 Shiqin Li
Shiqin Li Bingbing Shi
Bingbing Shi Xinli Liu
Xinli Liu Han-Xiang An
Han-Xiang An- Department of Medical Oncology, Xiang'an Hospital of Xiamen University, Xiamen, China
Hundreds of DNA repair proteins coordinate together to remove the diverse damages for ensuring the genomic integrity and stability. The repair system is an extensive network mainly encompassing cell cycle arrest, chromatin remodeling, various repair pathways, and new DNA fragment synthesis. Acetylation on DNA repair proteins is a dynamic epigenetic modification orchestrated by lysine acetyltransferases (HATs) and lysine deacetylases (HDACs), which dramatically affects the protein functions through multiple mechanisms, such as regulation of DNA binding ability, protein activity, post-translational modification (PTM) crosstalk, and protein–protein interaction. Accumulating evidence has indicated that the aberrant acetylation of DNA repair proteins contributes to the dysfunction of DNA repair ability, the pathogenesis and progress of cancer, as well as the chemosensitivity of cancer cells. In the present scenario, targeting epigenetic therapy is being considered as a promising method at par with the conventional cancer therapeutic strategies. This present article provides an overview of the recent progress in the functions and mechanisms of acetylation on DNA repair proteins involved in five major repair pathways, which warrants the possibility of regulating acetylation on repair proteins as a therapeutic target in cancers.
Introduction
Several mechanistically distinct DNA repair pathways have evolved to restore a myriad of DNA damages induced by exogenous and endogenous stressors, such as radiation, chemical, and biological toxins, as well as reactive oxygen species (ROS) generated during cellular metabolism. Defective DNA repair pathways fail to remove DNA damages, which leads to genomic instability, malignant transformation, and cancer susceptibility. Thus, repressing the activity of DNA repair proteins in cancers might increase sensitivity to other therapeutic regimens (1–3).
Lysine acetylation is one of the most frequently occurring post-translational modifications (PTMs) and has a profound effect in extensive biological processes. Two groups of enzymes, lysine acetyltransferases (HATs) and lysine deacetylases (HDACs), function antagonistically to control the balance between acetylation and deacetylation. Histone protein is the first well-established example of biological functional protein acetylation (4). Acetylation on histones neutralizes the positive charge of the lysine residue, resulting in the de-structure of the chromatin–histone complex. Transcriptional and repair machineries are able to access into the DNA template sites where they modulate the gene expressions or fulfill the damage repair processes (5). Over the past few decades, advances in mass spectrometry allowed the identification of over 2,000 acetylated non-histone proteins participated in widespread cellular processes including DNA repair (6). There is compelling evidence that reversible lysine acetylation is engaged to regulate the functions of repair proteins (6, 7).
Modulating the acetylation of repair proteins has been confirmed as a promising approach for cancer treatment. Alterations in the transcriptional level of various HDACs were observed in numerous cancers, such as colorectal, gastric, esophagus, breast, ovary, lung, pancreas, thyroid, prostate, and oral cancers, as well as malignant melanoma (8). Several HDACIs (belinostat, romidepsin, panobinostat, vorinostat, and chidamide) that obtained the US Food and Drug Administration (FDA) or the China FDA approval have already been applied singly or with other therapeutic drugs in the clinical treatment of various hematologic and few solid tumors. However, the occurrence of high adverse reaction rate and the potential possibility for promoting the pathogenesis or expediting the progression of cancers severely limit the clinical application of HDACI for lacking HDAC subtype specificity (9, 10). Hence, illuminating the acetylation status at specific lysine sites of DNA repair proteins and figuring out the upstream HATs and HDACs as well as downstream substrates are a great help in better understanding the pathogenesis mechanism and developing new targeted therapeutics for clinical treatment of cancers.
HAT and HDAC in Humans
HATs are a group of enzymes that catalyze the transfer of the acetyl moiety from acetyl-coenzyme A to the lysine residues of the side-chain epsilon amino group. Based on sequence similarities, canonical HATs include three families: GNAT family (GCN5 and PCAF), CBP family (CBP and p300), and MYST family (Tip60, MOZ, MORF, HBO1, and MOF) (11, 12). NAT10, ATAT1, ESCO1, ESCO2, and HAT1 are included as non-canonical HATs (12).
Eighteen kinds of HDACs in eukaryocytes promote the counteraction of lysine acetylation. HDACs are categorized into two major groups: Zn2+-dependent HDACs including class I (HDAC1, 2, 3, and 8), II (HDAC4, 5, 7, and 9), IV (HDAC11), and NAD+-dependent sirtuin deacetylases including SIRT 1–SIRT7 (6, 12). Although precise functions and substrates of these HATs/HDACs are not thoroughly characterized, acetylation on repair proteins has been implicated in the modulation of DNA repair ability via different repair pathways in cancer cells.
Acetylation on Repair Protein in Major DNA Repair Pathway
Five major pathways including mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR), and NHEJ are devoted to remedying types of DNA lesions (13). The regulation mechanism of acetylation/deacetylation on DNA repair proteins and acetylation-accompanied functions in tumorigenesis and therapeutic response of cancers are summarized in each repair pathway (Table 1).
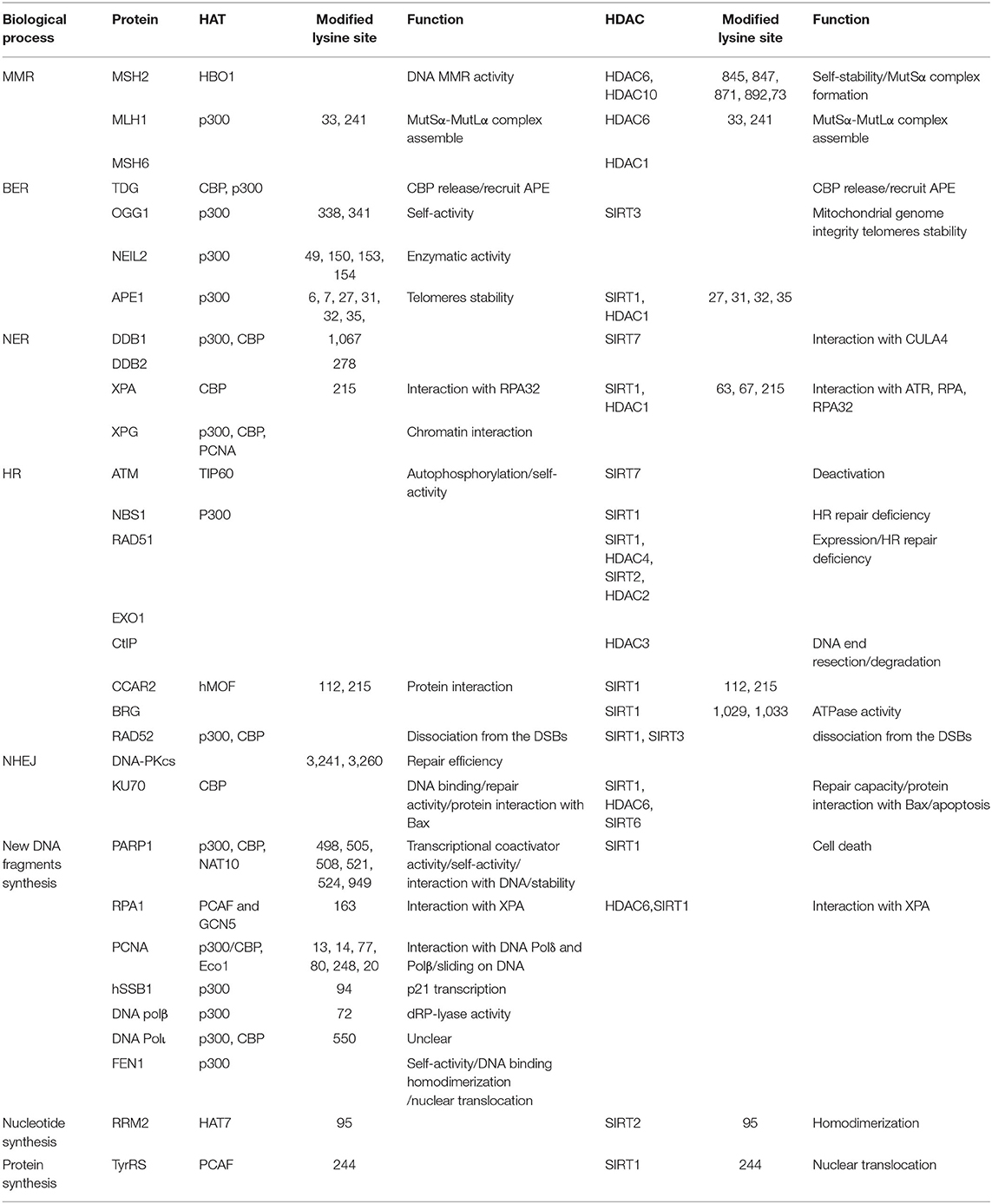
Table 1. HAT and HDAC mediating the acetylation of repair proteins and exact lysine sites.
Mismatch Repair
Base pair mismatch errors that occur during DNA replication are recognized and rectified by the MMR system. Defect of this mutation circumvention system might result in microsatellite instability and increased cancer risks (14). Base–base mispairing and base insertions/deletions are recognized by MutS homolog 2 (MSH2)–MSH6 (MutSα) and MSH2–MSH3 (MutSβ) heterodimeric complexes, respectively (15, 16). Multiple mass spectrum-based studies have identified a diverse number of acetyl-lysine sites on MMR proteins (MSH2, MSH3, MSH6, MLH1, and PMS2) (17, 18). Dysregulation of MSH2 has been pointed out to generate genomic instability, resulting in the development of prostate (19), colorectal (20), and hepatic (21) cancers. A previous study found that the levels of MutSα were controlled through MSH2 acetylation in vivo. HDAC6 directly catalyzed the deacetylation of MSH2 at Lys845/847/871/892, which downregulated the stability of MSH2 by promoting ubiquitination and turnover of MSH2. MutSα complex conformation was disrupted, thereby leading to decreased MMR activity. These experiments might provide an interpretation for HDAC6-induced cellular tolerance to DNA-damaging agents (22) (Figure 1A). Depletion of HDAC6 was able to abolish the drugs resistance in non-small cell lung cancer (NSCLC) cells (23), whereas, another study reported that HDAC10 stimulated MMR activity by deacetylation of MSH2 at Lys73. The counteraction of deacetylation on this residue was possibly catalyzed by HAT bound to ORC1 (HBO1) (24). Further studies are necessary to uncover the role of Lys73 deacetylation on MSH2. In addition, an Oncomine database analysis revealed that HDAC10 is probably used as a favorable predictor of prognosis in patients with colon carcinoma (25), which potentially related to increased MMR activity induced by the deacetylation of MSH2.
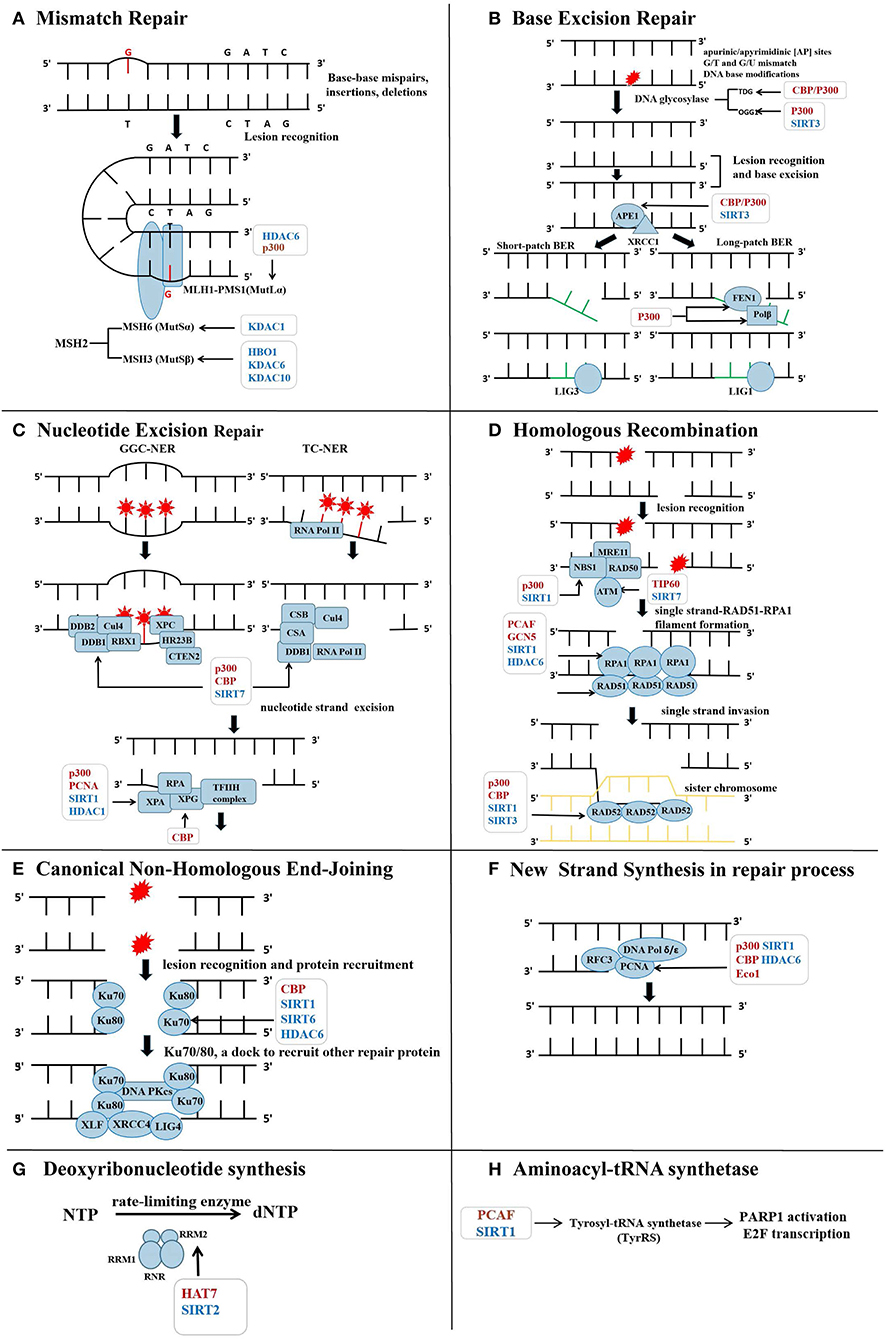
Figure 1. Lysine acetyltransferases (HATs) and lysine deacetylases (HDACs) in the regulation of repair protein across the DNA repair process. (A) Acetylation on MutS homolog 2 (MSH2) and MLH1, respectively, affects their assembly with MSH6 or PMS1 into complex, influencing the lesion recognition in mismatch repair (MMR). (B) During base excision repair (BER), base excision and new short DNA fragment synthesis are modulated by several HATs and HDACs. Collectively, acetylation on the repair proteins within BER is favorable to BER efficiency. (C) Acetylation of DNA-binding protein 1 (DDB1), Xeroderma pigmentosum group A (XPA), and XPG within nucleotide excision repair (NER) is conductive to ensure efficient NER process. Specific HAT and HDAC are highlighted in the figure during the injury recognition and resection process. (D) Acetylation on ataxia-telangiectasia-mutated (ATM) help to transmit the double strand breaks (DSBs) damage signals. p300 is recruited to the DSBs sites and acetylates NBS1 in the MRN complex. Acetylation on Recombinant DNA repair protein 51 (RAD51) further promotes homologous recombination (HR). Acetylation on RAD52 acted as a signal to guide its dissociation from the DSBs. (E) Ku70 acetylation increases DSBs repair activity and protects the cell from apoptosis. Deacetylated DNA PKcs decreases DSBs repair capacity. (F) Acetylation of proliferating cell nuclear antigen (PCNA) promotes its binding to DNA polymerase (DNA Pol) and DNA strands and the formation of new fragments. (G) Acetylation on RRM2 disrupts the homodimerization of itself, leading to decline in ribonucleotide reductase (RNR) activity and reduction in deoxynucleotide triphosphate (dNTP) pool. (H) Nuclear translocation of tyrosyl necessitates acetylation modification to assist against DNA damage through activating the transcription factor E2F1 and poly (ADP-ribose) polymerase-1 (PARP1) as well as subsequent downstream DNA repair genes.
MLH1-PMS2 (MutLα) heterodimeric complex with a latent endonuclease activity is another indispensable molecule in correcting the mismatched bases. A previous study revealed that p300 and HDAC6 were capable of acetylating and deacetylating MLH1, respectively. The HDAC6 deacetylated MLH1 at Lys33 and Lys241 both in vivo and in vitro, leading to the disintegration of MutSα-MutLα complex. The DNA damage sensor role of the MutSα-MutLα complex in DNA damage malfunctioned, which impaired MMR activity and induced DNA damage tolerance (26) (Figure 1A). Additionally, proteomics-based substrate trapping identified MSH6 as a substrate of HDAC1 (27). MSH2–MSH6 heterodimeric complex acted as a bidirectional ATPase with ability to change ADP and ATP ratio in DNA damage repair (28). MSH6 mutation might associate with hereditary non-polyposis colorectal and endometrial cancer (29, 30). However, whether acetylation modification on MSH6 interferes with ATPase activity and MMR efficiency still needs further elucidation.
Base Excision Repair
DNA base lesions, including base losses and bulky modifications, are restored by the BER pathway (31, 32). BER in eukaryotic cell is mainly initiated by 11 kinds of damage-specific DNA glycosylases [uracil DNA glycosylase (UNG), single-strand-selective monofunctional uracil glycosylase 1 (SMUG1), methyl CpG-binding domain protein 4 (MBD4), thymine DNA glycosylase (TDG), 8-OxoG DNA glycosylase (OGG1), MutY glycosylase homolog (MYH), endonuclease III homolog (NTH1), methylpurine DNA glycosylase (MPG), Nei-like DNA Glycosylase 1 (NEIL1), NEIL2, and NEIL1] (33, 34). In addition, AP endonuclease 1 (APE1), ligases (LIG3), X-ray repair cross complementing 3 (XRCC3), and DNA polymerase (DNA Pol) are also required for conducting the following excision–repair process (35). Acetylations on SMUG1, MYH, NTH1, MPG, NEIL1, and NEIL3 were unclear by now, whereas acetylation on UNG (Lys5), MBD4 (Lys232/234/239), and NEIL2 (Lys49/150/153/154) (18, 36) have been identified. To probe into the function of acetylation on these glycosylases may add to an understanding of precisely regulating the BER pathway.
TDG, an enzyme responsible for the removal of thymine moieties from G/T mismatches, generates an abasic site during BER. CBP/p300 was recruited to DNA damage sites by TDG, which potentiated CREB-binding protein (CBP)/p300-dependent transcription and turned into the substrate of CBP/p300. The release of CBP from DNA trinary complexes and the recruitment of APE were both regulated by Ac-TDG, which exhibited a vital role in maintaining genomic stability (37, 38).
Another glycosylase OGG1 is known to remove the 8-oxo-7,8-dihydroguanine generated in DNA oxidative damage. The activity of OGG1 was increased after acetylated by p300 at Lys338 and Lys341, which enhanced the rate of BER via reducing affinity for the abasic site under the oxidative stress. Class I HDACs probably catalyzed the reverse deacetylation (39). Another study documented that deacetylation of OGG1 catalyzed by Sirt3 was required in the repair of mtDNA damages induced by oxidative stress (40). Given that oxidative stress and defective mitochondria are a vital driving force for tumorigenesis and tumor development (41, 42) (Figure 1B), the acetylation of OGG1 may be a potential target for cancer therapy.
NEIL2 is charged for repairing oxidative lesions on cytosine and pyrimidine (43). An experimental study conducted in HCT116 cells reported that p300 catalyzed acetylation of NEIL2 at Lys49/150/153/154 in oxidative damage. Moreover, the reversible acetylation of Lys49 was able to decrease both glycosylase and AP lyase activities and regulate the repair activity of NEIL2. In contrast, K153 acetylation was irrelevant to the enzymatic activity (36). Thereby, the acetylation status of essential glycosylases may be considered as an indicator of cancer prognosis.
APE1 is a multifunctional protein involved in BER and the transcription of repair genes. p300, sirtuin (SIRT1), and HDAC1 were reported to mediate the acetylation and deacetylation of APE1, respectively (44, 45). Higher levels of Ac-AP endonuclease 1 (APE1) that exhibited an enhanced DNA repair efficiency for abasic sites have been observed in several primary cancer types, such as colonal, pancreatic, and NSCL cancers, compared with adjacent normal tissues. Acetylation on Lys6/7/27/31/32 of APE1 prevented its degradation and resulted in decreased sensitivity to DNA-damaging agents, which sustained the tumor cell proliferation (46, 47). In addition to increase BER activity, acetylation on APE1 also might impede tumor development through maintaining telomerase stability. Since loss of telomerase stability is a hallmark of cancer cells, exposed telomerase tends to aberrant end-joining reactions, which results in chromosomal fusions and translocations. Human telomerases are guanine-rich regions containing a G-quadruplex structure, which is a hotspot for oxidation forming 8-oxoguanine. As shown by pathogenetic lysine mutation experiments, acetylation at Lys27/31/32/35 on APE1 promoted the cleavage of abasic sites in different G4 structures and maintained the telomerase stability (48) (Figure 1B). Hence, the acetylation of APE1 may underlie a promising target in cancer therapy.
LIG3 is considered as a critical ligase in nuclear and mitochondria BER (49). However, the molecular signaling pathway in regulation of LIG3 acetylation across the process of DNA repair remains to be elucidated. Since LIG3 has been demonstrated as a crucial anti-solid and -hematologic tumor target (50–52), additional works are required to be performed to investigate the connection between LIG3 acetylation and repair efficiency in cancer.
Nucleotide Excision Repair
Two NER pathways have been described, namely, global genome coupled (GGC)-NER pathway, which handles the removal of helix-distorting DNA lesions throughout the genome, and the transcription coupled (TC)-NER pathway, which deals with transcription-blocking lesions in actively transcribed DNA by RNA polymerase II (RNAP II) (53). GGC-NER is activated by the recognition of damage-induced DNA helix distortions, while the stalling of RNAP II initiates TC-NER at damage sites (54). NER mainly consists of lesion recognition, dual incision on the 3′ and 5′ sides of lesion chains, new DNA fragment synthesis, and gap ligation.
Cullin protein 4 (Cul4)-DNA-binding protein (DDB) complex [RING box-domain protein (RBX1), Cul4, DDB1, and DDB2)] and Xeroderma pigmentosum complementation group C (XPC) complex [(XPC, Rad23 homolog B (HR23B), and Centrin 2 (CETN2)] take charge of damage recognition in GGR-NER. Upon UV light exposure, the DDB complex (DDB1 and DDB2 heterodimer) was recruited to the UV-induced lesions and initiated GGC-NER. DNA repair activity was promoted after the DDB1 subunit combined with p300 and CBP independent of DDB2 in vivo (55–57). A proteomic study identified that Lys278 on DDB2 and Lys1067 on DDB1 were possibly acetylated in vivo (18). Another study revealed that the interaction between DDB1 and CUL4 was disrupted once DDB1 was deacetylated by SIRT7, which inhibited the activity of the CUL4-ring E3 ubiquitin ligase (CRL4) complex under nucleolar stress induced by actinomycin or 5-fluorouracil. Subsequently, two substrates of CRL4, large tumor suppressor homolog 1 (LATS1) and p73, are accumulated to mediate cell apoptosis (58) (Figure 1C). However, no evidence supported that p300 or CBP mediated the acetylation of DDB1. Although other proteins within Cul4–DDB and XPC complexes all have identified acetyl-lysine sites (59), the corresponding functions of acetylation on these proteins need to be further investigated. Moreover, XPB and XPD subunits in the multisubunit factor Holo–TFIIH complex mainly facilitate DNA duplex unwinding in GGC-NER (60). Whether acetylation plays a role in regulating the functions of XPB and XPD over the repair process also needs to be illuminated.
CSB and Cul4–CSA complex (RBX1, CSA, DDB1) are responsible for damage recognition in TC-NER. XPA interacts directly with all NER core factors at the damage site, aside from XPG, thus, functioning as a scaffold for the excision of the damaged oligonucleotide (61, 62). Following UV-C radiation, XPA was deacetylated by SIRT1 (63). The hypoacetylation state of XPA increased the combination between XPA and RPA, which ensures the following efficient NER. Furthermore, deacetylation on XPA (Lys63/67) by SIRT1 influenced the binding between XPA and RPA32, whereas Lys215 that is located in the ATR-binding region of XPA was deacetylated to enhance TC-NER activity by promoting the bond between ataxia-telangiectasia and Rad3 related (ATR) and XPA (63). By contrast, acetylation of XPA at Lys215 mediated by CBP might play a negative role in regulating XPA–protein interaction, which attenuated TC-NER capacity (64). XPG protein equipped with 3′ endonuclease activity is another core TC-NER factor that is necessary for DNA incision and lesion removal. UV-C irradiation increased the contact between XPG, p300, and CBP, which mediated the acetylation of XPG. Ac-XPG was preferentially released from chromatin (65) (Figure 1C).
Homologous Recombination
HR mainly restores the lethal DSBs with high fidelity and is usually limited to the S and G2 phases (66). Timely repair of the DSBs restores the structure of DNA double-strand, protectively avoiding large-scale rearrangement of chromosomes (66). Repair proteins involved in the HR process mainly include ATR, ATM, CtIP, cell cycle and apoptosis regulator protein 2 (CCAR2), EXO, MRE11–RAD50–NBS1 (MRN complex), RAD51, RAD52, RPA, DNA Pol, and DNA ligase.
ATR and ATM are two closely related phosphatidylinositol 3′ kinase-like kinases that are activated upon DNA damage, resulting in phosphorylation of various key proteins involved in many kinds of biological processes including cell cycle, DNA repair, and apoptosis (67). Acetylation of ATM has been demonstrated as a critical regulation in self-activation and phosphorylation of various factors in DDR and repair. Acetylation of ATM by Tip60 was a prerequisite for its autophosphorylation and subsequent activation (68). On the other hand, SIRT7 catalyzed deacetylation at K3016 on ATM, promoting the dephosphorylation and deactivation of ATM after repair processes were accomplished (69). However, the effect of the acetylation on ATR was still not investigated.
CtBP-interacting protein (CtIP), exonuclease 1 (EXO1), and MRN complex participate in DNA-end resection and creates stretches of ssDNA coated by replication protein A (RPA) (70). CtIP is contributed to license HR and hamper NHEJ by activating DNA end resection. A study in yeast showed that HDACI valproic acid induced the acetylation and degradation of Sae2 (homologous to CtIP of human) by promoting autophagy (71). Autophagy is performed as a double-edged sword self-degradative system: it participates in the development of MDR and protects cancer cells from chemotherapeutics but can also kill MDR cancer cells escaping from apoptosis pathways. Autophagy induced by anticancer drugs was also able to activate apoptosis signaling pathway in multidrug resistance (MDR) cells, facilitating MDR reversal (72, 73). Hence, a bridge constructed by acetylation of Sae2 and autophagy might be an orientation for the treatment of tumors. The human homologous protein of Sae2, CtIP, is a major player in the choice between NHEJ and HR. At least four acetylation lysine sites were identified on CtIP (74). A previous study has confirmed that the deacetylation of CtIP facilitated DNA end resection (75) HDACI SFN inhibited the activity of HDAC3 in HCT116 cells and promoted the degradation of HDAC3. The interaction between HDAC3 and CtIP was broken due to the turnover of HDAC3, which facilitated the acetylation and degradation of CtIP. HR repair activity was impeded (76). Therefore, acetylation of CtIP perhaps can turn into a potential target for modulating the HR activity in cancer treatment. Moreover, additional studies are required to establish whether acetylation of CtIP has crosstalk with the autophagy pathway.
In a study by using a genome-wide human siRNA library, a CtIP antagonist named CCAR2 was found to inhibit the initiation and limit the extent of DNA end resection, which favored the process of NHEJ (77). Human CCAR2 was discovered as a negative regulator for the deacetylase activity of SIRT1 (78). Acetylation of CCAR2 on Lys54/97 induced by HDACI sulforaphane (SFN) diminished its interactions with HDAC3 and β-catenin. Treatment with the BET (acetyl-lysine reading proteins) inhibitor JQ1 synergized with SFN suppressed tumor development effectively in a preclinical model of colorectal cancer (79). Another study identified that once Lys112 and Lys215 on CCAR2 were acetylated by MYST family HAT hMOF, the interaction of CCAR2–SIRT1 was suppressed, resulting in an increased activity of SIRT1, which further deacetylated CCAR2 to stabilize the SIRT1–CCAR2 complex. Thereby, the function of SIRT1 was limited due to the negative feedback regulation. DNA damage inhibited the binding between hMOF and CCAR2, whereas increased the combination of SIRT1 and CCAR2, which might lead to p53 acetylation and p53-dependent apoptosis (80, 81). Hence, DNA damage-induced acetylation of CCAR2 is critically linked to the p53 apoptosis pathway and might have an effect on the choice of HR and NHEJ in a CtIP-dependent or -independent manner, thereby affecting the therapy response of cancers.
Although an article published in 2009 revealed that EXO1 underwent acetylation modification after treatment with HDACI, the functional alteration of EXO1 induced by acetylation is still unclear (18). In responses to DDR, HAT Tat-interacting protein of 60 kDa (Tip60) was recruited around DNA double-strand breaks (DSBs) to mediate histone acetylation. Other repair factors, such as Rad51 and vital DNA damage sensor MRN complex, were mobilized to anchor on damage sites, forming physical interaction and driving HR (82). Acetylation of ATM catalyzed by Tip60 was critical for its autophosphorylation and activation (68, 83). p300 phosphorylated by ATM interacted with NBS1 and increased its stability, leading to enhanced DNA repair ability (84) (Figure 1D).
Rad51 plays an indispensable role in catalyzing homologous pairing and strand-invasion. A previous study showed that SIRT1 interacted and deacetylated HR repair machinery proteins, including NBS1 and Rad51. Inhibition of SIRT1 impaired HR repair activity, which sensitized the lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis in lung cancer xenograft model (85). Another HDACI PCI-24781 caused a complete inhibition of RAD51 subnuclear repair foci in response to IR and a significant reduction in the transcription of genes associated with HR, including RAD51. Impaired HR realized a favorable antitumor outcome in combination with poly (ADP-ribose) polymerase (PARP)-inhibitors in HCT116 colon cancer cells (86). In addition, overexpressed HDAC2 and SIRT2 as well as nearly depleted SIRT3 were observed in the aggressive basal-like breast cancer cells. HDAC2 might indirectly influence the expression of RAD51 via Mir-182. Experimental study in vivo had demonstrated that decreased HR efficiency could lead to breast cancer progression (87) However, whether RAD51 was acetylated by HDAC2 or SIRT2 was not discussed in detail.
Rad52 is a key factor in driving post-invasion steps of both crossover and non-crossover HR pathways. It was recruited to RAD51–ssDNA filament and promoted RAD51 dissociation from DNA (88). Acetylation of RAD52 was regarded as a signal for guiding its dissociation from the DSBs. SIRT1–SIRT3 competed with CBP/P300 to catalyze the deacetylation of RAD52 at the DSBs sites. Accumulated unacetylated RAD52 facilitated the dissociation of RAD51 from the ssDNA filament prematurely, resulting in a reduced ability of HR (88) (Figure 1D).
Non-homologous End Joining
NHEJ, a non-homologous DSBs repair pathway, including c-NHEJ and alt-NHEJ, undertakes up to 90% of DSBs without sister chromosomes as replication templates. c-NHEJ is carried out mainly by Ku70/Ku80 heterodimer, DNA–PK, LIG4, XRCC4, and XLF, whereas CtIP, PARPs, LIG1/3, and XRCC1 are responsible for the process of the alt-NHEJ pathway (89, 90).
Ku70/Ku80 heterodimer is one of the initial sensors for DSBs and promptly binds to the margin sites in c-NHEJ. Acetylation of Ku70 by CBP in vivo disrupted the interaction between Ku70 and apoptotic protein Bax preventing apoptosis. Furthermore, increased acetylation of Ku70 following UV radiation exposure strengthened the combination of Ku70 and DNA, which impaired DSBs repair ability. In contrast, SIRT1 was able to enhance DNA repair capacity via catalyzing the deacetylation of Ku70 (91). Reduction of CBP also resulted in an increased DNA repair activity due to decreased acetylation of Ku70 (92). Another study also found that primary green tea polyphenol, epigallocatechin-3-gallate, upregulated the Ku70 acetylation in lung cancer A549 cells. Hyperacetylation of Ku70 blocked the binding between Ku70 and Bax, resulting in the apoptosis of lung cancer cells (93). In neuroblastoma cells, deacetylation of Ku70 by HDAC6 influenced the Ku70–Bax interaction, thus promoting cell death (94, 95). In hepatocellular carcinoma, deacetylation of Ku70 by SIRT6 attenuated Bax-mediated apoptosis (96) (Figure 1E). Collectively, regulating the acetylation of Ku70 will be a promising target for cancer therapy. Together with DNA–PKcs forming DNA–PK complex, Ku70/Ku80 heterodimer served as a docking site for the other c-NHEJ proteins (97, 98). Deacetylation of DNA–PKcs (Lys3241 and Lys3260) in vivo has been confirmed to decrease the capacity of DSBs and increase the radiosensitivity of Chinese hamster ovary cells. However, which HAT catalyzed the acetylation of DNA–PKcs needs further validation (99). As a docking site for other c-NHEJ proteins, Ku70/80 heterodimer was known to interact with the LIG4–XRCC4–XLF complex, which plays an essential role in DNA end bridging and ligation (100, 101). However, the influence of acetylation modification on these proteins in c-NHEJ still needs further investigation.
Alt-NHEJ shares similar initiating steps with HR pathways, including the end resection of 5′ ends of the DSBs. PARP1 competed with Ku70 for binding to DNA DSB sites and together with LIG1/3 initiated end-joining in the alt-NHEJ pathway (102). Previous studies discovered that LIG1, LIG3, and PARP1 protein were upregulated in tumorigenic neuroblastoma cells. Inhibition of LIG1 and LIG3 led to DSB accumulation and cell death in neuroblastoma, suggesting the alt-NHEJ pathway as a critical function in cancer cell survival and progression (103) HDACI differentially acetylated DNA repair factors to inhibit NHEJ activity in cancer cells (104). A most recent study demonstrated that the efficiency of DSBs repair by alt-NHEJ and HR, but not c-NHEJ, was increased in mammalian immortalized cells treated with HDACI TSA and PCI-2478. Immunoprecipitation experiments detected that the acetylation levels of c-NHEJ factors (Ku70 and Ku80) and alt-NHEJ factor PARP1 were increased along with a decreased DSBs binding activity. However, no changes in LIG3 and HR factors (Rad51and Rad52) were shown in acetylation levels compared with the untreated group. One explanation for the increased efficiency of HR and alt-NHEJ was that acetylation of PARP1 increased the same end resection in the initial step of these two pathways, while the increased end resection suppressed the c-NHEJ pathway (105). Another study published in 2016 also observed a decreased activity of c-NEHJ with increased acetylation levels of Ku70, Ku80, and PARP1 in acute leukemia cells treated with HDACi. However, vital components in alt-NHEJ including LIG3 and WRN were not affected by HDACI treatment. Instead, the acetylation of PARP1 showed an anomalous persistent binding ability to DNA breaks. Moreover, the activity of alt-NHEJ was not shown to be increased (104). Hence, the molecular mechanism of acetylation on PARP1 in conducting the DNA repair pathway requires deeper investigation (Figure 1E).
PARP1 has been known as a multifaceted and pleiotropic protein across multiple repair pathways. PARPi can cause synthetic lethal effect in BRCA-deficient cancer cells and has already been applied in the clinical therapy of breast, ovarian, prostate, and pancreatic cancers (106–108). PARylation of PARP1 is a prerequisite for contacting XRCC1, Polβ, and LIG3 in damage sites. In addition, PARP1 also facilitated the detection of DNA strand breaks and the choice of repair pathway choice through interaction and modification of DNA repair factors in alt-NHEJ, c-NHEJ, as well as HR (109–115). More than 20 acetylation sites have been identified on PARP1 (18, 116). PTMs including PARylation, acetylation, and ubiquitination exist with mutual competition and crosstalks on PARP1, which is critical for regulating protein functions. Lys498, Lys505, Lys508, Lys521, and Lys524 were related to the regulation of transcriptional coactivator activity of PARP1 (117). Among these lysine sites, Lys498, Lys521, and Lys524 in the automodification domain of PARP1 were targets for PARylation and acetylation at the same time. Dynamic and transient PARylation on PARP1 modulated the recruitment and dissociation of the critical DNA repair proteins in the DNA damage sites (118), whereas acetylation on these lysine sites of PARP1 impeded the access of repair factors (117). In human breast cancer cells, PARP1 was revealed to mediate PARylation of MORC2 in DNA damage sites following DDR (119), which stimulated the ATPase and chromatin remodeling activities of MORC2 and protected the cell from death. MORC2 conversely promoted HAT NAT10-mediated acetylation of PARP1 at Lys949, which blocked the degradation of itself. thereby, the genome repair ability was increased. However, which repair pathway involved in the acetylation of PARP1 was not deeply investigated (120). Moreover, PARP1 also served as a scaffold protein in the damage sites for the functional interaction between BRG (an active subunit of the SWI/SNF chromatin-remodeling complex) and SIRT1. SIRT1 deacetylated BRG at Lys1029 and Lys1033, which stimulated the ATPase activity of BRG, thus remodeling chromatin and promoting HR (121). As described above for the known effects of acetylation on PARP1, it seems that acetylation on different lysine sites plays an opposite role in effecting the efficiency of repair. Hence, precisely regulating the acetylation of lysine sites on these proteins will be more expected.
Mutual Proteins Throughout Multiple Repair Pathway During DDR
Different repair pathways have an intersection of some proteins, such as ribonucleotide reductase (RNR), tyrosyl-tRNA synthetase (TyrRS), single-stranded DNA binding proteins (SSBs), proliferating cell nuclear antigen (PCNA), DNA and Pol, involved in several mutual biological processes.
Nucleotide Synthesis
Deoxynucleotide triphosphate (dNTP) metabolism and balance are critical in carcinogenesis. In DNA repair process, the requirement for dNTP was increased since new DNA fragments need to be synthesized. Blocked dNTP synthesis can affect the formation of new DNA fragments and interference DNA repair (122). RNR composed of two large and two small subunits (RRM1 and RRM2) is the rate-limiting enzyme that converts ribonucleotide diphosphates to deoxyribonucleotides (123). In lung cancer H1299 cells and xenografts, SIRT2 specifically deacetylated RRM2 at Lys95 following DDR induced by IR or camptothecin. RNR was activated to ensure sufficient raw materials for DNA synthesis. In contrast, HAT7 induced acetylation of RRM2 at Lys95, which impaired the activity of RNR via disrupting RRM2 homodimerization. Consequently, the acetylation of RRM2 at Lys95 suppressed tumor growth both in vitro and in vivo (124) (Figure 1G).
Protein Synthesis
TyrRS, one of the 20 aminoacyl-tRNA synthetases, plays a central role in protein synthesis (125). p300/CBP-associated factor (PCAF) and SIRT1 reciprocally regulated the acetylation of TyrRS. Oxidative stress was demonstrated to induce an increased level of PCAF and decreased level of SIRT1, which sequentially led to hyperacetylation at Lys244 on TyrRS. Hyperacetylated TyrRS was promoted to translocate into the nucleus where it protected against DNA damage by activating the transcription factor E2F1 and PARP1 as well as subsequent downstream DNA repair genes. The activity of hyperacetylated TyrRS itself was decreased (126). In addition, acetylation of multiple aminoacyl-tRNAs has been reported in the proteomic data (127) (Figure 1H). The significance of acetylation on these tRNA synthetases remains to be elucidated.
New DNA Fragments Synthesis
Several DNA damage events generate the exposure of ssDNA. Naked ssDNA is susceptible to suffer further damage induced by various physical and chemical factors. Hence, before repair machinery assembled onto the damage sites, ssDNA needed to be protected by the SSB protein family (128). In humans, four kinds of SSB proteins have been identified, namely, mitochondrial SSB (mtSSB), hSSB1, hSSB2, and RPA (129). Evidence indicated that acetylation of RPA1 (Lys163) catalyzed by PCAF and GCN5 enhanced the interaction between XPA and RPA1 after UV-damage, leading to retention of XPA and eventual efficient repair, whereas HDAC6 and SIRT1 removed this acetyl group from RPA1 after the repair process was accomplished. In addition, UV-induced acetylation of RPA1 was also critical for efficient removal of CPDs and 6-4PPs (130, 131) (Figure 1D). hSSB1 has been implicated in BER (132), HR (128), and NHEJ (128). The combination between hSSB and p300 was required for efficient transcriptional activation of the p53 target gene p21 in p53 wild-type cancer cell lines after exposure to IR (133). Another study uncovered that K94 acetylation on hSSB1, which was mediated by p300, SIRT4, and HDAC10, impaired ubiquitin-mediated degradation by proteasomes. Moreover, hSSB1-K94R mutant had reduced cell survival in response to DNA damage induced by radiation or chemotherapy drugs. C646, an inhibitor of the p300/CBP, significantly enhanced the chemosensitivity of cancer cells to etoposide, adriamycin, and camptothecin. A positive correlation between the level of hSSB1 and p300 was also observed in clinical colorectal cancer samples with immunohistochemistry (134). As for mtSSB and hSSB2, further investigations are needed in understanding the role of acetylation modification on them in the DNA repair process and cancers.
PCNA acts as a master coordinator responsible for timely recruiting DNA replication and repair factors to accurately erase DNA lesions (135). A study showed that following UV irradiation, P300 and CBP attached to the C-terminal domain of PCNA and acetylated PCNA at Lys13, Lys14, Lys77, Lys80, and Lys248 in human embryonic fibroblasts and HeLa cells. The interaction between ac-PCNA, DNA pol δ, and pol β was enhanced. As there was inhibited ubiquitination and consequent degradation of PCNA, DNA synthesis activity and repair efficiency were increased. Mutation of these lysine sites increased the cell sensitivity to UV irradiation (18, 136). Except for acetylation-related self-degradation, another study complemented that in the S-phase, yeast cells, exposed to the DNA-damaging agent, Lys20 acetylation on PCNA catalyzed by HAT Eco1 (homologous to ESCO1 in human), regulated its sliding on DNA strands, thereby, favoring sister-chromatid cohesion in HR (137) (Figure 1F). Whether this regulatory mechanism exists in mammals needs further investigation.
DNA Pol catalyzes the polymerization of deoxynucleotides during DNA replication and DNA repair, which ensures the maintenance of the genetic information and faithful transmission through generations. In total, there are four major families (A, B, X, and Y family) of polymerases including 15 human polymerases that work in the five major pathways and in other three special repair pathways including trans-lesion DNA synthesis, DNA interstrand crosslink repair, and V(D)J recombination (138). Dysregulated expression and mutation of polymerases have been linked with the pathogenesis of cancers (139, 140). In 2002, Hasan et al. discovered that DNA Polβ in the X family formed a complex with the p300 and was acetylated at Lys72 both in vitro and in vivo. The dRP-lyase activity of Polβ was impaired along with a severely reduced BER (141). Moreover, Polβ catalyzed the formation of a 5′ flap from the 3′ incision during the long-patch BER pathway. Finally, FEN1 was responsible for incising the 5′ flap to complete the repair process (142). Inhibiting the expression of FEN1 restrained the progression and increased the chemo-sensitivity of breast cancer cells (143) (Figure 1B). Acetylation of FEN1 by p300 reduced the DNA binding activity and inhibited its endonuclease activity, which impaired flap cleavage and repair ability (144, 145). A recent study in 2019 pointed out that Polι of the Y family, a non-canonical polymerase involved in trans-lesion DNA synthesis, was acetylated at Lys550 by p300/CBP in response to SN2 alkylating agents (methyl methanesulfonate and dimethyl sulfate) (146). Further studies are needed to clarify the role of acetylation on Polι and other DNA Pols in DNA repair.
Summary
The dysfunction of repair proteins caused by aberrant acetylation modification is strongly connected with pathogenesis, development, and drug resistance of cancers. Abnormal expressions of HDACs have been detected in a variety of tumors. Based on the favorable therapeutic response in clinical trials, several HDACIs have been approved in the treatment of hematologic tumors and a few solid tumors. However, the alteration of acetylation on repair proteins in many types of solid tumors is less focused, and the affected downstream signaling pathways are also unclear. Digging deeper into the regulations of acetylation on DNA repair proteins may shed light on the pathogenesis of tumors and contribute to the discovery of new drugs targeting this PTM for better therapeutic effects of cancers.
Author Contributions
SL collected the published data, wrote the paper, as well as made the figures. BS and XL participated in the discussion of the content of the review. H-XA conceived and revised this review. All authors contributed to the article and approved the submitted version.
Funding
This project is supported by the grant Peking Medical and Health Foundation (No. 20180519).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. (2015) 66:129–43. doi: 10.1146/annurev-med-081313-121208
2. O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. (2015) 60:547–60. doi: 10.1016/j.molcel.2015.10.040
3. Saez GT. DNA injury and repair systems. Int J Mol Sci. (2018) 19:E1902. doi: 10.3390/ijms19071902
4. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. (1964) 51:786–94. doi: 10.1073/pnas.51.5.786
5. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. (2009) 10:32–42. doi: 10.1038/nrg2485
6. Gil J, Ramirez-Torres A, Encarnacion-Guevara S. Lysine acetylation and cancer: a proteomics perspective. J Proteomics. (2017) 150:297–309. doi: 10.1016/j.jprot.2016.10.003
7. Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. (2013) 288:29036–45. doi: 10.1074/jbc.M113.486753
8. Roos WP, Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. (2016) 44:10017–30. doi: 10.1093/nar/gkw922
9. McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res. (2018) 138:183–211. doi: 10.1016/bs.acr.2018.02.006
10. Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol. (2005) 125:1045–52. doi: 10.1111/j.0022-202X.2005.23925.x
11. Avvakumov N, Côté J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. (2007) 26:5395–407. doi: 10.1038/sj.onc.1210608
12. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
13. Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. (2004) 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723
14. Modrich P. Mismatch repair, genetic stability, and cancer. Science. (1994) 266:1959–60. doi: 10.1126/science.7801122
15. Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. (1995) 268:1909–12. doi: 10.1126/science.7604264
16. Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. (1998) 273:19895–901. doi: 10.1074/jbc.273.31.19895
17. Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. (2013) 10:634–7. doi: 10.1038/nmeth.2518
18. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. (2009) 325:834–40. doi: 10.1126/science.1175371
19. Das S, Salami SS, Spratt DE, Kaffenberger SD, Jacobs MF, Morgan TM. Bringing prostate cancer germline genetics into clinical practice. J Urol. (2019) 202:223–30. doi: 10.1097/JU.0000000000000137
20. Gelsomino F, Barbolini M, Spallanzani A, Pugliese G, Cascinu S. The evolving role of microsatellite instability in colorectal cancer: a review. Cancer Treat Rev. (2016) 51:19–26. doi: 10.1016/j.ctrv.2016.10.005
21. Eso Y, Takai A, Matsumoto T, Inuzuka T, Horie T, Ono K, et al. MSH2 dysregulation is triggered by proinflammatory cytokine stimulation and is associated with liver cancer development. Cancer Res. (2016) 76:4383–93. doi: 10.1158/0008-5472.CAN-15-2926
22. Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha. Mol Cell. (2014) 55:31–46. doi: 10.1016/j.molcel.2014.04.028
23. Wang L, Xiang S, Williams KA, Dong H, Bai W, Nicosia SV, et al. Depletion of HDAC6 enhances cisplatin-induced DNA damage and apoptosis in non-small cell lung cancer cells. PLoS ONE. (2012) 7:e44265. doi: 10.1371/journal.pone.0044265
24. Radhakrishnan R, Li Y, Xiang S, Yuan F, Yuan Z, Telles E, et al. Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of MutS homolog 2. J Biol Chem. (2015) 290:22795–804. doi: 10.1074/jbc.M114.612945
25. Tao X, Yan Y, Lu L, Chen B. HDAC10 expression is associated with DNA mismatch repair gene and is a predictor of good prognosis in colon carcinoma. Oncol Lett. (2017) 14:4923–9. doi: 10.3892/ol.2017.6818
26. Zhang M, Hu C, Moses N, Haakenson J, Xiang S, Quan D, et al. HDAC6 regulates DNA damage response via deacetylating MLH1. J Biol Chem. (2019) 294:5813–26. doi: 10.1074/jbc.RA118.006374
27. Nalawansha DA, Zhang Y, Herath K, Pflum MKH. HDAC1 substrate profiling using proteomics-based substrate trapping. ACS Chem Biol. (2018) 13:3315–24. doi: 10.1021/acschembio.8b00737
28. Mazur DJ, Mendillo ML, Kolodner RD. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol Cell. (2006) 22:39–49. doi: 10.1016/j.molcel.2006.02.010
29. Offit K. MSH6 mutations in hereditary nonpolyposis colon cancer: another slice of the pie. J Clin Oncol. (2004) 22:4449–51. doi: 10.1200/JCO.2004.06.940
30. Cerretelli G, Ager A, Arends MJ, Frayling IM. Molecular pathology of Lynch syndrome. J Pathol. (2020) 250:518–31. doi: 10.1002/path.5422
31. Lindahl T. Instability and decay of the primary structure of DNA. Nature. (1993) 362:709–15. doi: 10.1038/362709a0
32. Lindahl T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc Natl Acad Sci USA. (1974) 71:3649–53. doi: 10.1073/pnas.71.9.3649
33. Jacobs AL, Schar P. DNA glycosylases: in DNA repair and beyond. Chromosoma. (2012) 121:1–20. doi: 10.1007/s00412-011-0347-4
34. Wallace SS. DNA glycosylases search for and remove oxidized DNA bases. Environ Mol Mutagen. (2013) 54:691–704. doi: 10.1002/em.21820
35. Carter RJ, Parsons JL. Base excision repair, a pathway regulated by posttranslational modifications. Mol Cell Biol. (2016) 36:1426–37. doi: 10.1128/MCB.00030-16
36. Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. (2004) 32:3033–9. doi: 10.1093/nar/gkh632
37. Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. (2002) 9:265–77. doi: 10.1016/S1097-2765(02)00453-7
38. Hardeland U, Bentele M, Jiricny J, Schar P. The versatile thymine DNA-glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. (2003) 31:2261–71. doi: 10.1093/nar/gkg344
39. Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. (2006) 26:1654–65. doi: 10.1128/MCB.26.5.1654-1665.2006
40. Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS, et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. (2013) 4:e731. doi: 10.1038/cddis.2013.254
42. Yuzefovych LV, Kahn AG, Schuler MA, Eide L, Arora R, Wilson GL, et al. Mitochondrial DNA repair through OGG1 activity attenuates breast cancer progression and metastasis. Cancer Res. (2016) 76:30–4. doi: 10.1158/0008-5472.CAN-15-0692
43. Dutta A, Yang C, Sengupta S, Mitra S, Hegde ML. New paradigms in the repair of oxidative damage in human genome: mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell Mol Life Sci. (2015) 72:1679–98. doi: 10.1007/s00018-014-1820-z
44. Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. (2003) 22:6299–309. doi: 10.1093/emboj/cdg595
45. Sengupta S, Mantha AK, Mitra S, Bhakat KK. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene. (2011) 30:482–93. doi: 10.1038/onc.2010.435
46. Bhakat KK, Sengupta S, Adeniyi VF, Roychoudhury S, Nath S, Bellot LJ, et al. Regulation of limited N-terminal proteolysis of APE1 in tumor via acetylation and its role in cell proliferation. Oncotarget. (2016) 7:22590–604. doi: 10.18632/oncotarget.8026
47. Sengupta S, Mantha AK, Song H, Roychoudhury S, Nath S, Ray S, et al. Elevated level of acetylation of APE1 in tumor cells modulates DNA damage repair. Oncotarget. (2016) 7:75197–209. doi: 10.18632/oncotarget.12113
48. Burra S, Marasco D, Malfatti MC, Antoniali G, Virgilio A, Esposito V, et al. Human AP-endonuclease (Ape1) activity on telomeric G4 structures is modulated by acetylatable lysine residues in the N-terminal sequence. DNA Repair. (2019) 73:129–43. doi: 10.1016/j.dnarep.2018.11.010
49. Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, et al. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. (2011) 471:240–4. doi: 10.1038/nature09773
50. Caracciolo D, Di Martino MT, Amodio N, Morelli E, Montesano M, Botta C, et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia. (2019) 33:487–98. doi: 10.1038/s41375-018-0238-2
51. Hu Y, Lin J, Fang H, Fang J, Li C, Chen W, et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia. (2018) 32:2250–62. doi: 10.1038/s41375-018-0104-2
52. Li D, Suzuki H, Liu B, Morris J, Liu J, Okazaki T, et al. DNA repair gene polymorphisms and risk of pancreatic cancer. Clin Cancer Res. (2009) 15:740–6. doi: 10.1158/1078-0432.CCR-08-1607
53. Spivak G. Nucleotide excision repair in humans. DNA Repair. (2015) 36:13–8. doi: 10.1016/j.dnarep.2015.09.003
54. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. (2014) 15:465–81. doi: 10.1038/nrm3822
55. Zhu Q, Wani AA. Nucleotide excision repair: finely tuned molecular orchestra of early pre-incision events. Photochem Photobiol. (2017) 93:166–77. doi: 10.1111/php.12647
56. Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T, et al. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat Res. (2001) 486:89–97. doi: 10.1016/S0921-8777(01)00082-9
57. Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. (2002) 30:2588–98. doi: 10.1093/nar/30.11.2588
58. Mo Y, Lin R, Liu P, Tan M, Xiong Y, Guan KL, et al. SIRT7 deacetylates DDB1 and suppresses the activity of the CRL4 E3 ligase complexes. FEBS J. (2017) 284:3619–36. doi: 10.1111/febs.14259
59. Weinert BT, Schölz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. (2013) 4:842–51. doi: 10.1016/j.celrep.2013.07.024
60. Rimel JK, Taatjes DJ. The essential and multifunctional TFIIH complex. Protein Sci. (2018) 27:1018–37. doi: 10.1002/pro.3424
61. Lagerwerf S, Vrouwe MG, Overmeer RM, Fousteri MI, Mullenders LH. DNA damage response and transcription. DNA Repair. (2011) 10:743–50. doi: 10.1016/j.dnarep.2011.04.024
62. Fadda E. Role of the XPA protein in the NER pathway: a perspective on the function of structural disorder in macromolecular assembly. Comput Struct Biotechnol J. (2016) 14:78–85. doi: 10.1016/j.csbj.2015.11.007
63. Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. (2010) 39:247–58. doi: 10.1016/j.molcel.2010.07.006
64. Jarrett SG, Carter KM, Bautista RM, He D, Wang C, D'Orazio JA. Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J Biol Chem. (2018) 293:19025–37. doi: 10.1074/jbc.RA118.003940
65. Tillhon M, Cazzalini O, Nardo T, Necchi D, Sommatis S, Stivala LA, et al. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair. (2012) 11:844–52. doi: 10.1016/j.dnarep.2012.08.001
66. Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. (2018) 293:10524–35. doi: 10.1074/jbc.TM118.000372
67. Jin MH, Oh DY. ATM in DNA repair in cancer. Pharmacol Ther. (2019) 203:107391. doi: 10.1016/j.pharmthera.2019.07.002
68. Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA. (2005) 102:13182–7. doi: 10.1073/pnas.0504211102
69. Tang M, Li Z, Zhang C, Lu X, Tu B, Cao Z, et al. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci Adv. (2019) 5:eaav1118. doi: 10.1126/sciadv.aav1118
70. Maizels N, Davis L. Initiation of homologous recombination at DNA nicks. Nucleic Acids Res. (2018) 46:6962–73. doi: 10.1093/nar/gky588
71. Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. (2011) 471:74–9. doi: 10.1038/nature09803
72. Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye WC, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer. (2017) 36:52. doi: 10.1186/s40880-017-0219-2
73. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. (2017) 17:528–42. doi: 10.1038/nrc.2017.53
74. Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell. (2014) 54:1022–33. doi: 10.1016/j.molcel.2014.04.011
75. Xie J, Peng M, Guillemette S, Quan S, Maniatis S, Wu Y, et al. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. (2012) 8:e1002786. doi: 10.1371/journal.pgen.1002786
76. Okonkwo A, Mitra J, Johnson GS, Li L, Dashwood WM, Hegde ML, et al. Heterocyclic analogs of sulforaphane trigger DNA damage and impede DNA repair in colon cancer cells: interplay of HATs and HDACs. Mol Nutr Food Res. (2018) 62:e1800228. doi: 10.1002/mnfr.201800228
77. López-Saavedra A, Gómez-Cabello D, Domínguez-Sánchez MS, Mejías-Navarro F, Fernández-Ávila MJ, Dinant C, et al. A genome-wide screening uncovers the role of CCAR2 as an antagonist of DNA end resection. Nat Commun. (2016) 7:12364. doi: 10.1038/ncomms12364
78. Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. (2008) 451:583–6. doi: 10.1038/nature06500
79. Rajendran P, Johnson G, Li L, Chen YS, Dashwood M, Nguyen N, et al. Acetylation of CCAR2 establishes a BET/BRD9 acetyl switch in response to combined deacetylase and bromodomain inhibition. Cancer Res. (2019) 79:918–27. doi: 10.1158/0008-5472.CAN-18-2003
80. Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. (2008) 451:587–90. doi: 10.1038/nature06515
81. Zheng H, Yang L, Peng L, Izumi V, Koomen J, Seto E, et al. hMOF acetylation of DBC1/CCAR2 prevents binding and inhibition of SirT1. Mol Cell Biol. (2013) 33:4960–70. doi: 10.1128/MCB.00874-13
82. Chailleux C, Tyteca S, Papin C, Boudsocq F, Puget N, Courilleau C, et al. Physical interaction between the histone acetyl transferase Tip60 and the DNA double-strand breaks sensor MRN complex. Biochem J. (2010) 426:365–71. doi: 10.1042/BJ20091329
83. Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. (2007) 27:8502–9. doi: 10.1128/MCB.01382-07
84. Jang ER, Choi JD, Lee JS. Acetyltransferase p300 regulates NBS1-mediated DNA damage response. FEBS Lett. (2011) 585:47–52. doi: 10.1016/j.febslet.2010.11.034
85. Chen G, Zhang B, Xu H, Sun Y, Shi Y, Luo Y, et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene. (2017) 36:6863–72. doi: 10.1038/onc.2017.297
86. Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA. (2007) 104:19482–7. doi: 10.1073/pnas.0707828104
87. Shan W, Jiang Y, Yu H, Huang Q, Liu L, Guo X, et al. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am J Cancer Res. (2017) 7:1213–26.
88. Yasuda T, Kagawa W, Ogi T, Kato TA, Suzuki T, Dohmae N, et al. Novel function of HATs and HDACs in homologous recombination through acetylation of human RAD52 at double-strand break sites. PLoS Genet. (2018) 14:e1007277. doi: 10.1371/journal.pgen.1007277
89. Biehs R, Steinlage M, Barton O, Juhász S, Künzel J, Spies J, et al. DNA Double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell. (2017) 65:671–84. doi: 10.1016/j.molcel.2016.12.016
90. Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. (2008) 4:e1000110. doi: 10.1371/journal.pgen.1000110
91. Jeong J, Juhn K, Lee H, Kim SH, Min BH, Lee KM, et al. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. (2007) 39:8–13. doi: 10.1038/emm.2007.2
92. Subramanian C, Hada M, Opipari AW Jr, Castle VP, Kwok RP. CREB-binding protein regulates Ku70 acetylation in response to ionization radiation in neuroblastoma. Mol Cancer Res. (2013) 11:173–81. doi: 10.1158/1541-7786.MCR-12-0065
93. Li M, Li JJ, Gu QH, An J, Cao LM, Yang HP, et al. EGCG induces lung cancer A549 cell apoptosis by regulating Ku70 acetylation. Oncol Rep. (2016) 35:2339–47. doi: 10.3892/or.2016.4587
94. Subramanian C, Jarzembowski JA, Opipari AW Jr, Castle VP, Kwok RP. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia. (2011) 13:726–34. doi: 10.1593/neo.11558
95. Subramanian C, Opipari AW Jr, Bian X, Castle VP, Kwok RP. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. (2005) 102:4842–7. doi: 10.1073/pnas.0408351102
96. Tao NN, Ren JH, Tang H, Ran LK, Zhou HZ, Liu B, et al. Deacetylation of Ku70 by SIRT6 attenuates Bax-mediated apoptosis in hepatocellular carcinoma. Biochem Biophys Res Commun. (2017) 485:713–9. doi: 10.1016/j.bbrc.2017.02.111
97. Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair. (2014) 17:21–9. doi: 10.1016/j.dnarep.2014.02.020
98. Davis AJ, Lee KJ, Chen DJ. The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J Biol Chem. (2013) 288:7037–46. doi: 10.1074/jbc.M112.434498
99. Mori E, Davis AJ, Hasegawa M, Chen DJ. Lysines 3241 and 3260 of DNA-PKcs are important for genomic stability and radioresistance. Biochem Biophys Res Commun. (2016) 477:235–40. doi: 10.1016/j.bbrc.2016.06.048
100. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. (2006) 124:301–13. doi: 10.1016/j.cell.2005.12.031
101. Sulkowski PL, Scanlon SE, Oeck S, Glazer PM. PTEN regulates nonhomologous end joining by epigenetic induction of NHEJ1/XLF. Mol Cancer Res. (2018) 16:1241–54. doi: 10.1158/1541-7786.MCR-17-0581
102. Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. (2010) 38:5706–17. doi: 10.1093/nar/gkq379
103. Newman EA, Lu F, Bashllari D, Wang L, Opipari AW, Castle VP. Alternative NHEJ pathway components are therapeutic targets in high-risk neuroblastoma. Mol Cancer Res. (2015) 13:470–82. doi: 10.1158/1541-7786.MCR-14-0337
104. Robert C, Nagaria PK, Pawar N, Adewuyi A, Gojo I, Meyers DJ, et al. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. (2016) 45:14–23. doi: 10.1016/j.leukres.2016.03.007
105. Li G, Zhang X, Wang H, Liu D, Li Z, Wu Z, et al. Increasing CRISPR/Cas9-mediated homology-directed DNA repair by histone deacetylase inhibitors. Int J Biochem Cell Biol. (2020) 125:105790. doi: 10.1016/j.biocel.2020.105790
106. Maintenance olaparib new standard in pancreatic cancer? Cancer Discov. (2019) 9:Of6. doi: 10.1158/2159-8290.CD-NB2019-065
107. Mateo J, Lord CJ, Serra V, Tutt A, Balmana J, Castroviejo-Bermejo M, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. (2019) 30:1437–47. doi: 10.1093/annonc/mdz192
108. Lin KY, Kraus WL. PARP inhibitors for cancer therapy. Cell. (2017) 169:183. doi: 10.1016/j.cell.2017.03.034
109. Leppard JB, Dong Z, Mackey ZB, Tomkinson AE. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol. (2003) 23:5919–27. doi: 10.1128/MCB.23.16.5919-5927.2003
110. Prasad R, Lavrik OI, Kim SJ, Kedar P, Yang XP, Vande Berg BJ, et al. DNA polymerase beta -mediated long patch base excision repair. Poly(ADP-ribose)polymerase-1 stimulates strand displacement DNA synthesis. J Biol Chem. (2001) 276:32411–4. doi: 10.1074/jbc.C100292200
111. Liu Y, Kadyrov FA, Modrich P. PARP-1 enhances the mismatch-dependence of 5'-directed excision in human mismatch repair in vitro. DNA Repair. (2011) 10:1145–53. doi: 10.1016/j.dnarep.2011.08.012
112. Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. (2012) 199:235–49. doi: 10.1083/jcb.201112132
113. Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. (2006) 25:1305–14. doi: 10.1038/sj.emboj.7601015
114. Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJL, et al. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol Cell. (2016) 61:547–62. doi: 10.1016/j.molcel.2016.01.019
115. Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. (2017) 18:610–21. doi: 10.1038/nrm.2017.53
116. Wu Q, Cheng Z, Zhu J, Xu W, Peng X, Chen C, et al. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci Rep. (2015) 5:9520. doi: 10.1038/srep09520
117. Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. (2005) 280:40450–64. doi: 10.1074/jbc.M507553200
118. Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. (2009) 37:3723–38. doi: 10.1093/nar/gkp229
119. Li DQ, Nair SS, Ohshiro K, Kumar A, Nair VS, Pakala SB, et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep. (2012) 2:1657–69. doi: 10.1016/j.celrep.2012.11.018
120. Zhang L, Li DQ. MORC2 regulates DNA damage response through a PARP1-dependent pathway. Nucleic Acids Res. (2019) 47:8502–20. doi: 10.1093/nar/gkz545
121. Chen Y, Zhang H, Xu Z, Tang H, Geng A, Cai B, et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. (2019) 47:8563–80. doi: 10.1093/nar/gkz592
122. Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. (2015) 34:2011–21. doi: 10.1038/onc.2014.155
123. Nordlund P, Reichard P. Ribonucleotide reductases. Ann Rev Biochem. (2006) 75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443
124. Chen G, Luo Y, Warncke K, Sun Y, Yu DS, Fu H, et al. Acetylation regulates ribonucleotide reductase activity and cancer cell growth. Nature Commun. (2019) 10:3213. doi: 10.1038/s41467-019-11214-9
125. Richardson CJ, First EA. A continuous tyrosyl-tRNA synthetase assay that regenerates the tRNA substrate. Anal Biochem. (2015) 486:86–95. doi: 10.1016/j.ab.2015.05.008
126. Cao X, Li C, Xiao S, Tang Y, Huang J, Zhao S, et al. Acetylation promotes TyrRS nuclear translocation to prevent oxidative damage. Proc Natl Acad Sci USA. (2017) 114:687–92. doi: 10.1073/pnas.1608488114
127. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. (2010) 327:1000–4. doi: 10.1126/science.1179689
128. Croft LV, Bolderson E, Adams MN, El-Kamand S, Kariawasam R, Cubeddu L, et al. Human single-stranded DNA binding protein 1 (hSSB1, OBFC2B), a critical component of the DNA damage response. Semin Cell Dev Biol. (2019) 86:121–8. doi: 10.1016/j.semcdb.2018.03.014
129. Richard DJ, Bolderson E, Khanna KK. Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit Rev Biochem Mol Biol. (2009) 44:98–116. doi: 10.1080/10409230902849180
130. He H, Wang J, Liu T. UV-induced RPA1 acetylation promotes nucleotide excision repair. Cell Rep. (2017) 20:2010–25. doi: 10.1016/j.celrep.2017.08.016
131. Zhao M, Geng R, Guo X, Yuan R, Zhou X, Zhong Y, et al. PCAF/GCN5-mediated acetylation of RPA1 promotes nucleotide excision repair. Cell Rep. (2017) 20:1997–2009. doi: 10.1016/j.celrep.2017.08.015
132. Paquet N, Adams MN, Ashton NW, Touma C, Gamsjaeger R, Cubeddu L, et al. hSSB1 (NABP2/OBFC2B) is regulated by oxidative stress. Sci Rep. (2016) 6:27446. doi: 10.1038/srep27446
133. Xu S, Wu Y, Chen Q, Cao J, Hu K, Tang J, et al. hSSB1 regulates both the stability and the transcriptional activity of p53. Cell Res. (2013) 23:423–35. doi: 10.1038/cr.2012.162
134. Wu Y, Chen H, Lu J, Zhang M, Zhang R, Duan T, et al. Acetylation-dependent function of human single-stranded DNA binding protein 1. Nucleic Acids Res. (2015) 43:7878–87. doi: 10.1093/nar/gkv707
135. Slade D. Maneuvers on PCNA rings during DNA replication and repair. Genes. (2018) 9:416. doi: 10.3390/genes9080416
136. Cazzalini O, Sommatis S, Tillhon M, Dutto I, Bachi A, Rapp A, et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. (2014) 42:8433–48. doi: 10.1093/nar/gku533
137. Billon P, Li J, Lambert JP, Chen Y, Tremblay V, Brunzelle JS, et al. Acetylation of PCNA sliding surface by Eco1 promotes genome stability through homologous recombination. Mol Cell. (2017) 65:78–90. doi: 10.1016/j.molcel.2016.10.033
138. Garcia-Diaz M, Bebenek K. Multiple functions of DNA polymerases. Crit Rev Plant Sci. (2007) 26:105–22. doi: 10.1080/07352680701252817
139. Park VS, Pursell ZF. POLE proofreading defects: contributions to mutagenesis and cancer. DNA Repair. (2019) 76:50–9. doi: 10.1016/j.dnarep.2019.02.007
140. Wood RD, Doublié S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair. (2016) 44:22–32. doi: 10.1016/j.dnarep.2016.05.003
141. Hasan S, El-Andaloussi N, Hardeland U, Hassa PO, Bürki C, Imhof R, et al. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Mol Cell. (2002) 10:1213–22. doi: 10.1016/S1097-2765(02)00745-1
142. Balakrishnan L, Brandt PD, Lindsey-Boltz LA, Sancar A, Bambara RA. Long patch base excision repair proceeds via coordinated stimulation of the multienzyme DNA repair complex. J Biol Chem. (2009) 284:15158–72. doi: 10.1074/jbc.M109.000505
143. Lu X, Liu R, Wang M, Kumar AK, Pan F, He L, et al. MicroRNA-140 impedes DNA repair by targeting FEN1 and enhances chemotherapeutic response in breast cancer. Oncogene. (2020) 39:234–47. doi: 10.1038/s41388-019-0986-0
144. Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA. Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J Biol Chem. (2010) 285:4398–404. doi: 10.1074/jbc.M109.086397
145. Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, et al. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol Cell. (2001) 7:1221–31. doi: 10.1016/S1097-2765(01)00272-6
146. McIntyre J, Sobolewska A, Fedorowicz M, McLenigan MP, Macias M, Woodgate R, et al. DNA polymerase ι is acetylated in response to S(N)2 alkylating agents. Sci Rep. (2019) 9:4789. doi: 10.1038/s41598-019-41249-3
Abbreviations
.
Keywords: DNA repair, acetylation, deacetylation, cancer, acetyltransferase, deacetylase
Citation: Li S, Shi B, Liu X and An H-X (2020) Acetylation and Deacetylation of DNA Repair Proteins in Cancers. Front. Oncol. 10:573502. doi: 10.3389/fonc.2020.573502
Received: 17 June 2020; Accepted: 17 September 2020;
Published: 22 October 2020.
Edited by:
Dario Palmieri, The Ohio State University, United StatesReviewed by:
Vandna Kukshal, Washington University School of Medicine in St. Louis, United StatesPraveen Rajendran, Texas A&M Health Science Center, United States
Copyright © 2020 Li, Shi, Liu and An. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Han-Xiang An, YW5oYW54aWFuZ0B4bXUuZWR1LmNu