in mitochondria, tricarboxylic acid recycling, β-oxidation of fatty acid, and the metabolism of ketones and triglycerides also produces energy. The energy supply of normal cells is mostly provided by the aerobic oxidation of glucose, while in the tumor-associated microenvironment it is mainly provided through aerobic glycolysis (1). Aerobic glycolysis results in a small amount of ATP and a large number of intermediate products required for cell proliferation (6, 100, 101). Recent research showed that SREBP-1 plays an important role in the regulation of lipid metabolism and contributes substantially to glucose metabolism (102); there exists an interactive relationship between SREPBs and glucose. SREPBs are necessary for intracellular glycolysis and oxidative phosphorylation in natural killer cells (103). SREBP-1a can trans-regulate the promoter of the PFKFB gene (104), which is a 6-phosphate fructose-2-kinase/fructose-2,6-bisphosphate enzyme catalyzing the synthesis of fructose-2,6-diphosphate and degradation in gluconeogenesis. SREBP-1 is also involved in the generation of glycogen under special situation. The lack of SREBP-1 reduces the levels of glycogen and lower the activity of glycogen synthase mRNA (105). On the contrary, glucose exerts a reverse regulatory effect on SREBPs. Kinetic experiments showed that exogenous glucose can upregulate SREBP-1c precursors and ribosomes within 30 min following the translocation of SREBP-1c to the nucleus. Glucose rapidly stimulates SREBP-1c maturation through the Janus kinase/signal transducer and activator of transcription pathway (106). Meanwhile, the protein SCAP binding SREBP in the ER is also stimulated by glucose, leading to glycosylation of SCAP and further promoting the release of mature SREBPs (107). Collectively, these studies have shown that SREBPs, especially SREBP-1 and SREBP-1c, play important roles in regulating glucose metabolism. Aerobic glycolysis is widespread in tumor cells. Current studies indicate that SREBPs may affect the progression of tumors by altering glucose metabolism (108). Some natural drugs, such as silibinin, can affect glucose uptake through SREBP-phosphatidylinositol 3 kinase-protein kinase B (109), and paeoniflorin can promote β-oxidation and glycogen production (94) to regulate energy metabolism. These compounds may be the potential candidates for the treatment of tumors through regulation of energy metabolism.
Regulating FAO as Supplementary Energy
Aerobic glycolysis is an inefficient oxidation method associated with limited energy supply. Therefore, tumor cells have to take various measures, such as increasing the rate of glycolysis to produce lactic acid (110) and strengthening fatty acid oxidation (FAO) (111) to produce more ATP. FAO is an important auxiliary production mode of the body. In the case of nutritional deficiency, FA replaces glucose to provide sufficient energy for the body. Meanwhile, FAO is also the preferred way of energy supply for the heart, skeletal muscle, and kidneys (112). Studies have found changes in FAO in various types of tumors. In triple-negative breast tumors, FAO is extremely active, and blocking FAO may greatly influence energy metabolism, reduce proliferation, and inhibit growth in vivo in tumor cells (113). Similar results were also reported in models of prostate cancer, multiple myeloma, and leukemia (114–117). Moreover, FAO was also the main energy provider in metastatic tumors (111). As the primary energy supplier for some cancer cells, the process of FAO is affected by SREBPs. The basis of FAO is long-chain FA, which can be obtained through food or synthesized endogenously. SREBPs (especially SREBP-1) upregulate a large number of enzymes that catalyze the synthesis of FA, such as FASN, and stearoyl coenzyme α desaturating enzyme in a variety of human tumors (118–120). Silencing of SREBP-1 or SREBP-2 in established tumor cell lines and tumor cells of patient origin led to an overall transformation of cell metabolism, including glycolysis, mitochondrial respiration, and reduction of the levels of FAO (108). In the natural drug family ursodeoxycholic acid can regulate the expression of SREBPs and the occurrence of FAO, thereby improving inflammatory response, angiogenesis, and macrophage differentiation (121). These biological processes exert marked effects in tumors and affect tumor progression. Therefore, natural drugs may be worthy of study in regulating SREBPs and pointing to the process of FAO in blocking the development of tumors.
Providing the Necessary Lipids for the Proliferation of Tumor Cells
Lipids are particularly important for maintaining the biosynthesis of cell membranes and coordinating numerous biological processes (13, 50, 122). Phospholipids are widely involved in the construction of cell membrane modules (123), while cholesterol is one of the main components of lipid rafts that can be used as the tissue center for the assembly of signaling molecules (12). This is necessary for cell division, metabolism, and proliferation (124, 125). Unlike normal cells that obtain lipids from the blood in the form of dietary free FAs, tumor cells show a strong de novo synthesis of lipids to support their growth (126).
A large amount of lipid production in tumor cells is mediated by SREBPs. SREBPs regulate the production of sterols, especially cholesterol. Studies have shown that tumor cells possess high cholesterol levels, derived by increasing the uptake of low-density lipoprotein (LDL), reducing the outflow of cholesterol, and accelerating endogenous synthesis of cholesterol and FAs (Figure 2) (127–130). SREBPs are involved in almost all the pathways associated with high cholesterol. They increase the levels of cholesterol in cells by increasing the intake of LDL and synthesis of cholesterol. Inhibition of low-density lipoprotein receptor (LDLR) can promote glioblastoma cell death (46). In addition, they promote the transcription of enzymes, such as 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), that can directly induce the synthesis of cholesterol (131). Meanwhile, miR-33 which is embedded in introns of, and co-transcribed with SREBPs can also prevent loss of cholesterol by inhibiting the expression of cassette subfamily transporters (e.g., ABCA1 and ABCG1) to antagonize the binding between transporters and cholesterol (132). Overaccumulation of unesterified cholesterol can be toxic to cells. Cholesterol esters, which mainly exist as cytosolic lipid droplets, can be a safe form to store cholesterol. Increased lipid droplets have been found in glioblastoma and some other types of tumor (34, 133). Tumor cells maintain a large amount of cholesterol by combining cholesterol and FA to the formation of cholesterol ester to escape the monitoring of cholesterol (134). Abnormal levels of lipids are intimately related to carcinogenesis and cancer metastasis (135). In liver cancer, obesity caused by the accumulation of lipids accelerates the progression of hepatitis and liver cancer. It was also found that an increase in total cholesterol can lead to the development of gastric cancer (136). Natural drugs have great potential in maintaining lipid homeostasis. Schisandra polysaccharide can improve the production of lipids by downregulating SREBP-2/HMGCR (137). Xanthohumol is a natural inhibitor of SREBPs that competes with sphingosine-1-phosphate to antagonize the activation of SREBPs inhibiting the synthesis of cholesterol (81). Similarly, betulin, ursodeoxycholic acid, and other natural drugs possess unique properties, inhibiting SREBPs to regulate lipid homeostasis (82, 138). The production of lipids, especially cholesterol, can be limited by natural drugs through the SREBPs pathway, suggesting that the effects of natural drugs in altering the production of lipids may be applied to the treatment of tumors.
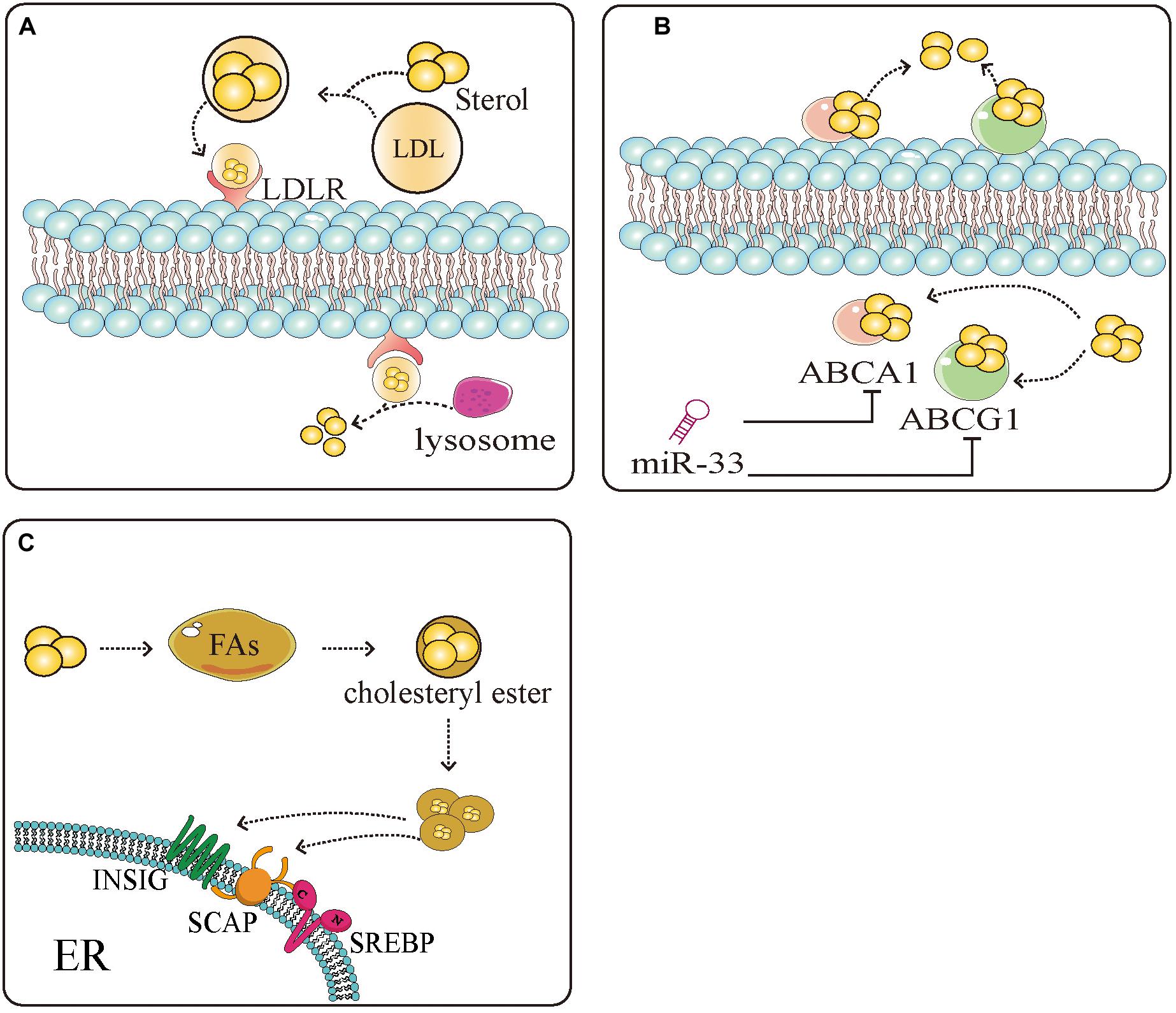
Figure 2. Sterol regulatory element-binding protein (SREBPs) mediate the uptake, efflux, and storage of cholesterol. (A) Extracellular cholesterol is carried by low density lipoprotein (LDL) and binds to LDL receptors (LDLR) on the cell surface. After binding, cholesterol is transported to the cells and decomposed by the lysosome into intracellular cholesterol. (B) Intracellular cholesterol binds to the sterol transporter ATP binding cassette subfamily A member 1 (ABCA1)/ATP binding cassette subfamily G member 1 (ABCG1) and is transferred to the extracellular space to complete the uptake and discharge of cholesterol. miR-33 which is embedded in introns of, and co-transcribed with SREBPs can inhibit the expression of ABCA1 and ABCG1, thus inhibiting the reversal of cholesterol. (C) Excess cholesterol in the cells binds to SREBP-mediated fatty acids and esterifies them into cholesterol esters to avoid the negative regulation of cholesterol. These measures provide a large amount of energy and nutrients and protection for tumor cell proliferation.
Currently, there is insufficient evidence regarding the regulation of phospholipid production by SREBPs. However, recent studies have provided clues concerning the interaction between SREBPs and phospholipids. In small intestinal tumors, phospholipid remodeling can change the expression of SREBPs to regulate the generation of cholesterol, and affect the occurrence and development of intestinal tumors (139). Low levels of phosphatidylcholine lead to the maturation of SREBP-1 in nematode or mammalian models (140), and phospholipids rich in eicosapentaenoic acid inhibit the SREBP-1c-mediated production of fat. These findings suggest a relationship between phospholipids, cholesterol, and SREBPs.
Provide Protection for Tumor Cells
Sterol regulatory element-binding protein-mediated regulation of lipids provides considerable protection for tumor cells. Improving drug resistance and regulating cell cycle are common ways. Research has found that statins exert a good effect on some prostate tumors (141, 142); however, this effect can be blocked by SREBPs. Studies have shown that SREBPs may lower the sensitivity of statins in the treatment of prostate tumors by upregulating HMGCR and other lipid metabolism genes (143). Hence, inhibition of HMGCR and SREBPs would increase responsiveness to drugs (144). The same drug resistance mechanism related to SREBPs and lipid metabolism was also found in breast tumors, multiple myeloma, glioblastoma, lung tumors, and liver tumors (22, 58, 77, 145–147), as well as anti-tumor drugs, including cisplatin (148), rapamycin, epidermal growth factor receptor-targeted inhibitors and docetaxel (57, 147, 149). Natural drugs combined with traditional anti-tumor drugs to increase the sensitivity of cells to treatment are very promising. In the treatment of hepatocellular carcinoma, emodin has been used to increase the sensitivity of cells to sorafenib by regulating SREBP-2 (150). Although the evidence is promising, further research studies are warranted to investigate the mechanism of SREBPs involved in drug resistance.
In addition to participating in the development of drug resistance, SREBP-mediated metabolism of glycolipids also protects tumor cells in multiple aspects. SREBP-2-mediated synthesis of sterol protects against oxidative stress by reducing lipid peroxidation to maintain membrane integrity (151). SREBP-2 also occupies the promoter of autophagy-related genes to activate autophagy (152). Cells can remove damaged proteins and organelles through autophagy, while recapturing energy and essential substances through the same process during periods of nutrient deficiency (153). SREBPs can also promote tumorigenesis by activating the mevalonate pathway (154). SREBPs lead to the de novo synthesis of lipids; however, excess lipids do not cause negative feedback regulation with SREBPs, because lipids are stored in cells in the form of lipid droplets. Lipid droplets have been observed in many tumors and may become a potential biomarkers in tumor (133). Reduction of LD formation by inhibiting SOAT1 can effectively suppress tumor growth (53). These lipids can help in the late stage of cell metabolism and counteract the lipid toxicity induced by the accumulation of intracellular FA (155, 156).
Sterol regulatory element-binding proteins can also directly or indirectly regulate the cell cycle to facilitate the proliferation of tumor cells. Indirectly, the SREBP-mediated synthesis of FAs and cholesterol has a certain impact on the cell cycle. Unsaturated FAs increase the expression of cyclin D1 and cell proliferation by activating β-catenin in renal clear cell carcinoma (157). Cholesterol is also essential for cell cycle progression, and cholesterol deficiency leads to cell cycle arrest at the G2/M phase (124). Directly, SREBP-1 contains a binding site in the host cell factor C1 gene, through which it stimulates the expression of key genes involved in cell cycle control and participates in the cell cycle and fibroin A adjustment (158). Studies have also shown that silencing SREBP-1 can directly lead to cell arrest at the G1 phase in human HeLa, U2OS, and MCF-7 cells, thereby attenuating cell growth (159). However, different subtypes of SREBPs appear to play different roles in the regulation of the cell cycle. Unlike SREBP-1, excess SREBP-1a causes cell cycle disorder, resulting in the accumulation of cyclin-dependent kinase inhibitors, such as p27, p21, and p16. Moreover, overexpression of SREBP-1a activates a novel SREBP binding site in the promoter of the gene p21 (waf1/cip1) that activates cyclin-dependent kinase inhibitors, leading to inhibition of cell growth and cell cycle arrest at the G1 phase (160, 161). SREBP-1c also promotes cell cycle progression by enhancing the expression of its target gene pituitary tumor-transforming 1 (162). Pituitary tumor-transforming 1 prevents premature chromosome segregation by inhibiting the activity of isolated enzymes, and promotes cell cycle disorders by regulating cell cycle genes (163). Previous research shed light on the effects of SREBPs on the regulation of tumor cell cycle. However, further studies area warranted to completely elucidate the mechanism involved in this process.
Potential in Modulating Inflammation and Immunity
The immune environment and inflammatory environment are hotspots of the tumor microenvironment. This environment can be shaped in many ways; and metabolism plays a unique role (164). As a transcription factor that modulates a large number of metabolism-related genes, SREBPs show great potential in shaping the tumor microenvironment. Although current research has focused on cardiovascular and adipose diseases, we predict that the same results may be achieved in tumors. Here, we review the known studies of SREBP with the aim to provide assistance for future treatment. We first focused on macrophages because they are important cells for intervention in inflammation and immunity. As one of the important immune cells of the body, macrophages show powerful phagocytosis and SREBPs exert control by influencing the expressions of target genes. As early as the 1970s, changes in the composition of membrane FAs have been shown to affect phagocytosis by macrophages (165). The phagocytic immunity of macrophages depends on the direct interaction between the plasma membrane and the actin cytoskeleton (166). SREBP-1a can regulate a variety of lipid components involved in the actin cytoskeleton network and cytoplasmic membrane to change the phagocytic function of macrophages (47). Moreover, macrophages exhibit significant plasticity and can be transformed into M1 and M2 phenotypes. The polarization change between M1 and M2 is an important intervention point for many diseases, including tumors. The two main phenotypes of macrophages showed distinct metabolic characteristics, and SREBPs are mainly responsible for the activation of M1 macrophages (Figure 3). M1 macrophages are known to rely on aerobic glycolysis. This metabolic adaptation favors rapid ATP production to sustain their phagocytic function and provides metabolic precursors to feed the pentose phosphate pathway. The levels of ATP citrate lyase (ACLY) and FAS are important in M1 macrophage activation. The increase of ACLY was found in activated M1 macrophages, and the silencing of ACLY was sufficient to reduce the expression of inflammatory mediators (167). Meanwhile, fatty acid synthase (FAS) deletion in macrophages prevented macrophage recruitment and inflammatory response in diabetic mice (168). Both ACLY and FAS are important partners of SREBP. Therefore, SREBP1-a was found to be highly expressed in the LPS-induced M1 macrophage model, and the deficiency of SREBP 1-a would lead to deficiency of innate immune response (169). On the contrary, M2 macrophages have an enhanced fatty acid oxidation (FAO) and oxidative phosphorylation (170). Blocking FAO by drugs inhibits IL-4-induced M2 polarization (171). The regulation of SREBPs involves a variety of lipids, which are the raw materials of FAO. In conclusion, SREBP can influence macrophage polarization and phagocytosis by conditioning its target genes. The consequence is inflammation and immune changes.
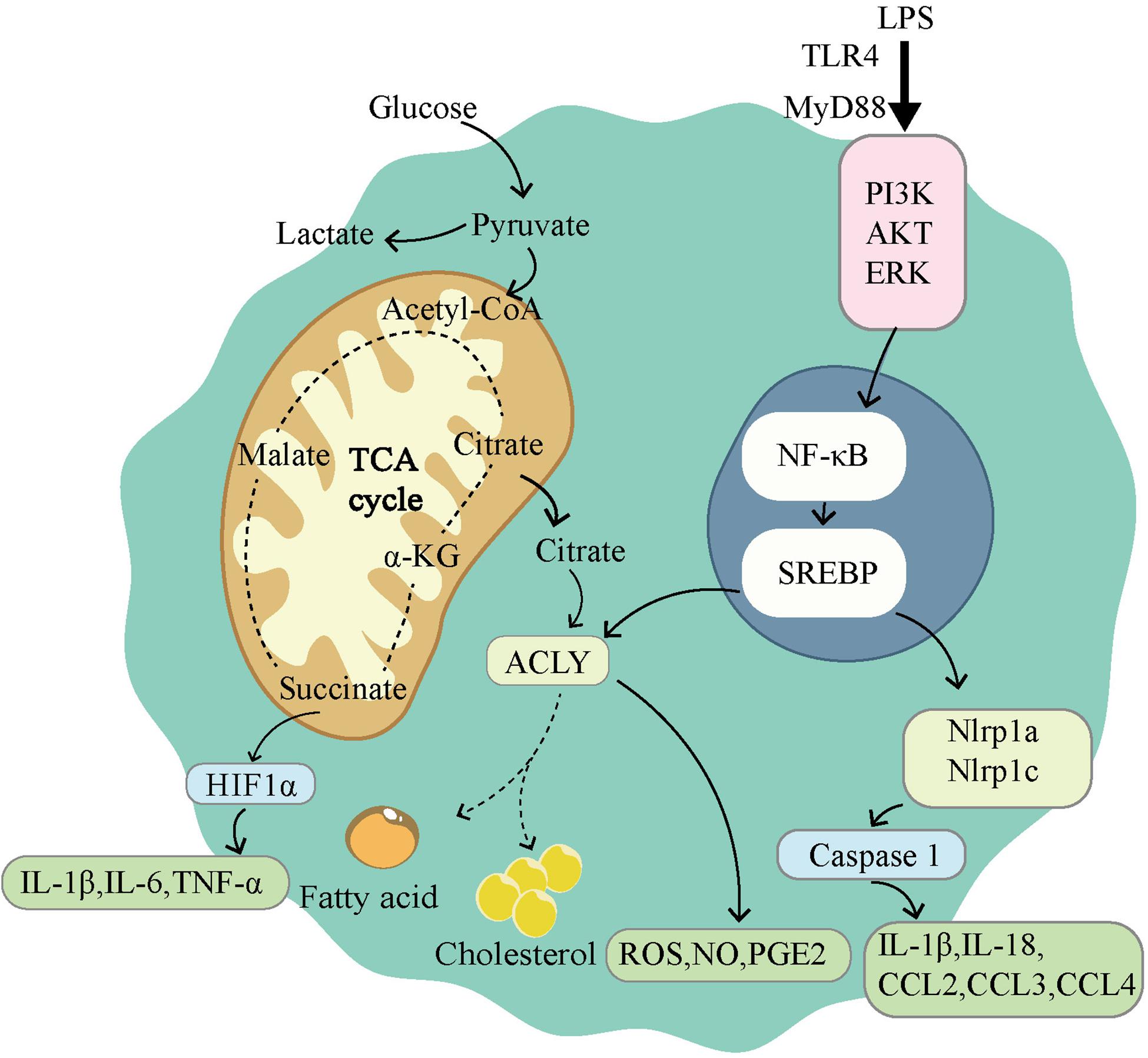
Figure 3. Sterol regulatory element-binding protein (SREBPs) is involved in the activation of M1-type macrophages. M1 macrophages use glycolysis as the main way of energy supply. The tricarboxylic acid cycle is disrupted in M1 macrophages, leading to the accumulation of citrate and succinate. Succinate can activate HIF1α, which in turn leads to the release of proinflammatory factors. LPS induces the activation of NF-κB through TLR4 dependent and independent pathways. Activation of NF-κB induces the expression of SREBPs, promoting lipid synthesis and accumulation. ACLY, as a downstream gene of SREBP, participates in lipid synthesis and drives the release of ROS, NO, PGE2. SREBPs also activate Nlrp1a and Nlrp1c, which lead to the release of proinflammatory factors.
The effect of SREBPs was not restricted to macrophages alone. SREBPs is also widely involved in T-cell function and specific functions of innate and adaptive immunity (172). SREBPs are required for the metabolic reprogramming of mitotic signaling in response to CD8+ T cells. Loss of SREBPs in CD8+ T cells renders them ineffective for blast, resulting in decreased proliferative capacity in vitro and attenuated clonal expansion during viral infection (173). In dendritic cells, the accumulation of cholesterol accelerates the development of autoimmunity at the transcriptional level via the nod-like receptor 3 (NLRP3) isoform (174, 175). NLRP3 is an important molecule involved in the inflammatory response and innate immune response of the body. The immune response affected by NLRP3 in vivo or in vitro requires the participation of the SCAP-SREBP2 complex from the ER to the Golgi translocation process. Therefore, SCAP-SREBP2 plays an important role as a signaling hub for the integrated metabolism of cholesterol by macrophages and inflammation (176). In triple-negative breast cancer, the natural drug berberine inhibits the expression of NLRP3 (177).
In T-cells, FA metabolism is important in the development, differentiation, distribution, and function of different subsets of T cells (178). Cholesterol and phospholipids can be enriched around the immune synapse by lipid rafts to regulate the immune function of T cells (179). The aforementioned studies have shown the special role of lipids in immunity. SREBPs, as important regulators of lipid metabolism, act as a bridge between natural drugs and the treatment of tumors.
Conclusion and Future Prospects
Sterol regulatory element-binding proteins, as key molecules for the traditional regulation of cellular lipid metabolism, have greatly expanded their range of capabilities in recent years (Figure 4). In the previous decades, research on SREBPs has gradually deepened. However, the mechanism through which SREBPs regulate lipid metabolism, affect other biological processes, and other factors targeting tumor cells remain to be fully understood. Although there are numerous drugs against SREBPs, their efficacy is limited and insufficient to transform the clinical treatment of tumors. This is attributed to the complex regulatory mechanisms of SREBPs. For example, SREBPs are well established as traditional lipid-regulating molecules, previous studies have suggested that almost all lipids are regulated by SREBPs. Nevertheless, current studies have found that the production of lipids also involves other pathways, such as protein kinase B. Induction of FA production is a mechanism independent of SREBP-1-mediated FA synthesis (180). This suggests that tumor cells can use a variety of ways to meet their energy requirements. At the same time, the regulation of target genes downstream of SREBPs can also be independent. For example, FASN can also regulate the production of lipids independently of SREBPs (54). The upstream mechanism regulating SREBPs is also diverse. Current research suggests that the feedback mechanism of ER is the main cause of dissociation of SREBPs and entry to the nucleus; however, the increase in SREBPs can also be cholesterol-insensitive. This suggests that, although SREBPs are extremely important in the metabolism of lipids, they are not required in special cases.
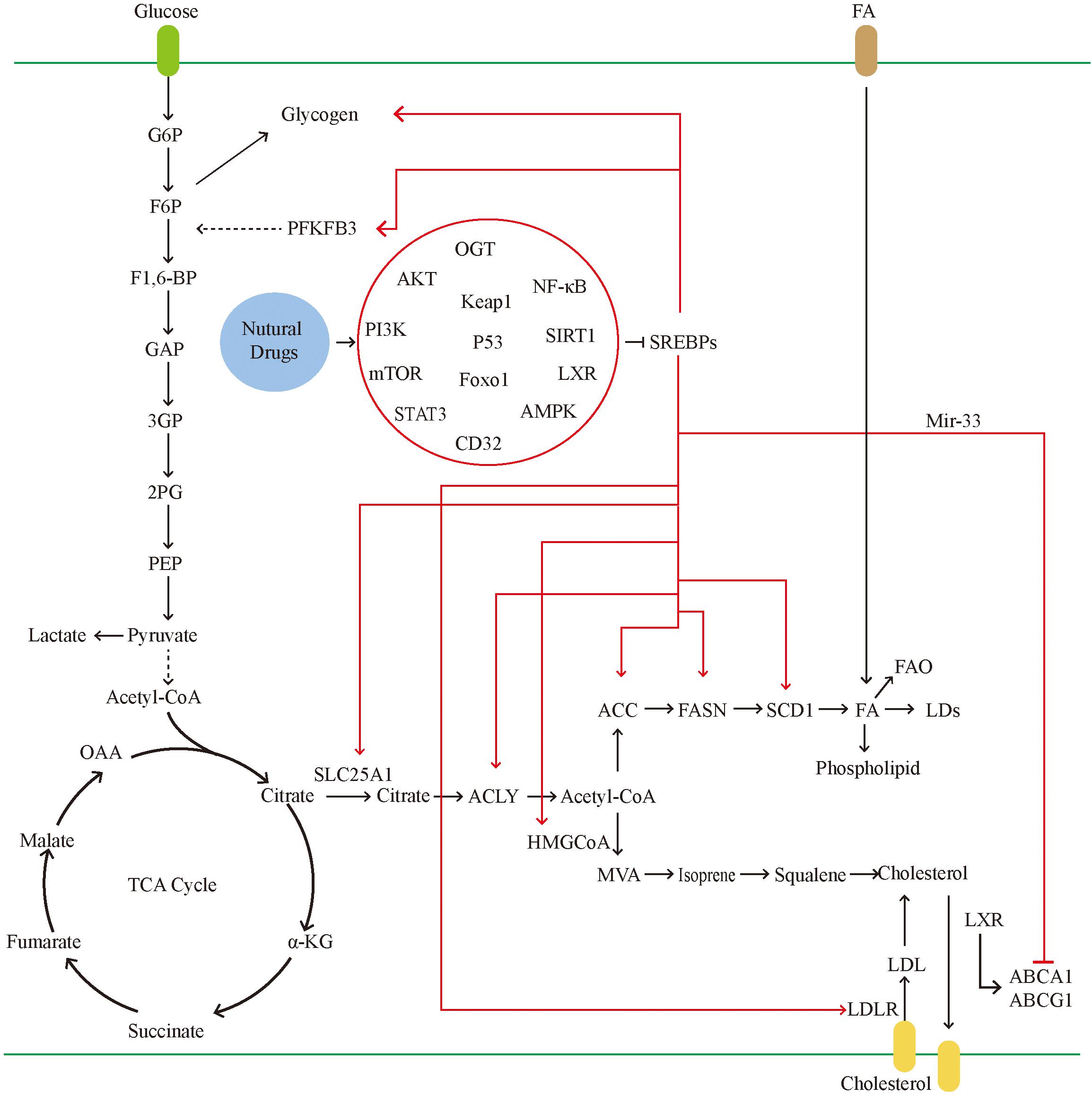
Figure 4. In tumor cells, the glucose uptake is significantly increased. SREBPs can promote glycogen production and accelerate glycolysis by activating the PFKFB. glycolysis leads to the massive production of pyruvate. Pyruvate can be converted into lactic acid and also participates in the tricarboxylic acid cycle. The tricarboxylic acid cycle produces citrate, which can be used as raw materials for lipid synthesis and cholesterol synthesis. Citrate transports mitochondria into the cytoplasm through SLC25A1. In tumors, activated SREBPs can promote the activity of multiple fatty acid synthases and accelerate the formation of fatty acids. Excess fatty acids can be converted into LDs and phospholipids. On the one hand, it is used for the proliferation of tumor cells and on the other hand to avoid lipotoxicity. SREBPs promote cholesterol absorption by activating LDLR, and mir-33, which is co-transcribed with SREBPs, can also inhibit the gene expression of transporters, thereby inhibiting cholesterol efflux. Natural drugs ultimately inhibit the activity of SREBPs through a variety of ways, including inhibiting gene transcription, reducing the transport of SREBPs, and reducing the maturation of SREBPs protein.
Primarily, the growth of tumor cells requires energy, glucose, environment, and other factors. SREBPs can provide abundant energy supply, abundant material reserves, excellent growth environment, and special protection for tumor cells. Many of these effects are dependent on the lipid metabolism regulated by SREBPs. Such complicated biological effects are strictly controlled by numerous mechanisms in the body; however, in tumor cells, this control is uncoordinated. Many drugs have demonstrated the unique ability and advantages of SREBPs.
To achieve better clinical results, we must re-examine the functions of SREBPs, and study the upstream and downstream mechanisms involved in these processes. It is also necessary to identify the specific mechanisms through which tumor cells regulate the elevation of SREBPs and ways to suppress this elevation. Combining targeting SREBPs with chemotherapy and immunotherapy is also a direction worthy of further consideration (27). For this purpose, natural medicine has already taken the lead, especially in the area of regulating lipid and glucose metabolism disorders, which is the pivotal cause of tumorigenesis. The signal pathways connecting lipid and glucose metabolism with tumors are the key targets of natural drugs or traditional Chinese medicines. Compounds exerting effects on the regulation of lipid or glucose metabolism, as well as inhibiting tumor growth, are candidates for the development of innovative anti-tumor drugs.
Author Contributions
TJ prepared and edited the manuscript. ZL was responsible for modifying the manuscript. GZ provided some critical useful suggestions. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 81874455 and 81573962 to GZ and 81503328 to ZL), Zhejiang TCM Science and Technology Program (No. 2019ZB034 to ZL), and Research Program of Zhejiang Chinese Medical University (No. 2018ZG28 to ZL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Charlesworth Co., for editing the language.
References
2. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (2018) 38:27. doi: 10.1186/s40880-018-0301-4
3. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
4. Cairns RA, Harris I, McCracken S, Mak TW. Cancer cell metabolism. Cold Spring Harb Symp Quant Biol. (2011) 76:299–311.
6. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
7. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
8. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. (2015) 17:351–9. doi: 10.1038/ncb3124
9. Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. (2014) 15:243–56. doi: 10.1038/nrm3772
10. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. (2012) 491:364–73. doi: 10.1038/nature11706
11. Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. (2000) 16:459–81. doi: 10.1146/annurev.cellbio.16.1.459
13. Spector AA, Yorek MA. Membrane lipid composition and cellular function. J Lipid Res. (1985) 26:1015–35.
14. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
15. Min X, Wen J, Zhao L, Wang K, Li Q, Huang G, et al. Role of hepatoma-derived growth factor in promoting de novo lipogenesis and tumorigenesis in hepatocellular carcinoma. Mol Oncol. (2018) 12:1480–97. doi: 10.1002/1878-0261.12357
16. Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. (2014) 20:2619–26. doi: 10.2174/13816128113199990486
17. Zhu Z, Zhao X, Zhao L, Yang H, Liu L, Li J, et al. p54(nrb)/NONO regulates lipid metabolism and breast cancer growth through SREBP-1A. Oncogene. (2016) 35:1399–410. doi: 10.1038/onc.2015.197
18. Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? J Cell Biochem. (2004) 91:47–53. doi: 10.1002/jcb.10708
19. Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab. (2012) 16:414–9. doi: 10.1016/j.cmet.2012.09.002
20. Menendez JA, Lupu R. Oncogenic properties of the endogenous fatty acid metabolism: molecular pathology of fatty acid synthase in cancer cells. Curr Opin Clin Nutr Metab Care. (2006) 9:346–57. doi: 10.1097/01.mco.0000232893.21050.15
21. Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer. (2005) 41:1316–22. doi: 10.1016/j.ejca.2004.12.037
22. Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. (2012) 23:65–72. doi: 10.1016/j.tem.2011.10.004
23. Bedi S, Hostetler HA, Rider SD Jr. Mutations in liver x receptor alpha that impair dimerization and ligand dependent transactivation. Nucl Recept Res. (2017) 4:101302.
24. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. (1997) 89:331–40. doi: 10.1016/s0092-8674(00)80213-5
25. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Investig. (2002) 109:1125–31. doi: 10.1172/jci0215593
26. Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem. (2000) 275:32379–82. doi: 10.1074/jbc.r000017200
27. Cheng X, Li J, Guo D. SCAP/SREBPs are central players in lipid metabolism and novel metabolic targets in cancer therapy. Curr Top Med Chem. (2018) 18:484–93. doi: 10.2174/1568026618666180523104541
28. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. (2006) 124:35–46. doi: 10.1016/j.cell.2005.12.022
29. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Investig. (2004) 114:147–52. doi: 10.1172/jci200422422
30. Chujo Y, Fujii N, Okita N, Konishi T, Narita T, Yamada A, et al. Caloric restriction-associated remodeling of rat white adipose tissue: effects on the growth hormone/insulin-like growth factor-1 axis, sterol regulatory element binding protein-1, and macrophage infiltration. Age. (2013) 35:1143–56. doi: 10.1007/s11357-012-9439-1
31. Sato R, Inoue J, Kawabe Y, Kodama T, Takano T, Maeda M. Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J Biol Chem. (1996) 271:26461–4. doi: 10.1074/jbc.271.43.26461
32. Shin DJ, Osborne TF. Thyroid hormone regulation and cholesterol metabolism are connected through sterol regulatory element-binding protein-2 (SREBP-2). J Biol Chem. (2003) 278:34114–8. doi: 10.1074/jbc.m305417200
33. Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes Dev. (2009) 23:2578–91. doi: 10.1101/gad.1854309
34. Guo D, Bell EH, Chakravarti A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. (2013) 2:289–99. doi: 10.2217/cns.13.20
35. Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. (2002) 99:12753–8. doi: 10.1073/pnas.162488899
36. Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. (2002) 110:489–500.
37. Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. (2005) 280:26483–90. doi: 10.1074/jbc.m504041200
38. Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. (2006) 3:15–24. doi: 10.1016/j.cmet.2005.11.014
39. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. (2008) 8:512–21. doi: 10.1016/j.cmet.2008.10.008
40. DeBose-Boyd RA, Ye J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci. (2018) 43:358–68. doi: 10.1016/j.tibs.2018.01.005
41. Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, et al. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. (1998) 2:505–14. doi: 10.1016/s1097-2765(00)80150-1
42. Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, et al. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell. (1997) 1:47–57. doi: 10.1016/s1097-2765(00)80006-4
43. Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA. (1999) 96:11041–8. doi: 10.1073/pnas.96.20.11041
44. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. (2000) 14:2819–30. doi: 10.1101/gad.844900
45. Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, et al. Cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell. (2016) 30:683–93.
46. Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. (2011) 1:442–56. doi: 10.1158/2159-8290.cd-11-0102
47. Lee JH, Phelan P, Shin M, Oh BC, Han X, Im SS, et al. SREBP-1a-stimulated lipid synthesis is required for macrophage phagocytosis downstream of TLR4-directed mTORC1. Proc Natl Acad Sci USA. (2018) 115:E12228–34.
48. Li X, Chen YT, Josson S, Mukhopadhyay NK, Kim J, Freeman MR, et al. MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS One. (2013) 8:e70987. doi: 10.1371/journal.pone.0070987
49. Ru P, Hu P, Geng F, Mo X, Cheng C, Yoo JY, et al. Feedback loop regulation of SCAP/SREBP-1 by miR-29 modulates EGFR signaling-driven glioblastoma growth. Cell Rep. (2017) 18:1076–7. doi: 10.1016/j.celrep.2017.01.016
50. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology – divergent pathophysiology. Nat Rev Endocrinol. (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
51. Järvinen R, Knekt P, Hakulinen T, Rissanen H, Heliövaara M. Dietary fat, cholesterol and colorectal cancer in a prospective study. Br J Cancer. (2001) 85:357–61. doi: 10.1054/bjoc.2001.1906
52. Morioka S, Sai K, Omori E, Ikeda Y, Matsumoto K, Ninomiya-Tsuji J. TAK1 regulates hepatic lipid homeostasis through SREBP. Oncogene. (2016) 35:3829–38. doi: 10.1038/onc.2015.453
53. Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin Cancer Res. (2016) 22:5337–48. doi: 10.1158/1078-0432.ccr-15-2973
54. Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. (2013) 73:2850–62. doi: 10.1158/0008-5472.can-13-0382-t
55. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. (2018) 50:206–18. doi: 10.1038/s41588-017-0027-2
56. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther. (2014) 13:855–66. doi: 10.1158/1535-7163.mct-13-0797
57. Li X, Wu JB, Chung LW, Huang WC. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget. (2015) 6:41018–32. doi: 10.18632/oncotarget.5879
58. Li J, Yan H, Zhao L, Jia W, Yang H, Liu L, et al. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget. (2016) 7:52392–403. doi: 10.18632/oncotarget.10721
59. Siqingaowa, Sekar S, Gopalakrishnan V, Taghibiglou C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem Biophys Res Commun. (2017) 488:136–40. doi: 10.1016/j.bbrc.2017.05.023
60. Gao S, Shi Z, Li X, Li W, Wang Y, Liu Z, et al. Fatostatin suppresses growth and enhances apoptosis by blocking SREBP-regulated metabolic pathways in endometrial carcinoma. Oncol Rep. (2018) 39:1919–29.
61. Yao L, Chen S, Li W. Fatostatin inhibits the development of endometrial carcinoma in endometrial carcinoma cells and a xenograft model by targeting lipid metabolism. Arch Biochem Biophys. (2020) 684:108327. doi: 10.1016/j.abb.2020.108327
62. Blanchet M, Seidah NG, Labonté P. SKI-1/S1P inhibition: a promising surrogate to statins to block hepatitis C virus replication. Antiviral Res. (2012) 95:159–66. doi: 10.1016/j.antiviral.2012.05.006
63. Caruana BT, Skoric A, Brown AJ, Lutze-Mann LH. Site-1 protease, a novel metabolic target for glioblastoma. Biochem Biophys Res Commun. (2017) 490:760–6. doi: 10.1016/j.bbrc.2017.06.114
64. Guan M, Fousek K, Jiang C, Guo S, Synold T, Xi B, et al. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin Cancer Res. (2011) 17:1796–806. doi: 10.1158/1078-0432.ccr-10-3216
65. Guan M, Su L, Yuan YC, Li H, Chow WA. Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Sci Rep. (2015) 5:9698.
66. Hill EJ, Roberts C, Franklin JM, Enescu M, West N, MacGregor TP, et al. Clinical trial of oral nelfinavir before and during radiation therapy for advanced rectal cancer. Clin Cancer Res. (2016) 22:1922–31. doi: 10.1158/1078-0432.ccr-15-1489
67. Driessen C, Kraus M, Joerger M, Rosing H, Bader J, Hitz F, et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: a phase I trial (SAKK 65/08). Haematologica. (2016) 101:346–55. doi: 10.3324/haematol.2015.135780
68. Rengan R, Mick R, Pryma D, Rosen MA, Lin LL, Maity AM, et al. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a report of toxicities and clinical response. J Thorac Oncol. (2012) 7:709–15. doi: 10.1097/jto.0b013e3182435aa6
69. Wilson JM, Fokas E, Dutton SJ, Patel N, Hawkins MA, Eccles C, et al. A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother Oncol. (2016) 119:306–11. doi: 10.1016/j.radonc.2016.03.021
70. Huang LH, Chung HY, Su HM. Docosahexaenoic acid reduces sterol regulatory element binding protein-1 and fatty acid synthase expression and inhibits cell proliferation by inhibiting pAkt signaling in a human breast cancer MCF-7 cell line. BMC Cancer. (2017) 17:890. doi: 10.1186/s12885-017-3936-7
71. Bedikian AY, DeConti RC, Conry R, Agarwala S, Papadopoulos N, Kim KB, et al. Phase 3 study of docosahexaenoic acid-paclitaxel versus dacarbazine in patients with metastatic malignant melanoma. Ann Oncol. (2011) 22:787–93. doi: 10.1093/annonc/mdq438
72. Gucalp A, Zhou XK, Cook ED, Garber JE, Crew KD, Nangia JR, et al. A randomized multicenter phase II study of docosahexaenoic acid in patients with a history of breast cancer, premalignant lesions, or benign breast disease. Cancer Prev Res. (2018) 11:203–14. doi: 10.1158/1940-6207.capr-17-0354
73. Nakahara M, Fujii H, Maloney PR, Shimizu M, Sato R. Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J Biol Chem. (2002) 277:37229–34. doi: 10.1074/jbc.m206749200
74. Pearson T, Caporaso JG, Yellowhair M, Bokulich NA, Padi M, Roe DJ, et al. Effects of ursodeoxycholic acid on the gut microbiome and colorectal adenoma development. Cancer Med. (2019) 8:617–28. doi: 10.1002/cam4.1965
75. Banerjee B, Shaheen NJ, Martinez JA, Hsu CH, Trowers E, Gibson BA, et al. Clinical study of ursodeoxycholic acid in Barrett’s Esophagus patients. Cancer Prev Res. (2016) 9:528–33. doi: 10.1158/1940-6207.capr-15-0276
76. Bordbar M, Shakibazad N, Fattahi M, Haghpanah S, Honar N. Effect of ursodeoxycholic acid and vitamin E in the prevention of liver injury from methotrexate in pediatric leukemia. Turk J Gastroenterol. (2018) 29:203–9. doi: 10.5152/tjg.2018.17521
77. Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. (2009) 2:ra82. doi: 10.1126/scisignal.2000446
78. Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem. (1997) 272:3137–40. doi: 10.1074/jbc.272.6.3137
79. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, et al. A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. (2007) 13:5183–94. doi: 10.1158/1078-0432.ccr-07-0161
80. Stevens JF, Page JE. Xanthohumol and related prenylflavonoids from hops and beer: to your good health! Phytochemistry. (2004) 65:1317–30. doi: 10.1016/j.phytochem.2004.04.025
81. Miyata S, Inoue J, Shimizu M, Sato R. Xanthohumol improves diet-induced obesity and fatty liver by suppressing sterol regulatory element-binding protein (SREBP) activation. J Biol Chem. (2015) 290:20565–79. doi: 10.1074/jbc.m115.656975
82. Tang JJ, Li JG, Qi W, Qiu WW, Li PS, Li BL, et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. (2011) 13:44–56. doi: 10.1016/j.cmet.2010.12.004
83. Frew Q, Rennekampff HO, Dziewulski P, Moiemen N, Zahn T, Hartmann B. Betulin wound gel accelerated healing of superficial partial thickness burns: results of a randomized, intra-individually controlled, phase III trial with 12-months follow-up. Burns. (2019) 45:876–90. doi: 10.1016/j.burns.2018.10.019
84. Huyke C, Reuter J, Rödig M, Kersten A, Laszczyk M, Scheffler A, et al. Treatment of actinic keratoses with a novel betulin-based oleogel. A prospective, randomized, comparative pilot study. J Dtsch Dermatol Ges. (2009) 7:128–33. doi: 10.1111/j.1610-0387.2008.06865.x
85. Nambiar DK, Deep G, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1. Oncotarget. (2014) 5:10017–33.
86. Wah Kheong C, Nik Mustapha NR, Mahadeva S. A randomized trial of silymarin for the treatment of nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. (2017) 15:1940–9.e1948.
87. Braun DL, Rauch A, Aouri M, Durisch N, Eberhard N, Anagnostopoulos A, et al. Lead-In with silibinin prior to triple-therapy translates into favorable treatment outcomes in difficult-to-treat HIV/Hepatitis C Coinfected patients. PLoS One. (2015) 10:e0133028:1–11. doi: 10.1371/journal.pone.0133028
88. Lazzeroni M, Guerrieri-Gonzaga A, Gandini S, Johansson H, Serrano D, Cazzaniga M, et al. A presurgical study of oral silybin-phosphatidylcholine in patients with early breast cancer. Cancer Prev Res. (2016) 9:89–95. doi: 10.1158/1940-6207.capr-15-0123
89. Giorgi VS, Peracoli MT, Peracoli JC, Witkin SS, Bannwart-Castro CF. Silibinin modulates the NF-κb pathway and pro-inflammatory cytokine production by mononuclear cells from preeclamptic women. J Reprod Immunol. (2012) 95:67–72. doi: 10.1016/j.jri.2012.06.004
90. Oh JH, Lee JI, Karadeniz F, Seo Y, Kong CS. 3,5-Dicaffeoyl-epi-quinic acid isolated from edible halophyte inhibits adipogenesis via AMPK/MAPK pathway in 3T3-L1 adipocytes. Evid Based Complement Alternat Med. (2018) 2018:8572571.
91. Chen J, Yue J, Liu J, Liu Y, Jiao KL, Teng MY, et al. Salvianolic acids improve liver lipid metabolism in ovariectomized rats via blocking STAT-3/SREBP1 signaling. Chin J Nat Med. (2018) 16:838–45. doi: 10.1016/s1875-5364(18)30125-0
92. Liu P, Hu YY, Liu C, Zhu DY, Xue HM, Xu ZQ, et al. Clinical observation of salvianolic acid B in treatment of liver fibrosis in chronic hepatitis B. World J Gastroenterol. (2002) 8:679–85. doi: 10.3748/wjg.v8.i4.679
93. Ji H, Ahn K, Cho H, Kim HE, Kim Y, Kim O. Sanguisorba officinalis L. extracts activate Wnt/beta-catenin pathway, and subsequently control adipo-osteogenic differentiation. Biochem Biophys Res Commun. (2018) 504:352–8. doi: 10.1016/j.bbrc.2018.08.196
94. Li YC, Qiao JY, Wang BY, Bai M, Shen JD, Cheng YX. Paeoniflorin ameliorates fructose-induced insulin resistance and hepatic steatosis by activating LKB1/AMPK and AKT pathways. Nutrients. (2018) 10:1024. doi: 10.3390/nu10081024
95. Chen L, Qi H, Jiang D, Wang R, Chen A, Yan Z, et al. The new use of an ancient remedy: a double-blinded randomized study on the treatment of rheumatoid arthritis. Am J Chin Med. (2013) 41:263–80. doi: 10.1142/s0192415x13500195
96. Guo D, Wang Y, Wang J, Song L, Wang Z, Mao B, et al. RA-XII suppresses the development and growth of liver cancer by inhibition of lipogenesis via SCAP-dependent SREBP Supression. Molecules. (2019) 24:1829. doi: 10.3390/molecules24091829
97. Wang Y, Guo D, He J, Song L, Chen H, Zhang Z, et al. Inhibition of fatty acid synthesis arrests colorectal neoplasm growth and metastasis: anti-cancer therapeutical effects of natural cyclopeptide RA-XII. Biochem Biophys Res Commun. (2019) 512:819–24. doi: 10.1016/j.bbrc.2019.03.088
98. Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. (2017) 292:7189–207. doi: 10.1074/jbc.m116.774471
99. Yamanaka R, Tabata S, Shindo Y, Hotta K, Suzuki K, Soga T, et al. Mitochondrial Mg(2+) homeostasis decides cellular energy metabolism and vulnerability to stress. Sci Rep. (2016) 6:30027.
100. Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. (2012) 22:585–600. doi: 10.1016/j.ccr.2012.09.020
101. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. (2011) 43:869–74. doi: 10.1038/ng.890
102. Gosmain Y, Dif N, Berbe V, Loizon E, Rieusset J, Vidal H, et al. Regulation of SREBP-1 expression and transcriptional action on HKII and FAS genes during fasting and refeeding in rat tissues. J Lipid Res. (2005) 46:697–705. doi: 10.1194/jlr.m400261-jlr200
103. Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. (2017) 18:1197–206. doi: 10.1038/ni.3838
104. Meton I, Egea M, Anemaet IG, Fernandez F, Baanante IV. Sterol regulatory element binding protein-1a transactivates 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene promoter. Endocrinology. (2006) 147:3446–56. doi: 10.1210/en.2005-1506
105. Ruiz R, Jideonwo V, Ahn M, Surendran S, Tagliabracci VS, Hou Y, et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem. (2014) 289:5510–7. doi: 10.1074/jbc.m113.541110
106. Guillet-Deniau I, Pichard AL, Kone A, Esnous C, Nieruchalski M, Girard J, et al. Glucose induces de novo lipogenesis in rat muscle satellite cells through a sterol-regulatory-element-binding-protein-1c-dependent pathway. J Cell Sci. (2004) 117(Pt 10):1937–44. doi: 10.1242/jcs.01069
107. Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, et al. Glucose-mediated n-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell. (2015) 28:569–81. doi: 10.1016/j.ccell.2015.09.021
108. Wen YA, Xiong X, Zaytseva YY, Napier DL, Vallee E, Li AT, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. (2018) 9:265.
109. Liu Y, Yu Q, Chen Y. Effect of silibinin on CFLAR-JNK pathway in oleic acid-treated HepG2 cells. Biomed Pharmacothe. (2018) 108:716–23. doi: 10.1016/j.biopha.2018.09.089
110. Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol. (2010) 8:88. doi: 10.1186/1741-7007-8-88
111. Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science. (2019) 363:644–9. doi: 10.1126/science.aav0173
112. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
113. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. (2016) 22:427–32. doi: 10.1038/nm.4055
114. Itkonen HM, Brown M, Urbanucci A, Tredwell G, Ho Lau C, Barfeld S, et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget. (2017) 8:38264–75.
115. Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. (2015) 126:1925–9. doi: 10.1182/blood-2014-12-617498
116. Tirado-Velez JM, Joumady I, Saez-Benito A, Cozar-Castellano I, Perdomo G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PLoS One. (2012) 7:e46484. doi: 10.1371/journal.pone.0046484
117. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. (2011) 25:1041–51. doi: 10.1101/gad.1987211
118. Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta. (2010) 1805:141–52. doi: 10.1016/j.bbcan.2010.01.005
119. Peck B, Schug ZT, Zhang Q, Dankworth B, Jones DT, Smethurst E, et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. (2016) 4:6.
120. Zaytseva YY, Harris JW, Mitov MI, Kim JT, Butterfield DA, Lee EY, et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget. (2015) 6:18891–904. doi: 10.18632/oncotarget.3783
121. Chen YS, Liu HM, Lee TY. Ursodeoxycholic acid regulates hepatic energy homeostasis and white adipose tissue macrophages polarization in leptin-deficiency obese mice. Cells. (2019) 8:253. doi: 10.3390/cells8030253
122. Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. (2011) 147:173–84. doi: 10.1016/j.cell.2011.08.034
123. Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. (2007) 67:8180–7. doi: 10.1158/0008-5472.can-07-0389
124. Martínez-Botas J, Suárez Y, Ferruelo AJ, Gómez-Coronado D, Lasuncion MA. Cholesterol starvation decreases p34(cdc2) kinase activity and arrests the cell cycle at G2. FASEB J. (1999) 13:1359–70. doi: 10.1096/fasebj.13.11.1359
125. Martínez-Botas J, Ferruelo AJ, Suárez Y, Fernández C, Gómez-Coronado D, Lasunción MA. Dose-dependent effects of lovastatin on cell cycle progression. Distinct requirement of cholesterol and non-sterol mevalonate derivatives. Biochim Biophys Acta. (2001) 1532:185–94. doi: 10.1016/s1388-1981(01)00125-1
126. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. (2006) 9:358–65. doi: 10.1097/01.mco.0000232894.28674.30
127. Pitroda SP, Khodarev NN, Beckett MA, Kufe DW, Weichselbaum RR. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc Natl Acad Sci USA. (2009) 106:5837–41. doi: 10.1073/pnas.0812029106
128. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. (2012) 10:133–42. doi: 10.1158/1541-7786.mcr-11-0206
129. Lee BH, Taylor MG, Robinet P, Smith JD, Schweitzer J, Sehayek E, et al. Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Cancer Res. (2013) 73:1211–8. doi: 10.1158/0008-5472.can-12-3128
130. Rudling MJ, Angelin B, Peterson CO, Collins VP. Low density lipoprotein receptor activity in human intracranial tumors and its relation to the cholesterol requirement. Cancer Res. (1990) 50:483–7.
131. Horton JD. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans. (2002) 30(Pt 6):1091–5. doi: 10.1042/bst0301091
132. Lai L, Azzam KM, Lin WC, Rai P, Lowe JM, Gabor KA, et al. MicroRNA-33 regulates the innate immune response via ATP binding cassette transporter-mediated remodeling of membrane microdomains. J Biol Chem. (2016) 291:19651–60. doi: 10.1074/jbc.m116.723056
133. Geng F, Guo D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern Med Rev. (2017) 3.
134. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. (2006) 22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656
135. Long J, Zhang CJ, Zhu N, Du K, Yin YF, Tan X, et al. Lipid metabolism and carcinogenesis, cancer development. Am J Cancer Res. (2018) 8:778–91.
136. Zhu YH, Jeong S, Wu M, Jin ZY, Zhou JY, Han RQ, et al. Dietary intake of fatty acids, total cholesterol, and stomach cancer in a chinese population. Nutrients. (2019) 11:1730. doi: 10.3390/nu11081730
137. Wang CM, Yuan RS, Zhuang WY, Sun JH, Wu JY, Li H, et al. Schisandra polysaccharide inhibits hepatic lipid accumulation by downregulating expression of SREBPs in NAFLD mice. Lipids Health Dis. (2016) 15:195.
138. Hu J, Hong W, Yao KN, Zhu XH, Chen ZY, Ye L. Ursodeoxycholic acid ameliorates hepatic lipid metabolism in LO2 cells by regulating the AKT/mTOR/SREBP-1 signaling pathway. World J Gastroenterol. (2019) 25:1492–501. doi: 10.3748/wjg.v25.i12.1492
139. Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell. (2018) 22:206–20.e204.
140. Smulan LJ, Ding W, Freinkman E, Gujja S, Edwards YJK, Walker AK. Cholesterol-independent SREBP-1 maturation is linked to ARF1 inactivation. Cell Rep. (2016) 16:9–18. doi: 10.1016/j.celrep.2016.05.086
141. Marcella SW, David A, Ohman-Strickland PA, Carson J, Rhoads GG. Statin use and fatal prostate cancer: a matched case-control study. Cancer. (2012) 118:4046–52. doi: 10.1002/cncr.26720
142. Van Rompay MI, Solomon KR, Nickel JC, Ranganathan G, Kantoff PW, McKinlay JB. Prostate cancer incidence and mortality among men using statins and non-statin lipid-lowering medications. Eur J Cancer. (2019) 112:118–26. doi: 10.1016/j.ejca.2018.11.033
143. Longo J, Mullen PJ, Yu R, van Leeuwen JE, Masoomian M, Woon DTS, et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol Metab. (2019) 25:119–30. doi: 10.1016/j.molmet.2019.04.003
144. Gobel A, Breining D, Rauner M, Hofbauer LC, Rachner TD. Induction of 3-hydroxy-3-methylglutaryl-CoA reductase mediates statin resistance in breast cancer cells. Cell Death Dis. (2019) 10:91.
145. Casella C, Miller DH, Lynch K, Brodsky AS. Oxysterols synergize with statins by inhibiting SREBP-2 in ovarian cancer cells. Gynecol Oncol. (2014) 135:333–41. doi: 10.1016/j.ygyno.2014.08.015
146. Clendening JW, Pandyra A, Li Z, Boutros PC, Martirosyan A, Lehner R, et al. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood. (2010) 115:4787–97. doi: 10.1182/blood-2009-07-230508
147. Meena AS, Sharma A, Kumari R, Mohammad N, Singh SV, Bhat MK. Inherent and acquired resistance to paclitaxel in hepatocellular carcinoma: molecular events involved. PLoS One. (2013) 8:e61524. doi: 10.1371/journal.pone.0061524
148. Zheng L, Li L, Lu Y, Jiang F, Yang XA. SREBP2 contributes to cisplatin resistance in ovarian cancer cells. Exp Biol Med. (2018) 243:655–62. doi: 10.1177/1535370218760283
149. Kelsey I, Zbinden M, Byles V, Torrence M, Manning BD. mTORC1 suppresses PIM3 expression via miR-33 encoded by the SREBP loci. Sci Rep. (2017) 7:16112.
150. Kim YS, Lee YM, Oh TI, Shin DH, Kim GH, Kan SY, et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism. Int J Mol Sci. (2018) 19:3127. doi: 10.3390/ijms19103127
151. Soundararajan R, Wishart AD, Rupasinghe HP, Arcellana-Panlilio M, Nelson CM, Mayne M, et al. Quercetin 3-glucoside protects neuroblastoma (SH-SY5Y) cells in vitro against oxidative damage by inducing sterol regulatory element-binding protein-2-mediated cholesterol biosynthesis. J Biol Chem. (2008) 283:2231–45. doi: 10.1074/jbc.m703583200
152. Seo YK, Jeon TI, Chong HK, Biesinger J, Xie X, Osborne TF. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. (2011) 13:367–75. doi: 10.1016/j.cmet.2011.03.005
153. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
154. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. (2016) 16:718–31. doi: 10.1038/nrc.2016.76
155. Shatz O, Holland P, Elazar Z, Simonsen A. Complex relations between phospholipids, autophagy, and neutral lipids. Trends Biochem Sci. (2016) 41:907–23. doi: 10.1016/j.tibs.2016.08.001
156. Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids. (2010) 82:243–50. doi: 10.1016/j.plefa.2010.02.005
157. Kim H, Rodriguez-Navas C, Kollipara RK, Kapur P, Pedrosa I, Brugarolas J, et al. Unsaturated fatty acids stimulate tumor growth through stabilization of beta-catenin. Cell Rep. (2015) 13:495–503. doi: 10.1016/j.celrep.2015.09.010
158. Motallebipour M, Enroth S, Punga T, Ameur A, Koch C, Dunham I, et al. Novel genes in cell cycle control and lipid metabolism with dynamically regulated binding sites for sterol regulatory element-binding protein 1 and RNA polymerase II in HepG2 cells detected by chromatin immunoprecipitation with microarray detection. FEBS J. (2009) 276:1878–90. doi: 10.1111/j.1742-4658.2009.06914.x
159. Bengoechea-Alonso MT, Ericsson J. Cdk1/cyclin B-mediated phosphorylation stabilizes SREBP1 during mitosis. Cell Cycle. (2006) 5:1708–18. doi: 10.4161/cc.5.15.3131
160. Inoue N, Shimano H, Nakakuki M, Matsuzaka T, Nakagawa Y, Yamamoto T, et al. Lipid synthetic transcription factor SREBP-1a activates p21WAF1/CIP1, a universal cyclin-dependent kinase inhibitor. Mol Cell Biol. (2005) 25:8938–47. doi: 10.1128/mcb.25.20.8938-8947.2005
161. Nakakuki M, Shimano H, Inoue N, Tamura M, Matsuzaka T, Nakagawa Y, et al. A transcription factor of lipid synthesis, sterol regulatory element-binding protein (SREBP)-1a causes G(1) cell-cycle arrest after accumulation of cyclin-dependent kinase (cdk) inhibitors. FEBS J. (2007) 274:4440–52. doi: 10.1111/j.1742-4658.2007.05973.x
162. Lee JH, Jeon YG, Lee KH, Lee HW, Park J, Jang H, et al. RNF20 suppresses tumorigenesis by inhibiting the SREBP1c-PTTG1 axis in kidney cancer. Mol Cell Biol. (2017) 37:e00265-17.
163. Vlotides G, Eigler T, Melmed S. Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev. (2007) 28:165–86. doi: 10.1210/er.2006-0042
164. Cildir G, Akincilar SC, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med. (2013) 19:487–500. doi: 10.1016/j.molmed.2013.05.001
165. Schroit AJ, Gallily R. Macrophage fatty acid composition and phagocytosis: effect of unsaturation on cellular phagocytic activity. Immunology. (1979) 36:199–205.
166. Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. (2014) 262:193–215. doi: 10.1111/imr.12212
167. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun. (2013) 440:105–11. doi: 10.1016/j.bbrc.2013.09.037
168. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. (2016) 539:294–8. doi: 10.1038/nature20117
169. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
170. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
171. Zhang Q, Wang H, Mao C, Sun M, Dominah G, Chen L, et al. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol Immunol. (2018) 94:27–35. doi: 10.1016/j.molimm.2017.12.011
172. Jande SS, Bélanger LF. Fine structural study of rat molar cementum. Anat Rec. (1970) 167:439–63. doi: 10.1002/ar.1091670406
173. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. (2013) 14:489–99. doi: 10.1038/ni.2570
174. Ito A, Hong C, Oka K, Salazar JV, Diehl C, Witztum JL, et al. Cholesterol accumulation in CD11c(+) immune cells is a causal and targetable factor in autoimmune disease. Immunity. (2016) 45:1311–26. doi: 10.1016/j.immuni.2016.11.008
175. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. (2017) 25:1294–304.e1296.
176. Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. (2018) 49:842–56.e847.
177. Yao M, Fan X, Yuan B, Takagi N, Liu S, Han X, et al. Berberine inhibits NLRP3 inflammasome pathway in human triple-negative breast cancer MDA-MB-231 cell. BMC Complement Altern Med. (2019) 19:216. doi: 10.1186/s12906-019-2615-4
178. Shen H, Shi LZ. Metabolic regulation of TH17 cells. Mol Immunol. (2019) 109:81–7. doi: 10.1016/j.molimm.2019.03.005
179. Zech T, Ejsing CS, Gaus K, de Wet B, Shevchenko A, Simons K, et al. Accumulation of raft lipids in T-cell plasma membrane domains engaged in TCR signalling. EMBO J. (2009) 28:466–76. doi: 10.1038/emboj.2009.6
Keywords: natural drug, sterol regulatory protein, lipid metabolism, glucose metabolism, microenvironment, cytoprotection
Citation: Jiang T, Zhang G and Lou Z (2020) Role of the Sterol Regulatory Element Binding Protein Pathway in Tumorigenesis. Front. Oncol. 10:1788. doi: 10.3389/fonc.2020.01788
Received: 24 August 2019; Accepted: 11 August 2020;
Published: 08 September 2020.
Edited by:
Huizi Jin, Shanghai Jiao Tong University, ChinaReviewed by:
Nor Eddine Sounni, University of Liège, BelgiumDeliang Guo, The Ohio State University, United States
Copyright © 2020 Jiang, Zhang and Lou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangji Zhang, emdqQHpjbXUuZWR1LmNu; Zhaohuan Lou, emhhb2h1YW5sb3VAemNtdS5lZHUuY24=