 Wang Liu
Wang Liu Jean C. Li1
Jean C. Li1 Benyi Li
Benyi Li- 1Department of Urology, The University of Kansas Medical Center, Kansas City, KS, United States
- 2Institute of Molecular Pathology, The Affiliated Hospital, Guangdong Medical University, Zhanjiang, China
- 3Sungen Bioscience Institute, Shantou, China
Alternol and its oxidate isomer Alteronol are small compounds isolated from the fermentation of a mutant fungus obtained from Taxus brevifolia bark. Preclinical studies showed their potent anti-cancer activities, including attenuating cellular survival pathways, altering protein levels of cell cycle regulators, activating xanthine dehydrogenase to cause accumulation of cellular reactive oxygen species and disrupting cell metabolism by disturbing four Krebs cycle enzymes specifically in malignant cells while having no significant effect on benign cells. In cancer cell culture models, Alternol or Alteronol exert their anti-cancer effect by inducing cell cycle arrest and triggering apoptotic cell death. In mice xenograft models, Alternol or Alteronol potently suppresses tumor growth with no obvious toxicity to the host with a wide therapeutic index over 30-fold. In conclusion, Alternol or Alteronol possess a great potential and feasibility to be developed as an effective anti-tumor therapeutic.
Introduction
Alternol (Formula C20H16O6, MW 352.3) and Alteronol (Formula C20H14O6, MW 350.3) were purified from the fermented extracts of a mutant fungus Alternaria alternate var.monosporus (1). The wild-type of this fungus was isolated from the bark of Taxus brevifolia, the same source for paclitaxel purification (2) and was then mutagenized via UV irradiation to produce high quantity of paclitaxel (3, 4). A dimeric binaphthyl chemical (named as Alterfungin) was highly yielded from the mutant fungal extract (5). Chemical structure analysis revealed that Alterfungin is a chiral isomer of Cladosporol (6), a secondary metabolite originally isolated from a hyperparasite of rust fungi Cladosporium tenuissimum (Figure 1). Interestingly, Cladosporol was also purified late on from a fermentation broth of the paclitaxel-producing fungus Alternaria alternate var.monosporus (7) and exhibited a moderate anti-cancer effect on multiple human cancer cell lines in vitro and in vivo (7–11). Later on, Alterfungin was re-named as Alternol (12) and its oxidate derivative was named as Alteronol (13). As the isomers, Alternol and Alteronol share the same physical and chemical properties with only one difference at the position 4 due to oxidation; the hydroxyl group on Alternol is replaced by a carbonyl group in Alteronol (Figure 1). In the past 13 years, Alternol and Alteronol have been tested in a variety of cancer cell lines in vitro and animal xenograft models in vivo for their anti-cancer potential (Table 1). Accumulating data demonstrated that these compounds selectivity inhibit cancer cell proliferation in vitro and suppress tumor growth in vivo without obvious toxicity to benign cells or host animals. This review summarizes the research findings of their anti-cancer effects and underlying mechanisms.
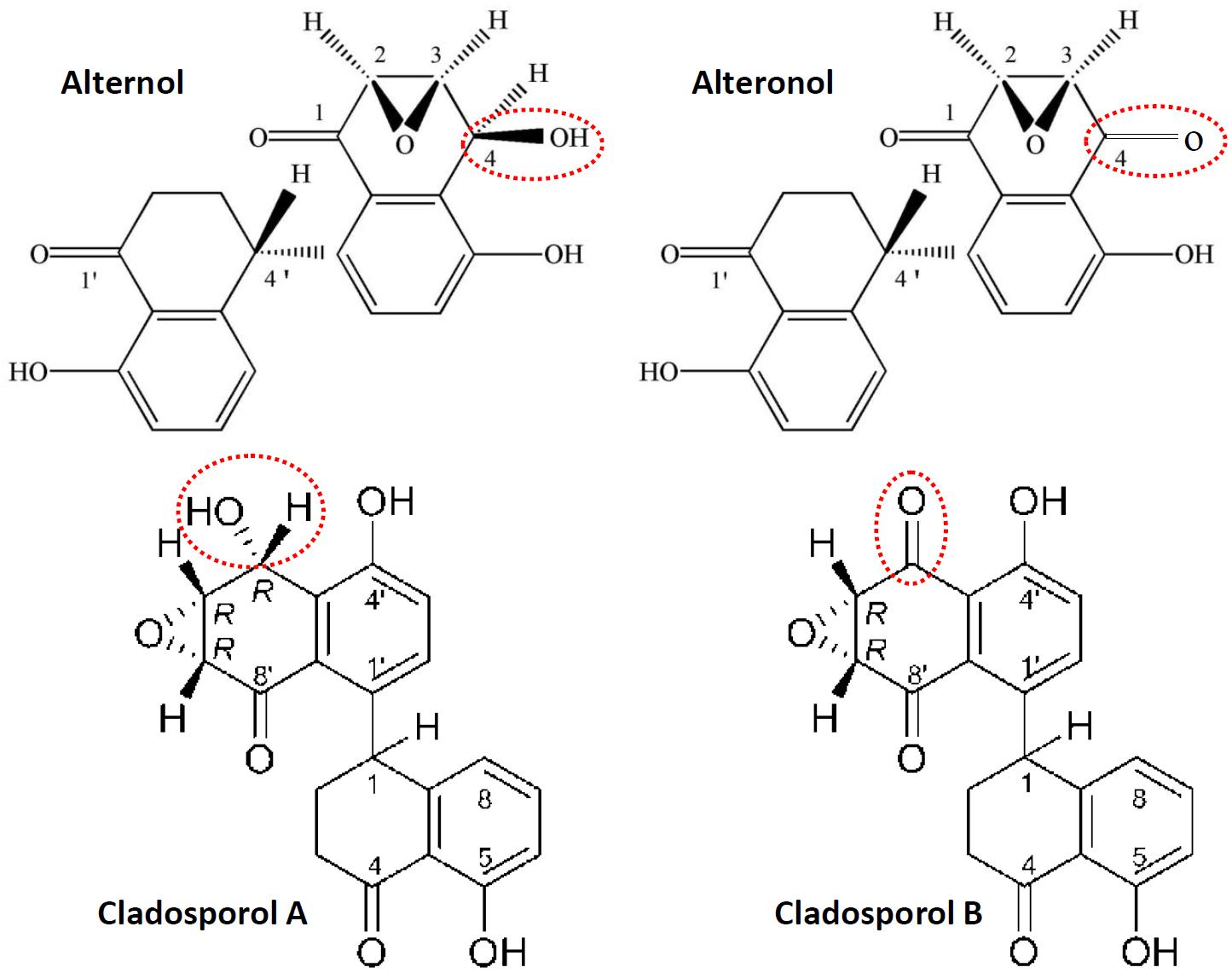
Figure 1. Chemical structures for Alternol, Alteronol and Cladosporols. The differences between these two pairs of isomer compounds were highlighted in red-dotted circle, indicating the oxidate site in Alteronol and Cladosporol B.
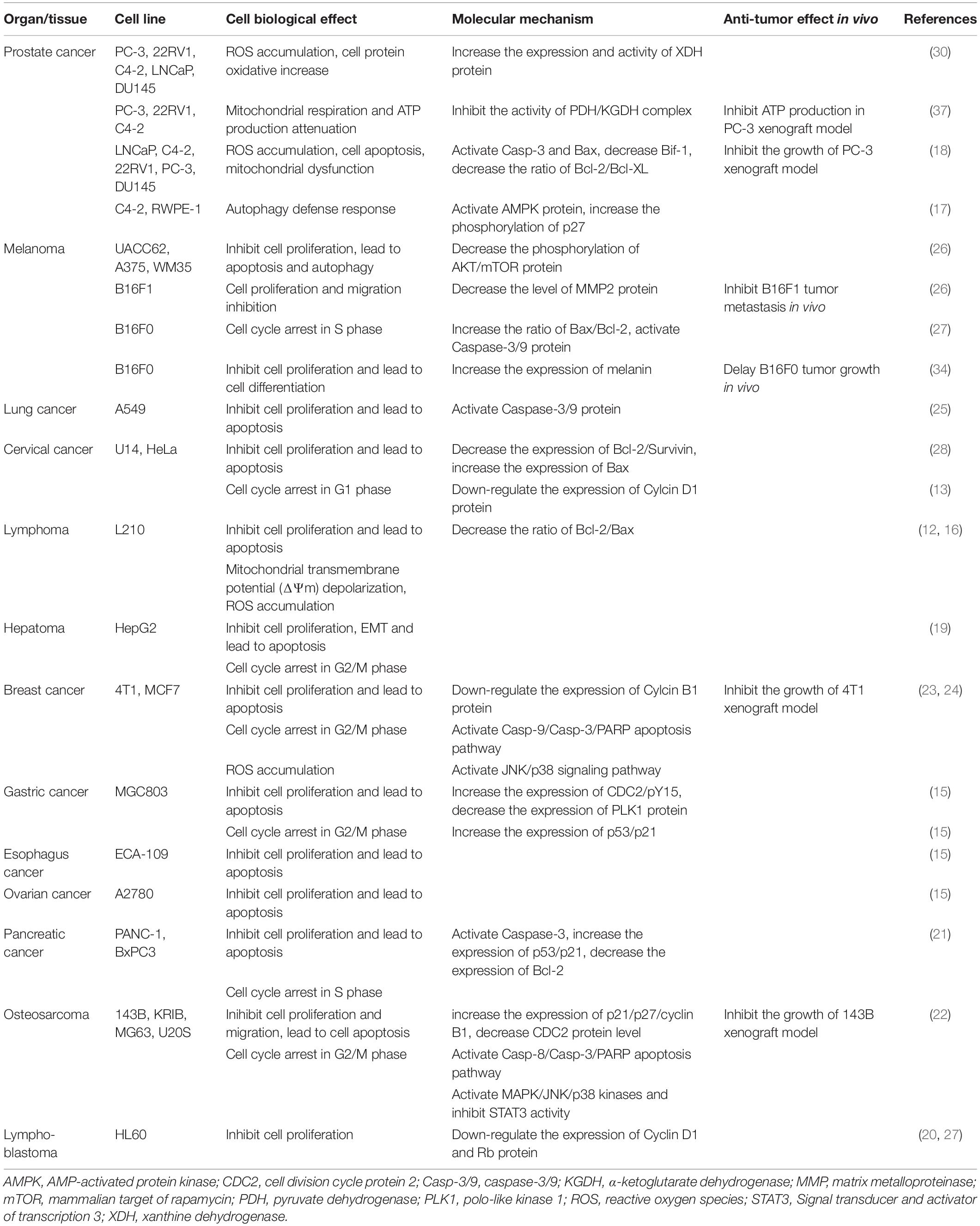
Table 1. Summary of current studies for Alternol/Alteronol effect in vitro and in vivo.
Inhibition of Cancer Cell Proliferation
Malignant tumor growth is always accompanied with rapid cell proliferation, inactivation of cell cycle checkpoints and aberrant expression of cyclin proteins (14). To investigate its anti-cancer effect, Alternol was first applied to human gastric cancer cell MGC-803 and murine leukemia L1210 cells in vitro, and the results showed a prominent inhibition of cell proliferation (15, 16). These anti-cancer effects by Alternol and Alteronol were extended to a variety of human cancer types, including prostate (17, 18), liver (19), cervical (13), leukemia (20), pancreatic (21), osteosarcoma (22), breast (23, 24), lung (25) and melanoma (26). Dose-response experiments determined that the concentration range of 50% growth inhibition (GI50) was between 2 and 10 μM at 24–48 h treatment (12, 13, 15–24, 26, 27). However, lung cancer A549 cells were not sensitive to Alternol (GI50 at 37 μM) (25) compared to other cell lines and prostate cancer DU145 cells are resistant to Alternol due to lack of Bax protein expression (18).
Flow cytometry analysis revealed that Alternol induced G2/M cell cycle arrest, which was related to a significant reduction of polo-like kinase 1 (PLK1) protein, a major regulator of G2/M transition, in parallel with reduced CDC25C and elevated Wee1 protein levels (15). In murine melanoma B16F0 and B16F10 cells, Alternol induced S phase cell cycle arrest, but had a lesser effect on human embryonic kidney 293T cells (27). Similar S phase arrest was also reported in human pancreatic cancer PANC-1 and BxPC3 cells (21). Further analysis determined that Alternol treatment increased CDK inhibitory protein p21cip1/waf1 expression and reduced the expression of proliferating cell nuclear antigen (PCNA) and cyclin-dependent kinase 2 (CDK2) proteins, leading to S phase cell cycle arrest (27). In human cervical cancer HeLa cells, Alteronol inhibited cell proliferation by causing G1 phase cell cycle arrest, which was associated with reduced expression of CDK2, CDK4, cyclin D1 and an increased p21cip1/waf1 expression (13). Interestingly, Cladosporol also caused a similar effect on G1 phase arrest and increased p21cip1/waf1 gene expression in multiple human colon cancer cells upon activating PPARγ-related pathway (8–10). However, in human breast T47D and 4T1 cells, Alteronol inhibited cell proliferation via G2 phase arrest, possibly due to increased p21cip1/waf1 expression and decreased expression of CDC2 and cyclin B1 (23, 24). Also, a significant synergistic effect of Alteronol plus Adriamycin was observed in murine breast cancer 4T1 cell (24). These studies indicate that Alternol and Alteronol inhibit cancer cell proliferation by inducing cell cycle arrest via cell-specific mechanisms (Figure 2 and Table 1).
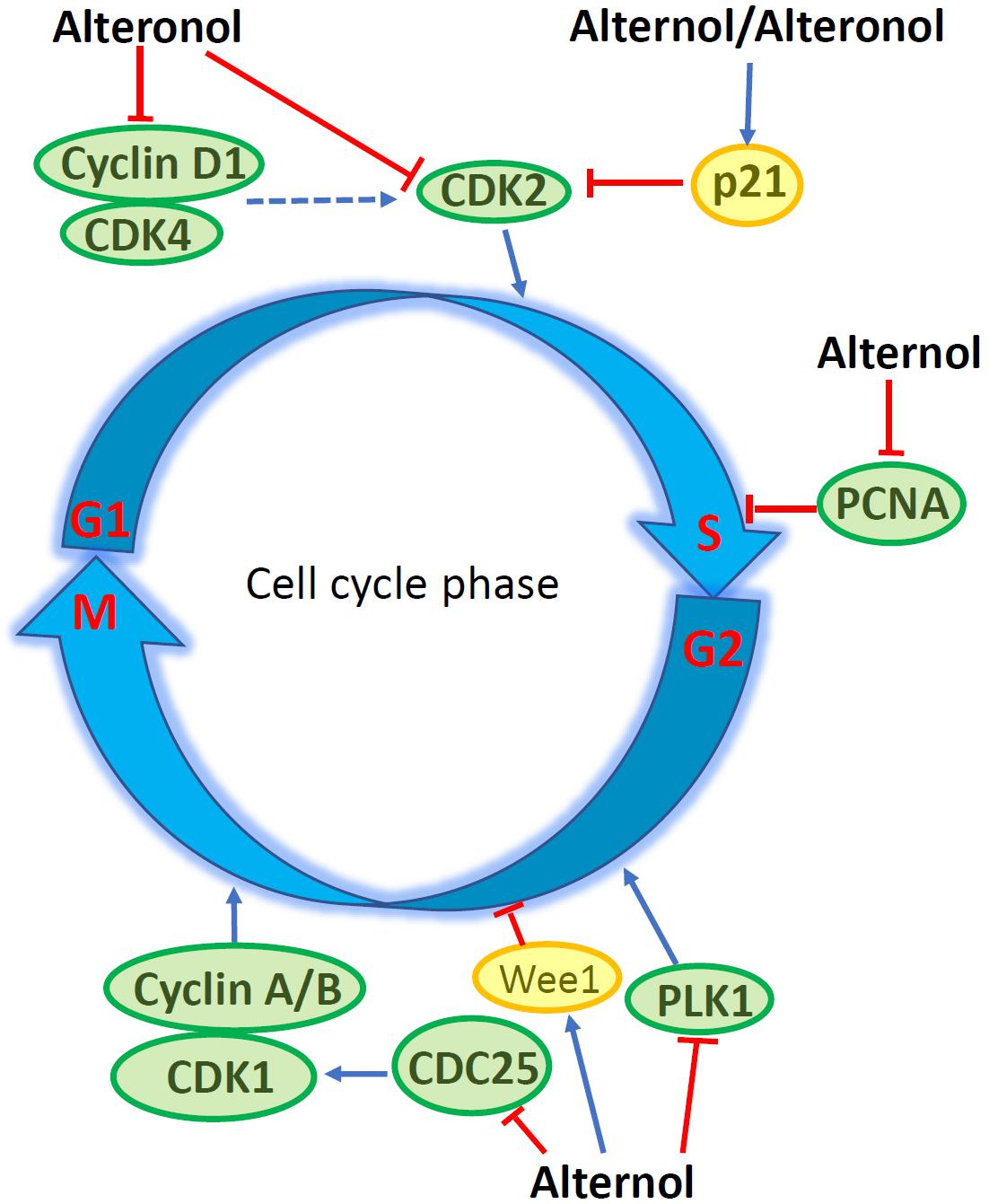
Figure 2. Alternol/Alteronol induce cell cycle arrest in cancer cells by attenuating the expression of cell cycle regulators, including down-regulation of CDK2/4, Cyclin D1, PCNA, PLK1 and CDC25 expression and up-regulation of Wee1 and p21cip1/waf1 expression.
Induction of Apoptotic Cell Death
Apoptosis is one of many forms of programmed cell death and dysregulation of apoptosis is one of the basic characteristics in malignant tumors (14). Alternol-induced apoptosis was first reported in L1210 and MGC803 cells, accompanied with cellular reactive oxygen species (ROS) accumulation and a reduction of Bcl-2/Bax ratio (12, 15, 16). This apoptotic effect was later confirmed in many other cancer cell types (18, 21–24), except prostate cancer DU145 and leukemia HL60 cells, which showed apoptotic resistance (18, 20). Alternol- or Alteronol-induced apoptosis is mechanistically caused by the disturbance of pro- and anti-apoptotic Bcl-2 family proteins and the damage of mitochondrial membrane potential, leading to cytochrome c release and caspase activation (18, 21, 23, 24). In addition, Alternol reduced survivin expression in parallel to Bcl-2 reduction in murine cervical cancer U14 cells (28), and Alteronol increased p53 expression in breast cancer cells (23). Most importantly, Alternol selectively induced apoptosis in prostate cancer LNCaP, C4-2, PC-3 and 22RV1 cells in a time- and dose-dependent manner, without a significant effect on benign prostatic RWPE-1 and BPH1 cells (18). In depth analysis revealed that ROS-dependent Bax protein activation is a major mechanism in Alternol-induced apoptosis in prostate cancer cells (18). These data indicate that Alternol or Alteronol triggers an intrinsic apoptotic pathway to induce cancer cell death but sparing benign cells (Figure 3 and Table 1).
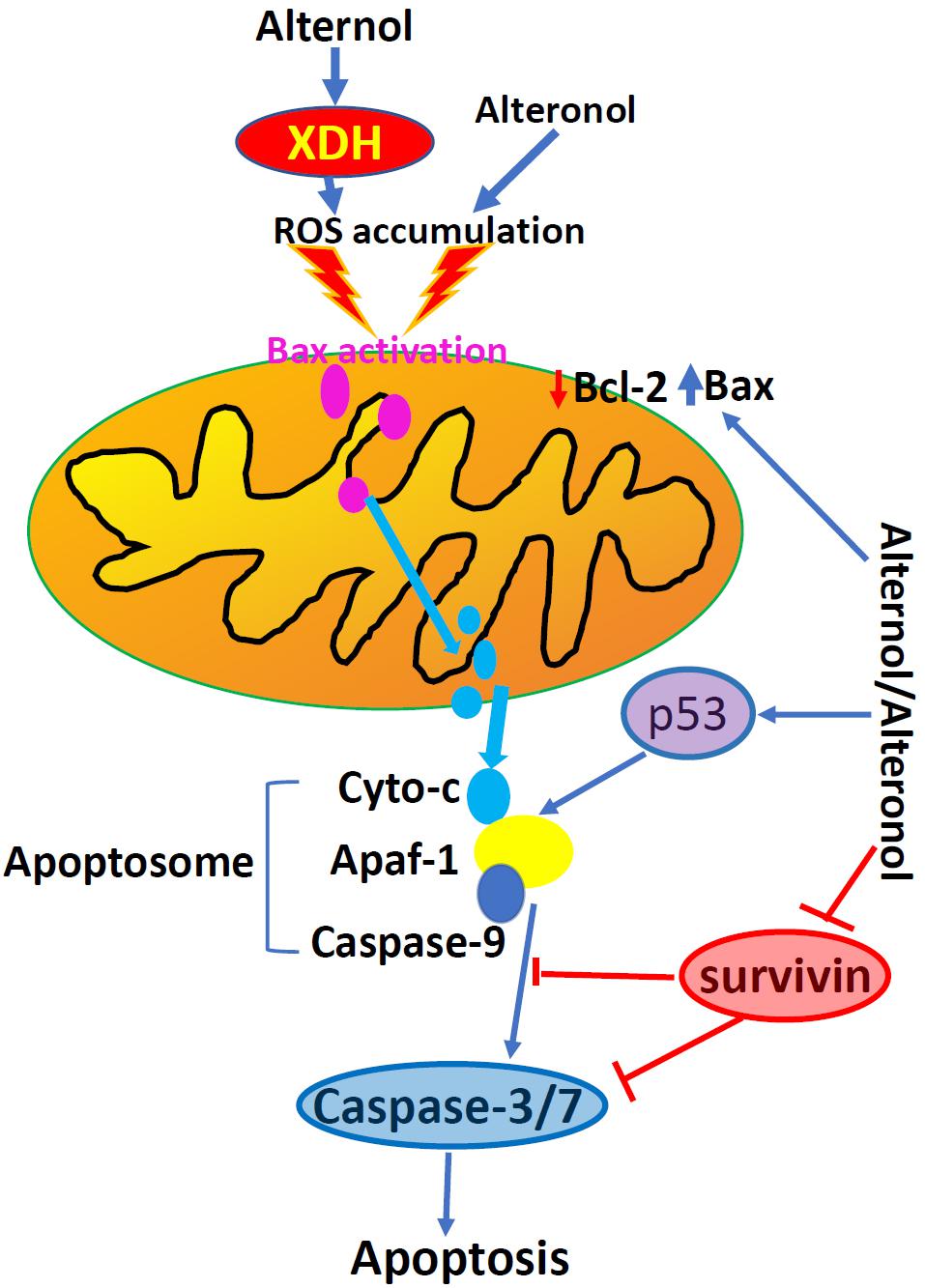
Figure 3. Alternol and Alteronol induce ROS accumulation by activating cytosolic XDH, resulting in Bax activation, cytochrome c release and apoptosis. Alternol/Alteronol also reduces the expression of anti-apoptotic Bcl-2 and Survivin proteins but enhances the expression of pro-apoptotic p53 protein, leading to intrinsic apoptotic cell death. Pink dots denote activated Bax proteins and light-blue dots denote cytochrome c proteins.
Owing to rapid cell growth, cancer cells generate excessive ROS compared to benign cells. This has been used as a therapeutic hit for anti-cancer drug development (29). Although Alternol-induced ROS accumulation was first reported in L1210 cells (12), and the causative role of ROS accumulation in Alternol-induced apoptosis was only demonstrated later in prostate cancer cells (18). Alternol-induced ROS accumulation peaked at 4 h after drug treatment, and Alternol-induced apoptosis was abrogated by ROS scavengers N-acetylcysteine (N-Ac) and dihydrolipoic acid, which was supported by studies from different groups (22, 23). A similar effect was also reported using Alteronol and Cladosporol in breast cancer cells (11, 23, 24).
The major source of cellular ROS accumulation induced by Alternol treatment was recently defined as the cytoplasmic xanthine dehydrogenase (XDH, also called xanthine oxidase, XO) using pharmacological inhibitors and genetic approaches (30). In prostate cancer cells, Alternol treatment only moderately increased mitochondrial superoxide formation that was significantly lower than the total cellular ROS level, indicating that the mitochondrial ROS source was not the major contributor. Also, total cellular ROS level or cell death after Alternol treatment was not reduced by pre-treatment with mitochondria-specific antioxidant MitoQ, inhibitors for NADPH oxidase (NOX) or nitric oxide synthase (NOS). Conversely, XDH-specific small chemical inhibitors or gene silencing reduced total cellular ROS levels and protected cells from apoptosis induced by Alternol. Further analysis revealed that Alternol treatment significantly enhanced XDH oxidative activity and induced a profound cellular protein oxidation in malignant but not in benign cells. Meanwhile, the study also discovered that benign cells had a dramatic increase of antioxidant superoxide dismutase (SOD) and catalase activities compared to malignant cells after Alternol treatment, indicating a potential mechanism for Alternol’s low toxicity to benign cells. In addition, an in-silicon docking analysis suggested that Alternol interacts with the XDH protein at two amino acid residues K755 and R787 within its catalytic molybdenum binding domain (30). Therefore, Alternol is considered as an XDH agonist, leading to excessive ROS generation, cellular stress and apoptosis.
Although the direct consequence from Alternol-induced ROS accumulation was apoptotic cell death in cancer cells (18, 22, 23), multiple cellular signal kinases including MAPK, JNK and p38 were activated via a ROS-dependent mechanism (22). In addition, STAT3 activity was inhibited in Alternol-treatment osteosarcoma cells independent of ROS accumulation (22). The significance of these alterations requires further investigation.
Attenuation of Cellular Autophagy Flux
Autophagy flux is an essential cellular machinery that regenerates nutrients by digesting damaged cellular proteins or organelles under energy stress condition (31). Cellular energy sensing AMPK and growth promoting AKT/mTOR pathways are the major modulators of autophagy flux (32). Early studies showed that Alternol treatment at 0.5 μM concentration induced a significant elevation of autophagy flux in benign prostate RWPE-1 but not in malignant prostate C4-2 cells (17). Autophagy response was evidenced by increased biosynthesis and processing of the LC3B protein, a key player in autophagy flux. Autophagy activation was associated with less cell death accompanied with increased AMPK activation in RWPE-1 cells. Consistently, inhibition of AMPK activity in RWPE-1 cells enhanced Alternol-induced cell death. These data indicate a pro-survival role of autophagy flux in benign cell after Alternol treatment.
On the other hand, a recent report showed that Alteronol at 1–2 μM concentrations induced autophagy response in malignant melanoma A375 and UACC62 cells, as evidenced by LC3B processing and cellular re-distribution, SQSTM1/p62 protein degradation and autophagic vacuole formation (26). Alternol-induced autophagy response was associated with reduced AKT/mTOR activation after Alteronol treatment. Autophagy inhibition with 3-MA or autophagy disruption by Bif-1 knockout enhanced Alteronol-induced cell death in A375 and UACC62 cells. In addition, Cladosporol was also reported to induce autophagy response via a ROS-dependent mechanism in breast cancer cells (11). These studies suggest that Alternol or Alteronol are capable of autophagy induction, leading to a protective effect on cell death.
Inhibition of Cancer Cell Motility
Metastasis is the sole cause of cancer-related casualty and the metastatic potential is mainly defined by cancer cell motility (14). Matrix metalloproteinases (MMPs) are enzymes that degrade extracellular matrix and basement membrane, key factors in cancer metastasis (33). Alternol was shown to inhibit HepG2 cell migration and invasion, which was associated with reduced MMP-9 expression and a reversal of epithelial-to-mesenchymal transition (EMT) (19). Similarly, Alteronol was shown to inhibit cell invasion/migration in vitro and lung metastasis in vivo in murine melanoma B16F10 and B16F1 cells through a mechanism related to MMP2 reduction plus tissue inhibitor of metalloproteinases-2 (TIMP-2) induction (34). Consistently, in human melanoma A375 and UACC62 cells, Alteronol was also found to reduce cell invasion/migration in vitro, possibly through a TGFβ/Smad3 signal pathway-related epithelial-mesenchymal transition (EMT) mechanism (26). Not surprisingly, Cladosporol was found to reduce β-catenin protein level via a PPARγ-dependent proteasome degradation and to enhance E-cadherin gene expression, two strong regulators of cancer metastasis (9).
Alteration of Cancer Cell Energy Metabolism
Metabolic reprogramming is a malignant hallmark and targeting metabolic pathway has been a hotspot in anti-cancer drug development (35, 36). To identify the protein targets, a recent report used a biotin labeled Alternol coupled with avidin beads to pull down cellular proteins that bond with Alternol (37). The eluted proteins were processed using Mass-Spectrometry approach and fourteen identified proteins were verified in western blot assays. Among them are four enzymes involved in the Krebs cycle, including the E2 component dihydrolipoyllysine-residue acetyltransferase (DLAT) of pyruvate dehydrogenase complex (PDHC), the E2 component dihydrolipoyllysine-residue succinyltransferase (DLST) of α-ketoglutarate dehydrogenase complex (KGDHC), fumarate hydratase (FH) and malate dehydrogenase-2 (MDH2). In prostate cancer cells, PDHC or KGDHC activities at the basal condition were significantly higher than that in benign prostate BPH1 cells, while Alternol treatment reduced PDHC and KGDHC activities in cancer cells to the levels close to that in BPH1 cells. Although FH and MDH2 activities were comparable among prostate cancer and benign cell lines at the basal condition, interestingly, Alternol enhanced their activities in prostate cancer cells but not in BPH1 cells. Further analysis using metabolomic approaches revealed that Krebs cycle intermediates, including citric acid, succinic acid, fumaric acid and malic acid, were much higher in malignant cells compared to benign cells under basal condition. Alternol treatment remarkably reduced the levels of malic acid, fumaric acid, and isocitric acid and mitochondrial respiration in prostate cancer cells. Consequently, mitochondrial respiration and ATP production were drastically reduced after Alternol treatment in prostate cancer PC-3 cells in vitro or in PC-3 cell-derived xenograft tissues but not in BPH1 cells or host liver tissues (37). These studies demonstrated that malignant cells posse a higher metabolic activity for energy production and that Alternol specifically interferes with the Krebs cycle enzymes, resulting in reduced ATP production and energy crisis in malignant cells and xenograft tissue.
Conclusion
Alternol and Alteronol are relatively new compounds with potent anti-cancer effects via multiple mechanisms, including cell cycle arrest, cell motility reduction, intrinsic apoptosis, ROS stress, and metabolic disruption (Figure 4 and Table 1). Most interestingly, Alternol was found to interact with four Krebs cycle enzymes, resulting in the disruption of ATP production and energy crisis specifically in cancer cells or xenograft tumors without affecting benign or host tissues. This malignant tissue selectivity provides a huge safety advantage over current clinical chemotherapy that targets all proliferating cells.
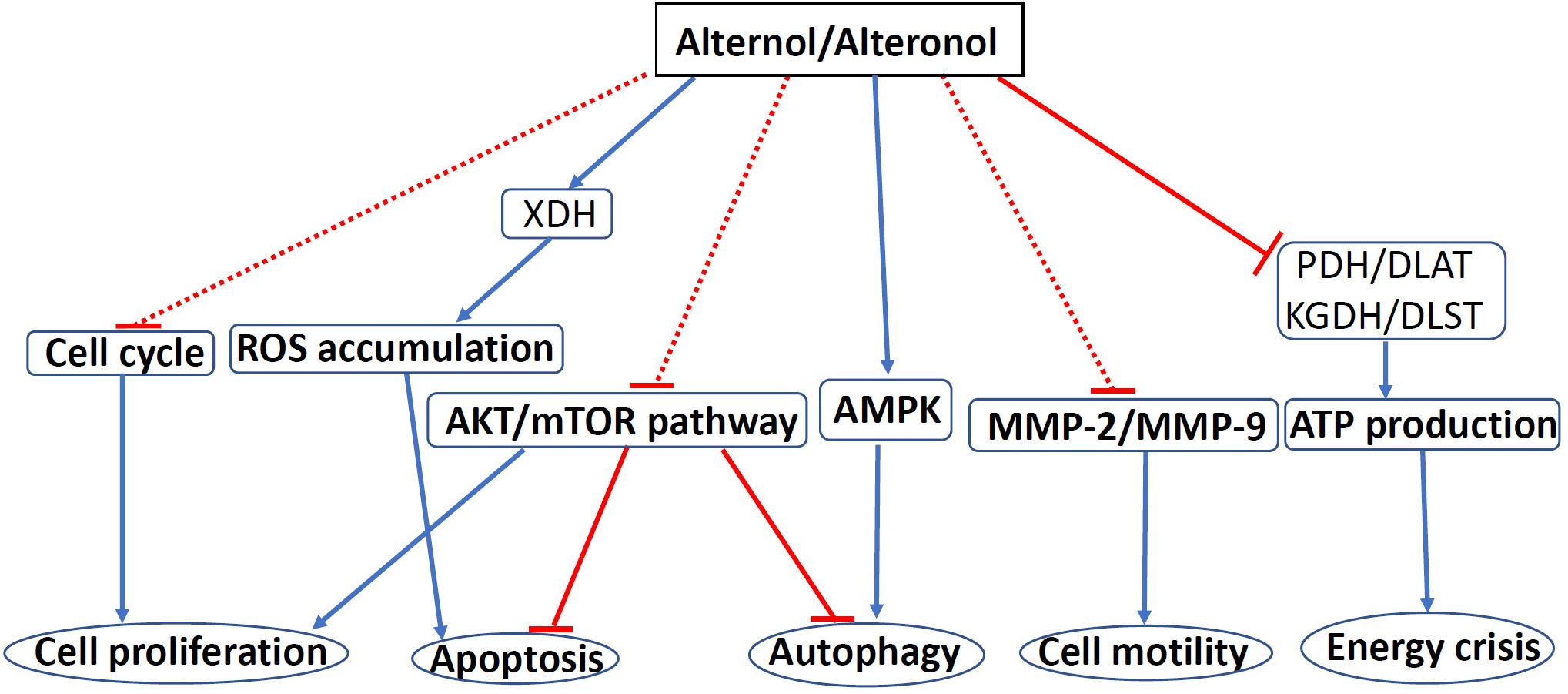
Figure 4. The mechanisms of Alternol/Alteronol-induced anti-cancer effects. Alternol and Alteronol reduce cell proliferation via cell cycle arrest, trigger apoptotic cell death via ROS accumulation and AKT/mTOR inactivation and attenuate cell motility by down-regulating MMP-2/9 expression. Alternol enhances autophagy flux via AMPK activation/AKT-mTOR inactivation and causes energy crisis by inhibiting Krebs’ cycle enzymes PDH/DLAT and KGDH/DLST complexes. Blue solid arrows denote a direct stimulating effect. Red solid lines denote a direct suppressive effect. Red dotted lines denote an indirect suppressive effect. Abbreviations: AMPK, AMP-activated protein kinase; DLAT, dihydrolipoyllysine-residue acetyltransferase; DLST, dihydrolipoyllysine-residue succinyltransferase; KGDH, a-ketoglutarate dehydrogenase; MMP, matrix metalloproteinase; mTOR, mammalian target of rapamycin; PDH, pyruvate dehydrogenase; ROS, reactive oxygen species; XDH, xanthine dehydrogenase.
Author Contributions
WL, JL, and BL wrote the draft. BL, JC, JHu, and JHo revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by KU Valk Foundation (to BL) and a grant from the National Natural Science Foundation of China (#81572610 to JHu). The authors declare that this study received Alternol and Alteronol from SungenBio Inc. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
JC was employed by the company SungenBio Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Duan L, Chen H, Chen J, Hong H, Li W. The fermentation process of the new anticancer compound Alternol. J Shenyang Pharm Univ. (2009) 26: 316–23.
2. Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. (1971) 93:2325–7. doi: 10.1021/ja00738a045
3. Cheng S, Wang R, Chen J, Duan L, Qiu X. Isolation, purification and identification of paclitaxel from the fermentated extra of fungus ST026 (Alternaria alternate var.monosporus). J Chin Pharm Univ. (2011) 42:570–2.
4. Duan L, Chen HR, Chen JP, Li WP, Hong L. Screening for the high-yield paclitaxel producing strain Alternaria alternate var.monosporus. Chinese J Antibiotics. (2008) 34:650–2.
5. Chen J, Duan LL, Chen HR, Lin H, Li WP, Luo JG, et al. Studies on the antifungal activities of Alterfungin and its derivatives. Chin J Antibiotics. (2009) 34:15–7.
6. Nasini G, Arnone A, Assante G, Bava A, Moricca S, Ragazzi A. Secondary mould metabolites of Cladosporium tenuissimum, a hyperparasite of rust fungi. Phytochemistry. (2004) 65:2107–11. doi: 10.1016/j.phytochem.2004.03.013
7. Chen J, Qiu X, Wang R, Duan L, Chen S, Luo L, et al. Inhibition of human gastric carcinoma cell growth in vitro and in vivo by cladosporol isolated from the paclitaxel-producing strain Alternaria alternata var. monosporus. Biol Pharm Bull. (2009) 32:2072–4. doi: 10.1248/bpb.32.2072
8. Zurlo D, Leone C, Assante G, Salzano S, Renzone G, Scaloni A, et al. Cladosporol a stimulates G1-phase arrest of the cell cycle by up-regulation of p21(waf1/cip1) expression in human colon carcinoma HT-29 cells. Mol Carcinog. (2013) 52:1–17. doi: 10.1002/mc.20872
9. Zurlo D, Assante G, Moricca S, Colantuoni V, Lupo A. Cladosporol A, a new peroxisome proliferator-activated receptor gamma (PPARgamma) ligand, inhibits colorectal cancer cells proliferation through beta-catenin/TCF pathway inactivation. Biochim Biophys Acta. (2014) 1840:2361–72. doi: 10.1016/j.bbagen.2014.04.007
10. Zurlo D, Ziccardi P, Votino C, Colangelo T, Cerchia C, Dal Piaz F, et al. The antiproliferative and proapoptotic effects of cladosporols A and B are related to their different binding mode as PPARgamma ligands. Biochem Pharmacol. (2016) 108:22–35. doi: 10.1016/j.bcp.2016.03.007
11. Koul M, Kumar A, Deshidi R, Sharma V, Singh RD, Singh J, et al. Cladosporol A triggers apoptosis sensitivity by ROS-mediated autophagic flux in human breast cancer cells. BMC Cell Biol. (2017) 18:26. doi: 10.1186/s12860-017-0141-0
12. Liu ZZ, Chen JP, Zhao SL, Li CL. [Apoptosis-inducing effect of alternol on mouse lymphocyte leukemia cells and its mechanism]. Yao Xue Xue Bao. (2007) 42:1259–65.
13. Yao Y, Zhang B, Chen N, Liu L, Yishan W, Li C, et al. Alteronol inhibits proliferation in HeLa cells through inducing a G1-phase arrest. J Pharm Pharmacol. (2012) 64:101–7. doi: 10.1111/j.2042-7158.2011.01375.x
15. Liu X, Wang J, Sun B, Zhang Y, Zhu J, Li C. Cell growth inhibition, G2M cell cycle arrest, and apoptosis induced by the novel compound Alternol in human gastric carcinoma cell line MGC803. Invest New Drugs. (2007) 25:505–17. doi: 10.1007/s10637-007-9057-4
16. Liu ZZ, Zhu J, Sun B, Liu S, Geng S, Liu X, et al. Alternol inhibits proliferation and induces apoptosis in mouse lymphocyte leukemia (L1210) cells. Mol Cell Biochem. (2007) 306:115–22. doi: 10.1007/s11010-007-9560-0
17. Yeung ED, Morrison A, Plumeri D, Wang J, Tong C, Yan X, et al. Alternol exerts prostate-selective antitumor effects through modulations of the AMPK signaling pathway. Prostate. (2012) 72:165–72. doi: 10.1002/pros.21417
18. Tang Y, Chen R, Huang Y, Li G, Huang Y, Chen J, et al. Natural compound Alternol induces oxidative stress-dependent apoptotic cell death preferentially in prostate cancer cells. Mol Cancer Ther. (2014) 13:1526–36. doi: 10.1158/1535-7163.MCT-13-0981
19. Zhu XL, Wang YL, Chen JP, Duan LL, Cong PF, Qu YC, et al. Alternol inhibits migration and invasion of human hepatocellular carcinoma cells by targeting epithelial-to-mesenchymal transition. Tumour Biol. (2014) 35:1627–35. doi: 10.1007/s13277-013-1224-y
20. Liu LL, Chen N, Yuan X, Yao Y, Zhang B, Zheng QS, et al. [The mechanism of alteronol inhibiting the proliferation of human promyelocytic leukemia HL-60 cells]. Yao Xue Xue Bao. (2012) 47:1477–82.
21. Cong PF, Qu Y-C, Chen J-P, Duan L-L, Lin C-J, Zhu X-L, et al. Growth inhibition and apoptosis induction by alternol in pancreatic carcinoma cells. World J Gastroenterol. (2015) 21:4526–35. doi: 10.3748/wjg.v21.i15.4526
22. Zuo D, Zhou Z, Wang H, Zhang T, Zang J, Yin F, et al. Alternol, a natural compound, exerts an anti-tumour effect on osteosarcoma by modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol Med. (2017) 21:208–21. doi: 10.1111/jcmm.12957
23. Ren B, Li D, Si L, Ding Y, Han J, Chen X, et al. Alteronol induces cell cycle arrest and apoptosis via increased reactive oxygen species production in human breast cancer T47D cells. J Pharm Pharmacol. (2018) 70:516–24. doi: 10.1111/jphp.12879
24. Ren B, Ye L, Gong J, Ren H, Ding Y, Chen X, et al. Alteronol enhances the anti-tumor activity and reduces the toxicity of high-dose adriamycin in breast cancer. Front Pharmacol. (2019) 10:285. doi: 10.3389/fphar.2019.00285
25. Yang F, Ran F, Guo D, Yu B, Zhang B, Zheng Q. Alternol induced apoptosis in human lung cancer A549 cells. J Shihezhi University (Natural Science Edition). (2013) 31:365–70.
26. Bao Y, Ding Z, Zhao P, Li J, Chen P, Zheng J, et al. Autophagy inhibition potentiates the anti-EMT effects of alteronol through TGF-beta/Smad3 signaling in melanoma cells. Cell Death Dis. (2020) 11:223. doi: 10.1038/s41419-020-2419-y
27. Liu L, Zhang B, Yuan X, Wang P, Sun X, Zheng Q. Alternol induces an S-phase arrest of melanoma B16F0 cells. Cell Biol Int. (2014) 38:374–80. doi: 10.1002/cbin.10226
28. Su X, Gong P, Wang D, Liu W, Yang X, Zheng Q. Effect of Anterol on the anticancer of mouse cervical carcinoma U14 cells. Mordern Oncol. (2012) 20:1321–4.
29. Perillo B, Donato M. Di, Pezone A, Zazzo E. Di, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. (2020) 52:192–203. doi: 10.1038/s12276-020-0384-2
30. Xu H, Li C, Mozziconacci O, Zhu R, Xu Y, Tang Y, et al. Xanthine oxidase-mediated oxidative stress promotes cancer cell-specific apoptosis. Free Radic Biol Med. (2019) 139:70–9. doi: 10.1016/j.freeradbiomed.2019.05.019
31. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
32. Xiang H, Zhang J, Lin C, Zhang L, Liu B, Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. (2020) 10:569–81. doi: 10.1016/j.apsb.2019.10.003
33. Agren MS, Auf dem Keller U. Matrix metalloproteinases: how much can they do? Int J Mol Sci. (2020) 21:2678. doi: 10.3390/ijms21082678
34. Wang Z, Wang D, Liu L, Guo D, Yu B, Zhang B, et al. Alteronol inhibits the invasion and metastasis of B16F10 and B16F1 melanoma cells in vitro and in vivo. Life Sci. (2014) 98:31–8. doi: 10.1016/j.lfs.2013.12.213
35. Torresano L, Nuevo-Tapioles C, Santacatterina F, Cuezva JM. Metabolic reprogramming and disease progression in cancer patients. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165721. doi: 10.1016/j.bbadis.2020.165721
36. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
Keywords: Alternol, Alteronol, apoptosis, cell cycle, radical oxygen species, Cladosporol
Citation: Liu W, Li JC, Huang J, Chen J, Holzbeierlein J and Li B (2020) Alternol/Alteronol: Potent Anti-cancer Compounds With Multiple Mechanistic Actions. Front. Oncol. 10:568110. doi: 10.3389/fonc.2020.568110
Received: 31 May 2020; Accepted: 25 August 2020;
Published: 03 November 2020.
Edited by:
Feng-Yao Tang, China Medical University, TaiwanReviewed by:
Meng-Shih Weng, Fu Jen Catholic University, TaiwanJing-Hsien Chen, Chung Shan Medical University, Taiwan
Copyright © 2020 Liu, Li, Huang, Chen, Holzbeierlein and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benyi Li, YmxpQGt1bWMuZWR1