 Yoshie Yoshikawa
Yoshie Yoshikawa Kozo Kuribayashi
Kozo Kuribayashi Toshiyuki Minami
Toshiyuki Minami Masaki Ohmuraya
Masaki Ohmuraya Takashi Kijima
Takashi Kijima- 1Department of Genetics, Hyogo College of Medicine, Nishinomiya, Japan
- 2Department of Respiratory Medicine and Hematology, Hyogo College of Medicine, Nishinomiya, Japan
Malignant pleural mesothelioma (MPM) is strongly associated with occupational or environmental asbestos exposure and arises from neoplastic transformation of mesothelial cells in the pleural cavity. The only standard initial treatment for unresectable MPM is combination chemotherapy with cisplatin (CDDP) and pemetrexed (PEM). Further, CDDP/PEM is the only approved regimen with evidence of prolonged overall survival (OS), although the median OS for patients treated with this regimen is only 12 months after diagnosis. Thus, the development of new therapeutic strategies has been investigated for approximately 20 years. In contrast to recent advances in personalized lung cancer therapies, diagnostic and prognostic biomarker research has just started in mesothelioma. Epigenetic alterations include DNA methylation, histone modifications, and other chromatin-remodeling events. These processes are involved in numerous cellular processes including differentiation, development, and tumorigenesis. Epigenetic modifications play an important role in gene expression and regulation related to malignant MPM phenotypes and histological subtypes. An immune checkpoint PD-1 inhibitor, nivolumab, was approved as second-line therapy for patients who had failed initial chemotherapy, based on the results of the MERIT study. Various clinical immunotherapy trials are ongoing in patients with advanced MPM. In this review, we describe recent knowledge on epigenetic alterations, which might identify candidate therapeutic targets and immunotherapeutic regimens under development for MPM.
Introduction
Malignant pleural mesothelioma (MPM) remains one of the most incurable malignancies, with a very poor prognosis and a 7.9-month median survival time (MST) from diagnosis to death (1). Even after the widespread prevention of asbestos use, mortality rates have not decreased substantially in most countries. Further, the use of asbestos is not banned worldwide, and is still collected and used in many countries (e.g., China, Russia, and India) (2). Thus, the incidence of mesothelioma is not expected to peak before 2020 because asbestos was widely used in most developed countries until 1970 and the long latency (about 30 to 50 years) of asbestos exposure (3).
When asbestos fibers reach the lung periphery and the pleura, they cause local chronic inflammation and carcinogenesis. Common markers of carcinogenic processes include overexpression of growth factors such as VEGF, inactivating mutations of tumor suppressor genes (BAP1, CDKN2A, NF2, and TP53), extensive chromosomal deletions, and epigenetic alterations, which are essential for genetic abnormalities in malignant mesothelioma (4) (Table 1). These factors result in enhanced cell proliferation, apoptosis resistance, and local tumor immune evasion, and are the targets of several new therapeutic approaches (16).
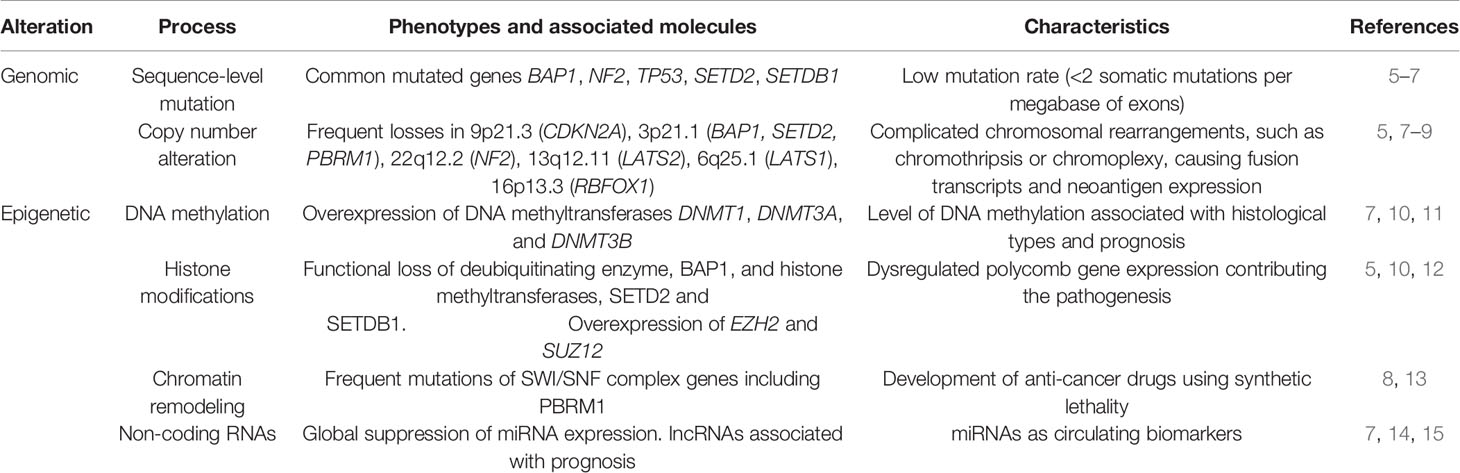
Table 1 Summary of the biomarkers for diagnosis or as therapeutic targets for malignant mesothelioma.
Immune mechanisms may be important therapeutic targets in MPM, as demonstrated by tumor-infiltrating lymphocytes (17, 18), spontaneous regression of mesothelioma (19, 20), and tumor responses to immunotherapy (21). Indeed, immunotherapy with immune checkpoint inhibitors (ICIs) has dramatically improved the prognosis of various solid tumor types, including melanoma and non-small cell lung cancer (22). Several clinical trials show that ICIs may also have promising antitumor effects in MPM (23).
Conversely, no validated biomarker is currently available for these agents, and even the representative biomarker PD-L1 has limited significance. In fact, recent clinical trials have shown that the role of ICIs in MPM is still debated, especially when considering single agent use (24).
Mutations
Recently, various gene mutations were identified in MPM via technological advances in genetic analysis. However, unlike other cancer types, active genetic mutation types such as epidermal growth factor receptor (EGFR) mutations in lung adenocarcinoma are not well understood in MPM. However, inactivating mutations of multiple tumor suppressor genes have been identified (5).
The most frequent tumor suppressor gene mutation in MPM is an inactivating mutation of CDKN2A (cyclin-dependent kinase inhibitor 2A gene), which occurs in approximately 70% or more MPM cases (25). CDKN2A has 3 exons and codes for 2 proteins: pl6/INK4a and p14/ARF.
pl6/INK4a is a cyclin-dependent kinase (CDK) inhibitor that inhibits pRB phosphorylation by CDK 4/6-cyclin D and arrests the cell cycle at G1. p14/ARF inhibits p53 degradation by MDM2 activity and stabilizes p53 function. When inactivating CDKN2A mutations render the pl6 and pl4 proteins nonfunctional, pRB and p53 lose their ability to regulate the cell cycle, leading to constitutive cell growth. Thus, CDKN2A is strongly associated with MPM development (6).
Neurofibromin2 (NF2) is a tumor suppressor gene located on the long arm of chromosome 22. Inactivating mutations in NF2 are reported in approximately 40-50% of MPMs (26). NF2 encodes the Merlin protein, and deletion of the Merlin protein results in constant yes-associated protein (YAP) activation by Hippo pathway inactivation (26, 27). YAP controls cell proliferation, survival, differentiation, organ size maintenance, tissue homeostasis, and plays a central role in the Hippo pathway (28). Merlin negatively regulates mTOR, and deletion of Merlin leads to mTOR signaling activation (29).
BRCA1-associated protein 1 (BAP1) is a tumor suppressor gene located on the short arm of chromosome 3, and the overall frequency of BAP1 alterations, including single-nucleotide variants, small indels, several exons to whole gene deletions, and structural variants, is reported to be approximately 57% in MPMs (7, 30). BAP1 encodes deubiquitinating enzymes and is involved in the regulation of expression and transcription of many genes. Further, BAP1 is involved in DNA repair, especially double-strand breaks (31). Germline mutations in BAP1 are reported in familial intraocular/cutaneous malignant melanoma. Similar mutations occur in familial MPM (32), drawing attention to the relationship between BAP1 and MPM.
Complicated chromosomal rearrangements such as chromothripsis and chromoplexy are two of the most characteristic genome alterations. Multiple noncontiguous minute deletions in 3p21 carrying BAP1, SETD2, and PBRM1 (8) and fusion transcripts caused by gene fusions including 3p21 region and NF2 have been reported (5). These chromosomal rearrangements produce neoantigen expression (9).
In a study, 2.4% of mesothelioma patients were diagnosed with microsatellite instability based on whole exome sequencing data, which revealed somatic mutations in the repair genes MSH2, MSH6, MLH1, PMS2, EXO1, POLD1, and POLE (33). Another study reported DNA mismatch repair (MMR) protein expression in all 159 MPM samples analyzed using IHC (34). Therefore, further studies on MMR molecules are warranted.
DNA Methylation
Epigenetic modifications cause phenotypic changes that do not involve alterations in the DNA sequence. These changes significantly affect gene regulation and expression as they occur in various cellular processes including differentiation, development, and tumorigenesis. Epigenetic mechanisms include DNA methylation, histone modifications, and other chromatin-remodeling events. Non-coding RNAs also work as regulators in epigenetics.
DNA methylation is the main epigenetic modulation. Methylation occurs naturally on cytosine bases at CpG sequences. Clusters of CpG dinucleotides (CpG islands) (35) are located in promoters of approximately 60% of genes, and additional CpG dinucleotides are dispersed throughout the genome. The former mostly are unmethylated, and the latter are typically hypermethylated in normal cells (36, 37). DNA methyltransferases, including DNMT1, 3A, and 3B, mediate transfer of a methyl group from S-adenosyl-methionine to the 5’ position of cytosine at CpG dinucleotides. Methylation in CpG islands inhibits binding of methylation-sensitive transcription factors and silences genes. Indeed, transcriptionally silencing tumor suppressor genes causes tumorigenesis. Genome-wide DNA hypomethylation increases chromosomal fragility (36, 37).
Studies analyzing DNA methylation indicate that silencing tumor suppressor genes through site-specific DNA hypermethylation and genome-wide hypomethylation are observed in MPM. Goto et al. examined the methylation status of 6,157 CpG islands using methylated CpG island amplification-microarray in 20 MPM cases and compared them to 20 pulmonary adenocarcinomas (38). Approximately 387 genes (6.3%) were hypermethylated in MPM in comparison with 544 genes (8.8%) in lung adenocarcinomas. These results show that epithelial-type MPM with less methylation tends to have longer survival rates. Hypermethylation of three genes (TMEM30B, KAZALD1, and MAPK13) distinguish MPM from adenocarcinoma. Christensen et al. identified significant differences among the epigenetic profiles of MPM compared to normal pleura (39). Methylation of 1505 CpG loci associated with 803 cancer-related genes was studied in 158 MPM cases and 18 normal pleura samples using methylation arrays. This report showed that methylation status is significantly related with asbestos body burden and patient survival.
To develop the non-invasive methods for diagnosis, circulating biomarkers can be detected in liquid samples such as plasma, serum, or urine using a method that has attracted considerable attention. Guarrera et al. analyzed DNA methylation levels in the whole blood in 163 MPM cases and 137 non-tumor controls using a methylation array (40). Differential methylation levels between MPMs and controls were observed mainly in the immune system–related genes. The gene showing the most hypomethylated single-CpG was FOXK1.
TCGA data suggest that DNMT1, DNMT3A, and DNMT3B overexpression is correlated with shorter MPM patient survival (10). Blum et al. applied “a deconvolution approach” to extract the molecular profile characterizing epithelioid and sarcomatoid MPM (11). They integrated epigenetic data and showed a correlation of gene expression levels with their DNA methylation levels. Furthermore, the methylation level of CpG sites is associated with histology: the differentially methylated CpG sites associated with epithelioid or sarcomatoid MPM are non-CpG islands and CpG islands, respectively.
Clinical efforts made previously to restrain DNMT gene activity in MPM have been unsuccessful. Yogelzang et al. (41) reported a 17% overall response rate in 41 MPM patients who received 120-h continuous infusion of dihydro-5-azacytidine, a DNA methyltransferase inhibitor. It is interesting to note that one of the complete responders was free from disease for six years after treatment.
Histone Modifications
Gene expression is controlled by loosening and compacting DNA wrapped around core histones (H2A, H2B, H3, and H4) through histone modification, three major types of which are known. Acetylation/deacetylation, methylation/demethylation, and ubiquitination/deubiquitination are major modifications in normal and cancer cells. Covalent modification is carried out at lysine-rich tails of core histones that extend out from the nucleosome. Histone acetylation increases the negative charge of the histone and resultantly repulses DNA, thus activating gene expression (42). Histone lysine methylation can activate or repress gene expression at some sites. Histone H3K9 (histone H3 at lysine 9) and H3K27 methylation are associated with transcriptional repression, while H3K4, H3K36, or H3K79 methylations are linked with gene activation (43). Histone H2A monoubiquitination is frequently correlated with gene silencing, while H2B monoubiquitination is mostly associated with transcriptional activation (44).
Lysine acetylation is catalyzed by histone acetyltransferase (HAT), which transfers the acetyl group from acetyl-CoA to the epsilon-amino group of a core histone protein lysine residue (45). Histone deacetylases (HDACs) are either Zn2+-dependent histone deacetylases or NAD+-dependent sirtuin deacetylases, which remove acetyl groups (46). Histone lysine methylation is mediated by numerous enzymes called histone lysine methyltransferase (KMTs) that mediate mono-, di-, and trimethylation of specific residues of lysine. On the contrary, histone demethylation is mediated by histone demethylases (KDMs) (43). Ubiquitin (Ub) conjugation reactions are catalyzed by the sequential action of Ub-activating (E1), Ub-conjugating (E2), and Ub-ligating (E3) enzymes. Deubiquitinating enzymes (DUBs) catalyze Ub removal from targeted substrates (44). E3 ubiquitin ligases, a large protein family, and DUBs have been well known as important regulators of numerous cellular processes (47).
BAP1 encodes DUB, and it is one of the most frequently altered genes in MPM (5, 48, 49). BAP1 assembles DUB complexes with the transcription regulators Additional Sex Combs-like (ASXL1, ASXL2, and ASXL3) similar to the Drosophila ortholog Calypso binding to the Polycomb group protein, ASX (50). These proteins form the Polycomb repressive deubiquitinase (PR-DUB) complex that cleaves Ub from histone H2A in nucleosomes. PR-DUB activity is essential for the regulation of H2A ubiquitination and Polycomb group-dependent gene silencing.
Polycomb repressive complex 2, formed by EZH2, SUZ12, and EED protein subunits, induces gene silencing through the addition of methyl groups to histone H3 at lysine 27, leading to trimethylated histone H3 lysine 27 (H3K27me3) (51, 52). EZH2 and SUZ12 overexpression were identified in asbestos-associated MPM cell lines (10), and EZH2 was overexpressed in approximately 85% of MPMs compared to normal pleura (52). Gene expression of EZH2 and SUZ12 is associated with MPM patient survival (10). LaFave et al. reported that BAP1 loss in mice results in increased H3K27me3 and EZH2 expression (12). Additionally, mesothelioma cells that lack BAP1 are sensitive to the inhibitor EZH2. This finding could provide a novel therapeutic approach against malignancies with BAP1 mutation.
The gene SETD2 encodes a member of the SET domain family, which are histone methyltransferases specific for H3K36. This gene is frequently mutated in MPM (5, 8). In addition to SETD2, Bueno et al. observed somatic mutations of the histone methyltransferase family genes SETDB1, SETD5, ASH1L, PRDM2, and KMT2D, the histone demethylase gene KDM2B, and the histone acetyltransferase gene CREBBP in MPMs (5).
HDAC inhibitors have emerged as anti-cancer drugs to treat hematological and solid malignancies. They could lead to cancer cell cycle arrest, differentiation, cell death, reduce angiogenesis, and modulate immune responses (46). However, these drugs have never provided sufficient therapeutic effects in MPM with the use of a single agent. Krug et al. randomized 661 patients to receive either vorinostat (n=329) or placebo (n=332). In this trial, vorinostat given as a second- or third-line therapy did not improve OS [30.7 weeks (95% CI: 26.7–36.1) compared to 27.1 weeks (95% CI: 23.1–31.9) for patients receiving placebo]. Thus, vorinostat would not be suitable as a therapy for patients with advanced MPM (53).
Chromatin Remodeling
Chromatin remodeling complexes are evolutionarily conserved multi-unit protein complexes that involve four subfamilies of ATP-dependent nucleosome-remodeling complexes: switch/sucrose non-fermentable (SWI/SNF), imitation switch, chromodomain helicase DNA-binding, and chromatin-remodeling ATPase INO80 (54). These complexes regulate chromatin structure to enable specific interactions with particular transcriptional activators, repressors, and histone modifiers. The SWI/SNF complex is the most well characterized tumor suppressor (55). Mammalian SWI/SNF complexes are composed of 12–15 subunits, and mutations in subunit genes including ARID1A, SMARCA4, ARID1B, ARID2, and PBRM1 occur across a wide spectrum of human cancers (56). In MPM, we found frequent multiple minute simultaneous biallelic deletions on chromosome 3p21. The top four frequently altered genes are associated with epigenetic modifications: BAP1, SETD2, PBRM1, and SMARCC1 (8). The last two genes belong to SWI/SNF complexes. We also detected sequence-level somatic mutations in ARID2 and SMARCA4 (13). The functional loss of these genes may lead to chromosomal instability because of their role in transcriptional regulation, DNA repair, and regulation of chromatin architecture and topology (57).
Synthetic lethality refers to cellular or organismal death that occurs when two genes are simultaneously perturbed (58). This phenomenon might be useful against tumor suppressor gene products in cancer-drug discovery. ARID1A, a SWI/SNF complex subunit, is frequently mutated in cancer. There are two mammalian homologs with an ARID domain, ARID1B and ARID2. Loss of ARID1B in ARID1A-deficient backgrounds destabilizes SWI/SNF complexes and impairs cancer and primary cell proliferation (59). This could be a conserved function underlying the synthetic lethality between ARID1A and ARID1B. Further, this finding increases information about synthetic lethality among chromatin remodeling subunits (60). To develop a new generation of anti-cancer drugs for MPM, synthetic lethality could be an attractive research field.
NON-CODING RNAs IMMUNOTHERAPY WITH ICIs IN MPM
Non-coding RNAs are a cluster of RNAs that are not translated into a protein and play an important role in post-transcriptional gene silencing. Based on their size, RNAs are characterized into short chain non-coding RNAs, including siRNA, miRNA, and piRNA, and long non-coding RNAs (lncRNAs) (61). There is a global suppression of miRNA expression in MPM: for example, let-7 family, miR-31, miR-34 family, including miR-34b and miR-34c, and miR-15 family, including miR-15a and miR-16. Lo Russo et al. reviewed the biological roles of miRNAs in MPM and listed miRNAs with diagnostic and/or prognostic value (14). Numerous cell functions such as apoptosis, cell proliferation, migration, invasion, and cell death are modulated by miRNA. As regulators of biological reactions, the response of MPM toward platinum-based chemotherapy is strongly affected by miRNAs and lncRNAs (62). It is reported that increased programmed death-ligand 1 (PD-L1) expression is associated with downregulated miRNAs, including miR-15b, miR-16, miR-193a-3p, miR-195, and miR-200c (63). As therapeutic targets, the phase I clinical trial (NCT02369198) using TargomiRs, delivering an miR-16-based miRNA mimic in the minicell-based formulation, was performed, and acceptable safety was observed. This trial showed a partial response in 1 of 22 patients (64). It is increasing the probability of non-coding RNAs in serum or plasma as circulating MPM biomarkers (15). Downregulated expression of miR-126 in serum from MPM patients was confirmed by several groups, but the discrimination power between MPM patients and healthy controls is not so high (15). A combination of circulating biomarkers is expected to overcome the poor sensitivity and specificity of single markers.
The biomarkers or therapeutic targets described above are summarized in Table 1. Recent insights about epigenetic mechanisms and dysregulated gene expression in MPM provide new opportunities with therapeutically challenging MPM.
Systemic MPM Treatment
To date, only cisplatin-pemetrexed combination therapy has been approved as frontline chemotherapy for unresectable MPM. This treatment was established as the standard of care in 2003, with no new modality (65). Platinum doublets, including pemetrexed, have been the international standard of care for MPM for approximately 20 years as first-line chemotherapy (66). However, the median OS for pemetrexed-cisplatin combination therapy does not exceed 13–16 months, according to data from randomized phase 3 trials (67). Thus, improved medical treatment is essential for improved outcomes.
Immunotherapy With ICIs in MPM
Since the introduction of ICIs, immunotherapy has made dramatic advances in various cancers. There are now three clinically available ICIs that block each of the following immunosuppressive molecules: cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death-1 (PD-1), and PD-1 ligand-1 (PD-L1) (68). However, the PD-L1 positivity ratio and tumor proportion score (TPS) are 20%–40% in MPM. PD-L1 binds and inhibits PD-1 on T cells. Further, PD-L1 is a spontaneously occurring factor that diminishes the anti-tumor immune response, and its expression is associated with poor prognosis in mesothelioma (69). The median OS of patients who are PD-L1-positive is 5.0 months compared to 14.5 months in patients who are PD-L1-negative. Thus, PD-L1 positivity is an independent risk factor for survival, with a median OS to risk ratio of 1.71 (95% CI: 1.03–2.78, p = 0.04) (70). Therefore, several therapies targeting the immune tumor microenvironment to restore antitumor immune responses have been studied in the MPM setting.
MPM has a low PD-L1 positivity rate and TPS, hence clinical trials have evaluated the antitumor activity of ICI. Though most evaluations were performed for patients with relapsed MPM after standard chemotherapy, ICIs exerted some promising results as salvage therapy (71). Three representative trials are shown below.
● KEYNOTE-028: PembrolizumabM
● This is a phase Ib study of Pembrolizumab alone in patients with PD-L1 TPS ≥1%. The efficacy was achieved with an objective response rate (ORR) of 20%, disease control rate (DCR) of 76%, median PFS of 5.8 months, and MST of 18.0 months (72).
● The NivoMes trial and MERIT: Nivolumab
● The efficacy of Nivolumab alone after second-line therapy was evaluated in a single-arm phase II study both overseas and in Japan. In the NivoMes study, conducted at a single institution in the Netherlands, ORR was 24% and DCR was 47%, median PFS was 2.6 months, 6-month survival was 74%, and MST was 11.8 months (73). On the other hand, in the MERIT trial, conducted as a multicenter trial in Japan, the ORR was 29%, the DCR was 68%, the median PFS was 6.1 months, the 6-month survival rate was 85%, and the MST was 17.3 months, showing favorable results (74).
Based on the results of MERIT, nivolumab was approved for regulatory use in patients with recurrent MPM following chemotherapy in Japan, ahead of all other countries. In addition, the antitumor effects of anti-CTLA-4 antibodies, such as ipilimumab and tremelimumab, have been evaluated in combination with nivolumab, an anti-PD-1 antibody, or durvalumab, an anti-PD-L1 antibody. Further, the possibility of ICI combination therapy has been researched. Specifically, results from three clinical trials are currently under review: NIBIT-MESO: (tremelimumab and durvalumab), MAPS-2: (ipilimumab and nivolumab combination or nivolumab monotherapy), and INITIATE: (ipilimumab and nivolumab). These studies have confirmed that combination therapy with anti-CTLA-4 and anti-PD-1/PD-L1 antibodies results in an ORR of 26 ~ 29% and a DCR of 50 ~ 68% in patients with recurrent MPM (75–77).
Conclusion
Thus, when MPM treatment has stagnated for more than 15 years, immunotherapy with ICI is generally well tolerated and is expected to markedly improve the outcome of patients with MPM. In contrast, ICI does not necessarily provide the expected antitumor effect in all cases; the recent negative results of the PROMISE-meso trial suggest that the role of these agents should be reconsidered in pretreated MPM in unselected patients. These data should be included as they reinforce the need for biomarkers to select patients who can benefit from the treatment, especially considering the limited role of PD-L1 expression observed in this trial. The solution to these challenges lies with the investigation of reliable predictive biomarkers, which will allow the selection of appropriate candidates for improving the benefits of ICI therapy. The role of chemo-immunotherapy combinations (78) and dual blockage immune checkpoint inhibition (77) seems more promising, but the best combination of ICIs has not yet been determined. Because MPM is also characterized as an extremely inflammatory tumor, further studies focusing on MPM cells and epigenetic alterations may lead to the identification of reliable biomarkers.
Author Contributions
KK and TM wrote the MPM treatment and Immune checkpoint inhibitors section of the manuscript. YY and MO wrote the Epigenetic alteration part of the manuscript. KK and TK provided a critical review of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Editorial support in the form of medical rewriting was provided by Cactus Communications.
References
1. Kuribayashi K, Doi H, Kijima T. Types of surgery post-neoadjuvant chemotherapy for pleural mesothelioma. Expert Rev Respir Med (2019) 13(12):1189–94. doi: 10.1080/17476348.2019.1679119
2. Abdel-Rahman O. Global trends in mortality from malignant mesothelioma: Analysis of WHO mortality database (1994-2013). Clin Respir J (2018) 12(6):2090–100. doi: 10.1111/crj.12778
3. Scherpereel A, Wallyn F, Albelda SM, Munck C. Novel therapies for malignant pleural mesothelioma. Lancet Oncol (2018) 19(3):e161–e72. doi: 10.1016/S1470-2045(18)30100-1
4. Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet (2005) 366(9483):397–408. doi: 10.1016/S0140-6736(05)67025-0
5. Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet (2016) 48(4):407–16. doi: 10.1038/ng.3520
6. Guo G, Chmielecki J, Goparaju C, Heguy A, Dolgalev I, Carbone M, et al. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res (2015) 75(2):264–9. doi: 10.1158/0008-5472.CAN-14-1008
7. Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov (2018) 8(12):1548–65. doi: 10.1158/2159-8290.CD-18-0804
8. Yoshikawa Y, Emi M, Hashimoto-Tamaoki T, Ohmuraya M, Sato A, Tsujimura T, et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci U S A (2016) 113(47):13432–7. doi: 10.1073/pnas.1612074113
9. Mansfield AS, Peikert T, Smadbeck JB, Udell JBM, Garcia-Rivera E, Elsbernd L, et al. Neoantigenic Potential of Complex Chromosomal Rearrangements in Mesothelioma. J Thorac Oncol (2019) 14(2):276–87. doi: 10.1016/j.jtho.2018.10.001
10. McLoughlin KC, Kaufman AS, Schrump DS. Targeting the epigenome in malignant pleural mesothelioma. Transl Lung Cancer Res (2017) 6(3):350–65. doi: 10.21037/tlcr.2017.06.06
11. Blum Y, Meiller C, Quetel L, Elarouci N, Ayadi M, Tashtanbaeva D, et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat Commun (2019) 10(1):1333. doi: 10.1038/s41467-019-09307-6
12. LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med (2015) 21(11):1344–9. doi: 10.1038/nm.3947
13. Yoshikawa Y, Sato A, Tsujimura T, Otsuki T, Fukuoka K, Hasegawa S, et al. Biallelic germline and somatic mutations in malignant mesothelioma: multiple mutations in transcription regulators including mSWI/SNF genes. Int J Cancer (2015) 136(3):560–71. doi: 10.1002/ijc.29015
14. Lo Russo G, Tessari A, Capece M, Galli G, de Braud F, Garassino MC, et al. MicroRNAs for the Diagnosis and Management of Malignant Pleural Mesothelioma: A Literature Review. Front Oncol (2018) 8:650. doi: 10.3389/fonc.2018.00650
15. Ferrari L, Carugno M, Mensi C, Pesatori AC. Circulating Epigenetic Biomarkers in Malignant Pleural Mesothelioma: State of the Art and critical Evaluation. Front Oncol (2020) 10:445. doi: 10.3389/fonc.2020.00445
16. Andujar P, Lacourt A, Brochard P, Pairon JC, Jaurand MC, Jean D. Five years update on relationships between malignant pleural mesothelioma and exposure to asbestos and other elongated mineral particles. J Toxicol Environ Health B Crit Rev (2016) 19(5-6):151–72. doi: 10.1080/10937404.2016.1193361
17. Anraku M, Cunningham KS, Yun Z, Tsao MS, Zhang L, Keshavjee S, et al. Impact of tumor-infiltrating T cells on survival in patients with malignant pleural mesothelioma. J Thorac Cardiovasc Surg (2008) 135(4):823–9. doi: 10.1016/j.jtcvs.2007.10.026
18. Yamada N, Oizumi S, Kikuchi E, Shinagawa N, Konishi-Sakakibara J, Ishimine A, et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol Immunother (2010) 59(10):1543–9. doi: 10.1007/s00262-010-0881-6
19. Moser JC, Peikert T, Roden AC, Midthun DE, Mansfield AS. Spontaneous Regression of Malignant Pleural Mesothelioma in a Patient with New-Onset Inflammatory Arthropathy. Ann Am Thorac Soc (2015) 12(9):1416–7. doi: 10.1513/AnnalsATS.201506-315LE
20. Robinson BW, Robinson C, Lake RA. Localised spontaneous regression in mesothelioma – possible immunological mechanism. Lung Cancer (2001) 32(2):197–201. doi: 10.1016/s0169-5002(00)00217-8
21. Alley EW, Katz SI, Cengel KA, Simone CB 2nd. Immunotherapy and radiation therapy for malignant pleural mesothelioma. Transl Lung Cancer Res (2017) 6(2):212–9. doi: 10.21037/tlcr.2017.04.01
22. Lievense LA, Sterman DH, Cornelissen R, Aerts JG. Checkpoint Blockade in Lung Cancer and Mesothelioma. Am J Respir Crit Care Med (2017) 196(3):274–82. doi: 10.1164/rccm.201608-1755CI
23. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
24. Popat S, Curioni-Fontecedro A, Dafni U, Shah R, O’Brien M, Pope A, et al. A multicentre randomised phase III trial comparing pembrolizumab vs single-agent chemotherapy for advanced pre-treated malignant pleural mesothelioma: the European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann Oncol (2020) 22:1734–45. doi: 10.1016/j.annonc.2020.09.009
25. Illei PB, Rusch VW, Zakowski MF, Ladanyi M. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin Cancer Res (2003) 9(6):2108–13.
26. Thurneysen C, Opitz I, Kurtz S, Weder W, Stahel RA, Felley-Bosco E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer (2009) 64(2):140–7. doi: 10.1016/j.lungcan.2008.08.014
27. Sekido Y. Inactivation of Merlin in malignant mesothelioma cells and the Hippo signaling cascade dysregulation. Pathol Int (2011) 61(6):331–44. doi: 10.1111/j.1440-1827.2011.02666.x
28. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev (2014) 94(4):1287–312. doi: 10.1152/physrev.00005.2014
29. Lopez-Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol Cell Biol (2009) 29(15):4235–49. doi: 10.1128/MCB.01578-08
30. Nasu M, Emi M, Pastorino S, Tanji M, Powers A, Luk H, et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol (2015) 10(4):565–76. doi: 10.1097/JTO.0000000000000471
31. Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer (2013) 13(3):153–9. doi: 10.1038/nrc3459
32. Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet (2011) 43(10):1022–5. doi: 10.1038/ng.912
33. Bonneville R, Krook M, Kautto E, Miya J, Wing M, Chen HZ, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol (2017) 1:1–15. doi: 10.1200/PO.17.00073
34. Cedres S, Ponce-Aix S, Iranzo P, Callejo A, Pardo N, Navarro A, et al. Analysis of mismatch repair (MMR) proteins expression in a series of malignant pleural mesothelioma (MPM) patients. Clin Transl Oncol (2020) 22(8):1390–8. doi: 10.1007/s12094-019-02275-9
35. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol (1987) 196(2):261–82. doi: 10.1016/0022-2836(87)90689-9
36. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol (2014) 6(5):a019133. doi: 10.1101/cshperspect.a019133
37. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol (2016) 8(9):a019505. doi: 10.1101/cshperspect.a019505
38. Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H, et al. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res (2009) 69(23):9073–82. doi: 10.1158/0008-5472.CAN-09-1595
39. Christensen BC, Houseman EA, Poage GM, Godleski JJ, Bueno R, Sugarbaker DJ, et al. Integrated profiling reveals a global correlation between epigenetic and genetic alterations in mesothelioma. Cancer Res (2010) 70(14):5686–94. doi: 10.1158/0008-5472.CAN-10-0190
40. Guarrera S, Viberti C, Cugliari G, Allione A, Casalone E, Betti M, et al. Peripheral Blood DNA Methylation as Potential Biomarker of Malignant Pleural Mesothelioma in Asbestos-Exposed Subjects. J Thorac Oncol (2019) 14(3):527–39. doi: 10.1016/j.jtho.2018.10.163
41. Yogelzang NJ, Herndon JE,2, Cirrincione C, Harmon DC, Antman KH, Corson JM, et al. Dihydro-5-azacytidine in malignant mesothelioma. A phase II trial demonstrating activity accompanied by cardiac toxicity. Cancer and Leukemia Group B. Cancer (1997) 79(11):2237–42. doi: 10.1002/(SICI)1097-0142(19970601)79:11<2237::AID-CNCR23>3.0.CO;2-W
42. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature (1997) 389(6649):349–52. doi: 10.1038/38664
43. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet (2012) 13(5):343–57. doi: 10.1038/nrg3173
44. Cao J, Yan Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol (2012) 2:26. doi: 10.3389/fonc.2012.00026
45. Di Martile M, Del Bufalo D, Trisciuoglio D. The multifaceted role of lysine acetylation in cancer: prognostic biomarker and therapeutic target. Oncotarget (2016) 7(34):55789–810. doi: 10.18632/oncotarget.10048
46. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci (2017) 18(7):1414. doi: 10.3390/ijms18071414
47. Hammond-Martel I, Yu H, Affar El B. Roles of ubiquitin signaling in transcription regulation. Cell Signal (2012) 24(2):410–21. doi: 10.1016/j.cellsig.2011.10.009
48. Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet (2011) 43(7):668–72. doi: 10.1038/ng.855
49. Yoshikawa Y, Sato A, Tsujimura T, Emi M, Morinaga T, Fukuoka K, et al. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci (2012) 103(5):868–74. doi: 10.1111/j.1349-7006.2012.02223.x
50. Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature (2010) 465(7295):243–7. doi: 10.1038/nature08966
51. Laugesen A, Hojfeldt JW, Helin K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb Perspect Med (2016) 6(9):a026575. doi: 10.1101/cshperspect.a026575
52. Kemp CD, Rao M, Xi S, Inchauste S, Mani H, Fetsch P, et al. Polycomb repressor complex-2 is a novel target for mesothelioma therapy. Clin Cancer Res (2012) 18(1):77–90. doi: 10.1158/1078-0432.CCR-11-0962
53. Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D, et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol (2015) 16(4):447–56. doi: 10.1016/S1470-2045(15)70056-2
54. Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol (2017) 18:407–22. doi: 10.1038/nrm.2017.26
55. Lu P, Roberts CW. The SWI/SNF tumor suppressor complex: Regulation of promoter nucleosomes and beyond. Nucleus (2013) 4(5):374–8. doi: 10.4161/nucl.26654
56. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet (2013) 45(6):592–601. doi: 10.1038/ng.2628
57. Hodges C, Kirkland JG, Crabtree GR. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med (2016) 6(8):a026930. doi: 10.1101/cshperspect.a026930
58. Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett (2011) 585(1):1–6. doi: 10.1016/j.febslet.2010.11.024
59. Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med (2014) 20(3):251–4. doi: 10.1038/nm.3480
60. Caumanns JJ, Wisman GBA, Berns K, van der Zee AGJ, de Jong S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim Biophys Acta Rev Cancer (2018) 1870(2):176–84. doi: 10.1016/j.bbcan.2018.07.005
61. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet (2011) 12(12):861–74. doi: 10.1038/nrg3074
62. Biersack B. Relations between approved platinum drugs and non-coding RNAs in mesothelioma. Noncoding RNA Res (2018) 3(4):161–73. doi: 10.1016/j.ncrna.2018.08.001
63. Kao SC, Cheng YY, Williams M, Kirschner MB, Madore J, Lum T, et al. Tumor Suppressor microRNAs Contribute to the Regulation of PD-L1 Expression in Malignant Pleural Mesothelioma. J Thorac Oncol (2017) 12(9):1421–33. doi: 10.1016/j.jtho.2017.05.024
64. van Zandwijk N, Pavlakis N, Kao SC, Linton A, Boyer MJ, Clarke S, et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol (2017) 18(10):1386–96. doi: 10.1016/S1470-2045(17)30621-6
65. Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol (2003) 21(14):2636–44. doi: 10.1200/JCO.2003.11.136
66. [NCCN] NCCN. NCCN malignant pleural mesothelioma guidelines (version 2. 2019). National Comprehensive Cancer Network (2019). Available at: https://wwwnccnorg/professionals/physician_gls/pdf/mpm_blockspdf.
67. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet (2016) 387(10026):1405–14. doi: 10.1016/S0140-6736(15)01238-6
68. Berger KN, Pu JJ. PD-1 pathway and its clinical application: A 20year journey after discovery of the complete human PD-1 gene. Gene (2018) 638:20–5. doi: 10.1016/j.gene.2017.09.050
69. Mansfield AS, Roden AC, Peikert T, Sheinin YM, Harrington SM, Krco CJ, et al. B7-H1 expression in malignant pleural mesothelioma is associated with sarcomatoid histology and poor prognosis. J Thorac Oncol (2014) 9(7):1036–40. doi: 10.1097/JTO.0000000000000177
70. Cedres S, Ponce-Aix S, Zugazagoitia J, Sansano I, Enguita A, Navarro-Mendivil A, et al. Analysis of expression of programmed cell death 1 ligand 1 (PD-L1) in malignant pleural mesothelioma (MPM). PloS One (2015) 10(3):e0121071. doi: 10.1371/journal.pone.0121071
71. Forde PM, Scherpereel A, Tsao AS. Use of Immune Checkpoint Inhibitors in Mesothelioma. Curr Treat Options Oncol (2019) 20(2):18. doi: 10.1007/s11864-019-0613-x
72. Alley EW, Lopez J, Santoro A, Morosky A, Saraf S, Piperdi B, et al. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol (2017) 18(5):623–30. doi: 10.1016/S1470-2045(17)30169-9
73. Quispel-Janssen J, van der Noort V, de Vries JF, Zimmerman M, Lalezari F, Thunnissen E, et al. Programmed Death 1 Blockade With Nivolumab in Patients With Recurrent Malignant Pleural Mesothelioma. J Thorac Oncol (2018) 13(10):1569–76. doi: 10.1016/j.jtho.2018.05.038
74. Okada M, Kijima T, Aoe K, Kato T, Fujimoto N, Nakagawa K, et al. Clinical Efficacy and Safety of Nivolumab: Results of a Multicenter, Open-label, Single-arm, Japanese Phase II study in Malignant Pleural Mesothelioma (MERIT). Clin Cancer Res (2019) 25(18):5485–92. doi: 10.1158/1078-0432.CCR-19-0103
75. Calabro L, Morra A, Giannarelli D, Amato G, D’Incecco A, Covre A, et al. Tremelimumab combined with durvalumab in patients with mesothelioma (NIBIT-MESO-1): an open-label, non-randomised, phase 2 study. Lancet Respir Med (2018) 6(6):451–60. doi: 10.1016/S2213-2600(18)30151-6
76. Scherpereel A, Mazieres J, Greillier L, Lantuejoul S, Do P, Bylicki O, et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol (2019) 20(2):239–53. doi: 10.1016/S1470-2045(18)30765-4
77. Disselhorst MJ, Quispel-Janssen J, Lalezari F, Monkhorst K, de Vries JF, van der Noort V, et al. Ipilimumab and nivolumab in the treatment of recurrent malignant pleural mesothelioma (INITIATE): results of a prospective, single-arm, phase 2 trial. Lancet Respir Med (2019) 7(3):260–70. doi: 10.1016/S2213-2600(18)30420-X
78. Nowak AK, Lesterhuis WJ, Kok PS, Brown C, Hughes BG, Karikios DJ, et al. Durvalumab with first-line chemotherapy in previously untreated malignant pleural mesothelioma (DREAM): a multicentre, single-arm, phase 2 trial with a safety run-in. Lancet Oncol (2020) 21(9):1213–23. doi: 10.1016/S1470-2045(20)30462-9
Keywords: chromatin modification, histone methylation, asbestos, mutations, immunotherapy
Citation: Yoshikawa Y, Kuribayashi K, Minami T, Ohmuraya M and Kijima T (2020) Epigenetic Alterations and Biomarkers for Immune Checkpoint Inhibitors–Current Standards and Future Perspectives in Malignant Pleural Mesothelioma Treatment. Front. Oncol. 10:554570. doi: 10.3389/fonc.2020.554570
Received: 22 April 2020; Accepted: 13 November 2020;
Published: 14 December 2020.
Edited by:
Hongchuan Jin, Zhejiang University, ChinaCopyright © 2020 Yoshikawa, Kuribayashi, Minami, Ohmuraya and Kijima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kozo Kuribayashi, a3VyaXJpbkBoeW8tbWVkLmFjLmpw
†These authors have contributed equally to this work