 Bolun Jiang
Bolun Jiang Junchao Guo
Junchao Guo- Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Pancreatic ductal adenocarcinoma (PDAC) is a malignancy with one of the worst prognoses worldwide and has an overall 5-year survival rate of only 9%. Although chemotherapy is the recommended treatment for patients with advanced PDAC, its efficacy is not satisfactory. The dense dysplastic stroma of PDAC is a major obstacle to the delivery of chemotherapy drugs and plays an important role in the progression of PDAC. Therefore, stroma-targeting therapy is considered a potential treatment strategy to improve the efficacy of chemotherapy and patient survival. While several preclinical studies have shown encouraging results, the anti-tumor potential of the PDAC stroma has also been revealed, and the extreme depletion might promote tumor progression and undermine patient survival. Therefore, achieving a balance between stromal abundance and depletion might be the further of stroma-targeting therapy. This review summarized the current progress of stroma-targeting therapy in PDAC and discussed the double-edged sword of its therapeutic effects.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) ranks seventh as the cause of cancer-associated mortality worldwide and has an overall 5-year survival rate of only 9% (1, 2). In the United States, PDAC is projected to become the second deadliest cancer by 2030 (3). With the acceleration of industrialization and population aging, the incidence of PDAC has also increased in China (4). In the past few decades, the treatment of PDAC has not made substantial progress. Surgery remains the only treatment that might achieve a cure. However, about 80% of PDAC patients are unable to access surgery at the time of diagnosis (5). Because of the early recurrence and metastasis of PDAC, even the patients who have already undergone radical surgery present a 5-year survival rate of only 25% (5). Therefore, PDAC has emerged as a major public health problem that needs to be addressed urgently.
For the majority of patients with advanced PDAC, chemotherapy is the generally recommended treatment. The standardized chemotherapy regimens include the use of the cytosine nucleoside analog gemcitabine or a more potent but highly toxic four-drugs regimen, FOLFIRINOX (i.e., 5-fluorouracil, leucovorin, oxaliplatin, and irinotecan) (6). In addition, chemotherapy is also applied as a neoadjuvant treatment for some PDAC patients who are suitable for surgery to lower the risk of recurrence (7). However, the efficacy of these regimens is not satisfactory. One of the main features of PDAC is its dense abundant stroma, which increases the interstitial fluid pressure (IFP). The compression of intratumoral vasculature impedes the efficiency of chemotherapeutic agent delivery and forms a hypoxic microenvironment, which promotes tumor progression (8, 9). Therefore, reversing the negative effects of PDAC stroma might improve the efficacy of chemotherapy and patient survival (8).
PDAC Stroma and Its Role in Disease Progression
In PDAC, tumor cells account for less than 20% of the total tumor volume, while the stroma components occupy more than 70% (10). The dense desmoplasia stroma of PDAC consists of several cellular components (e.g., fibroblasts, stellate cells, immune cells, and pericytes), acellular components (e.g., fibrin, collagen, hyaluronic acid, fibronectin, growth factors, and cytokines), and biophysical components (e.g., low pH, hypoxia, and high tumor IFP) (11). These components interact mutually to promote the progression of PDAC (12).
During the progression of PDAC, pro-inflammatory cytokines secreted by neoplastic cells stimulate fibroblasts and pancreatic stellate cells (PSCs), which produce extracellular matrix (ECM) and increase fibrotic stromal deposition (13, 14). The solid stress generated by the dense stroma and lymphatic obstruction causes intratumoral IFP to increase, which leads to vascular compression, tissue perfusion reduction, and a hypoxic microenvironment (15, 16). In fact, approximately 80% of intratumoral blood vessels are dysfunctional, poorly fenestrated, and covered with a thick layer of pericytes, which hinder the effective accumulation of chemotherapeutic agents. In addition, the PDAC stroma potentially alters the pharmacokinetics and pharmacodynamics of chemotherapeutic drugs (8). For example, the high expression level of cytidine deaminase in the stroma shortened the circulating half-life of gemcitabine (17). Furthermore, the fibrotic microenvironment of PDAC results in an inhibitory effect on the innate and adaptive immune systems, reducing cytotoxic T cells and increasing M2 macrophages, N2 neutrophils, and T-regulatory cells (Tregs) at tumor sites. Growth factors and cytokines secreted by PSCs also promote the formation of a tumor immunosuppressive microenvironment (18). In summary, the dense proliferative stroma of PDAC promotes tumor progression and metastasis via various routes (11). Therefore, overcoming the tumor-promoting effects of PDAC stromal barriers has become an imperative issue.
Stroma-Targeting Therapy in PDAC
In order to overcome the physical and biological barriers to effective PDAC treatments, several strategies based on targeting the stroma have been designed to improve the efficacy of chemotherapeutic agents and reverse the stroma’s impact on tumor progression (Table 1).
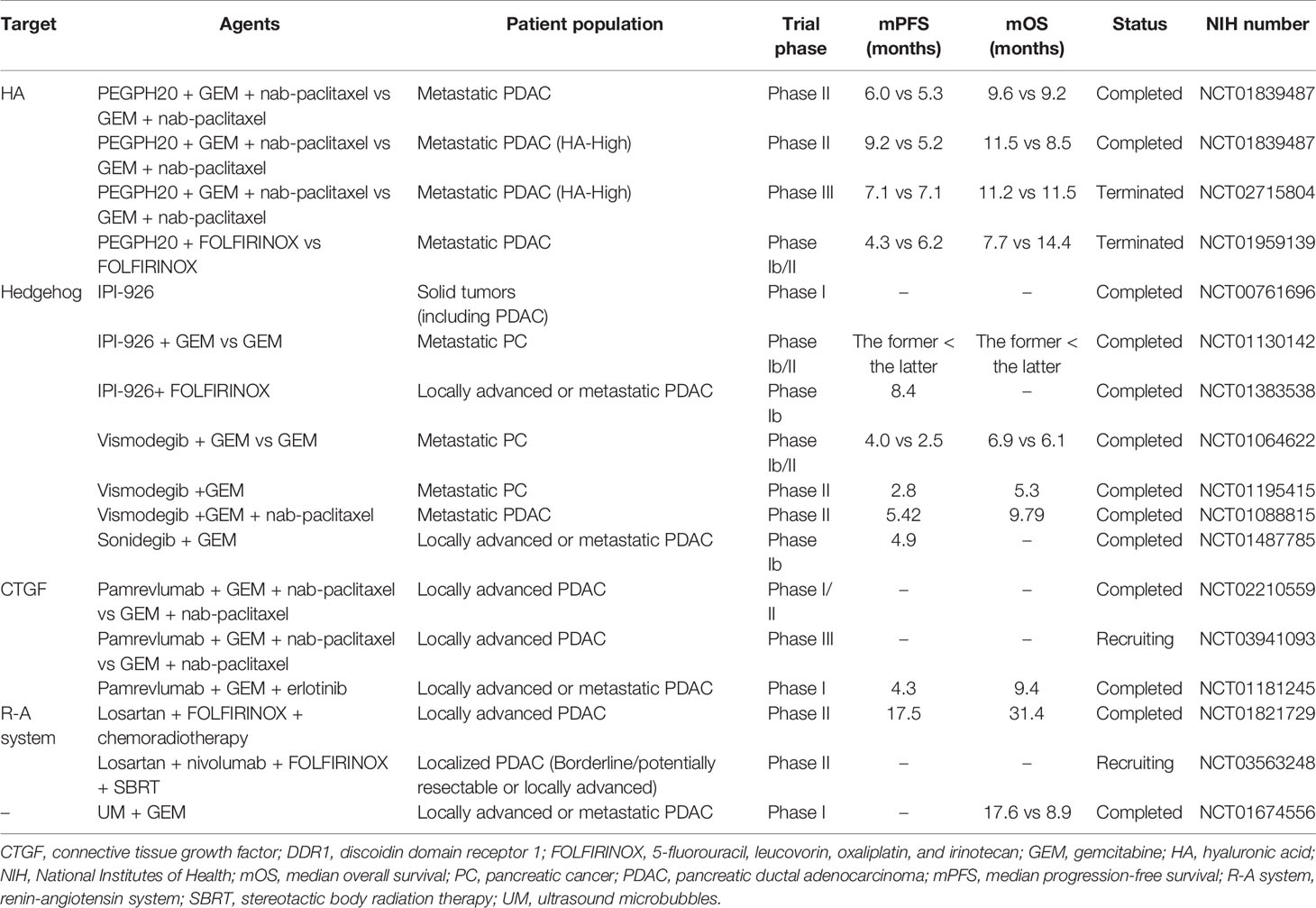
Table 1 Clinical trials assessing stroma-targeting therapy in PDAC.
Depletion of the Stroma in PDAC
One of the main components of PDAC stroma is hyaluronic acid (HA) or hyaluronan (Figure 1). HA is a complex glycosaminoglycan secreted abundantly by neoplastic cells (19–21), and it is a core polymer in the assembly of multiple hydrophilic matrix proteoglycans (22). HA binds to cell surface receptors to maintain tumor cell survival and activate downstream signaling pathways related to tumor proliferation, migration, and invasion (23–25). In addition, its ability to absorb and retain water increases IFP (24, 26). Therefore, HA is considered a potential therapeutic target in PDAC. Pegylated hyaluronidase (PEGPH20) is a pegylated nanoscale complex of recombinant human hyaluronidase (27, 28). Previous studies showed that PEGPH20 induced the degradation of HA, remodeled tumor vasculature, and improved chemotherapeutic drug efficacy (22, 27, 29). A phase II clinical study HALO-109-202 (NCT01839487), involving 279 patients with metastatic PDAC, showed that PEGPH20 combined with Abraxane (an albumin-bound paclitaxel nanocomplex) and gemcitabine nearly doubled the progression-free survival and improved the overall survival in patients with high level HA (30). However, a subsequent phase III clinical study HALO-109-301 (NCT02715804) showed that PEGPH20 combined with Abraxane and gemcitabine did not significantly extend the overall survival of PDAC patients. Therefore, Halozyme announced the termination of further research and development of PEGPH20 in November 2019.
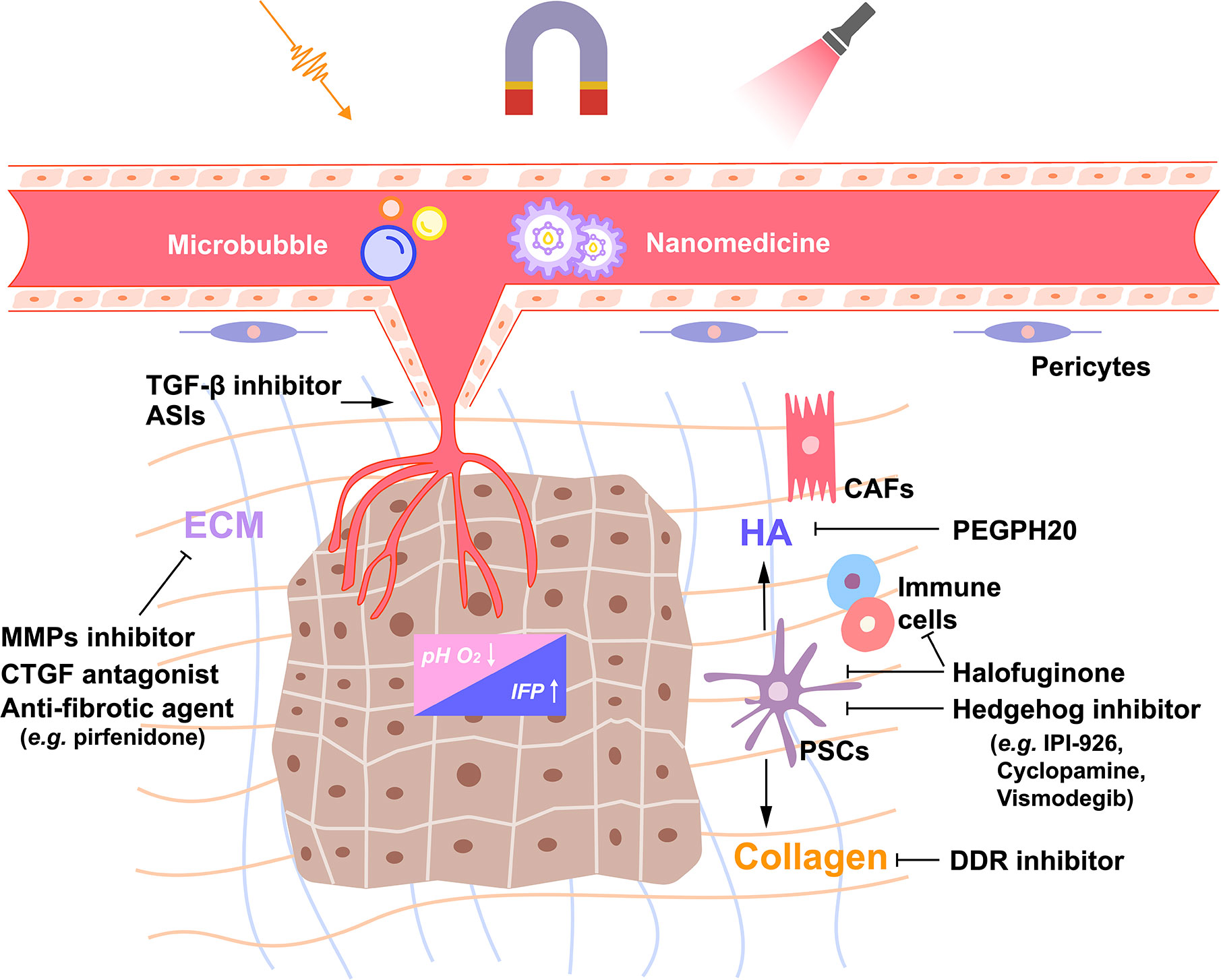
Figure 1 Schematic of the extracellular matrix network and stroma-targeting strategies in pancreatic ductal adenocarcinoma (PDAC). The dense desmoplasia stroma of PDAC consists of several cellular, acellular and biophysical components, which interact mutually to promote the growth and metastasis of PDAC. Inactivating pancreatic stellate cells (PSCs) or a certain population of cancer-associated fibroblasts (CAFs) by Hedgehog inhibitors and halofuginone or altering the products these CAFs produce, such as hyaluronic acid (HA) and collagen, by PEGPH20 or discoidin domain receptor (DDR) inhibitor might be potential strategies to reverse the dense dysplastic stroma of PDAC. In addition, remodeling the tumor vasculature by transforming growth factor-β (TGF-β) inhibitors or angiotensin system inhibitors (ASIs) decreases the interstitial fluid pressure (IFP), reverses acidosis, and the hypoxic environment, and enhances the efficacy of drug delivery. Furthermore, nano-drug delivery systems have been designed to respond to environmental or external stimuli which triggers drug release. T-mark represents inhibition; arrow, activation. ECM, extracellular matrix; MMPs, matrix metalloproteinase; CTGF, connective tissue growth factor.
Another major component of the tumor ECM is collagen (Figure 1). PDAC stroma is rich in fibrillar collagens, which support tumor cell survival and promote tumor progression through discoidin domain receptor 1 and 2 (DDR1 and 2). High expression of DDR1 was identified as an independent risk factor for poor prognosis in patients with PDAC (31). In fact, the selective small molecule inhibitor of DDR1 reduced collagen deposition and improved the response to chemotherapy in PDAC mouse models (32). Preclinical studies are currently investigating the effects of DDR1 inhibition on the immune microenvironment of PDAC. Recently, the potent antifibrotic ability of halofuginone (a natural quinazolinone alkaloid febrifugine analog) was observed in a genetically engineered mouse model of PDAC (33). Halofuginone directly inhibited the activation of PSCs, thereby reducing the deposition of several ECM components, including collagen and HA. The decrease of the intratumoral IFP resulted in the improved delivery of chemotherapeutic agents, which was visually proven by exploiting the autofluorescence of doxorubicin. In addition, halofuginone helped increase immune cell infiltration and shift them closer to cytokeratin positive tumor cells, suggesting that stroma-targeting therapy might improve the efficacy of immunotherapy to some extent in PDAC.
The Hedgehog (Hh) signaling pathway is generally hyperactive in PDAC (Figure 1) (34). It is known that the Hh signaling pathway activates PSCs through paracrine effects and regulates stroma deposition (35). There are several strategies intended to treat PDAC by inhibiting the Hh signaling pathway in order to eliminate the tumor stroma (36). Cyclopamine is a natural steroidal alkaloid that inhibits the Hh signaling pathway via binding to Smoothened (SMO) (37). It reduced fibronectin content and enhanced tumor vascularization in a PDAC xenograft mouse model. When combined with paclitaxel-loaded nanoparticles, cyclopamine increased tumor growth inhibition by 63.3% (38). In addition, a preclinical study showed that IPI-926, a small molecule that inhibited the Hh pathway, increased blood vessel density and gemcitabine drug concentration in PDAC (39). In a phase I clinical trial, IPI-926 was well tolerated by PDAC patients (40). However, a phase II clinical trial of IPI-926 failed to show significant therapeutic benefits in PDAC patients (41, 42). Similarly, Vismodegib, another Hh pathway inhibitor, combined with gemcitabine did not significantly improve the median overall survival and progression-free survival in patients with metastatic PDAC compared with gemcitabine alone (43). In fact, while several treatments targeting the Hh signaling pathway showed uplifting results in preclinical models (44), few of them improved patient survival, and their use was often accompanied by drug-related toxicity (45). In addition to the above-mentioned drugs, other stroma-targeting drugs include metalloproteinase inhibitors (46–48), connective tissue growth factor (CTGF) antagonists (e.g., pamrevlumab) (49, 50), and anti-fibrotic agents (e.g., pirfenidone) (Figure 1) (51, 52).
Remodeling Tumor Vasculature
The dense fibrotic stroma surrounding the blood vessels and the proliferating cancer-associated fibroblasts (CAFs) damage intratumoral vasculature, resulting in a hypoxic microenvironment, which promotes PDAC invasion, metastasis, and the acquisition of chemoresistance phenotypes (Figure 1) (53–55). Therefore, remodeling PDAC blood vessels not only improves drug delivery but also overcomes the hypoxic microenvironment.
The transforming growth factor-β (TGF-β) signaling pathway is involved in the adhesion of pericytes to tumor vascular endothelial cells (56). TGF-β receptor kinase inhibitors or monoclonal antibodies enhanced vascular access and promoted the perfusion of chemotherapeutic agents to PDAC tumor sites (56–58). In addition, the repurposing of antihypertensive drugs targeting the renin-angiotensin pathway can reprogram CAFs towards normalizing ECM by inhibiting TGF-β (59, 60). In tumors, Angiotensin II (Ang II) activates TGF-β and promotes CAFs to express CTGF by binding to Ang II type 1 receptor (AT1R) (61). Moreover, Ang II stimulates proliferation of PSCs by activating protein kinase C and EGF-ERK signaling pathways (61, 62). Several angiotensin system inhibitors (ASIs) have been used to target the PDAC stroma (59, 63). A phase II clinical study evaluated the efficacy of neoadjuvant chemoradiotherapy with FOLFIRINOX combined with losartan in patients with locally advanced PDAC. The therapy was associated with a high proportion (69%) of patients achieving R0 resection (64). Interestingly, the lack of Angiotensin II type 2 receptor (AT2R) in pancreatic fibroblasts also leads to tumor cell proliferation (65). Therefore, the role of the Ang II signaling pathway in the PDAC stroma needs to be further investigated before its use in the clinic.
In addition to the pharmacological interference, focused ultrasound combined with microbubbles was investigated to determine if it improved vascular patency. Microbubbles are restrained within the vascular compartment because of their diameter. Focused ultrasound causes the microbubbles to disintegrate, which releases acoustic forces that are capable of inducing thrombolysis and increasing drug penetration (66). Moreover, appropriate ultrasound energy induces the destruction of the microbubbles and generates acoustic pressure that temporarily increases the permeability of blood vessels and cell membranes (66). The efficacy of ultrasound microbubble delivery of gemcitabine was evaluated in a PDAC murine model. The study showed that the ultrasound microbubbles treatment group presented significant tumor growth inhibition (67). In addition, the use of nab-paclitaxel combined with ultrasound microbubbles significantly inhibited the activity of PDAC tumor cells in vitro and reduced the tumor volume in a subcutaneous PDAC mouse model (68). Furthermore, a clinical trial evaluated the efficacy of focused ultrasound combined with microbubble delivery of gemcitabine in PDAC patients. The combination-treated patients tolerated more gemcitabine chemotherapeutic cycles, and half of them showed maximum tumor diameter decrease. In addition, the median survival was prolonged by 8.7 months (69).
Nano-Drug Delivery Systems in PDAC
The dense stroma of PDAC limits the application of chemotherapeutic drugs to efficiently target neoplastic cells through the enhanced permeability and retention (EPR) effect (15, 70). Nanomedicine, the result of the formulation of drugs into nano-size delivery carriers, such as liposomes and polymer nanoparticles, delivers chemotherapeutic agents through mechanisms that are different from those used to deliver conventional non-encapsulated molecular drugs (71). By optimizing the particle size, surface charge, and specific ligand modification of nanocarriers, nanomedicine improves the ability of nanocarriers to penetrate the stromal barrier in PDAC tumor sites (72). A study showed that both 30-nm and 100-nm nanoparticles penetrated into the hyperpermeable colon cancer model, but only the 30-nm nanoparticle penetrated the stroma-rich PDAC model and achieved anti-tumor efficacy in the murine model (73). Different charges on ECM components make the nano-drug delivery heterogeneous in spatial distribution, compromising the drug delivery efficiency (74). Therefore, active targeted drug delivery with modified specific ligands has been designed to address the issues (75, 76). Several studies showed that nanoparticle albumin–bound paclitaxel (Nab-paclitaxel) improved the delivery efficiency of paclitaxel. In addition, nab-paclitaxel combined with gemcitabine improved the overall survival and disease-free survival in patients with advanced PDAC (77), and this combination therapy strategy has been approved by the Food and Drug Administration (FDA). Several nab-paclitaxel-based combination therapy strategies are currently in the clinical evaluation stage (78, 79). A recent meta-analysis showed that nanoparticles improved the efficacy of various anti-tumor drugs while reducing their toxicity in patients with PDAC (80).
To further improve the efficiency of nano-delivery drugs, smart nanoparticles have been designed to respond to environmental or external stimuli, which triggers drug release after passive or active tumor accumulation (81). The PDAC tumor microenvironment exhibits some unique physiological characteristics, such as the low pH, cancer cells lysosomes (81), and a high level of reducing substances in tumor cells (e.g., glutathione) (82). Therefore, a nano-drug delivery system responsive to the stimuli from the internal environment of the tumor has been designed. In addition, researchers have also designed nano-drug delivery systems triggered by the external stimuli. Thermal, magnetic, and mechanical waves (including ultrasound) via image guidance have been applied to trigger the nano-drug delivery system to release the loaded drug. Furthermore, a study combined the internal and external stimuli and designed a new nano-drug carrier to respond to a three-dimensional trigger (ultrasonic, acid-sensitivity and reduced glutathione) (Figure 1) (83).
Stroma-Targeting Therapy as a Double-Edged Sword
The tumor suppressive properties of the PDAC stroma have been revealed gradually. Several studies, using genetic or pharmacological approaches to eliminate the PDAC stroma in preclinical models, showed that stromal depletion promoted tumor cell proliferation, invasion, and metastasis and reduced survival (84, 85). PDAC tumor cells in the primary lesion showed loss of differentiation, epithelial-to-mesenchymal transition (EMT), and enhanced cancer stem cell-like phenotypes after stromal ablative treatment. In addition, severe weight loss, acidosis, and cachexia were observed in PDAC mouse models (84–87). These findings suggest that stroma depletion might activate dormant neoplastic cells and induce their metastatic potential, thereby promoting PDAC progression.
Prominently, different subtypes of CAFs that exhibit distinct characteristics and activity levels have been identified in breast cancer (88) and PDAC (89). In PDAC, CAFs have been classified into several subpopulations on the basis of the expression patterns of various fibroblast markers (89, 90). One subgroup is characterized by the α-SMA expression and generally considered as myofibroblastic CAFs, while another is classified as inflammatory CAFs for the secretion of multiple inflammatory chemokines (e.g., IL-6). Studies have assessed the effects of these CAFs subgroups on tumor microenvironment and immune escape in PDAC. For example, the targeted inhibition of IL-6, which is produced by inflammatory CAFs, improved the efficacy of anti-PD-L1 checkpoint inhibitors in PDAC. However, the genetic deletion of Hedgehog resulted in the reduction of stroma deposition and myofibroblastic CAFs, inducing more aggressive and de-differentiated tumor phenotypes (85). Similarly, another study determined that genetic deletion of myofibroblastic CAFs induced more aggressive PDAC tumor phenotypes, manifested by EMT, cancer stemness, chemotherapy resistance, and tumor immune evasion (84). These findings suggest that inflammatory CAFs might be the potential therapeutic target, while myofibroblastic CAFs support tumor suppression in PDAC. Recently, Biffi et al. identified that TGFβ and interleukin 1 (IL1) as tumor-secreted ligands promoted CAFs heterogeneity, illuminating strategies to selectively target CAFs that support tumor growth (91).
Pericyte is an important cellular component related to the tumor vasculature in PDAC (92). One of the main biomarkers associated with vascular pericytes is platelet-derived growth factor receptor-β (PDGFR-β) (93). Its ligand, PDGF, was shown to induce pericyte-fibroblast transition (PFT), which makes pericyte an origin of CAFs, thereby promoting tumor invasion and metastasis. Pharmacological inhibition and genetic deletion of PDGFR-β reversed PDGF induced PFT (92). However, PDGFR-β inhibitors (e.g., imatinib and sunitinib) caused pericytes to detach from vascular endothelial cells, destroying vasculature integrity and promoting tumor metastasis (93). Therefore, non-discriminatory depletion of CAFs might remove partial tumor-suppressing CAFs subgroups. Although stroma-targeting therapy enhances the delivery of chemotherapeutic agents, it might also promote tumor chemoresistance and metastasis. In fact, the unsatisfactory results in clinical applications and the revelation that the PDAC stroma harbors tumor suppressive activity indicates that future research should focus on the tumor ECM biology. The various components in the PDAC stroma might have different effects on tumor biological behavior. Therefore, the pathophysiological analysis of distinct PDAC stromal cell might enlighten future stroma-targeting strategies (90).
Discussion and Perspective
The dense desmoplasia stroma of PDAC, which hinders the efficient delivery of chemotherapeutic agents, plays a critical role in tumor progression and metastasis. On the one hand, various stroma-targeting treatment strategies have been applied to improve drug efficacy and inhibit tumor progression. However, the clinical efficacy of these treatments has not been satisfactory in PDAC patients. On the other hand, several studies showed that extreme PDAC stromal depletion caused a deterioration in tumor phenotypes, supporting the perspective that the stromal response is a host response to inhibit tumor growth. Therefore, a stroma-targeting treatment might not be an optimal neoadjuvant therapy for patients with locally advanced PDAC.
It is worth noting that lack of taking the heterogeneity of PDAC stromal composition into account might be one reason why unsatisfactory results were observed in the clinical studies. Stroma-targeting therapy would be beneficial in sites with high density of the targeted stromal composition, while be a disadvantage in sites with low density. Therefore, it is crucial to access the quantitative degree and type of tumor stroma density (TSD), which might be a significant section of the clinical study design (94). The phase II study, HALO-109-202, was an example of stratification analysis based on the level of HA (30). In addition, the activation status of stroma could be identified, according to consensus clustering of expression levels of certain biomarkers (95). For instance, prior work found that A Disintegrin And Metalloproteases 12 (ADAM12) was a blood-borne proxy for stromal activation, the levels of which had prognostic significance and correlate with treatment benefit (96). In fact, the stroma is a reservoir of various potential biomarkers, which might be used to classify PDAC patient subgroups and facilitate the clinical application of stroma-targeting therapy.
In addition, the heterogeneity of stromal microenvironment between the primary tumor and metastases sites might result in distinct responses to stroma-targeting therapy (94). Previous studies showed that PDAC metastatic tumors had limited driver-mutation heterogeneity compared to the primary tumors (97), and TSD was various across metastatic sites and primary tumors (94). These observations suggested that the heterogeneity of stroma might be a function of host organ-site physiology and not inferior to cancer genomic or epigenomic variations between the primary and metastatic tumors. Furthermore, the effects of individual and ethnic-specific genetic variations on the stroma microenvironment of PDAC might need further investigation. Genomic analyses of single PDAC stroma cell might help identify inter- and intra-individual heterogeneity and optimize individualized stroma-targeting therapy in order to maximize patient treatment benefits (98–100).
Pharmacological agents that alter the tumor microenvironment might help evade the negative effect of stromal depletion. For example, the inhibition of PDAC tumor-associated macrophages by clodronate liposomes prevented the establishment of a pre-metastatic microenvironment in organs of metastasis, markedly reducing metastasis formation (101). Therefore, clodronate liposomes might be a candidate for combination strategies with stroma-targeting therapy. Furthermore, although the proliferative stroma of PDAC usually exerts an immunosuppressive effect, stromal depletion might exacerbate the immunosuppression effect in some cases (84). Therefore, the combination of immune checkpoint inhibitors and stroma-targeting therapy might reduce the negative effects of stromal ablation and further enhance the efficacy of chemotherapy in PDAC patients.
In summary, stroma-targeting therapy in PDAC has emerged as a double-edged sword. On the one hand, it enhances the delivery of chemotherapeutic agents to tumor sites. On the other hand, it eliminates the physical and biochemical barriers that potentially inhibit tumor progression. Therefore, it is imperative to understand the complex role of the PDAC stroma in order to improve the current stroma-targeting therapies through appropriately adjusting the balance between stroma deposition and ablation.
Author Contributions
BJ and JG managed the study design. BJ, JL, and YW conducted the literature search. CL performed the data extraction. BJ drafted the manuscript. LZ, LY, and JG reviewed the manuscript and made critical revisions for important intellectual content. JG provided funding support. All authors contributed to the article and approved the submitted version.
Funding
The present study was supported by the National Natural Science Foundation of China (Grant No. 81972324) and the China Academy of Medical Sciences Innovation Fund for Medical Sciences (Grant No. 2016-I2M-3-019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Nicole Clarke, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin (2015) 65(2):87–108. doi: 10.3322/caac.21262
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
3. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res (2014) 74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155
4. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin (2016) 66(2):115–32. doi: 10.3322/caac.21338
5. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet (2016) 388(10039):73–85. doi: 10.1016/S0140-6736(16)00141-0
6. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med (2018) 379(25):2395–406. doi: 10.1056/NEJMoa1809775
7. Maeda S, Unno M, Yu J. Adjuvant and neoadjuvant therapy for pancreatic cancer. J Pancreatol (2019) 2(3):100–6. doi: 10.1097/JP9.0000000000000028
8. Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol (2012) 9(8):454–67. doi: 10.1038/nrgastro.2012.115
9. Dougan SK. The Pancreatic Cancer Microenvironment. Cancer J (2017) 23(6):321–5. doi: 10.1097/PPO.0000000000000288
10. Yang F, Jin C, Subedi S, Lee CL, Wang Q, Jiang Y, et al. Emerging inorganic nanomaterials for pancreatic cancer diagnosis and treatment. Cancer Treat Rev (2012) 38(6):566–79. doi: 10.1016/j.ctrv.2012.02.003
11. Kota J, Hancock J, Kwon J, Korc M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett (2017) 391:38–49. doi: 10.1016/j.canlet.2016.12.035
12. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut (2011) 60(6):861–8. doi: 10.1136/gut.2010.226092
13. Xu Z, Pothula SP, Wilson JS, Apte MV. Pancreatic cancer and its stroma: a conspiracy theory. World J Gastroenterol (2014) 20(32):11216–29. doi: 10.3748/wjg.v20.i32.11216
14. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol (2017) 10(1):76. doi: 10.1186/s13045-017-0448-5
15. Adiseshaiah PP, Crist RM, Hook SS, McNeil SE. Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer. Nat Rev Clin Oncol (2016) 13(12):750–65. doi: 10.1038/nrclinonc.2016.119
16. Meng H, Nel AE. Use of nano engineered approaches to overcome the stromal barrier in pancreatic cancer. Adv Drug Deliv Rev (2018) 130:50–7. doi: 10.1016/j.addr.2018.06.014
17. Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab Dispos (1992) 20(6):849–55.
18. Thind K, Padrnos LJ, Ramanathan RK, Borad MJ. Immunotherapy in pancreatic cancer treatment: a new frontier. Therap Adv Gastroenterol (2017) 10(1):168–94. doi: 10.1177/1756283X16667909
19. Fries H, Elsasser HP, Mahlbacher V, Neumann K, Kern HF. Localisation of hyaluronate (HA) in primary tumors and nude mouse xenografts of human pancreatic carcinomas using a biotinylated HA-binding protein. Virchows Arch (1994) 424(1):7–12. doi: 10.1007/BF00197386
20. Knudson CB, Knudson W. Hyaluronan-binding proteins in development, tissue homeostasis, and disease. FASEB J (1993) 7(13):1233–41. doi: 10.1096/fasebj.7.13.7691670
21. Mahlbacher V, Sewing A, Elsasser HP, Kern HF. Hyaluronan is a secretory product of human pancreatic adenocarcinoma cells. Eur J Cell Biol (1992) 58(1):28–34.
22. Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther (2010) 9(11):3052–64. doi: 10.1158/1535-7163.MCT-10-0470
23. Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle (2008) 7(21):3448–60. doi: 10.4161/cc.7.21.7014
24. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6(8):583–92. doi: 10.1038/nrc1893
25. Toole BP, Slomiany MG. Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Semin Cancer Biol (2008) 18(4):244–50. doi: 10.1016/j.semcancer.2008.03.009
26. Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer (2013) 108(1):1–8. doi: 10.1038/bjc.2012.569
27. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62(1):112–20. doi: 10.1136/gutjnl-2012-302529
28. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther (2015) 14(2):523–32. doi: 10.1158/1535-7163.MCT-14-0580
29. Whatcott CJ, Han H, Von Hoff DD. Orchestrating the Tumor Microenvironment to Improve Survival for Patients With Pancreatic Cancer: Normalization, Not Destruction. Cancer J (2015) 21(4):299–306. doi: 10.1097/PPO.0000000000000140
30. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol (2018) 36(4):359–66. doi: 10.1200/JCO.2017.74.9564
31. Huo Y, Yang M, Liu W, Yang J, Fu X, Liu D, et al. High expression of DDR1 is associated with the poor prognosis in Chinese patients with pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res (2015) 34(1):88–. doi: 10.1186/s13046-015-0202-1
32. Aguilera KY, Huang H, Du W, Hagopian MM, Wang Z, Hinz S, et al. Inhibition of Discoidin Domain Receptor 1 Reduces Collagen-mediated Tumorigenicity in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther (2017) 16(11):2473–85. doi: 10.1158/1535-7163.MCT-16-0834
33. Elahi-Gedwillo KY, Carlson M, Zettervall J, Provenzano PP. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res (2019) 79(2):372–86. doi: 10.1158/0008-5472.CAN-18-1334
34. Rucki AA, Zheng L. Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies. World J Gastroenterol (2014) 20(9):2237–46. doi: 10.3748/wjg.v20.i9.2237
35. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature (2008) 455(7211):406–10. doi: 10.1038/nature07275
36. Onishi H, Katano M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol (2014) 20(9):2335–42. doi: 10.3748/wjg.v20.i9.2335
37. Heretsch P, Tzagkaroulaki L, Giannis A. Cyclopamine and hedgehog signaling: chemistry, biology, medical perspectives. Angew Chem Int Ed Engl (2010) 49(20):3418–27. doi: 10.1002/anie.200906967
38. Zhang B, Jiang T, Shen S, She X, Tuo Y, Hu Y, et al. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials (2016) 103:12–21. doi: 10.1016/j.biomaterials.2016.06.048
39. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science (2009) 324(5933):1457–61. doi: 10.1126/science.1171362
40. Jimeno A, Weiss GJ, Miller WH Jr., Gettinger S, Eigl BJ, Chang AL, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res (2013) 19(10):2766–74. doi: 10.1158/1078-0432.CCR-12-3654
41. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med (2013) 19(11):1410–22. doi: 10.1038/nm.3389
42. Infinity Pharmaceuticals. Infinity reports update from phase 2 study of saridegib plus gemcitabine in patients with metastatic pancreatic cancer. Available at: http://www.businesswire.com/news/home/20120127005146/en/Infinity-Reports-Update-Phase-2-Study-Saridegib#.Vg2sAuGJMQM (Accessed January 27, 2012).
43. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol (2015) 33(36):4284–92. doi: 10.1200/JCO.2015.62.8719
44. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of Hedgehog Signaling Inhibits Pancreatic Cancer Invasion and Metastases: A New Paradigm for Combination Therapy in Solid Cancers. Cancer Res (2007) 67(5):2187–96. doi: 10.1158/0008-5472.CAN-06-3281
45. Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas (2016) 45(3):370–5. doi: 10.1097/MPA.0000000000000458
46. Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: a randomized trial. J Clin Oncol (2001) 19(15):3447–55. doi: 10.1200/JCO.2001.19.15.3447
47. Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JAC. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer (2002) 87(2):161–7. doi: 10.1038/sj.bjc.6600446
48. Moore MJ, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol (2003) 21(17):3296–302. doi: 10.1200/JCO.2003.02.098
49. Neesse A, Frese KK, Bapiro TE, Nakagawa T, Sternlicht MD, Seeley TW, et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci U S A (2013) 110(30):12325–30. doi: 10.1073/pnas.1300415110
50. Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest (2011) 121(10):4106–17. doi: 10.1172/JCI42754
51. Kozono S, Ohuchida K, Eguchi D, Ikenaga N, Fujiwara K, Cui L, et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res (2013) 73(7):2345–56. doi: 10.1158/0008-5472.CAN-12-3180
52. Ji T, Li S, Zhang Y, Lang J, Ding Y, Zhao X, et al. An MMP-2 Responsive Liposome Integrating Antifibrosis and Chemotherapeutic Drugs for Enhanced Drug Perfusion and Efficacy in Pancreatic Cancer. ACS Appl Mater Interfaces (2016) 8(5):3438–45. doi: 10.1021/acsami.5b11619
53. Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, et al. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia (2009) 11(5):497–508. doi: 10.1593/neo.81618
54. Guillaumond F, Iovanna JL, Vasseur S. Pancreatic tumor cell metabolism: focus on glycolysis and its connected metabolic pathways. Arch Biochem Biophys (2014) 545:69–73. doi: 10.1016/j.abb.2013.12.019
55. Bailey KM, Cornnell HH, Ibrahim-Hashim A, Wojtkowiak JW, Hart CP, Zhang X, et al. Evaluation of the “steal” phenomenon on the efficacy of hypoxia activated prodrug TH-302 in pancreatic cancer. PLoS One (2014) 9(12):e113586. doi: 10.1371/journal.pone.0113586
56. ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol (2007) 8(11):857–69. doi: 10.1038/nrm2262
57. Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A (2007) 104(9):3460–5. doi: 10.1073/pnas.0611660104
58. Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A (2012) 109(41):16618–23. doi: 10.1073/pnas.1117610109
59. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun (2013) 4:2516. doi: 10.1038/ncomms3516
60. Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med (2017) 9(410):eaan5616. doi: 10.1126/scitranslmed.aan5616
61. Hama K, Ohnishi H, Yasuda H, Ueda N, Mashima H, Satoh Y, et al. Angiotensin II stimulates DNA synthesis of rat pancreatic stellate cells by activating ERK through EGF receptor transactivation. Biochem Biophys Res Commun (2004) 315(4):905–11. doi: 10.1016/j.bbrc.2004.01.155
62. Hama K, Ohnishi H, Aoki H, Kita H, Yamamoto H, Osawa H, et al. Angiotensin II promotes the proliferation of activated pancreatic stellate cells by Smad7 induction through a protein kinase C pathway. Biochem Biophys Res Commun (2006) 340(3):742–50. doi: 10.1016/j.bbrc.2005.12.069
63. Masamune A, Hamada S, Kikuta K, Takikawa T, Miura S, Nakano E, et al. The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand J Gastroenterol (2013) 48(5):602–9. doi: 10.3109/00365521.2013.777776
64. Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY, et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol (2019) 5(7):1020–7. doi: 10.1001/jamaoncol.2019.0892
65. Doi C, Egashira N, Kawabata A, Maurya DK, Ohta N, Uppalapati D, et al. Angiotensin II type 2 receptor signaling significantly attenuates growth of murine pancreatic carcinoma grafts in syngeneic mice. BMC Cancer (2010) 10:67. doi: 10.1186/1471-2407-10-67
66. Kiessling F, Fokong S, Koczera P, Lederle W, Lammers T. Ultrasound microbubbles for molecular diagnosis, therapy, and theranostics. J Nucl Med (2012) 53(3):345–8. doi: 10.2967/jnumed.111.099754
67. Nesbitt H, Sheng Y, Kamila S, Logan K, Thomas K, Callan B, et al. Gemcitabine loaded microbubbles for targeted chemo-sonodynamic therapy of pancreatic cancer. J Control Release (2018) 279:8–16. doi: 10.1016/j.jconrel.2018.04.018
68. Bressand D, Novell A, Girault A, Raoul W, Fromont-Hankard G, Escoffre J-M, et al. Enhancing Nab-Paclitaxel Delivery Using Microbubble-Assisted Ultrasound in a Pancreatic Cancer Model. Mol Pharm (2019) 16(9):3814–22. doi: 10.1021/acs.molpharmaceut.9b00416
69. Dimcevski G, Kotopoulis S, Bjånes T, Hoem D, Schjøtt J, Gjertsen BT, et al. A human clinical trial using ultrasound and microbubbles to enhance gemcitabine treatment of inoperable pancreatic cancer. J Control Release (2016) 243:172–81. doi: 10.1016/j.jconrel.2016.10.007
70. Tanaka HY, Kano MR. Stromal barriers to nanomedicine penetration in the pancreatic tumor microenvironment. Cancer Sci (2018) 109(7):2085–92. doi: 10.1111/cas.13630
71. Kalyane D, Raval N, Maheshwari R, Tambe V, Kalia K, Tekade RK. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater Sci Eng C Mater Biol Appl (2019) 98:1252–76. doi: 10.1016/j.msec.2019.01.066
72. Ji T, Zhao Y, Ding Y, Nie G. Using functional nanomaterials to target and regulate the tumor microenvironment: diagnostic and therapeutic applications. Adv Mater (2013) 25(26):3508–25. doi: 10.1002/adma.201300299
73. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol (2011) 6(12):815–23. doi: 10.1038/nnano.2011.166
74. Lieleg O, Baumgärtel RM, Bausch AR. Selective filtering of particles by the extracellular matrix: an electrostatic bandpass. Biophys J (2009) 97(6):1569–77. doi: 10.1016/j.bpj.2009.07.009
75. Stylianopoulos T, Jain RK. Design considerations for nanotherapeutics in oncology. Nanomed Nanotechnol Biol Med (2015) 11(8):1893–907. doi: 10.1016/j.nano.2015.07.015
76. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol (2007) 2(12):751–60. doi: 10.1038/nnano.2007.387
77. Borazanci E, Von Hoff DD. Nab-paclitaxel and gemcitabine for the treatment of patients with metastatic pancreatic cancer. Expert Rev Gastroenterol Hepatol (2014) 8(7):739–47. doi: 10.1586/17474124.2014.925799
78. Hoy SM. Albumin-bound paclitaxel: a review of its use for the first-line combination treatment of metastatic pancreatic cancer. Drugs (2014) 74(15):1757–68. doi: 10.1007/s40265-014-0291-8
79. Vaz J, Ansari D, Sasor A, Andersson R. SPARC: A Potential Prognostic and Therapeutic Target in Pancreatic Cancer. Pancreas (2015) 44(7):1024–35. doi: 10.1097/MPA.0000000000000409
80. Au M, Emeto T, Power J, Vangaveti V, Lai H. Emerging Therapeutic Potential of Nanoparticles in Pancreatic Cancer: A Systematic Review of Clinical Trials. Biomedicines (2016) 4(3):20. doi: 10.3390/biomedicines4030020
81. van Elk M, Murphy BP, Eufrásio-da-Silva T, O’Reilly DP, Vermonden T, Hennink WE, et al. Nanomedicines for advanced cancer treatments: Transitioning towards responsive systems. Int J Pharm (2016) 515(1-2):132–64. doi: 10.1016/j.ijpharm.2016.10.013
82. Anajafi T, Scott MD, You S, Yang X, Choi Y, Qian SY, et al. Acridine Orange Conjugated Polymersomes for Simultaneous Nuclear Delivery of Gemcitabine and Doxorubicin to Pancreatic Cancer Cells. Bioconjug Chem (2016) 27(3):762–71. doi: 10.1021/acs.bioconjchem.5b00694
83. Yang P, Li D, Jin S, Ding J, Guo J, Shi W, et al. Stimuli-responsive biodegradable poly(methacrylic acid) based nanocapsules for ultrasound traced and triggered drug delivery system. Biomaterials (2014) 35(6):2079–88. doi: 10.1016/j.biomaterials.2013.11.057
84. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell (2015) 28(6):831–3. doi: 10.1016/j.ccell.2015.11.002
85. Rhim Andrew D, Oberstein Paul E, Thomas Dafydd H, Mirek Emily T, Palermo Carmine F, Sastra Stephen A, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
86. Lee JJ, Perera RM, Wang H, Wu D-C, Liu XS, Han S, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A (2014) 111(30):E3091–E100. doi: 10.1073/pnas.1411679111
87. Roberts KJ, Kershner AM, Beachy PA. The Stromal Niche for Epithelial Stem Cells: A Template for Regeneration and a Brake on Malignancy. Cancer Cell (2017) 32(4):404–10. doi: 10.1016/j.ccell.2017.08.007
88. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
89. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
90. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov (2019) 9(8):1102–23. doi: 10.1158/2159-8290.CD-19-0094
91. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov (2019) 9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710
92. Hosaka K, Yang Y, Seki T, Fischer C, Dubey O, Fredlund E, et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci U S A (2016) 113(38):E5618–E27. doi: 10.1073/pnas.1608384113
93. Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell (2012) 21(1):66–81. doi: 10.1016/j.ccr.2011.11.024
94. Torphy RJ, Wang Z, True-Yasaki A, Volmar KE, Rashid N, Yeh B, et al. Stromal Content Is Correlated With Tissue Site, Contrast Retention, and Survival in Pancreatic Adenocarcinoma. JCO Precis Oncol (2018) 2018. doi: 10.1200/PO.17.00121
95. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet (2015) 47(10):1168–78. doi: 10.1038/ng.3398
96. Veenstra VL, Damhofer H, Waasdorp C, van Rijssen LB, van de Vijver MJ, Dijk F, et al. ADAM12 is a circulating marker for stromal activation in pancreatic cancer and predicts response to chemotherapy. Oncogenesis (2018) 7(11):87. doi: 10.1038/s41389-018-0096-9
97. Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet (2017) 49(3):358–66. doi: 10.1038/ng.3764
98. Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology (2018) 155(6):1999–2013.e3. doi: 10.1053/j.gastro.2018.08.033
99. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531(7592):47–52. doi: 10.1038/nature16965
100. Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature (2015) 518(7540):495–501. doi: 10.1038/nature14169
Keywords: pancreatic ductal adenocarcinoma, stroma, chemotherapy, tumor microenvironment, nanotherapy
Citation: Jiang B, Zhou L, Lu J, Wang Y, Liu C, You L and Guo J (2020) Stroma-Targeting Therapy in Pancreatic Cancer: One Coin With Two Sides? Front. Oncol. 10:576399. doi: 10.3389/fonc.2020.576399
Received: 26 June 2020; Accepted: 25 September 2020;
Published: 15 October 2020.
Edited by:
Jorg Kleeff, University Hospital in Halle, GermanyReviewed by:
Maarten Fokke Bijlsma, Amsterdam University Medical Center (UMC), NetherlandsJinrong Zhu, Guangdong Pharmaceutical University, China
Copyright © 2020 Jiang, Zhou, Lu, Wang, Liu, You and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junchao Guo, Z2pjcHVtY2hAMTYzLmNvbQ==