 Georges Herbein1,2*
Georges Herbein1,2* Zeina Nehme1,3
Zeina Nehme1,3- 1Pathogens & Inflammation/EPILAB Laboratory, EA 4266, University of Franche-Comté, Université Bourgogne Franche-Comté (UBFC), Besançon, France
- 2Department of Virology, CHRU Besancon, Besançon, France
- 3Faculty of Sciences, Lebanese University, Beirut, Lebanon
Tumors are renowned as intricate systems that harbor heterogeneous cancer cells with distinctly diverse molecular signatures, sizes and genomic contents. Among those various genomic clonal populations within the complex tumoral architecture are the polyploid giant cancer cells (PGCC). Although described for over a century, PGCC are increasingly being recognized for their prominent role in tumorigenesis, metastasis, therapy resistance and tumor repopulation after therapy. A shared characteristic among all tumors triggered by oncoviruses is the presence of polyploidy. Those include Human Papillomaviruses (HPV), Epstein Barr Virus (EBV), Hepatitis B and C viruses (HBV and HCV, respectively), Human T-cell lymphotropic virus-1 (HTLV-1), Kaposi's sarcoma herpesvirus (KSHV) and Merkel polyomavirus (MCPyV). Distinct viral proteins, for instance Tax for HTLV-1 or HBx for HBV have demonstrated their etiologic role in favoring the appearance of PGCC. Different intriguing biological mechanisms employed by oncogenic viruses, in addition to viruses with high oncogenic potential such as human cytomegalovirus, could support the generation of PGCC, including induction of endoreplication, inactivation of tumor suppressors, development of hypoxia, activation of cellular senescence and others. Interestingly, chemoresistance and radioresistance have been reported in the context of oncovirus-induced cancers, for example KSHV and EBV-associated lymphomas and high-risk HPV-related cervical cancer. This points toward a potential linkage between the previously mentioned players and highlights PGCC as keystone cancer cells in virally-induced tumors. Subsequently, although new therapeutic approaches are actively needed to fight PGCC, attention should also be drawn to reveal the relationship between PGCC and oncoviruses, with the ultimate goal of establishing effective therapeutic platforms for treatment of virus-associated cancers. This review discusses the presence of PGCCs in tumors induced by oncoviruses, biological mechanisms potentially favoring their appearance, as well as their consequent implication at the clinical and therapeutic level.
Introduction
Tumors are perceived as composite systems that diversify at the molecular, cellular, and architectural levels, accounting for phenotypic and functional heterogeneity (1). With divergence at the genetic, epigenetic, transcriptomic, proteomic, and cellular levels tumor heterogeneity is regarded as an essential barrier hindering the development of curative anti-cancer therapies (2). In this perspective, giant multinucleated or large nucleated cells denoted as polyploid giant cancer cells (PGCC) appear to significantly contribute to the shaping and the composition of cancer genomes and tumor evolution, rendering them crucial therapeutic targets to fight therapy resistance (3). This paradigm establishes a new conceptual framework and relocates the attention on the centuries-old embryological theory of cancer beside the somatic mutation and the clonal selection theories (4). In this re-emerging consensus, it is speculated that PGCC act as keystone cancer sub-population and actuators of an endogenous mechanism of somatic cell de-differentiation or reprogramming, which set the stage for cancer initiation, therapeutic resistance and relapse in metastatic disease (5). This genomic reorganization is conceptualized by Niu et al. as a “giant cell cycle” depicted in four distinct but overlapping phases designed as initiation, self-renewal, termination, and stability, where a process of reductive division known as depolyploidization results in the formation of diploid progeny cells with novel or altered genotypes (6).
On the other hand, viral oncogenesis is a multistep process that rely on complex molecular mechanisms and multiplex interplay between the host and oncogenic viruses (7). The latter account for ~12% of human cancers and includes Human Papillomaviruses (HPV), Epstein Barr Virus (EBV), Hepatitis B and C viruses (HBV and HCV, respectively), Human T-cell lymphotropic virus-1 (HTLV-1), Kaposi's sarcoma herpesvirus (KSHV) and Merkel polyomavirus (MCPyV) (8). Although those pathogens hijack the cellular machinery to establish infection, replication and persistence, as well as to promote tumorigenesis through common pathways, targeted therapies and definitive curative clinical interventions are still lacking (9). This is due not only to limitation of equivalent animal models, but also due to the enigma associated with some mechanistic aspects of cancer induction by infectious agents and their disparate nature (10). In this context, dissecting the clonal sub-population of those tumors at the genetic level appears to be an essential clue to understand and decipher the aspects of cancer initiation and progression and the subsequent extrapolation toward prognosis and treatment. Genuinely, genomic instability, in the form of aneuploidy and most importantly polypoidy appear to be a shared characteristic among oncogenic viruses, where distinct viral proteins can pave the road, through various mechanisms, for the initial trigger of cellular transformation (11).
As a thorough causality between oncoviruses, polyploidy, and tumorigenesis has not yet been established, this review will discuss the presence or alternatively the induction of a polyploid giant cancer cell phenotype in the context of viral infections, as well as the molecular pathways that might mediated or favor this mechanism and its clinical implication in terms of therapy resistance.
Polyploidy and Oncoviruses
Although oncogenic viruses converge in common features from a broad perspective, divergent genomes, oncogenic factors, cellular tropism, and disease prevalence distinguish each of those infectious agents (12, 13). Nonetheless, polyploidy appears to be a crossroad and a biological equivalency trait shared among oncoviruses, where the latter could be regarded as an initiating engine that fuels phenotypic diversity. Such heterogeneity, characterized partly by the presence of proportion of giant cells with a highly enlarged nucleus or multiple nuclei in in vitro or in vivo systems is discussed in following section.
Human Papillomavirus (HPV)
Belonging to Papillomaviridae family, Human papillomavirus (HPV) is a small, double-stranded, circular DNA virus with a non-enveloped icosahedral capsid (14). Encompassing more than 200 types, HPVs have proven their tropism for cutaneous and mucosal epithelia (15). Besides, and based on their relative malignancy, HPVs are clustered into two types: low-risk (LR) HPV and high-risk (HR) HPV, where the former is linked to low-grade lesions as benign warts and the latter is interrelated to high-grade cervical lesions and cancers (16). Although HR HPV are further divided into 20 types (17), HPV-16 and−18 are the most studied ones as they account for 62.6 and 15.7% of cervical cancer cases respectively, with a special highlight on the two HPV-encoded oncoproteins E6 and E7 (18, 19). In this context, the detection of polyploidy is finely described. Indeed, tetraploidy is identified as an early and common event during cervical carcinogenesis (20). Further, an exceedingly significant correlation is established between HR-HPV infection, the presence and/or the development of a high-grade squamous intraepithelial lesion (HGSIL) and the detection of polyploidy (21, 22). It has been shown that infection of squamous intraepithelial lesions of the cervix with high-risk but not low-risk HPVs results in basal keratinocyte tetrasomy (23, 24). Expression of both E6 and E7 oncoproteins resulted in higher percentage of cells with >4n DNA content in neonate foreskin keratinocytes compared to control cells (25). In addition, spontaneous polyploidization was detected in HPV-16 E6-expressing fibroblasts at late passages (26), as well as in primary human keratinocytes (PHK) expressing E6 (27) and in the immortalized retinal pigment epithelial cells RPE1 in the presence of p53 through a p53-independent mechanism of E6-induced polyploidy (28). Not restricted to E6, HPV-16 E7 induces polyploidy formation in response to DNA damage (29) with the cell division cycle 6 (cdc6) protein as an important mediator (30). Polyploidization was also detected in human keratinocyte cell lines PHK16-I, PHK16-II, PHK16-L1, and PHK16-L2 and in primary mouse keratinocytes upon HPV-16 E7 expression (31), as well as in RPE1 cells expressing E7 (32). Interestingly, the expression of HPV-16 E5, a third viral protein with transforming potential upon independent expression (33), resulted not only in enlarged nuclei, but also in an increase in cellular DNA content and chromosomal number (34). Lastly, HPV-18 E7 transduction resulted in the emergence of cells with enlarged nuclei or alternatively binucleated or trinucleated cells in cultures of differentiated keratinocytes (35). Thus, the expression of HPV oncogenes could induce polyploidy as an early event during cervical carcinogenesis.
Epstein–Barr Virus (EBV)
Epstein–Barr virus (EPV) is a linear, double-stranded DNA virus classified in the family Herpesviridae, subfamily Gammaherpesvirinae (36). Although EBV can infect T lymphocytes or epithelial cells, it is well-thought-out to be a B-lymphotropic virus, where it establishes latency and induces proliferation of B lymphocytes (37). Based on the cell types for which EBV exhibits tropism, the virus is associated with a wide array of malignancies including several subset of lymphomas, for instance Hodgkin's lymphoma, diffuse large B-cell and Burkitt's lymphoma (BL), and a subset of carcinomas including gastric and nasopharyngeal carcinoma (NPC) (38). Genuinely, polyploidy is described in tissue biopsies of NPC where polynuclear giant cancer cells were detected, surrounded by an indefinite small nucleus-containing bodies, indicative of budding cells (39). In addition, increased volume and pronounced multinucleation with cells counting more than 12 nuclei were reported in a nasal mucosal neoplasm infected with EBV (40). Furthermore, EBV replication in epithelial NPC hybrid cells induced the formation of multinucleated giant cells (41). B cell infection with an EBV strain isolated from a nasopharyngeal carcinoma, predisposed to polyploidy with cells displaying several micronuclei or containing a single large polyploid nucleus (42). Besides, chromosomal integration of the viral genomic DNA into primary human B cells was associated with polyploidy in the generated lymphoblastoid cell lines (43). More specifically, expression of Epstein–Barr virus nuclear antigen 2 (EBNA2), a viral nuclear protein involved in EBV latency regulation and essential for B-cells immortalization (44) can induce micronuclei and multinucleated cell formation in the human laryngeal carcinoma HEp-2 and osteosarcoma U2-OSn cells (45). In line with this, transfection with EBNA3C, another viral latent protein crucial for B cells immortalization and with oncogenic property in primary rodent fibroblasts in co-operation with activated RAS (46), produced bi- and multi-nucleated cells in NIH3T3 fibroblasts and U2OS cells where distinct peaks corresponding to 6n and 8n and cells enclosing from 1 to 7 nuclei were detected (47). Lastly, stable expression of the latent membrane protein 1 (LMP1), a viral oncoprotein that provides essential survival signals (48) was associated with multinuclearity in a Burkitt's lymphoma cell line (49). This indicates that a close correlation between polyploidy and EBV infection or EBV-latent oncoproteins expression does exist.
Kaposi's Sarcoma-Associated Herpes Virus (KSHV)
Kaposi's sarcoma-associated herpes virus (KSHV) or human herpesvirus 8 (HHV-8) is a double stranded DNA gamma-2 herpesvirus (50). KSHV exhibits tropism toward a wide spectrum of cells including B cells, endothelial and epithelial cells, fibroblasts, keratinocytes, and dendritic cells (51). KSHV is identified as the etiological agent behind a heterogeneous group of malignancies, mainly Kaposi sarcoma (KS), a low-grade angiogenic vascular spindle cancer of endothelial cells and primary effusion lymphoma, in addition to multicentric Castleman disease, a non-cancer rare lymphoproliferative disease that can advance to plasmablastic lymphoma (52). In regards to polyploidy, KSHV infection of human umbilical vein endothelial cells (HUVECs), a model that mimics KSHV infection in KS tumor cells, induced a multinucleation state, associated with nuclei enlargement and shape irregularity (53). Activation of Rac1, a small GTPase suggested to be a key determinant of Kaposi sarcoma when triggered by host and viral genes, resulted in significant polyploidy in FVB/N transgenic mouse lines (54). On the other hand, the latency-associated nuclear antigen (LANA) is a multifunctional protein essential for efficient viral DNA replication and maintenance in latently infected tumor cells with a potential role in promoting oncogenesis (55). Constitutive expression of LANA in HeLa cells, a human cervical cancer cell line, in BJAB cells, a B cell line and in Rat1 fibroblasts induced a dramatic increase in the multinucleated phenotypes, characterized by cells with two or more polarized nuclei (56, 57). In addition, a KSHV cyclin D homolog denoted as K cyclin is recognized as a potential oncoprotein that interacts with cyclin-dependent kinases (CDKs) (58), phosphorylates a wide spectrum of substrates through complexing with CDK6 including p27 to trigger its degradation (59) and initiates DNA replication as well as cell cycle progression (60). Interestingly, K cyclin expression in mouse embryo fibroblasts yielded cells with enlarged nuclei as well as cells with multiple large multilobular ones (61) pointing toward a robust association linking KSHV to polyploidy.
Human T Cell Lymphotropic Virus (HTLV-1)
Acknowledged as the first human retrovirus to be discovered, human T cell lymphotropic virus (or T-cell leukemia virus) type 1 (HTLV-1) is a delta retrovirus belonging to Retroviridae family, subfamily Orthoretrovirinae (62). Although HTLV-1 is detected in multiple cell types, counting hematopoietic cells (e.g., monocytes, macrophages, dendritic cells) in addition to endothelial, synovial and glial cells, the virus preferentially infect CD4+ memory T cells, with both CD4+ and CD8+ T identified as viral reservoirs (63). In terms of induced malignancies, HTLV-1 is identified as the etiological agent behind adult T-cell leukemia/lymphoma (ATL), an aggressive lymphoproliferative non-Hodgkin's peripheral T-cell malignancy (64). In this context, HTLV-1 infection induces giant lymphoma cells with highly convoluted cerebriform nuclei (65). Distinctively, HTLV-1-encoded Tax is a pleiotropic oncoprotein indispensable for productive viral replication and direct or indirect activation of a variety of transcription pathways, along with T cells immortalization in vitro and transformation of rodent fibroblasts (66). Genuinely, Tax expression frequently prompts the generation of multinucleated polyploid cells (67). For instance, multinucleated giant cells with nuclei described as double, greatly enlarged, and/or highly lobulated were detected in mammalian cells highly expressing Tax (68, 69). Moreover, Tax-expressing HeLa cells exhibited enlarged cell sizes with greater than G2 DNA content compared to untransduced controls (70), with multinucleated cells showing giant lobulated nuclei (71). Likewise, HTLV-1 infected CD4+ clones displayed enlarged well-separated nuclei within binucleated and multinucleated cells, a morphological finding positively correlated with the degree of Tax expression (72). On the other hand, HTLV-1 accessory protein p30II, a multifunctional latency-maintenance factor, is also suggested to induce polyploidy in cooperation with c-Myc oncoprotein (73), although transfection of infectious HTLV-1 ACH. p30II mutant provirus in HT0180 clones was able to induce multinucleation (74). Being a pleiotropic sensor, c-Myc oncogene is considered as master regulator and mediator of multiple cellular signals and transcriptional responses (75). c-Myc was shown to promote S-phase cell cycle entry (76), endomitosis and polyploidy formation (77), in addition to neoplastic cellular transformation (78). HTLV-1 p30II distinctly enhances c-Myc transforming activity, engendering S-phase progression and polyploidy through the interaction with the Myc-associated transcriptional coactivators TRRAP/p434 and the histone acetyltransferase TIP60 and the stabilization of HTLV-1 p30II/Myc-TIP60 chromatin-remodeling complexes (73). Indeed, the retroviral p30II accessory protein is recruited on the E-box enhancer elements within the endogenous cyclin D2 promoter along with c-Myc and the acetyltransferases TIP60 and p300, where lysine-acetylation of the c-Myc oncoprotein contributes to HTLV-1-induced carcinogenesis (79). Hence, in the context of HTLV-1 infection, Tax expression, in addition to p30II, appear to play a crucial role in polyploidy induction.
Hepatitis B Virus (HBV)
Hepatitis B virus (HBV), the prototype member of the Hepadnaviridae family (80), is an enveloped partly double-stranded DNA virus with characteristics similar to retroviruses, where reverse transcriptase is employed in the replication cycle (81). Hepatocytes are the major target and the confirmed site of HBV replication as the latter is identified as a hepatotropic virus, by virtue of controversial data on HBV detection in bile ductular epithelium and other extrahepatic sites (82). Viral replication in liver can precipitate transient and chronic infections with complications ranging from acute hepatitis to liver failure with cirrhosis and hepatocellular carcinoma (HCC) (83). Although endoreduplication and hyperploidy were identified exclusively in peripheral blood cells isolated from HBV chronic carriers compared the HBV negative individuals (84), the correlation between polyploidy and oncogenesis is distinct in the context of HBV infection. As a matter of fact, hepatocytes undergo progressive polyploidization, a postnatal process examined as an indicator of terminal differentiation and cellular senescence (85). A converse interrelationship exists between HCC and cellular polyploidy as evidenced by the increased number of diploid mononucleated hepatocytes capacities during HCC pathogenesis, thus setting the stage for rapid proliferation with higher mutagenic risk (86). Despite this, it is noted that in HCC, nuclear ploidy is positively correlated with poor prognosis and HBV infection where mononuclear polyploid fractions are increased significantly in HCC related to HBV infection (87). In this perspective, the hepatitis B virus X protein (HBx, also denoted as pX) is a viral protein with pleiotropic biological functions indispensable for viral replication (88). Indeed, HBx promotes cell cycle progression, inhibits the expression of tumor suppressor genes, for instance p53, and modulates methyltransferases transcription, alongside multiple functions that underline its potential role in HCC pathogenesis and cellular transformation (89). It has been shown that full-length expression of HBx in transgenic mice with liver fibrosis resulted in aberrant hepatocytes ploidy (90). Further, in biopsies of hepatocellular carcinoma patients, although the percentage of binucleated hepatocytes was reduced, a significant increase in DNA polyploidy was confirmed in HCC patients with actual or previous HBV infection (91). Interestingly, the impact of the full-length HBx (FL-HBx) expression is divergent in the setting of normal liver maturation, proliferation, and in liver carcinogenesis. Whereas, FL-HBx delays hepatocytes binucleation during maturation, a greater percentage of population enriched with polyploid cell with a significantly higher fraction of ≥8n nuclei is detected in HBV-infected mice. Most importantly, FL-HBx transgenic mice exhibited an important increase of 4n hepatocytes during HCC initiation, along with an increase in HCC progenitor cell markers (92). This is endorsed by the fact that in the HBV-positive hepatoma cell lines HepG2.2.15 and 1.3ES2, a higher hyperploidy is detected compared to HBV-negative cells (93). Although a high fraction of pX-induced polyploid cells undergo apoptosis in poorly differentiated, immortalized hepatocytes, the surviving cells demonstrated two-fold increase in polyploidy, displayed characteristics of oncogenic transformation and an increase in the expression of proliferation genes know to be elevated in HCC, an effect repressed in the absence of pX (94). In line with this, pX expression in immortalized mouse hepatocytes also induced polyploidy with a 2.5-fold increase in cells containing >4n DNA and in the nuclear size (95), as well as the formation of multinucleated cells (96). In parallel to HBx, HBV large surface protein (LHBs), one of the three HBV viral surface proteins encoded by preS1, preS2, and S open reading frame (ORF) is identified to exhibit oncogenic properties that can potentially contribute to hepatocarcinogenesis, as well as the mutated/truncated pre-S2 from (97). LHBs expression in the immortalized hepatic progenitor cell line NeHepLxHT demonstrated a self-propagating cycles of hyperploidy, which accounted for intra-tumor heterogeneity (93). Pre-S2 mutant LHBs-expressing hepatocyte-derived carcinoma HUH7 cells also showed a significant number of multinucleated cells (98). Furthermore, a positive correlation between LHBs, hyperploidy and increased nuclear size was detected in hepatocytes of patients with chronic hepatitis B (93). Taken together, HBx appears to be a key player linking HBV and polyploidy.
Hepatitis C Virus (HCV)
Hepatitis C virus (HCV) is a small single-stranded enveloped RNA virus that belongs to the family Flaviviridae (99). With narrow host specificity and tissue tropism, HCV is mainly hepatotropic, although several cell types are suggested to support viral replication, for example dendritic and epithelial cells, lymph nodes and others (100). Persistence of HCV infection paves the way to cirrhosis, severe liver disease and eventually HCC (101), in addition to a strong potential correlation with non-Hodgkin's B-cell lymphoma (102). In this setting, polyploidy has been also described (103). Peripheral blood mononuclear cells (PBMCs) isolated from HCV-infected patients presented chromosomal polyploidy compared to PBMCs of healthy individuals (6). Furthermore, a higher polyploidy frequency is detected in human and mouse primary hepatocytes infected by the HCV strain JFH-1 with respect to non-infected cells (104). Additionally, expression of the viral core protein, previously shown to induce HCC in transgenic mice (105), induced an extensive polyploidy in human liver cell line HepG2, and embryonic kidney cell line HEK293. In line with this, primary splenocytes, hepatocytes, and embryo fibroblasts isolated from core protein-expressing transgenic mice displayed almost two-fold polyploidy increase relative to control mice (104).
Polyomaviruses (PyVs)
Polyomaviruses (PyVs) are a family of small, icosahedral, non-enveloped, double-stranded DNA viruses. Advances in DNA sequencing technologies allowed the identification of various human PyVs including Merkel cell polyomavirus (MCPyV), John Cunningham Polyomavirus (JCPyV) and BK polyomavirus (BKPyV) (106) in addition to simian virus 40 (SV40), an oncogenic prototypical primate closely related to JCPyV and BKPyV (107). Although strong evidence suggest an etiological role of JCPyV and BKPyV in multiple human malignancies, only MCPyV is recognized as an oncogenic virus by being the causative agent of a rare aggressive skin cancer known as Merkel cell carcinoma (MCC) (108). Expression of MCPyV small T (sT) antigen, an oncoprotein crucial for viral replication with transforming capacities in vitro (109) resulted in the formation of multinucleated cells along with an increase in the >4n population in WI38 human diploid fibroblastic cells (110). sT induction in C57BL/6 mice resulted in the formation of poorly-differentiated neoplasia pleomorphic nuclei varying in size and shape in conjunction with multiple nucleoli (111). In agreement with this, BKPyV infection of renal proximal tubule epithelial cells resulted in the accumulation of polyploid cells (112), an effect also seen upon large tumor antigen (TAg) expression (113). Alternatively, JCPyV infection of both cerebral hemispheres revealed giant multinucleated astrocytes with pyramidal neurons harboring enlarged nuclei (114). Transfection with two JCPyV strains, namely Mad-1 and−98 resulted in an increased ploidy in RKO cells, a human colon carcinoma-derived cell line (115). In line with this, astrocytes infection with Mad-1 JCPyV resulted in the appearance of a tetraploid population, which expanded with the time of infection (116). JCPyV-infected atypical astrocytes were deemed to be in the hypertetraploid range (117). Further, JCPyV-inoculated fetal brain cell cultures showed a significant upsurge in polyploid cells with relatively elevated level of endoreduplication, an effect shared with the simian papovavirus SV40 (118). Indeed, SV40 infection of primary and secondary hamster embryo cells and primary mouse cells induced the formation of tetraploid cells, in addition to cells with a higher ploidy, ranging from 16n and 32n to 64n (119). In line with this, SV40-infected monkey kidney cells CV-1 ensued the detection of a fraction of cells with a DNA content in the tetraploid–polyploid (10–12n) range (120). Distinctively, SV40 large T antigen (LT), a multifunctional key protein in driving viral replication and cellular transformation induction, plays a crucial role in SV40-induced polyploidy (121, 122). CV-1 cells infected with a temperature-sensitive mutant T antigen developed tetraploidy at 37°C but not at 40.5°C, which highlights the T antigen function in polyploidy induction (123). A shift from diploid to tetraploid and octaploid population in human fibroblast strain IMR9O infected with SV40 was positively correlated with a concomitant increase in number of T antigen-positive cells (124). Furthermore, expression of LT alone induced tetraploidy in the hTERT-immortalized foreskin fibroblast cell line BJ/tert (125) as well as the formation of an 8n population in IMR90 and BJ cell lines (126). To note that polyploidy induction in human diploid fibroblasts and embryonic kidney cells was correlated with the expression of the SV40 large T antigen but not with the small T antigen (127).
In this context, it should be noted that the presence of giant polyploid cells following infection with non-oncogenic viruses is also described in the literature (128). However, distinction in various aspects compared to oncogenic viruses is prominent. First, induction of the polyploid phenotype is not coupled to transformation (129). Second, cell-cell fusion due to the presence of viral fusion proteins that lead to syncytia formation is the main mechanism of polyploidy induction in non-oncogenic viruses (130, 131), which is in contrast to the oncogenic viruses-employed molecular mechanisms discussed in the succeeding section.
Several Biological Mechanisms Lead to PGCC Appearance in Oncoviruses Infections
Multiple cellular mechanisms are employed by oncoviruses to induce polyploidy. Several models have been suggested, ranging from spindle assembly and postmitotic checkpoint abrogation, re-replication and hypoxia, to centrosome duplication, inactivation of tumor suppressors, Myc activation and others. Since those mechanisms can result in the inhibition of some or all aspects of mitosis, this may enhance the occurrence of abortive cell cycle, or alternatively endoreplication, one of the most described mechanisms of PGCC formation (6). In the following section, biological mechanisms mobilized or activated by oncoviruses in parallel to polyploidy induction are discussed (Figure 1).
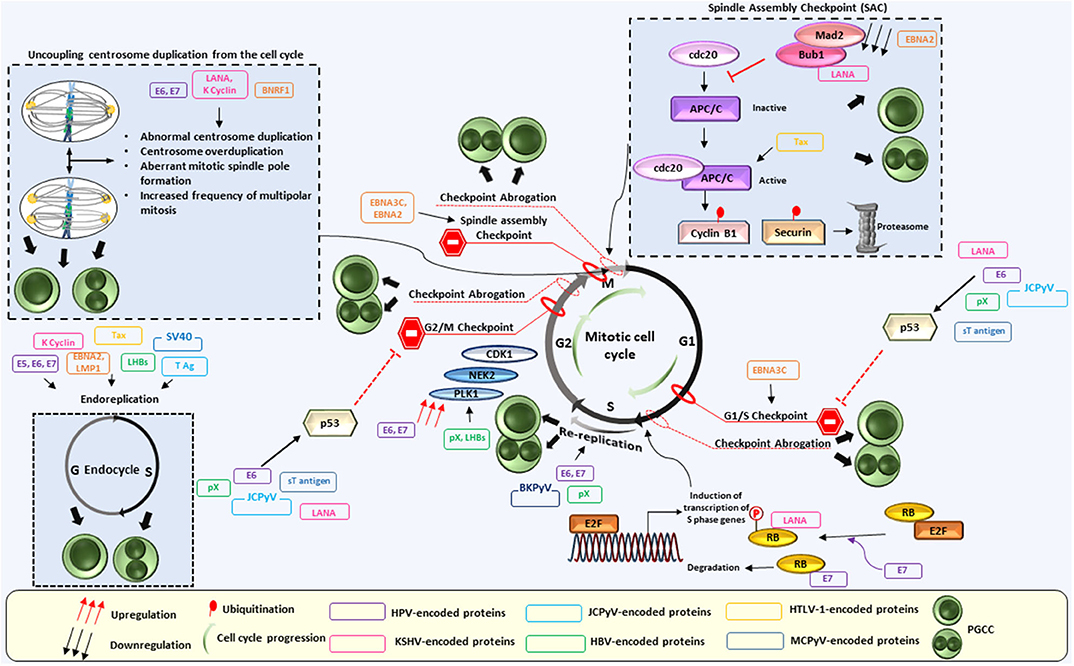
Figure 1. Interplay between oncoviral proteins and the cell cycle regulation leading to PGCC formation. Polyploidy has been described in the context of oncogenic viruses. Indeed, oncoviral proteins were shown to employ multiple cellular mechanisms to induce polyploidy. Those include centrosome overduplication and aberrant mitotic spindle pole formation, as well as abortive mitosis and cell cycle disruptions, The latter being dependent partly on p53 depletion or functional abolition, Rb inactivation or even through p53-independent mechanisms. Not limited to checkpoints abrogation, dysregulation of cell-cycle regulators activities such as polo-like kinase 1 (Plk1) also favor polyploidy induction, in addition to endoreplication and re-replication. Anaphase-promoting complex (APC), budding uninhibited by benzimidazoles 1 (Bub1), cell-division cycle protein 20 (cdc20), cyclin dependent kinase 1 (Cdk1), EBV nuclear antigen (EBNA), large HBV surface protein (LHBs), latency-associated nuclear antigen (LANA), latent membrane protein 1 (LMP1), mitotic arrest deficient 2 (Mad 2), NIMA-related protein kinase 2 (Nek2).
Aberrant Centrosome Numbers
Centrosome is a vital microtubule organizing center with multiple functional complexities. Each cell contains one centrosome or alternatively two centrosomes that support the formation of a bipolar mitotic spindle during mitosis, thus ensuring accurate chromosome segregation (132). Centrosome abnormalities can induce the formation of additional mitotic spindle poles and subsequently genetic instability (133). HPV-16 E7 oncoprotein demonstrated its ability to induce abnormal centrosome duplication and aberrant mitotic spindle pole formation, which consequently uncoupled centrosome duplication from the cell cycle division and triggered genomic instability (134). This effect was potentiated by HPV-16 E6 and reserved to high risk HPV-16 (134), although independent expression of E6 and E7 was also demonstrated to induce centrosome amplification (135). On the other hand, accumulation of abnormal centrosome number in E6-expressing cells was positively correlated with multinucleated cells characterized by nuclear enlargement and more than or equal to 3 nuclei/cell, with a cellular subpopulation ultimately enduring replicative senescence (136). Polyploid cells with single large nucleus were linked to the expression of the EBV major tegument protein BNRF1, capable of inducing centrosome overduplication and increasing the frequency of multipolar mitosis (42). In line with this, KSHV infection induced multipolar and monopolar spindles along abnormal centrosome duplication, resulting in multinucleated cells with enlarged nuclei (53). This effect could be linked to the KSHV-encoded latent protein LANA, whose expression alone increased abnormal centrosome activities as well as the number of centrosomes to more than three per cell (56), in addition to KSHV K cyclin, in turn linked to the presence of 3–40 centrosomes per cell (61). Beside aberrant centrosome numbers, Aurora kinase B (AURKB) has been described to play an important role during mitosis as a regulator of chromosome segregation and cytokinesis (137), where it localizes with the centrosome during early mitosis and acts as a component of the chromosome passenger complex (138). Interestingly, KSHV latently infected tumor cells showed serine protease-N terminus cleavage of AURKB, an N'-cleaved form of AURKB protein also shared in malignant tumor cells induced by other oncoviruses including EBV and HPV. This proteolytic cleavage might promote a transition from metaphase to telophase, thus promoting mitotic progress and tumorigenesis (139). In this specific context, KSHV-encoded LANA upregulates Aurora A kinase transcriptional expression through targeting the Sp1 cis-element within the promoter, and in turn, Aurora A expression was shown to be critical for LANA-induced p53 ubiquitylation and degradation (140).
Abortive Mitosis and Cell Cycle Disruptions
Described as “guardian of the genome,” p53 responds to endogenous and exogenous genotoxic attacks and DNA-damaging agents by inducing a cell cycle arrest at the G1/S checkpoint (141). Subsequently, loss of p53 expression or activity prompts a failure to induce a G1/S transition arrest and predisposes cells to genomic instability (142). Beside the cell cycle, p53 exerts a G1-like growth arrest upon mitotic spindle damage to prevent DNA re-replication and polyploidy formation (143). For instance, p53-deficient cells were shown to from tetraploid and octaploid cells (4) and overexpression of mutant-type p53 enriched the formation of giant myelomonocytic leukemic M1/2 cells that further accumulated after gamma-irradiation (144), suggesting that loss of wild-type p53 can accommodate for the emergence of PGCCs (145). In the context of oncogenic viruses, p53 depletion engendered a five-fold increase of polyploid cells in response to HBV pX expression, demonstrating that p53 antagonizes pX-induced polyploidy (95). Besides, p53 loss coupled to Merkel cell polyomavirus sT antigen expression induced poorly differentiated tumors characterized by pleomorphic nuclei with considerable disparities in size and shape along multiple nucleoli and hyperchromatism, associated with stimulation of progenitor cell proliferation (111). Polyploidy detected in the context of JCPyV infection was also correlated with functional abolition of p53 (117). On the other hand, HPV E6 oncoprotein was shown to interfere and inhibit all the p53-mediated functions (146). As a consequence, HPV16 E6-expressing cells abrogated the mitotic checkpoint, resulting in the loss of G2/M arrest and the appearance of a substantial 8n population (147). Interestingly, E7-expressing cells were also demonstrated to overcome the mitotic checkpoint by a p53-independent mechanism, possibly by deregulating Rb function and overexpressing Mdm2 (147), a cellular oncogene capable of inducing several rounds of S-phase replication without transition through mitosis (148). On the other hand, concomitant E6 and E7 expression induced polyploidy due to disruption of the spindle checkpoint and up-regulation of the G2-M proteins including Aurora-A, Nek2, cyclin-dependent kinase 1 (Cdk1) and polo-like kinase 1 (Plk1), the latter being dependent on p53 degradation and repression of pRb functions mediated by E6 and E7, respectively (149). Not limited to the abrogation of the spindle checkpoint, a p53-independent mechanism of E6-induced abrogation of the postmitotic G1-like checkpoint after adaptation of the mitotic stress is evidenced by the fact that E6 mutants defective in p53 degradation were also capable of inducing polyploidy (27). This could be elucidated by the reduced nuclear p21 localization and the active expression of Cdk1 that is mediated by E6-upreglated E2F1 upon microtubule disruption (28). This postmitotic checkpoint abrogation-induced polyploidy is also shared by E7 through its Rb-downregulation function (32). On the other hand, KSHV-encoded LANA was shown to deregulate both p53 and Rb pathways (150) and its expression can induce S-phase entry by protecting from the cell cycle arrest induced by the CDK4/6 inhibitor p16 INK4a (151). Subsequently, LANA-expressing cells showed multinucleation, abnormal spindle pole activities and an increased entry into S phase through LANA-mediated suppression of p53 transcription and transactivation activity, partly by repressing its endogenous promoter (56). In addition, by directly functioning as a component of the EC5S ubiquitin complex and interacting with the Cul5-Elongin BC complex, LANA expression in KSHV-infected B lymphoma cells induces polyubiquitylation and subsequently degradation of p53 and von Hippel–Lindau (VHL), another tumor suppressor (152). On the other hand, expression of EBV latent gene compromises the mitotic spindle assembly checkpoint and consequently averts metaphase arrest (149). Further, expression of EBNA3C, a protein with oncogenic activity comparable to E7 (46), induced cell cycle progression without cytokinesis in the absence of external mitogenic signals and not only deregulated the G1 checkpoint, but also disrupted the spindle assembly checkpoint, resulting in polyploidy (47), the latter effect also shared by EBNA2, another EBV protein with oncogenic activities (45). In addition, a prominent role of EBNA3C is pointed in this context as it can interact and transcriptionally regulate a wide array of cellular and viral transcriptional factors (153). For instance, EBNA3C N-terminal region was shown to complex with the N-terminal pRb binding domain and C-terminal domain of cyclin D1, which not only stabilizes cyclin D1 through the inhibition of its ubiquitin-mediated proteasomal degradation, but also enhances the functional kinase activity of cyclin D1/CDK6, thus facilitating the G1-S transition (154). Repression of p53 transcriptional activity and sparing p53-induced apoptosis in osteosarcoma cells was mediated by EBNA3C direct interaction of the latter N-terminal domain with p53 C-terminal DNA-binding and tetramerization domain (155). Also, EBNA3C directly interacts with the RNA helicase Gemin3, which stabilizes the latter and promotes formation of a complex with p53, subsequently blocking the DNA-binding affinity of p53 and p53-mediated transcriptional activity and apoptosis pathway (156). EBNA3C physically interacts with the oncoprotein Pim-1, enhancing Thr145 phosphorylation and proteosomal degradation of p21, thus enhancing cellular proliferation (157). Hence, a wide variety of oncoviruses-induced defects in distinct aspects of cell cycle regulation and checkpoints can give rise to abortive cell cycle and polyploidy.
Dysregulation of Cell-Cycle Regulators Activities
Cell cycle is a highly regulated multipronged process orchestrated by a suite of cell-cycle regulators (158). Dysregulation of any of these regulatory mechanisms can result in genomic instability and polyploidy (159). EBNA2 was shown to specifically downregulate the mitotic arrest deficient 2 (MAD2) and upregulate Plk1, which can result in the activation of the anaphase promoting complex/cyclosome (APC/C) and subsequently securin degradation, thus engendering a metaphase-anaphase transition and opening the door toward polyploidy (45). In line with this, expression of the HBV LHBs upregulated Plk1, resulting in a Plk1-dependent G2/M checkpoint override (93). Not limited to LHBs, HBS pX protein mediated the activation of Plk1 in G2 phase (160). This attenuated the DNA damage checkpoint through induction of clapsin proteasomal degradation and increased the inhibitory Tyr15-Cdc2 phosphorylation, as well as the suppression of p53-mediated apoptosis, promoting polyploidy generation (92). In line with this, pX-induced upregulation of Plk1 through p38 and ERK1/2 pathways can induce an attenuation of cell cycle checkpoint control, leading to an increase in polyploidy, DNA damage propagation and hepatocyte transformation, beside increased markers of cancer progenitor cells, such as AFP, Ly6D, and EpCam (90, 92). Indeed, Plk1 is crucial in pX protein-mediated oncogenic transformation, including pX-induced polyploidy, as Plk1 inhibition suppresses pX oncogenic potential (94). On the other hand, HTLV-1 oncoprotein Tax drastically reduced Pds1p/securin and Clb2p/cyclin B levels through the activation of the Cdc20p-associated anaphase-promoting complex APCCdc20p (161). Furthermore, Tax was shown to directly bind to TXBP181, a homolog of mitotic check-point MAD1 protein and target its mitotic checkpoint function by reducing its dimerization and stability, hence leading to the loss of the M checkpoint and the appearance of multinucleated cells (69). On the other hand, a key protein component of the spindle checkpoint, Bub1, was shown to interact with the KSHV-encoded LANA, which in turn promotes Bub1 ubiquitination and degradation via the APC/C E3-Ub ligase, thus favoring multinucleation (57).
Endoreplication
In contrast to the mitotic cycle that ensues the maintenance and the distribution of the same amount of genetic content between mother and daughter cells, endoreplication, or endoreduplication cycle consists only of G and S phases with no features of mitosis, a process termed endocycle, beside endomitosis, an abortive mitosis without cell division. This generates polyploidy with cells containing separate multiple nuclei or an enlarged, single nucleus harboring all the genetic content (162). Indeed, Zhang et al. reported endoreduplication as a potential mechanism of PGCC formation after cobalt chloride treatment (163). HPV16 E5 is capable of inducing endoreplication (34), a mechanism shared by E6 oncoprotein as evidenced by the presence of intracellular cytokinesis (164). Transduction with HPV-18 E7 pushed the cells to reenter another round of S phase resulting in two consecutive S phases without cytokinesis, hence ensuing multinucleated cells with enlarged nuclei (35). In addition, re-entering S phase and synthesizing DNA was also triggered in EBNA2-expressing cells (45) and LMP1 expression (49). Besides, DNA replication and cell nuclei division without concomitant cell division yielded large or multiple nuclei upon KSHV K cyclin expression (61). HTLV-1 Tax oncoprotein uncoupled DNA synthesis from cell division by activating G1/S entry and blocking mitosis, inducing the formation of multinucleated giant cells (161). Furthermore, cells expressing HBV LHBs demonstrated cytokinesis failure compared to controls (93). JCPyV T antigen was similarly shown to induce endoreplication, resulting in an excess of polyploidy (118). Endoreplication is also shared by SV40 with synchronous progression through the cell cycle into the first S phase, followed by a second S phase (120), an outcome dependent on SV40 large T antigen expression (121). This could be explained by an unappreciated in vivo interaction between large T antigen and nibrin (Nbs1), resulting in the formation of a T/Nbs1 complex that interferes with Nbs1 role in suppressing DNA replication re-initiation (126). Nonetheless, T antigen-induced tetraploidy was also shown to be dependent on the binding of the latter to the mitotic checkpoint serine/threonine kinase Bub1 (125).
Re-replication
In contrast to endoreplication, re-replication is a successive rounds of DNA synthesis within a given S phase without entering mitosis, leading consecutively to polyploidy (165). Polyploidy formation in HPV-16 E7-expressing cells was mediated through re-replication in response to DNA damage after being arrested at G2 checkpoint, as evidenced by the upregulation of the DNA replication initiation factor Cdt1, a gene responsible of triggering re-replication upon overexpression (29, 166). Another player in E7-induced re-replication is the DNA replication initiation factor known as the cell division cycle 6 (Cdc6) whose downregulation reduces E7-induced re-replication (30). Indeed, Cdc6 protein is expressed in both cervical squamous carcinoma and adenocarcinoma and is upregulated possibly due to E7-mediated release of E2F inhibition upon Rb binding, since Cdc6 is an E2F responsive gene (167). In addition, HBV pX-induced polyploid cells were shown to undergo DNA re-replication, beside aberrant mitotic spindles (94). Indeed, pX-mediated DNA re-replication was correlated with Cdt1 upregulation and Cdc6 expression, both required for pre-replicative complex assembly, along with suppressed expression of geminin, the main inhibitor of replication licensing in S and G2 phases (95). BKPyV was also demonstrated to induce multiple rounds of DNA replication within a single cell (112). This could be explained by a synergistic cooperation between ataxia telangiectasia, mutated (ATM) and ataxia telangiectasia, and Rad3-related (ATR), where ATM promotes efficient S-phase entry and mitosis block after complete DNA replication, and ATR arrests the cell cycle to prevent entry into mitosis in actively replicating cells, resulting in BKPyV-mediated polyploidization (113).
Hypoxia
Hypoxia is identified as a critical microenvironmental factor involved in tumor progression as well as maintenance and self-renewal of cancer stem cells. This is arbitrated by the hypoxia inducible factors (HIFs) and mediated through various mechanisms, including but not limited to activity enhancement of stem cell factors such as OCT-4, c-Myc, and Nanog (168). Hypoxia mimic was shown to underlie the formation of PGCC, the latter then contributing to the generation of cancer stem-like cells (163, 169). Notably, most oncogenic viruses are capable of deregulating cellular hypoxia-inducible factor 1 (HIF-1) signaling pathway and enhancing HIF-1 levels (170), the latter mediating a fundamental role in transcriptionally upregulating metabolic, angiogenic, and microenvironmental factors indispensable for oncogenesis (171). For instance, HPV-16 oncoproteins E6 and E7 significantly promote HIF-1a protein accumulation and enhanced activity in human cervical cancers (172). EBV LMP1, EBNA-1, EBNA-3, and EBNA-5 enhance the rate of HIF-1α synthesis, HIF-1α gene transcription and inhibit its breakdown, respectively (173–175). KSHV interferon regulatory factor 3 (vIRF-3) also called LANA-2 increases HIF-1α nuclear translocation and activity through direct binding (176). Mediated through several putative hypoxia response elements, LANA direct association with HIF-1α enhances HIF-1α mRNA level and transcriptional activities (177) and is compulsory for efficient viral replication in the hypoxic microenvironment through the inhibition of the cellular E3-ubiquitin ligase-mediated proteosomal degradation (178), as well as lytic and latent viral genome regulation through a unique SUMO-interacting motif during hypoxia (179). Furthermore, a KSHV-encoded G protein-coupled receptor (GPCR) stimulates HIF-1α through the phosphorylation of its regulatory/inhibitory domain, which enhances its transcriptional activity, resulting in transcriptional activation of the vascular endothelial growth factor (VEGF) promoter, a crucial angiogenic stimulator (180). This sheds the light on the potential therapeutic targeting of HIF-1α in the context of KSHV-induced PEL as HIF-1α activity is stimulated by KSHV infection and HIF-1α activates numerous KSHV genes. This is reinforced by the fact that HIF-1α suppression was associated with a significant inhibition of PEL growth and a reduction in the activation of KSHV lytic and latent genes (181). In addition, HBV HBx interferes with HIF-1α protein degradation (182) and enhances its synthesis (183). HCV core protein upregulates HIF-1α mRNA transcription (184), whereas HTLV1 Tax enhances its protein expression (185). Therefore, although employing divergent mechanisms, HIF-1 activation by enhancing its transcription, translation, or stabilization appears to be a common pathway among oncogenic viruses. Given that most of these studies outline the mechanisms underlying such activation and the subsequent downstream activation of key cancer-promoting genes, polyploidy assessment in the context of viral-induced HIF-1 activity is underestimated and future studies are undoubtedly indispensable to reveal the interrelationship between the triad of oncogenic virus, hypoxia and polyploidy.
Cellular Senescence
Typically considered in a senescent state, polyploid cells were generally examined as a tumor suppressor mechanism as senescence is characteristically considered a terminal cell fate (186). Nevertheless, PGCCs have been shown to escape senescence, generate daughter cells that could undergo mitotic cell divisions (187) and contribute to immortalization and transformation (188). In the context of latent KSHV infection, the viral v-cyclin protein was shown to induce senescence by deregulating the cell cycle and activating the DNA damage response (DDR) subsequent to aberrant host DNA replication. Interestingly, viral FLICE inhibitory protein (v-FLIP) can bypass senescence and facilitate the growth and division of latently infected cell population (189) that can comprise a subpopulation of polyploid cells as discussed previously. It is worthy to note that v-FLIP is present in several gamma-herpesviruses, including EBV, as well as in the tumorigenic human molluscipoxvirus (190). Hence, assessing the previously discussed v-FLIP function in the context of polyploidy induction could be of interest. On the other hand, HTLV-1 Tax induced senescence, accompanied by cells with enlarged nuclei or two nuclei, with a special emphasis on a small subpopulation that was found to circumvent Tax-induced rapid cellular senescence (70).
Telomere Dysfunction
As telomeres are essential for preserving chromosome stability, progressive telomere dysfunction triggers cytokinesis failure and engenders polyploidy (191). Expression of LMP1, the primary transforming gene product of EBV led to a significant increase in telomeric aggregates and a decrease of the total telomere number, with a significant increase of the number of multinucleated cells, as well as the nuclear volume. This perturbation of telomere protection was correlated with a suppression of shelterin proteins TRF1, TRF2, and POT1 responsible of preserving the telomere structure and its signaling functions (49, 192). In particular, TRF2 displacement from telomeres in EBV-infected cells was shown to significantly contribute to genomic instability (193).
Overall, oncoviruses deploy multiple mechanisms that could be regarded as precursor of polyploidy and driver of oncovirus-induced human cancers, tumor progression, and intratumoral heterogeneity. Interestingly, human cytomegalovirus (HCMV), a beta herpes virus with high oncogenic potential (194) shares most of the previously mentioned mechanisms of polyploidy induction. For instance, CMV infection not only induced supernumerary centrosomes, but also formation of abnormal mitotic spindles (195). In addition, several HCMV proteins were shown to interact with p53 (196). As an illustration, immediate early 2 protein demonstrated its ability to interact with p53 in vitro and in vivo, transcriptionally inactivating the latter (197). Besides, HCMV UL97 inactivates Rb by phosphorylation, whereas pp71 binds to the Rb family proteins and induces their proteasomal degradation (198). HCMV was also shown to activate Myc at the transcriptional level (199) as well as at the translational level (200) and to induce HIF-1α expression (201). In addition, multinucleated giant cells formed by cell fusion were detected following HCMV infection (202) as well as an up-modulation of the expression levels of the G2/M transition regulators, including the previously mentioned Plk1 (195). This emphasizes the fact that primary pathways and mechanisms of polyploid formation could be shared among oncogenic and potentially oncogenic viruses, and points toward the potential elaboration of an empirical hypothesis linking those viruses to polyploid induction and tumorigenesis.
New Therapeutic Approaches Are Actively Needed to Fight PGCC, but Also Oncoviruses
Recently, a growing number of evidence pointing toward polypoid giant cancer cells as key actuators of therapy resistance, metastasis, and relapse is emerging (203). For instance, PGCC purified from ovarian cancer cells were more resistant to treatment with cisplatin (163). Furthermore, giant cells selected from high metastatic human prostate cancer not only showed resistance to 5-fluorouracil (5-FU), doxorubicin and cisplatin, but also potently developed metastasis in lung, bone, and some major lymph nodes, including popliteal, inguinal, axillary, and cervical nodes (204). Large multinucleated cells in murine fibrosarcoma were more resistant to doxorubucin and established orthotopic subcutaneous tumors with spontaneous lung metastases (205). In addition, human colon cancer cells exposed to cycles of 5-FU, oxaliplatin, and irinotecan treatment to mimic the clinical therapeutic regimens demonstrated a resumption of proliferation and cancer re-population, a finding principally attributed to the progeny of the established polyploid giant cells (206). In the context of oncogenic viruses, although an explicit link with therapy resistance and PGCC is lacking, some interpretations could be drawn (Table 1).
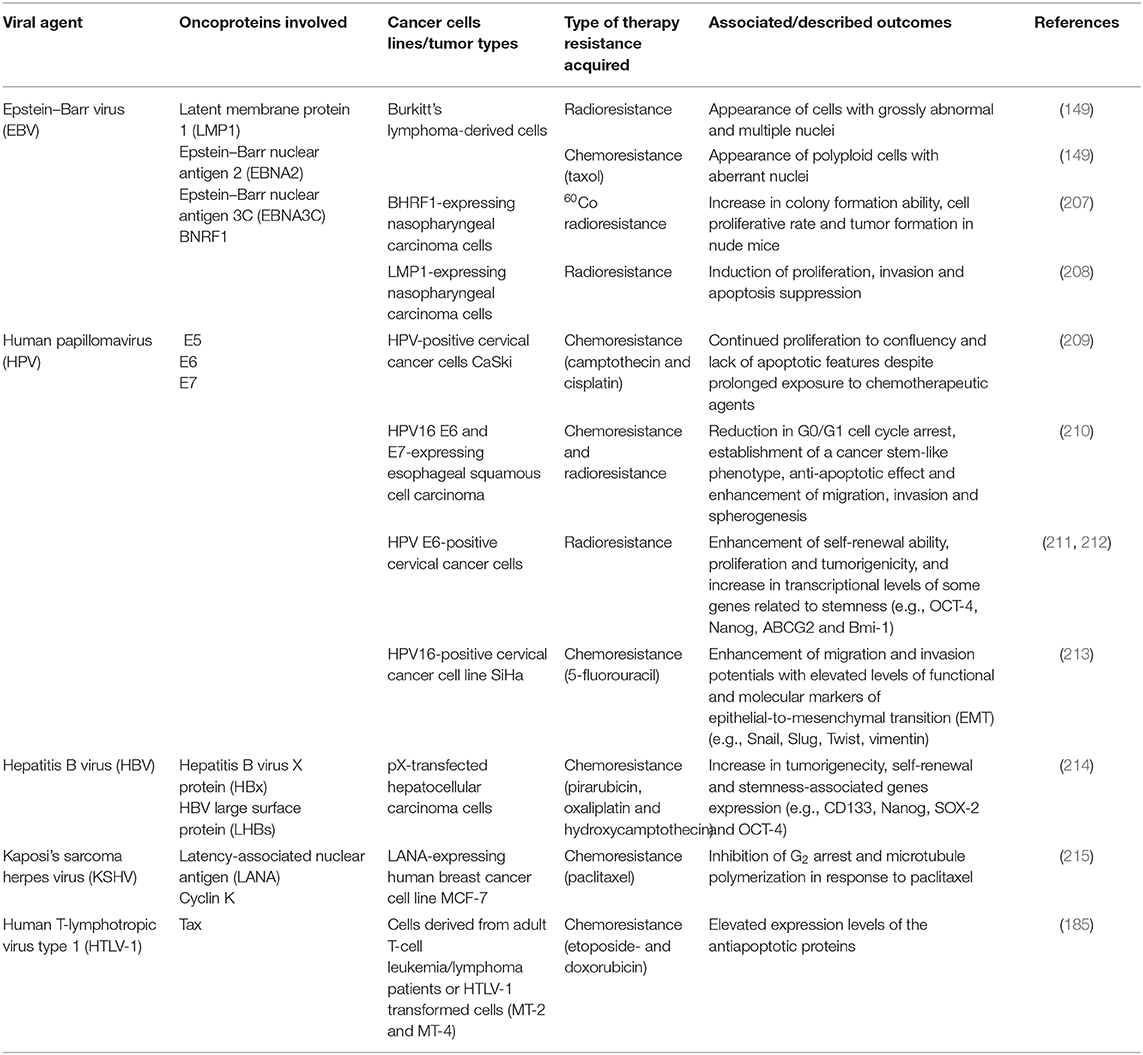
Table 1. Anti-tumor treatment, PGCC, and oncoviruses.
A Link Between Oncoviruses, PGCC and Therapy Resistance Is Observed in Several Cancers
It has been shown that EBV expression rescued Burkitt's lymphoma-derived cells from death after gamma-irradiation and resulted in the appearance of recovered cells with grossly abnormal and multiple nuclei, compared to rapid death in EBV-negative cells (149). Similar results were also reported at 72 h post-treatment with taxol, where treated cells became polyploid and developed aberrant nuclei in the EBV-positive population, versus a mass cellular death in EBV-negative controls (149). Further, nasopharyngeal carcinoma cells expressing EBV gene BHRF1 recovered faster after 60Co radiation with higher cell proliferative rate and colony formation ability, beside a greater tumor formation in nude mice compared to control groups (207). This is in line with the EBV LMP1-induced proliferation, invasion, apoptosis suppression, and radioresistance in NPC cells (208).
HPV-positive cervical cancer cells exhibit an inherent chemoresistance to both camptothecin and cisplatin (209). HPV16 E6 and E7 reduced G0/G1 cell cycle arrest, promoted a cancer stem-like phenotype and an anti-apoptotic effect, enhanced migration, invasion, spherogenesis, and increased chemoresistance and radioresistance after ionizing radiation in esophageal squamous cell carcinoma (210). A subpopulation of HPV-E6 positive cervical cancer cells enriched with CD71, a glycoprotein detected with poorly differentiated acute myeloid leukemia (216) and radioresistant glioma cells with cancer stem-like cells properties (217), demonstrated resistance to irradiation, an enhanced self-renewal ability, proliferation and tumorigenicity (211), in addition to higher transcriptional levels of some genes related to stemness including OCT-4, Nanog, ABCG2, and Bmi-1 (212). Furthermore, HPV16 E6 was shown to be actively involved in migration and invasion potentials, with elevated levels of functional and molecular markers of epithelial-to-mesenchymal transition (EMT) such as Snail, Slug, Twist, and vimentin, which could promote chemoresistance in cervical cancer (213).
Interestingly, hepatocellular carcinoma cells transfection with HBV pX protein contributes to the expansion of a subpopulation within the total HCC cell pool characterized by an increased tumorigenecity, self-renewal, stemness-associated genes expression such as CD133, Nanog, SOX-2, and OCT-4, and an enriched chemoresistance toward pirarubicin, oxaliplatin, and hydroxycamptothecin (214). This property is correspondingly shared in the setting of EBV-associated NPC (218).
KSHV latent protein LANA2 induces paclitaxel resistance, which suggests a potential correlation between LANA2 expression and the resistance to paclitaxel in the setting of primary effusion lymphoma (215).
HTLV-1-infected cells derived from adult T-cell leukemia/lymphoma patients or the HTLV-1 transformed cells MT-2 and MT-4 were highly resistant to etoposide and doxorubicin with elevated expression levels of the anti-apoptotic proteins mediated by Tax (185).
It is remarkable that many of the discussed tumors fall under the category of carcinoma or adenocarcinoma, where epithelial or epithelial-like cells are identified as a major system where PGCCs develop. Indeed, the latter are frequently described in similar settings, for instance prostate (219), colon (220) and breast cell lines (221), in addition to ovarian tumors (222), which could suggest that the polyploid phenotype could be interrelated to the origin of the cell or favored by the microenvironment.
Highlighting a Biological Model Where Oncoviruses, Through the Generation of PGCC, Could Favor the Establishment of Therapy Resistance, Metastasis and Relapse
Genuinely, a robust evidence relating polyploidy, cancer stem-cell phenotype and EMT do exist (3). For instance, upregulation of OCT4, SOX2, and Nanog in the setting of irradiated lymphoma cells was clearly detected in endopolyploid tumor cells, which can resist apoptosis, overcome cellular senescence and transfer this primitive phenotype to descendants through de-polyploidisation (223). Furthermore, reprogramming of differentiated breast cancer cells following ionizing radiation ensued only in a subpopulation of polyploid cells with re-expression of OCT-4, SOX-2, Nanog, and Klf4 (224). This is in line with expression of OCT-4, Nanog, SOX-2, and SSEA1 in PGCCs whose daughter cells demonstrate an increased resistance to paclitaxel (225). Interestingly, PGCCs and PGCC-derived daughter cells gained a mesenchymal phenotype as demonstrated by the enhanced expression of some markers such as Snail, Slug, Twist 2, vimentin, and others (163, 225). This not only indorses invasion and metastasis, but also may convey a drug refractory state due to therapy resistance (226). Taken together, it could be hypothesized that oncogenic viruses might, through the induction of polyploidy, play a central role in drug resistance, metastasis, and relapse of human cancer through the establishment of primitive cellular phenotypes. As the presence of such pathogens could compromise therapy, targeting those viruses with the purpose of blocking the formation of PGCC may potentially have some clinical implications as a promising anti-cancer therapeutic approach, in conjunction to the newly proposed therapeutic strategies to target polyploidy, for instance the pharmacological AMP kinase activity stimulators or modulators of metabolic pathways (Figure 2) (3, 227).
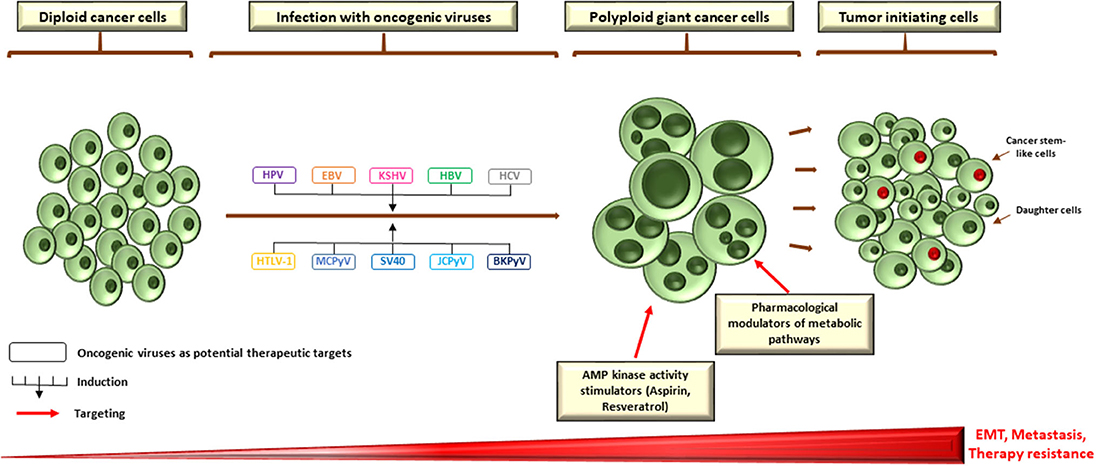
Figure 2. Potential therapeutic targets to block PGCC formation during infection with oncoviruses. As a growing body of evidence is pointing toward a crucial role of polypoid giant cancer cells in enhancing tumor evolution and the acquisition of therapy resistance, therapeutically targeting those cells is of utmost importance. Pharmacological modulators of metabolic pathways and AMP kinase activity stimulators appear to be promising candidates that specifically target polyploid tumor cells to counteract tumor repopulation. However, as oncogenic viruses might be mediators and/or inducers of PGCC formation, targeting those pathogens could also constitute a potential therapeutic strategy to block the formation of giant cells, which could disrupt the adaptive capacity of tumors in vivo and establish an effective strategy in the war against cancer. EBV nuclear antigen (EBNA), EMT (epithelial-to-mesenchymal transition) large HBV surface protein (LHBs), latency-associated nuclear antigen (LANA), latent membrane protein 1 (LMP1).
Perspectives and Conclusion
Overall, synthesizing the diverse literature on polyploidy converges to argument that an interrelationship might exist between infection with oncogenic pathogens, the polyploid phenotype, and tumorigenesis. This is reinforced by the fact that chromosomal changes occurred at the time of morphological transformation (119) and that the multinucleated cell phenotype was induced upon the expression of viral oncoproteins, rather than by trivial possibilities such as cell fusion (68, 228), the latter being also shared by other non-oncogenic pathogens (128). Nonetheless, and despite the prevalence and importance of polyploidy, the assessment of such phenotype is underestimated in the setting of oncoviruses. Several questions in terms of polyploidy generation as well as maintenance remained unanswered. Those include not only the contribution of each mechanism and the potential cooperation between the various signaling pathways, but also the estimated rates of polyploidy formation, in addition to the mechanism of stemness profile acquisition that arms this population with the advantage to propagate, form a subclonal populations and convey therapy resistance, although underassessed in this context. Addressing those questions is further sophisticated by the presence of viral proteins that might confer an additional layer of complexity to the tumor microenvironment. Conceptualizing the hypothesis outlined in this review warrants a collective effort to link the described findings to polyploidization and transformation. This will provide an enhanced mechanistic understanding of some molecular mechanisms and pave the road toward a novel perception of effective and innovative therapeutic targets. Thus, new modalities of treatment are encouraged not only to disrupt the viral replicative machinery and to block the immortalizing and transforming capacity of oncogenic viruses, but also to target PGCC, with the ultimate goal of rendering polyploidy a “druggable” phenotype, with a special emphasis on disease relapse and therapy resistance.
Author Contributions
ZN and GH wrote the article and draw the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the University of Franche-Comté—CR3300.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
APC/C, anaphase promoting complex/cyclosome; ATL, adult T-cell leukemia/lymphoma; ATM, ataxia telangiectasia, mutated; ATR, ataxia telangiectasia and Rad3-related; AURKB, aurora kinase B; BL, Burkitt's lymphoma; Cdc6, cell division cycle 6; CDK, cyclin-dependent kinase; DDR, DNA damage response; EBNA, Epstein–Barr virus nuclear antigen; EMT, epithelial-to-mesenchymal transition; GPCR, G protein-coupled receptor; HCC, hepatocellular carcinoma; HHV8, human herpesvirus 8; HGSIL, high-grade squamous intraepithelial lesion; HR, high-risk; KS, Kaposi sarcoma; LANA, latency-associated nuclear antigen; LMP1, latent membrane protein 1; LR, low-risk; MAD2, mitotic arrest deficient 2; MCC, Merkel cell carcinoma; NPC, nasopharyngeal carcinoma; ORF, open reading frame; PBMCs, peripheral blood mononuclear cells; PGCC, polyploid giant cancer cells; Plk1, polo-like kinase; VEGF, vascular endothelial growth factor; VHL, Von Hippel-Lindau; v-FLIP, viral FLICE inhibitory protein.
References
1. Gerashchenko TS, Denisov EV, Litviakov NV, Zavyalova MV, Vtorushin SV, Tsyganov MM, et al. Intratumor heterogeneity: nature and biological significance. Biochem Mosc. (2013) 78:1201–15. doi: 10.1134/S0006297913110011
2. McQuerry JA, Chang JT, Bowtell DDL, Cohen A, Bild AH. Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J Mol Med. (2017) 95:1167–78. doi: 10.1007/s00109-017-1587-4
3. Coward J, Harding A. Size Does Matter: why polyploid tumor cells are critical drug targets in the war on cancer. Front Oncol. (2014) 4:123. doi: 10.3389/fonc.2014.00123
4. Erenpreisa J, Salmina K, Huna A, Jackson TR, Vazquez-Martin A, Cragg MS. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience. (2014) 2:3–14. doi: 10.18632/oncoscience.108
5. Liu J. The dualistic origin of human tumors. Semin Cancer Biol. (2018) 53:1–16. doi: 10.1016/j.semcancer.2018.07.004
6. Niu N, Zhang J, Zhang N, Mercado-Uribe I, Tao F, Han Z, et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis. (2016) 5:e281. doi: 10.1038/oncsis.2016.75
7. Mui UN, Haley CT, Tyring SK. Viral oncology: molecular biology and pathogenesis. J Clin Med. (2017) 6:111. doi: 10.3390/jcm6120111
8. Luo GG, Ou JJ. Oncogenic viruses and cancer. Virol Sin. (2015) 30:83–4. doi: 10.1007/s12250-015-3599-y
9. Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. (2014) 15:266–82. doi: 10.1016/j.chom.2014.02.011
10. Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. (2009) 392:1–10. doi: 10.1016/j.virol.2009.06.001
11. Yasunaga J, Jeang K-T. Viral transformation and aneuploidy. Environ Mol Mutagen. (2009) 50:733–40. doi: 10.1002/em.20480
12. Krump NA, You J. Molecular mechanisms of viral oncogenesis in humans. Nat Rev Microbiol. (2018) 16:684–98. doi: 10.1038/s41579-018-0064-6
13. Chang Y, Moore PS, Weiss RA. Human oncogenic viruses: nature and discovery. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160264. doi: 10.1098/rstb.2016.0264
14. Luria L, Cardoza-Favarato G. Human Papillomavirus. Treasure Island, FL: StatPearls Publishing (2020).
15. zur Hausen H. Papillomaviruses in human cancers. Proc Assoc Am Physicians. (1999) 111:581–7. doi: 10.1046/j.1525-1381.1999.99723.x
16. Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev. (2003) 16:1–17. doi: 10.1128/CMR.16.1.1-17.2003
17. Kaspersen MD, Larsen PB, Ingerslev HJ, Fedder J, Petersen GB, Bonde J, et al. Identification of multiple HPV types on spermatozoa from human sperm donors. PLoS ONE. (2011) 6:e18095. doi: 10.1371/journal.pone.0018095
18. Wang C, Hai Y, Liu X, Liu N, Yao Y, He P, et al. Prediction of high-risk types of human papillomaviruses using statistical model of protein “sequence space.” Comput Math Methods Med. (2015) 2015:756345. doi: 10.1155/2015/756345
19. Tomaić V. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers. (2016) 8:95. doi: 10.3390/cancers8100095
20. Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, et al. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. (2006) 27:337–43. doi: 10.1093/carcin/bgi218
21. Lorenzato M, Clavel C, Masure M, Nou JM, Bouttens D, Evrard G, et al. DNA image cytometry and human papillomavirus (HPV) detection help to select smears at high risk of high-grade cervical lesions. J Pathol. (2001) 194:171–6. doi: 10.1002/path.874
22. Méhes G, Speich N, Bollmann M, Bollmann R. Chromosomal aberrations accumulate in polyploid cells of high-grade squamous intraepithelial lesions (HSIL). Pathol Oncol Res. (2004) 10:142–8. doi: 10.1007/BF03033742
23. Giannoudis A, Evans MF, Southern SA, Herrington CS. Basal keratinocyte tetrasomy in low-grade squamous intra-epithelial lesions of the cervix is restricted to high and intermediate risk HPV infection but is not type-specific. Br J Cancer. (2000) 82:424–8. doi: 10.1054/bjoc.1999.0937
24. Southern SA, Evans MF, Herrington CS. Basal cell tetrasomy in low-grade cervical squamous intraepithelial lesions infected with high-risk human papillomaviruses. Cancer Res. (1997) 57:4210–3.
25. Patel D, Incassati A, Wang N, McCance DJ. Human papillomavirus type 16 E6 and E7 cause polyploidy in human keratinocytes and up-regulation of G2-M-phase proteins. Cancer Res. (2004) 64:1299–306. doi: 10.1158/0008-5472.CAN-03-2917
26. Filatov L, Golubovskaya V, Hurt JC, Byrd LL, Phillips JM, Kaufmann WK. Chromosomal instability is correlated with telomere erosion and inactivation of G2 checkpoint function in human fibroblasts expressing human papillomavirus type 16 E6 oncoprotein. Oncogene. (1998) 16:1825–38. doi: 10.1038/sj.onc.1201711
27. Liu Y, Heilman SA, Illanes D, Sluder G, Chen JJ. p53-independent abrogation of a postmitotic checkpoint contributes to human papillomavirus E6-induced polyploidy. Cancer Res. (2007) 67:2603–10. doi: 10.1158/0008-5472.CAN-06-3436
28. Zhang W, Liu Y, Zhao N, Chen H, Qiao L, Zhao W, et al. Role of Cdk1 in the p53-independent abrogation of the postmitotic checkpoint by human papillomavirus E6. J Virol. (2015) 89:2553–62. doi: 10.1128/JVI.02269-14
29. Fan X, Liu Y, Heilman SA, Chen JJ. Human papillomavirus E7 induces rereplication in response to DNA damage. J Virol. (2013) 87:1200–10. doi: 10.1128/JVI.02038-12
30. Fan X, Zhou Y, Chen JJ. Role of Cdc6 in re-replication in cells expressing human papillomavirus E7 oncogene. Carcinogenesis. (2016) 37:799–809. doi: 10.1093/carcin/bgw059
31. Hashida T, Yasumoto S. Induction of chromosome abnormalities in mouse and human epidermal keratinocytes by the human papillomavirus type 16 E7 oncogene. J Gen Virol. (1991) 72(Pt 7):1569–77. doi: 10.1099/0022-1317-72-7-1569
32. Heilman SA, Nordberg JJ, Liu Y, Sluder G, Chen JJ. Abrogation of the postmitotic checkpoint contributes to polyploidization in human papillomavirus E7-expressing cells. J Virol. (2009) 83:2756–64. doi: 10.1128/JVI.02149-08
33. DiMaio D, Mattoon D. Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene. (2001) 20:7866–73. doi: 10.1038/sj.onc.1204915
34. Hu L, Potapova TA, Li S, Rankin S, Gorbsky GJ, Angeletti PC, et al. Expression of HPV16 E5 produces enlarged nuclei and polyploidy through endoreplication. Virology. (2010) 405:342–51. doi: 10.1016/j.virol.2010.06.025
35. Chien W-M, Noya F, Benedict-Hamilton HM, Broker TR, Chow LT. Alternative fates of keratinocytes transduced by human papillomavirus type 18 E7 during squamous differentiation. J Virol. (2002) 76:2964–72. doi: 10.1128/JVI.76.6.2964-2972.2002
36. IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Epstein–Barr virus. In: Biological Agents. Lyon, FR: International Agency for Research on Cancer (2012).
37. Thompson MP, Kurzrock R. Epstein–Barr virus and cancer. Clin Cancer Res. (2004) 10:803–21. doi: 10.1158/1078-0432.CCR-0670-3
38. Farrell PJ. Epstein–Barr virus and cancer. Annu Rev Pathol Mech Dis. (2019) 14:29–53. doi: 10.1146/annurev-pathmechdis-012418-013023
39. Jiang Q, Zhang Q, Wang S, Xie S, Fang W, Liu Z, et al. A fraction of CD133+ CNE2 cells is made of giant cancer cells with morphological evidence of asymmetric mitosis. J Cancer. (2015) 6:1236–44. doi: 10.7150/jca.12626
40. Gelardi M, Tomaiuolo M, Cassano M, Besozzi G, Fiorella ML, Calvario A, et al. Epstein–Barr virus induced cellular changes in nasal mucosa. Virol J. (2006) 3:6. doi: 10.1186/1743-422X-3-6
41. Sato H, Takimoto T, Tanaka S, Ogura H, Shiraishi K, Tanaka J. Cytopathic effects induced by Epstein–Barr virus replication in epithelial nasopharyngeal carcinoma hybrid cells. J Virol. (1989) 63:3555–9. doi: 10.1128/JVI.63.8.3555-3559.1989
42. Shumilov A, Tsai M-H, Schlosser YT, Kratz A-S, Bernhardt K, Fink S, et al. Epstein–Barr virus particles induce centrosome amplification and chromosomal instability. Nat Commun. (2017) 8:14257. doi: 10.1038/ncomms14257
43. Humme S, Reisbach G, Feederle R, Delecluse H-J, Bousset K, Hammerschmidt W, et al. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci USA. (2003) 100:10989–94. doi: 10.1073/pnas.1832776100
44. Lee JM, Lee K-H, Farrell CJ, Ling PD, Kempkes B, Park JH, et al. EBNA2 is required for protection of latently Epstein–Barr virus-infected B cells against specific apoptotic stimuli. J Virol. (2004) 78:12694–7. doi: 10.1128/JVI.78.22.12694-12697.2004
45. Pan S-H, Tai C-C, Lin C-S, Hsu W-B, Chou S-F, Lai C-C, et al. Epstein–Barr virus nuclear antigen 2 disrupts mitotic checkpoint and causes chromosomal instability. Carcinogenesis. (2009) 30:366–75. doi: 10.1093/carcin/bgn291
46. Parker GA, Crook T, Bain M, Sara EA, Farrell PJ, Allday MJ. Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene. (1996) 13:2541–9.
47. Parker GA, Touitou R, Allday MJ. Epstein–Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene. (2000) 19:700–9. doi: 10.1038/sj.onc.1203327
48. Kieser A, Sterz KR. The latent membrane protein 1 (LMP1). Curr Top Microbiol Immunol. (2015) 391:119–49. doi: 10.1007/978-3-319-22834-1_4
49. Lajoie V, Lemieux B, Sawan B, Lichtensztejn D, Lichtensztejn Z, Wellinger R, et al. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood. (2015) 125:2101–10. doi: 10.1182/blood-2014-08-594176
50. Dow DE, Cunningham CK, Buchanan AM. A review of human herpesvirus 8, the Kaposi's sarcoma-associated herpesvirus, in the pediatric population. J Pediatric Infect Dis Soc. (2014) 3:66–76. doi: 10.1093/jpids/pit051
51. Humans IWG on the E of CR to Kaposi Sarcoma Herpesvirus. In: Biological Agents. Lyon: International Agency for Research on Cancer (2012).
52. Goncalves PH, Ziegelbauer J, Uldrick TS, Yarchoan R. Kaposi-sarcoma herpesvirus associated cancers and related diseases. Curr Opin HIV AIDS. (2017) 12:47–56. doi: 10.1097/COH.0000000000000330
53. Pan H, Zhou F, Gao S-J. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. (2004) 64:4064–8. doi: 10.1158/0008-5472.CAN-04-0657
54. Ma Q, Cavallin LE, Yan B, Zhu S, Duran EM, Wang H, et al. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi's sarcoma. Proc Natl Acad Sci USA. (2009) 106:8683–8. doi: 10.1073/pnas.0812688106
55. Liu J, Martin HJ, Liao G, Hayward SD. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol. (2007) 81:10451–9. doi: 10.1128/JVI.00804-07
56. Si H, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J Virol. (2006) 80:697–709. doi: 10.1128/JVI.80.2.697-709.2006
57. Sun Z, Xiao B, Jha HC, Lu J, Banerjee S, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded LANA can induce chromosomal instability through targeted degradation of the mitotic checkpoint kinase Bub1. J Virol. (2014) 88:7367–78. doi: 10.1128/JVI.00554-14
58. Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, et al. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol. (1997) 71:4193–8. doi: 10.1128/JVI.71.6.4193-4198.1997
59. Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, et al. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin-cdk6 complex. EMBO J. (1999) 18:644–53. doi: 10.1093/emboj/18.3.644
60. Laman H, Coverley D, Krude T, Laskey R, Jones N. Viral cyclin–cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol Cell Biol. (2001) 21:624–35. doi: 10.1128/MCB.21.2.624-635.2001
61. Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. (2002) 2:229–41. doi: 10.1016/S1535-6108(02)00123-X
62. Afonso PV, Cassar O, Gessain A. Molecular epidemiology, genetic variability and evolution of HTLV-1 with special emphasis on African genotypes. Retrovirology. (2019) 16:39. doi: 10.1186/s12977-019-0504-z
63. Hoshino H. Cellular factors involved in HTLV-1 entry and pathogenicit. Front Microbiol. (2012) 3:222. doi: 10.3389/fmicb.2012.00222
64. Panfil AR, Martinez MP, Ratner L, Green PL. Human T-cell leukemia virus-associated malignancy. Curr Opin Virol. (2016) 20:40–6. doi: 10.1016/j.coviro.2016.08.009
65. Kikuchi M, Mitsui T, Takeshita M, Okamura H, Naitoh H, Eimoto T. Virus associated adult T-cell leukemia (ATL) in Japan: clinical, histological and immunological studies. Hematol Oncol. (1986) 4:67–81. doi: 10.1002/hon.2900040109
66. Currer R, Van Duyne R, Jaworski E, Guendel I, Sampey G, Das R, et al. HTLV tax: a fascinating multifunctional co-regulator of viral and cellular pathways. Front Microbiol. (2012) 3:406. doi: 10.3389/fmicb.2012.00406
67. Grassmann R, Aboud M, Jeang K-T. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene. (2005) 24:5976–85. doi: 10.1038/sj.onc.1208978
68. Liang M-H, Geisbert T, Yao Y, Hinrichs SH, Giam C-Z. Human T-lymphotropic virus type 1 oncoprotein tax promotes S-phase entry but blocks mitosis. J Virol. (2002) 76:4022–33. doi: 10.1128/JVI.76.8.4022-4033.2002
69. Jin DY, Spencer F, Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. (1998) 93:81–91. doi: 10.1016/S0092-8674(00)81148-4
70. Yang L, Kotomura N, Ho Y-K, Zhi H, Bixler S, Schell MJ, et al. Complex cell cycle abnormalities caused by human T-lymphotropic virus type 1 Tax. J Virol. (2011) 85:3001–9. doi: 10.1128/JVI.00086-10
71. Ducu RI, Dayaram T, Marriott SJ. The HTLV-1 tax oncoprotein represses Ku80 gene expression. Virology. (2011) 416:1–8. doi: 10.1016/j.virol.2011.04.012
72. Sibon D, Gabet A-S, Zandecki M, Pinatel C, Thête J, Delfau-Larue M-H, et al. HTLV-1 propels untransformed CD4+ lymphocytes into the cell cycle while protecting CD8+ cells from death. J Clin Invest. (2006) 116:974–83. doi: 10.1172/JCI27198
73. Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, et al. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol Cell Biol. (2005) 25:6178–98. doi: 10.1128/MCB.25.14.6178-6198.2005
74. Malu A, Hutchison T, Yapindi L, Smith K, Nelson K, Bergeson R, et al. The human T-cell leukemia virus type-1 tax oncoprotein dissociates NF-κB p65RelA-Stathmin complexes and causes catastrophic mitotic spindle damage and genomic instability. Virology. (2019) 535:83–101. doi: 10.1016/j.virol.2019.07.003
75. Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. (2014) 4:a014357. doi: 10.1101/cshperspect.a014357
76. Santoni-Rugiu E, Falck J, Mailand N, Bartek J, Lukas J. Involvement of Myc activity in a G(1)/S-promoting mechanism parallel to the pRb/E2F pathway. Mol Cell Biol. (2000) 20:3497–509. doi: 10.1128/0.20.10.3497-3509.2000
77. Guo Y, Niu C, Breslin P, Tang M, Zhang S, Wei W, et al. c-Myc-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Blood. (2009) 114:2097–106. doi: 10.1182/blood-2009-01-197947
78. Lombardi L, Newcomb EW, Dalla-Favera R. Pathogenesis of Burkitt lymphoma: expression of an activated c-myc oncogene causes the tumorigenic conversion of EBV-infected human B lymphoblasts. Cell. (1987) 49:161–70. doi: 10.1016/0092-8674(87)90556-3
79. Romeo MM, Ko B, Kim J, Brady R, Heatley HC, He J, et al. Acetylation of the c-MYC oncoprotein is required for cooperation with the HTLV-1 p30(II) accessory protein and the induction of oncogenic cellular transformation by p30(II)/c-MYC. Virology. (2015) 476:271–88. doi: 10.1016/j.virol.2014.12.008
80. Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol. (2007) 13:14–21. doi: 10.3748/wjg.v13.i1.14
81. Humans IWG on the E of CR to. HEPATITIS B VIRUS. In: Biological Agents. Lyon: International Agency for Research on Cancer (2012).
82. Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. (2000) 64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000
83. Liang TJ. Hepatitis B: the virus and disease. Hepatology. (2009) 49(5 Suppl.):S13–21. doi: 10.1002/hep.22881
84. Simon D, London T, Hann HW, Knowles BB. Chromosome abnormalities in peripheral blood cells of hepatitis B virus chronic carriers. Cancer Res. (1991) 51:6176–9.
85. Celton-Morizur S, Desdouets C. Polyploidization of liver cells. Adv Exp Med Biol. (2010) 676:123–35. doi: 10.1007/978-1-4419-6199-0_8
86. Wang M-J, Chen F, Lau JTY, Hu Y-P. Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis. (2017) 8:e2805. doi: 10.1038/cddis.2017.167
87. Bou-Nader M, Caruso S, Donne R, Celton-Morizur S, Calderaro J, Gentric G, et al. Polyploidy spectrum: a new marker in HCC classification. Gut. (2020) 69:355–64. doi: 10.1136/gutjnl-2018-318021
88. Lee H, Jeong H, Lee SY, Kim SS, Jang KL. Hepatitis B virus X protein stimulates virus replication via DNA methylation of the C-1619 in covalently closed circular DNA. Mol Cells. (2019) 42:67–78. doi: 10.14348/molcells.2018.0255
89. Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. (2011) 26(Suppl. 1):144–52. doi: 10.1111/j.1440-1746.2010.06546.x
90. Ahodantin J, Lekbaby B, Bou Nader M, Soussan P, Kremsdorf D. Hepatitis B virus X protein enhances the development of liver fibrosis and the expression of genes associated with epithelial-mesenchymal transitions and tumor progenitor cells. Carcinogenesis. (2020) 41:358–67. doi: 10.1093/carcin/bgz109
91. Gramantieri L, Melchiorri C, Chieco P, Gaiani S, Stecca B, Casali A, et al. Alteration of DNA ploidy and cell nuclearity in human hepatocellular carcinoma associated with HBV infection. J Hepatol. (1996) 25:848–53. doi: 10.1016/S0168-8278(96)80288-1
92. Ahodantin J, Bou-Nader M, Cordier C, Mégret J, Soussan P, Desdouets C, et al. Hepatitis B virus X protein promotes DNA damage propagation through disruption of liver polyploidization and enhances hepatocellular carcinoma initiation. Oncogene. (2019) 38:2645–57. doi: 10.1038/s41388-018-0607-3
93. Li T-N, Wu Y-J, Tsai H-W, Sun C-P, Wu Y-H, Wu H-L, et al. Intrahepatic hepatitis B virus large surface antigen induces hepatocyte hyperploidy via failure of cytokinesis. J Pathol. (2018) 245:502–13. doi: 10.1002/path.5102
94. Studach LL, Rakotomalala L, Wang W-H, Hullinger RL, Cairo S, Buendia M-A, et al. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology. (2009) 50:414–23. doi: 10.1002/hep.22996
95. Rakotomalala L, Studach L, Wang W-H, Gregori G, Hullinger RL, Andrisani O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J Biol Chem. (2008) 283:28729–40. doi: 10.1074/jbc.M802751200
96. Lee S, Tarn C, Wang W-H, Chen S, Hullinger RL, Andrisani OM. Hepatitis B virus X protein differentially regulates cell cycle progression in X-transforming versus nontransforming hepatocyte (AML12) cell lines. J Biol Chem. (2002) 277:8730–40. doi: 10.1074/jbc.M108025200
97. Musa J, Li J, Grünewald TG. Hepatitis B virus large surface protein is priming for hepatocellular carcinoma development via induction of cytokinesis failure. J Pathol. (2019) 247:6–8. doi: 10.1002/path.5169
98. Hsieh Y-H, Chang Y-Y, Su I-J, Yen C-J, Liu Y-R, Liu R-J, et al. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J Pathol. (2015) 236:337–47. doi: 10.1002/path.4531
99. Chevaliez S, Pawlotsky J-M. HCV genome and life cycle. In: Tan S-L, editor. Hepatitis C Viruses: Genomes and Molecular Biology. Norfolk : Horizon Bioscience (2006).
100. Humans IWG on the E of CR to. HEPATITIS C VIRUS. Biological Agents. Lyon: International Agency for Research on Cancer (2012).
101. Moosavy SH, Davoodian P, Nazarnezhad MA, Nejatizaheh A, Eftekhar E, Mahboobi H. Epidemiology, transmission, diagnosis, and outcome of Hepatitis C virus infection. Electron Physician. (2017) 9:5646–56. doi: 10.19082/5646
102. Zhu X, Jing L, Li X. Hepatitis C virus infection is a risk factor for non-Hodgkin lymphoma: a MOOSE-compliant meta-analysis. Medicine. (2019) 98:e14755. doi: 10.1097/MD.0000000000014755
103. McGivern DR, Lemon SM. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene. (2011) 30:1969–83. doi: 10.1038/onc.2010.594
104. Machida K, Liu J-C, McNamara G, Levine A, Duan L, Lai MMC. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J Virol. (2009) 83:12590–600. doi: 10.1128/JVI.02643-08
105. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. (1998) 4:1065–7. doi: 10.1038/2053
106. White MK, Gordon J, Khalili K. The rapidly expanding family of human polyomaviruses: recent developments in understanding their life cycle and role in human pathology. PLoS Pathog. (2013) 9:e1003206. doi: 10.1371/journal.ppat.1003206
107. Vilchez RA, Butel JS. Emergent human pathogen simian virus 40 and its role in cancer. Clin Microbiol Rev. (2004) 17:495–508. doi: 10.1128/CMR.17.3.495-508.2004
108. Prado JCM, Monezi TA, Amorim AT, Lino V, Paladino A, Boccardo E. Human polyomaviruses and cancer: an overview. Clinics (Saõ Paulo). (2018) 73(Suppl. 1):e558s. doi: 10.6061/clinics/2018/e558s
109. Tsang SH, Wang R, Nakamaru-Ogiso E, Knight SAB, Buck CB, You J. The oncogenic small tumor antigen of merkel cell polyomavirus is an iron-sulfur cluster protein that enhances viral DNA replication. J Virol. (2016) 90:1544–56. doi: 10.1128/JVI.02121-15
110. Kwun HJ, Wendzicki JA, Shuda Y, Moore PS, Chang Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene. (2017) 36:6784–92. doi: 10.1038/onc.2017.277
111. Shuda M, Guastafierro A, Geng X, Shuda Y, Ostrowski SM, Lukianov S, et al. Merkel cell polyomavirus small T antigen induces cancer and embryonic merkel cell proliferation in a transgenic mouse model. PLoS ONE. (2015) 10:e0142329. doi: 10.1371/journal.pone.0142329
112. Jiang M, Zhao L, Gamez M, Imperiale MJ. Roles of ATM and ATR-mediated DNA damage responses during lytic bk polyomavirus infection. PLoS Pathogens. (2012) 8:e1002898. doi: 10.1371/journal.ppat.1002898
113. Justice JL, Needham JM, Thompson SR. BK polyomavirus activates the DNA damage response to prolong S phase. J Virol. (2019) 93:e00130-19. doi: 10.1128/JVI.00130-19
114. Wüthrich C, Dang X, Westmoreland S, McKay J, Maheshwari A, Anderson MP, et al. Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol. (2009) 65:742–8. doi: 10.1002/ana.21619
115. Ricciardiello L, Baglioni M, Giovannini C, Pariali M, Cenacchi G, Ripalti A, et al. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. (2003) 63:7256–62.
116. Darbinyan A, White MK, Akan S, Radhakrishnan S, Valle LD, Amini S, et al. Alterations of DNA damage repair pathways resulting from JCV infection. Virology. (2007) 364:73–86. doi: 10.1016/j.virol.2007.02.015
117. Ariza A, Mate JL, Serrano S, Isamat M, Keysers U, Aracil C, et al. DNA Amplification in glial cells of progressive multifocal leukoencephalopathy: an image analysis study. J Neuropathol Exp Neurol. (1996) 55:729–33. doi: 10.1097/00005072-199606000-00007
118. Neel JV, Major EO, Awa AA, Glover T, Burgess A, Traub R, et al. Hypothesis: “Rogue cell” -type chromosomal damage in lymphocytes is associated with infection with the JC human polyoma virus and has implications for oncopenesis. Proc Natl Acad Sci USA. (1996) 93:2690–5. doi: 10.1073/pnas.93.7.2690
119. Lehman JM. Early chromosome changes in diploid Chinese hamster cells after infection with Simian virus 40. Int J Cancer. (1974) 13:164–72. doi: 10.1002/ijc.2910130203
120. Lehman JM, Laffin J, Friedrich TD. Simian virus 40 induces multiple S phases with the majority of viral DNA replication in the G2 and second S phase in CV-1 cells. Exp Cell Res. (2000) 258:215–22. doi: 10.1006/excr.2000.4927
121. Friedrich TD, Laffin J, Lehman JM. Simian virus 40 large T-antigen function is required for induction of tetraploid DNA content during lytic infection. J Virol. (1992) 66:4576–9. doi: 10.1128/JVI.66.7.4576-4579.1992
123. Friedrich TD, Laffin J, Lehman JM. Induction of tetraploid DNA content by simian virus 40 is dependent on T-antigen function in the G2 phase of the cell cycle. J Virol. (1994) 68:4028–30. doi: 10.1128/JVI.68.6.4028-4030.1994
124. Laffin J, Fogleman D, Lehman JM. Correlation of DNA content, p53, T antigen, and V antigen in simian virus 40-infected human diploid cells. Cytometry. (1989) 10:205–13. doi: 10.1002/cyto.990100212
125. Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, et al. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol. (2009) 83:117–27. doi: 10.1128/JVI.01515-08
126. Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, et al. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. (2004) 18:1305–16. doi: 10.1101/gad.1182804
127. Stewart N, Bacchetti S. Expression of SV40 large T antigen, but not small t antigen, is required for the induction of chromosomal aberrations in transformed human cells. Virology. (1991) 180:49–57. doi: 10.1016/0042-6822(91)90008-Y
128. White J, Matlin K, Helenius A. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol. (1981) 89:674–9. doi: 10.1083/jcb.89.3.674
129. Budka H. Multinucleated giant cells in brain: a hallmark of the acquired immune deficiency syndrome (AIDS). Acta Neuropathol. (1986) 69(3–4):253–8. doi: 10.1007/BF00688301
130. Muggeridge MI. Characterization of cell–cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J Gen Virol. (2000) 81(Pt 8):2017–27. doi: 10.1099/0022-1317-81-8-2017
131. Breuer W, Trustaedt M, Hermanns W. [Appearance of multinucleated giant cells in association with an adenovirus infection in two guinea pigs]. Berl Munch Tierarztl Wochenschr. (2006) 119:474–9.
132. Chavali PL, Pütz M, Gergely F. Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130468. doi: 10.1098/rstb.2013.0468
133. Srsen V, Merdes A. The centrosome and cell proliferation. Cell Div. (2006) 1:26. doi: 10.1186/1747-1028-1-26
134. Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci USA. (2000) 97:10002–7. doi: 10.1073/pnas.170093297
135. Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. (2002) 62:7075–82. doi: 10.7156/v3i2p043
136. Duensing S, Duensing A, Crum CP, Münger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. (2001) 61:2356–60.
137. Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. (2018) 13:7. doi: 10.1186/s13008-018-0040-6
138. Rannou Y, Troadec M-B, Petretti C, Hans F, Dutertre S, Dimitrov S, et al. Localization of aurora A and aurora B kinases during interphase: role of the N-terminal domain. Cell Cycle. (2008) 7:3012–20. doi: 10.4161/cc.7.19.6718
139. Zhu Q, Ding L, Zi Z, Gao S, Wang C, Wang Y, et al. Viral-mediated AURKB cleavage promotes cell segregation and tumorigenesis. Cell Rep. (2019) 26:3657–71.e5. doi: 10.1016/j.celrep.2019.02.106
140. Cai Q, Xiao B, Si H, Cervini A, Gao J, Lu J, et al. Kaposi's sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. (2012) 8:e1002566. doi: 10.1371/journal.ppat.1002566
141. Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. (2016) 6:a026104. doi: 10.1101/cshperspect.a026104
142. Li T, Liu X, Jiang L, Manfredi J, Zha S, Gu W. Loss of p53-mediated cell-cycle arrest, senescence and apoptosis promotes genomic instability and premature aging. Oncotarget. (2016) 7:11838–49. doi: 10.18632/oncotarget.7864
143. Meek DW. The role of p53 in the response to mitotic spindle damage. Pathol Biol. (2000) 48:246–54.
144. Peled A, Schwartz D, Elkind NB, Wolkowicz R, Li R, Rotter V. The role of p53 in the induction of polyploidity of myelomonocytic leukemic M1/2 cells. Oncogene. (1996) 13:1677–85.
145. Mirzayans R, Murray D. Intratumor heterogeneity and therapy resistance: contributions of dormancy, apoptosis reversal (anastasis) and cell fusion to disease recurrence. Int J Mol Sci. (2020) 21:1308. doi: 10.3390/ijms21041308
146. Thomas M, Pim D, Banks L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene. (1999) 18:7690–700. doi: 10.1038/sj.onc.1202953
147. Thomas JT, Laimins LA. Human papillomavirus oncoproteins E6 and E7 independently abrogate the mitotic spindle checkpoint. J Virol. (1998) 72:1131–7. doi: 10.1128/JVI.72.2.1131-1137.1998
148. Lundgren K, Montes de Oca Luna R, McNeill YB, Emerick EP, Spencer B, Barfield CR, et al. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. (1997) 11:714–25. doi: 10.1101/gad.11.6.714
149. Leao M, Anderton E, Wade M, Meekings K, Allday MJ. Epstein–Barr virus-induced resistance to drugs that activate the mitotic spindle assembly checkpoint in Burkitt's lymphoma cells. J Virol. (2007) 81:248–60. doi: 10.1128/JVI.01096-06
150. Wei F, Gan J, Wang C, Zhu C, Cai Q. Cell cycle regulatory functions of the KSHV oncoprotein LANA. Front Microbiol. (2016) 7:334. doi: 10.3389/fmicb.2016.00334
151. An F-Q, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, Renne R. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J Biol Chem. (2005) 280:3862–74. doi: 10.1074/jbc.M407435200
152. Cai Q-L, Knight JS, Verma SC, Zald P, Robertson ES. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. (2006) 2:e116. doi: 10.1371/journal.ppat.0020116
153. Kumar P, Murakami M, Kaul R, Saha A, Cai Q, Robertson ES. Deregulation of the cell cycle machinery by Epstein–Barr virus nuclear antigen 3C. Future Virol. (2009) 4:79–91. doi: 10.2217/17460794.4.1.79
154. Saha A, Halder S, Upadhyay SK, Lu J, Kumar P, Murakami M, et al. Epstein–Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. (2011) 7:e1001275. doi: 10.1371/journal.ppat.1001275
155. Yi F, Saha A, Murakami M, Kumar P, Knight JS, Cai Q, et al. Epstein–Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology. (2009) 388:236–47. doi: 10.1016/j.virol.2009.03.027
156. Cai Q, Guo Y, Xiao B, Banerjee S, Saha A, Lu J, et al. Epstein–Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. (2011) 7:e1002418. doi: 10.1371/journal.ppat.1002418
157. Banerjee S, Lu J, Qiliang C, Sun Z, Jha HC, Robertson ES. EBNA3C augments Pim-1 mediated phosphorylation and degradation of p21 to promote B-cell proliferation. PLoS Pathog. (2014) 10:e1004304. doi: 10.1371/journal.ppat.1004304
158. Hunt T, Nasmyth K, Novák B. The cell cycle. Philos Trans R Soc Lond B Biol Sci. (2011) 366:3494–7. doi: 10.1098/rstb.2011.0274
159. Wenzel ES, Singh ATK. Cell-cycle checkpoints and aneuploidy on the path to cancer. In Vivo. (2018) 32:1–5. doi: 10.21873/invivo.11197
160. Studach L, Wang W-H, Weber G, Tang J, Hullinger RL, Malbrue R, et al. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J Biol Chem. (2010) 285:30282–93. doi: 10.1074/jbc.M109.093963
161. Liu B, Liang M-H, Kuo Y, Liao W, Boros I, Kleinberger T, et al. Human T-lymphotropic virus type 1 oncoprotein tax promotes unscheduled degradation of Pds1p/Securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol Cell Biol. (2003) 23:5269–81. doi: 10.1128/MCB.23.15.5269-5281.2003
162. Shu Z, Row S, Deng W-M. Endoreplication: the good, the bad, and the ugly. Trends Cell Biol. (2018) 28:465–74. doi: 10.1016/j.tcb.2018.02.006
163. Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. (2014) 33:116–28. doi: 10.1038/onc.2013.96
164. Araldi RP, Mazzuchelli-de-Souza J, Modolo DG, Souza EB de, Melo TC de, Spadacci-Morena DD, et al. Mutagenic potential of Bos taurus papillomavirus type 1 E6 recombinant protein: first description. BioMed Research International. (2015) 2015:e806361. doi: 10.1155/2015/806361
165. Blow JJ, Hodgson B. Replication licensing—defining the proliferative state? Trends Cell Biol. (2002) 12:72–8. doi: 10.1016/S0962-8924(01)02203-6
166. Dorn ES, Chastain PD, Hall JR, Cook JG. Analysis of re-replication from deregulated origin licensing by DNA fiber spreading. Nucleic Acids Res. (2009) 37:60–9. doi: 10.1093/nar/gkn912
167. Bonds L, Baker P, Gup C, Shroyer KR. Immunohistochemical localization of cdc6 in squamous and glandular neoplasia of the uterine cervix. Arch Pathol Lab Med. (2002) 126:1164–8.
168. Li Z, Rich JN. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr Top Microbiol Immunol. (2010) 345:21–30. doi: 10.1007/82_2010_75
169. Lopez-Sánchez LM, Jimenez C, Valverde A, Hernandez V, Peñarando J, Martinez A, et al. CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PLoS ONE. (2014) 9:e99143. doi: 10.1371/journal.pone.0099143
170. Zhu C, Zhu Q, Wang C, Zhang L, Wei F, Cai Q. Hostile takeover: manipulation of HIF-1 signaling in pathogen-associated cancers (Review). Int J Oncol. (2016) 49:1269–76. doi: 10.3892/ijo.2016.3633
171. Cuninghame S, Jackson R, Zehbe I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology. (2014) 456–457:370–83. doi: 10.1016/j.virol.2014.02.027
172. Tang X, Zhang Q, Nishitani J, Brown J, Shi S, Le AD. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1 protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin Cancer Res. (2007) 13:2568–76. doi: 10.1158/1078-0432.CCR-06-2704
173. Wakisaka N, Kondo S, Yoshizaki T, Murono S, Furukawa M, Pagano JS. Epstein–Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1α. Mol Cell Biol. (2004) 24:5223–34. doi: 10.1128/MCB.24.12.5223-5234.2004
174. O'Neil JD, Owen TJ, Wood VHJ, Date KL, Valentine R, Chukwuma MB, et al. Epstein–Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J Gen Virol. (2008) 89(Pt 11):2833–42. doi: 10.1099/vir.0.2008/003392-0
175. Darekar S, Georgiou K, Yurchenko M, Yenamandra SP, Chachami G, Simos G, et al. Epstein–Barr virus immortalization of human B-cells leads to stabilization of hypoxia-induced factor 1 alpha, congruent with the Warburg effect. PLoS ONE. (2012) 7:e42072. doi: 10.1371/journal.pone.0042072
176. Shin YC, Joo C-H, Gack MU, Lee H-R, Jung JU. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. (2008) 68:1751–9. doi: 10.1158/0008-5472.CAN-07-2766
177. Cai Q, Lan K, Verma SC, Si H, Lin D, Robertson ES. Kaposi's sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: latency control under low oxygen conditions. J Virol. (2006) 80:7965–75. doi: 10.1128/JVI.00689-06
178. Singh RK, Lamplugh ZL, Lang F, Yuan Y, Lieberman P, You J, et al. KSHV-encoded LANA protects the cellular replication machinery from hypoxia induced degradation. PLoS Pathog. (2019) 15:e1008025. doi: 10.1371/journal.ppat.1008025
179. Cai Q, Cai S, Zhu C, Verma SC, Choi J-Y, Robertson ES. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog. (2013) 9:e1003750. doi: 10.1371/journal.ppat.1003750
180. Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, et al. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. (2000) 60:4873–80.
181. Shrestha P, Davis DA, Veeranna RP, Carey RF, Viollet C, Yarchoan R. Hypoxia-inducible factor-1 alpha as a therapeutic target for primary effusion lymphoma. PLoS Pathog. (2017) 13:e1006628. doi: 10.1371/journal.ppat.1006628
182. Yoo Y-G, Cho S, Park S, Lee M-O. The carboxy-terminus of the hepatitis B virus X protein is necessary and sufficient for the activation of hypoxia-inducible factor-1alpha. FEBS Lett. (2004) 577(1–2):121–6. doi: 10.1016/j.febslet.2004.10.004
183. Yoo Y-G, Oh SH, Park ES, Cho H, Lee N, Park H, et al. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. J Biol Chem. (2003) 278:39076–84. doi: 10.1074/jbc.M305101200
184. Abe M, Koga H, Yoshida T, Masuda H, Iwamoto H, Sakata M, et al. Hepatitis C virus core protein upregulates the expression of vascular endothelial growth factor via the nuclear factor-κB/hypoxia-inducible factor-1α axis under hypoxic conditions. Hepatol Res. (2012) 42:591–600. doi: 10.1111/j.1872-034X.2011.00953.x
185. Mühleisen A, Giaisi M, Köhler R, Krammer PH, Li-Weber M. Tax contributes apoptosis resistance to HTLV-1-infected T cells via suppression of Bid and Bim expression. Cell Death Dis. (2014) 5:e1575. doi: 10.1038/cddis.2014.536
186. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-Induced Senescence in Cancer. J Natl Cancer Inst. (2010) 102:1536–46. doi: 10.1093/jnci/djq364
187. Walen KH. Budded karyoplasts from multinucleated fibroblast cells contain centrosomes and change their morphology to mitotic cells. Cell Biol Int. (2005) 29:1057–65. doi: 10.1016/j.cellbi.2005.10.016
188. Zhang S, Mercado-Uribe I, Sood A, Bast RC, Liu J. Coevolution of neoplastic epithelial cells and multilineage stroma via polyploid giant cells during immortalization and transformation of mullerian epithelial cells. Genes Cancer. (2016) 7:60–72. doi: 10.18632/genesandcancer.102
189. Leidal AM, Cyr DP, Hill RJ, Lee PWK, McCormick C. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe. (2012) 11:167–80. doi: 10.1016/j.chom.2012.01.005
190. Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. (1997) 386:517–21. doi: 10.1038/386517a0
191. Pampalona J, Frías C, Genescà A, Tusell L. Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet. (2012) 8:e1002679. doi: 10.1371/journal.pgen.1002679
192. Knecht H, Mai S. LMP1 and dynamic progressive telomere dysfunction: a major culprit in EBV-associated Hodgkin's lymphoma. Viruses. (2017) 9:164. doi: 10.3390/v9070164
193. Lacoste S, Wiechec E, Dos Santos Silva AG, Guffei A, Williams G, Lowbeer M, et al. Chromosomal rearrangements after ex vivo Epstein–Barr virus (EBV) infection of human B cells. Oncogene. (2010) 29:503–15. doi: 10.1038/onc.2009.359
194. Herbein G. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses. (2018) 10:408. doi: 10.3390/v10080408
195. Hertel L, Mocarski ES. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of Pseudomitosis independent of US28 function. J Virol. (2004) 78:11988–2011. doi: 10.1128/JVI.78.21.11988-12011.2004
196. Kwon Y, Kim M-N, Choi EY, Kim JH, Hwang E-S, Cha C-Y. Inhibition of p53 transcriptional activity by human cytomegalovirus UL44. Microbiol Immunol. (2012) 56:324–31. doi: 10.1111/j.1348-0421.2012.00446.x
197. Tsai HL, Kou GH, Chen SC, Wu CW, Lin YS. Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J Biol Chem. (1996) 271:3534–40. doi: 10.1074/jbc.271.7.3534
198. Iwahori S, Umaña AC, VanDeusen HR, Kalejta RF. Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. J Biol Chem. (2017) 292:6583–99. doi: 10.1074/jbc.M116.773150
199. Boldogh I, AbuBakar S, Deng CZ, Albrecht T. Transcriptional activation of cellular oncogenes fos, jun, and myc by human cytomegalovirus. J Virol. (1991) 65:1568–71. doi: 10.1128/JVI.65.3.1568-1571.1991
200. Kumar A, Tripathy MK, Pasquereau S, Al Moussawi F, Abbas W, Coquard L, et al. The human cytomegalovirus strain DB activates oncogenic pathways in mammary epithelial cells. EBioMedicine. (2018) 30:167–83. doi: 10.1016/j.ebiom.2018.03.015
201. McFarlane S, Nicholl MJ, Sutherland JS, Preston CM. Interaction of the human cytomegalovirus particle with the host cell induces hypoxia-inducible factor 1 alpha. Virology. (2011) 414:83–90. doi: 10.1016/j.virol.2011.03.005
202. Garnett HM. Fusion of cytomegalovirus infected fibroblasts to form multinucleate giant cells. J Med Virol. (1979) 3:271–4. doi: 10.1002/jmv.1890030404
203. Amend SR, Torga G, Lin K-C, Kostecka LG, de Marzo A, Austin RH, et al. Polyploid giant cancer cells: unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate. (2019) 79:1489–97. doi: 10.1002/pros.23877
204. Zhang L, Wu C, Hoffman RM. Prostate cancer heterogeneous high-metastatic multi-organ-colonizing chemo-resistant variants selected by serial metastatic passage in nude mice are highly enriched for multinucleate giant cells. PLoS ONE. (2015) 10:e0140721. doi: 10.1371/journal.pone.0140721
205. Weihua Z, Lin Q, Ramoth AJ, Fan D, Fidler IJ. Formation of solid tumors by a single multinucleated cancer cell. Cancer. (2011) 117:4092–9. doi: 10.1002/cncr.26021
206. Was H, Czarnecka J, Kominek A, Barszcz K, Bernas T, Piwocka K. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol Ther. (2018) 19:63–75. doi: 10.1080/15384047.2017.1385675
207. Huang H, Pan XH, Zhou JH, Yu L, Kong XT, Zhou SM, et al. The effect of Epstein–Barr virus gene BHRF1 expression on radioresistance of nasopharyngeal carcinoma cells. ORL J Otorhinolaryngol Relat Spec. (1998) 60:329–33. doi: 10.1159/000027619
208. Zhang Z, Yu X, Zhou Z, Li B, Peng J, Wu X, et al. LMP1-positive extracellular vesicles promote radioresistance in nasopharyngeal carcinoma cells through P38 MAPK signaling. Cancer Medicine. (2019) 8:6082–94. doi: 10.1002/cam4.2506
209. Padilla LA, Leung BS, Carson LF. Evidence of an association between human papillomavirus and impaired chemotherapy-induced apoptosis in cervical cancer cells. Gynecol Oncol. (2002) 85:59–66. doi: 10.1006/gyno.2002.6604
210. Xi R, Pan S, Chen X, Hui B, Zhang L, Fu S, et al. HPV16 E6-E7 induces cancer stem-like cells phenotypes in esophageal squamous cell carcinoma through the activation of PI3K/Akt signaling pathway in vitro and in vivo. Oncotarget. (2016) 7:57050–65. doi: 10.18632/oncotarget.10959
211. Leung TH-Y, Tang HW-M, Siu MK-Y, Chan DW, Chan KK-L, Cheung AN-Y, et al. CD71+ population enriched by HPV-E6 protein promotes cancer aggressiveness and radioresistance in cervical cancer cells. Mol Cancer Res. (2019) 17:1867–80. doi: 10.1158/1541-7786.MCR-19-0068
212. Leung TH-Y, Chan KK-L, Ngan HY-S. HPV-E6 enriches CD71(+) population which promote radio-resistance in cervical cancer cells. Cancer Sci Ther. (2015) 17:1867–80.
213. Vishnoi K, Mahata S, Tyagi A, Pandey A, Verma G, Jadli M, et al. Human papillomavirus oncoproteins differentially modulate epithelial-mesenchymal transition in 5-FU-resistant cervical cancer cells. Tumour Biol. (2016) 37:13137–54. doi: 10.1007/s13277-016-5143-6
214. Wang C, Wang M, Cheng P, Huang H, Dong W, Zhang W, et al. Hepatitis B virus X protein promotes the stem-like properties of OV6+ cancer cells in hepatocellular carcinoma. Cell Death Dis. (2018) 8:e2560. doi: 10.1038/cddis.2016.493
215. Muñoz-Fontela C, Marcos-Villar L, Hernandez F, Gallego P, Rodriguez E, Arroyo J, et al. Induction of paclitaxel resistance by the Kaposi's sarcoma-associated herpesvirus latent protein LANA2. J Virol. (2008) 82:1518–25. doi: 10.1128/JVI.01704-07
216. Liu Q, Wang M, Hu Y, Xing H, Chen X, Zhang Y, et al. Significance of CD71 expression by flow cytometry in diagnosis of acute leukemia. Leuk Lymphoma. (2014) 55:892–8. doi: 10.3109/10428194.2013.819100
217. Kang MK, Hur BI, Ko MH, Kim CH, Cha SH, Kang SK. Potential identity of multi-potential cancer stem-like subpopulation after radiation of cultured brain glioma. BMC Neurosci. (2008) 9:15. doi: 10.1186/1471-2202-9-15
218. Wei-Man Lun S, Cheung S-T, Lo K-W. Cancer stem-like cells in Epstein–Barr virus-associated nasopharyngeal carcinoma. Chin J Cancer. (2014) 33:529–38. doi: 10.5732/cjc.014.10081
219. Lin K-C, Torga G, Sturm J, Pienta K, Austin R. the emergence of polyploid giant cancer cells as the reservoir of therapeutic resistance. APS. (2018) 2018:V46.003.
220. Fei F, Zhang M, Li B, Zhao L, Wang H, Liu L, et al. Formation of polyploid giant cancer cells involves in the prognostic value of neoadjuvant chemoradiation in locally advanced rectal cancer. J Oncol. (2019) 2019:e2316436. doi: 10.1155/2019/2316436
221. Fei F, Zhang D, Yang Z, Wang S, Wang X, Wu Z, et al. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J Exp Clin Cancer Res. (2015) 34:158. doi: 10.1186/s13046-015-0277-8
222. Zhang L, Ding P, Lv H, Zhang D, Liu G, Yang Z, et al. Number of polyploid giant cancer cells and expression of EZH2 are associated with VM formation and tumor grade in human ovarian tumor. Biomed Res Int. (2014) 2014:e903542. doi: 10.1155/2014/903542
223. Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, Klymenko T, et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp Cell Res. (2010) 316:2099–112. doi: 10.1016/j.yexcr.2010.04.030
224. Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells. (2012) 30:833–44. doi: 10.1002/stem.1058
225. Niu N, Mercado-Uribe I, Liu J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene. (2017) 36:4887–900. doi: 10.1038/onc.2017.72
226. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
227. Mirzayans R, Andrais B, Murray D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers. (2018) 10:118. doi: 10.3390/cancers10040118
Keywords: PGCC, oncoviruses, tumor heterogeneity, chemotherapy resistance, radiotherapy resistance
Citation: Herbein G and Nehme Z (2020) Polyploid Giant Cancer Cells, a Hallmark of Oncoviruses and a New Therapeutic Challenge. Front. Oncol. 10:567116. doi: 10.3389/fonc.2020.567116
Received: 29 May 2020; Accepted: 11 September 2020;
Published: 14 October 2020.
Edited by:
Shiwu Zhang, Nankai University, ChinaCopyright © 2020 Herbein and Nehme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georges Herbein, Z2Vvcmdlcy5oZXJiZWluQHVuaXYtZmNvbXRlLmZy