 Chunyue Feng
Chunyue Feng Xiaoli Huang1,2
Xiaoli Huang1,2 Xuekun Li
Xuekun Li Jianhua Mao
Jianhua Mao- 1The Children’s Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2 National Clinical Research Center for Child Health, Hangzhou, China
- 3Institute of Translational Medicine of Zhejiang University School of Medicine, Hangzhou, China
Epigenetic modifications including histone modifications and DNA and RNA modifications are involved in multiple biological processes and human diseases. One disease, kidney cancer, includes a common type of tumor, accounts for about 2% of all cancers, and usually has poor prognosis. The molecular mechanisms and therapeutic strategy of kidney cancer are still under intensive study. Understanding the roles of epigenetic modifications and underlying mechanisms in kidney cancer is critical to its diagnosis and clinical therapy. Recently, the function of DNA and RNA modifications has been uncovered in kidney tumor. In the present review, we summarize recent findings about the roles of epigenetic modifications (particularly DNA and RNA modifications) in the incidence, progression, and metastasis of kidney cancer, especially the renal cell carcinomas.
Introduction
Kidney cancer presents about 2% of all cancers and is the seventh most common cancer worldwide with 295,000 new cases being diagnosed annually (1). The most prevalent solid tumor of the kidney in adults is renal cell carcinoma (RCC), which accounts for about 90% of adult kidney cancer (2–4). RCC is a heterogeneous malignant tumor with more than ten histological subtypes, although it mainly stems from renal tubular epithelial cells. In addition to the high prevalence of kidney cancer in adults, this disease can also be diagnosed in children, where the main form is Wilms tumor (5). Because of the high malignancy rate and the unclear mechanisms of kidney cancer, current treatments, which include surgery, chemotherapy and radiation, cannot significantly inhibit tumor progression. In the past few years, targeted therapy has been shown to prolong survival of patients, but the overall survival rate still remains very low (4).
Epigenetic modifications including histone modifications, DNA and RNA modifications, and non-coding RNAs regulate gene expression at transcriptional, translational and posttranslational levels and therefore are involved in human diseases (6). DNA methylation at the 5’ position of cytosine (5-methylcytosine, 5mC) is an intensively studied type of epigenetic modification, and it plays a critical role in development and diseases (7). In addition, more than one hundred types of RNA modifications have been identified on mRNA, tRNA, etc. Among all RNA modifications, N6-methyladenosine (m6A) is the most common modification in eukaryotic mRNAs (8). RNA modification has been shown to play important roles in multiple biological processes and in diseases, as well as in DNA methylation (9). The dysfunction of epigenetic modifications leads to global changes in genomic structure and thus affects the expression of genes involved in cancer progression (10, 11).
During the past decade the important roles of epigenetic modifications have been revealed in kidney cancer (especially in RCC). Epigenetic alterations have been suggested as promising biomarkers for RCC diagnosis and potential therapeutic targets (3, 4, 11–14). In this review we summarize the landscape of main epigenetic modifications with a focus on DNA methylation and RNA methylation. We then discuss the function and underlying mechanisms of aberrant DNA and RNA modifications in kidney cancer.
DNA Modifications and Kidney Cancer
Diverse Modifications of DNA
DNA methylation mainly occurs at the fifth carbon atom of cytosine (5mC) in mammalian DNA and is catalyzed by DNA methyltransferases (DNMTs), which use S-adenosyl methionine (SAM) as a methyl group donor. Currently, there are five members of the DNMT family, which includes DNMT1, DNMT3a, and DNMT3b. DNMT1 displays a preference for hemi-methylated DNA at the CpG islands during DNA replication, whereas DNMT3a and DNMT3b are de novo methyltransferases. DNA methylation exhibits dynamic features of expression during embryonic and postnatal development, and the dysregulation of DNA methylation has been shown to result in changes in gene expression (15). In general, hypomethylation activates or increases gene expression, whereas hypermethylation leads to gene silencing or decreased gene expression (Figure 1).
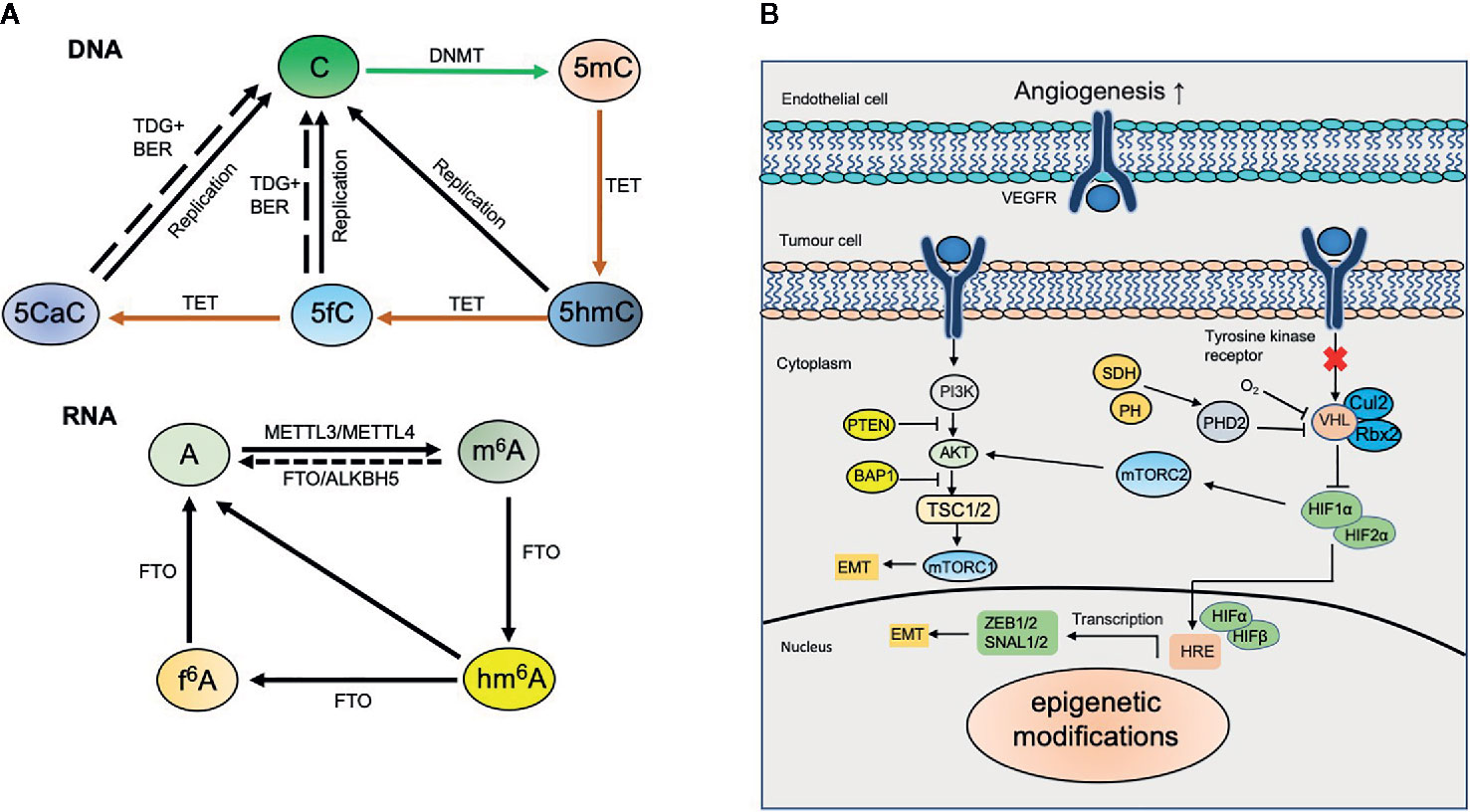
Figure 1 Dynamic modifications in kidney cancer. (A) Dynamic DNA and RNA modifications. DNA methyltransferases (DNMTs) including de novo methyltransferases DNMT3A, DNMT3B and maintenance methyltransferase DNMT1 convert unmodified cytosine (C) to 5‑methylcytosine (5mC). 5mC can be converted to 5‑hydroxymethylcytosine (5hmC) by ten-eleven translocation (TET) proteins‑mediated oxidation. TET proteins also catalyze the oxidation of 5hmC to 5‑formylcytosine (5fC) and 5‑carboxylcytosine (5caC). 5fC and 5caC can be further excised by thymine DNA glycosylase (TDG) coupled with base excision repair (BER) to generate unmodified cytosine. N6-methyladenosine (m6A) in mRNA is installed by methyltransferase-like protein 3 (METTL3) and METTL14, and erased by fat mass and obesity-associated protein (FTO) and α‑ketoglutarate-dependent dioxygenase alkB homologue 5 (ALKBH5). m6A can be further oxidized to N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (f6A) sequentially by FTO. (B) Epigenetic modifications involve in kidney cancer. Epigenetic modifications regulate diverse signaling pathways including HIF and PI3K-AKT and involve in kidney cancer.
For quite some time, 5mC has been considered as a stable epigenetic marker of DNA that cannot be further modified. However, in 2009 researchers found that 5mC can be oxidized to 5-hydroxymethylcytosine (5hmC) under the catalysis of ten-eleven-translocation (TET) family proteins (16–18). The TET family proteins consist of three members, TET1, TET2, and TET3, which share common typical characteristics of 2-oxoglutarate (2OG)- and Fell(II)-dependent dioxygenases (2OGFeDO) (19–21). TET enzymes can further oxidize 5-hmC to 5-carboxylcytosine (5caC) and 5-formylcytosine (5fC) (22, 23). Thymine-DNA glycosylase (TDG) is in charge of recognition and excision of 5caC and 5fC in mammals (22, 23). Under the catalysis of activation-induced cytidine deaminase (AID), 5mC can be transformed to 5-hydroxymethyluracil (5hmU) with a deamination reaction. In addition, previous studies have demonstrated that IDH1/2 can catalyze isocitrate to α-KG and can participate in the regulation of TETs and 5hmC (24). However, mutant IDH1/2 has been found to catalyze isocitrate to 2-hydroxyglutarate (2-HG), which is a competitive inhibitor of α-KG. IDH1/2 also can inhibit the transformation of 5mC to 5hmC by TETs; therefore, it plays a pivotal role in the regulation of 5hmC (24).
Current findings indicate that 5-hmC modification not only serves as an intermediate product, but also plays a pivotal role in development, aging, and diseases. Tissue/cell-specific distribution features and the content of 5hmC have been observed among varied tissues and organs. 5hmC is the most abundant in neuronal cells compared to other types of cells. In addition, 5hmC is mainly enriched at gene bodies, promoters, and distal regulatory regions of the genome. The enrichment of 5hmC at distinct genomic regions is correlated with gene expression, which can also be regulated by histone modifications (25). Abnormal distribution and/or level of 5hmC modification can induce disease. All of these findings suggest several important functions for dynamic DNA modifications.
The Function of DNA Methylation in Kidney Cancer
The aberrant level and distribution of DNA methylation have been revealed in various types of cancer including liver, colon, lung, and prostate cancer. These cancers are associated with the severity and metastatic potential of diseases (26). For example, DNA hypermethylation in cancer cells may be an alternative complementary mechanism, which triggers the silence of tumor-inhibiting genes and consequently results in tumorigenesis and metastasis (10, 27). In general, the global level of DNA methylation is decreased, while the acquisition of DNA methylation is observed at the promoter regions of some specific genes.
In studies of kidney cancer, Chen et al. applied the bisulfite sequencing method to map 5mC and found that the global level of 5mC is not changed (28). However, Mendoza-Pérez J et al. performed the analysis of 899 RCC cases and found that a low level of genomic DNA methylation (measured as 5mC%) in peripheral blood could significantly increase the risk of RCC (29). One possibility for these inconsistent results could be the ability of the methods used to distinguish DNA methylation and demethylation.
The Function of DNA Demethylation in Kidney Cancer
Mounting evidence has demonstrated that 5hmC plays an important function in a variety of tumors, such as acute myeloid leukemia, liver cancer, and melanoma (30, 31). Although the level of global 5mC is not altered, Chen et al. observed the decreased level of global 5hmC as well as the hypermethylation at gene body regions in kidney tumors (28). Their results also suggested that decreased 5hmC is correlated with the prognosis and survival. It has also been found that 5hmC is closely related with capsule invasion, vein invasion and clinical progress of RCC (32). RCC patients with high level of 5hmC show increased survival; therefore, 5hmC may serve as an independent prognostic and progression marker for RCC (32). Consistently, 5hmC hydroxymethylase TET1 can promote cell apoptosis and can inhibit cell proliferation and invasion, therefore inhibiting tumor growth in RCC (33). The inhibited expression of TET1 reduces 5hmC level at the promoter region of CCNY/CDK16 and consequently results in cell cycle arrest and inhibits self-renewal of renal cancer stem cells (34) (Figure 1).
The oxidation reaction of 5mC to 5hmC requires 2-ketoglutarate (2-KG) as co-substrates, which is generated by isocitrate dehydrogenase 1 (IDHs) during the tricarboxylic acid cycle (TCA). The down-regulated expression of IDH1 in kidney cancer contributes to the global loss of 5hmC in RCC (28). Consistently, ectopic expression of IDH1 and pharmacologically increasing intracellular 2-KG can restore the global levels of 5hmC, and consequently, can inhibit tumor growth (28, 35). IDH1 mutation leads to the increase of 2-hydroxyglutarate (2-HG), and the loss of 5hmC is partly mediated by the decrease 2-HG dehydrogenase (L2-HGDH), which has tumor inhibitory effects (36). The loss of L2HGDH is correlated with a worse prognosis, whereas the restoration of L2HGDH can increase 2-HG and can promote the accumulation of 5hmC in RCC cells (37). Ascorbic acid (AA), a cofactor for TET, can enhance the activities of TET enzymes and can restore the level of genomic 5hmC, thus reversing epigenetic aberrancy (38, 39). These findings suggest an interplay between DNA demethylation and metabolites that has an important role in kidney cancer (39, 40).
RNA Methylation and Kidney Cancer
Diverse Modifications of RNA and Molecular Mechanism of m6A Modification
To date, more than 110 types of RNA modifications have been identified, such as N1-methyladenosine (m1A), N6-methyladenosine (m6A), N6-methyl-2′-O-methyladenosine (m6Am), 5-methylcytosine (m5C), 5-hydroxymethylcytosine (hm5C) in messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), long non-coding RNAs (lncRNAs), etc (41). Among these modifications, m6A is the most abundant internal chemical modification in eukaryotic mRNA. In mammals, 0.1%–0.4% of adenosines (~3–5 m6A sites per mRNA) are modified by m6A, accounting for nearly half of total methylated ribonucleotides (42). m6A mainly enriches at the 3′ untranslated regions (3′UTRs), around the termination codons and the internal long exons (43).
m6A modification is mediated by three key elements called “writers”, “erasers”, and “readers” (44, 45). m6A modification is mainly catalyzed by the RNA methyltransferase complex (writers), including methyltransferase-like 3 and 4 (METTL3 and METTL14) and Wilms’ tumor 1-associated protein (WTAP) (46). METTL3 is in charge of m6A installation, while METTL14 participates in the interacting with target mRNA, and WTAP is responsible for the localization in the nuclear speckle (47). m6A modifications can be removed by RNA demethylases (erasers), including alkB homolog 5 (ALKBH5) and fat mass and obesity-associated protein (FTO, alpha-ketoglutarate dependent dioxygenase) (48). Both ALKBH5 and FTO belong to the alpha-ketoglutarate dependent dioxygenase family, which catalyze m6A demethylation in a Fe(II)-and alpha-ketoglutarate dependent manner. Similar to ALKBH5, alkB homolog 3 (ALKBH3) has been shown the demethylase activity for 1-methyladenine and 5-methylcytosine (49). m6A readers include the YTH domain family (YTHDF), insulin-like growth factor 2 mRNA binding protein 2 (IGF2BP), and HNRNPA2B1 (50). YTHDF proteins act as m6A readers, which can maintain the stability of m6A transcripts (51, 52) (Figure 1).
The dynamic and reversible m6A modification regulates various aspects of RNAs fate, such as nuclear exit, splicing, stability, efficiency of translation (41, 53); therefore, this modification has crucial roles in embryonic development, sex determination, neurogenesis, stress responses, and tumorigenesis in mammals (54, 55). Previous studies have shown that the dysregulation of m6A was induced, but was not limited to, the aberrant expression of its writers, erasers and readers. These result in profound outcomes in multiple biological processes, such as cell proliferation and fate determination, DNA damage response, embryogenesis, and heat shock responses, and therefore are involved in diseases (56–59). In addition, emerging evidence indicates that m6A modification plays a significant role in tumorigenesis and progression of a variety of cancers including breast cancer, gastric cancer, and pancreatic cancer (49, 55, 60–62).
The Function of m6A in Renal Cell Carcinoma
Although the function of m6A has been shown in several types of tumors, the important roles of m6A in RCC are still not completely known. Recent findings show that the level of global m6A decreases in RCC compared with adjacent non-tumor tissues (63), suggesting that the expression of m6A regulatory genes may be a biomarker for RCC. The protein level of m6A eraser FTO displays a significant decrease in RCC compared with normal tissues (64). Lower levels of m6A modification eraser FTO are usually associated with malignant prognosis whereas higher levels of FTO are associated with benign prognosis, suggesting that FTO may serve as a protective factor in RCC (65). Published findings about the role of ALKBH5 in RCC are controversial. Both increased and decreased expression of ALKBH5 in RCC have been reported (64, 66). In a retrospective study using TCGA database, Zhou et al. examined the alteration of m6A regulatory genes in clear cell renal cell carcinoma (ccRCC) and found that these m6A regulatory genes are significantly correlated with von Hippel-Lindau (VHL) and TP53, two key suppressors for RCC. This result suggests a relationship between m6A regulatory genes and the pathologic stage (63); however, it still lacks solid evidence about the roles of m6A writers METTL3 and METTL14 in RCC (Figure 1).
In human RCC tissues, mitochondrial enzyme methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is highly expressed, and the knockdown of MTHFD2 inhibits cell migration and invasion (67). High level of MTHFD2 is positively correlated with RCC grade, clinical stage, progression, and poor prognosis (68). Interestingly, MTHFD2 knockdown leads to a decrease of global m6A, and a hypomethylation of HIF-2α mRNA increases the translation of HIF-2α (67, 69), which in turn promotes the aerobic glycolysis (67). These findings establish a connection between m6A modification and MTHFD2-mediated one-carbon metabolism in RCC.
Conclusions
During the past several decades, significant progress has been made in understanding the function of epigenetic modifications in kidney cancer. However, the detailed molecular mechanisms underlying the kidney cancer carcinogenesis are still not completely known, and it has been challenging to explore the accurate diagnosis and effective treatment of kidney cancer. First, the interactions between DNA modifications, RNA modifications, and histone modifications in regulating gene expression in kidney cancer need to be determined. How these interactions cooperate to regulate diverse signaling pathways involved in kidney cancer requires further clarification. Second, the precise map of DNA and RNA modifications should be established in kidney cancer with high-throughput sequencing technologies. The identification of therapeutic targets relies on the analysis of high-throughput sequencing data. The therapeutic implications of epigenetic hallmarks are to be expected in kidney cancer considering the successful application of these hallmarks in other types of cancers.
Author Contributions
CF, XH, JM, and XL wrote the manuscript. All authors contributed to the article and approved the submitted version,
Funding
This work was supported in part by the Medicine & Health Technology Project of Zhejiang Province (2018RC007 to CF), Zhejiang Provincial Research Center for Cancer Intelligent Diagnosis and Molecular Technology (JBZX-202003 to JM) and the National Natural Science Foundation of China (grants 31571518, 31771395 to XL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Moch H. An overview of renal cell cancer: pathology and genetics. Semin Cancer Biol (2013) 23:3–9. doi: 10.1016/j.semcancer.2012.06.006
3. Joosten SC, Smits KM, Aarts MJ, Melotte V, Koch A, Tjan-Heijnen VC, et al. Epigenetics in renal cell cancer: mechanisms and clinical applications. Nat Rev Urol (2018) 15:430–51. doi: 10.1038/s41585-018-0023-z
4. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers (2017) 3:17009. doi: 10.1038/nrdp.2017.9
6. Wanner N, Bechtel-Walz W. Epigenetics of kidney disease. Cell Tissue Res (2017) 369:75–92. doi: 10.1007/s00441-017-2588-x
7. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet (2012) 13:484–92. doi: 10.1038/nrg3230
8. Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell (2019) 74:640–50. doi: 10.1016/j.molcel.2019.04.025
9. Deng X, Su R, Feng X, Wei M, Chen J. Role of N 6 -methyladenosine modification in cancer. Curr Opin Genet Dev (2018) 48:1–7. doi: 10.1016/j.gde.2017.10.005
10. Chi H-C, Tsai C-Y, Tsai M-M, Lin K-H. Impact of DNA and RNA Methylation on Radiobiology and Cancer Progression. Int J Mol Sci (2018) 19:555. doi: 10.3390/ijms19020555
11. de Cubas AA, Rathmell WK. Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol (2018) 15:599–614. doi: 10.1038/s41585-018-0052-7
12. Morris MR, Latif F. The epigenetic landscape of renal cancer. Nat Rev Nephrol (2017) 13:47–60. doi: 10.1038/nrneph.2016.168
13. Linehan WM, Ricketts CJ. The Cancer Genome Atlas of renal cell carcinoma: findings and clinical implications. Nat Rev Urol (2019) 16:539–52. doi: 10.1038/s41585-019-0211-5
14. Larkin J, Goh XY, Vetter M, Pickering L, Swanton C. Epigenetic regulation in RCC: opportunities for therapeutic intervention? Nat Rev Urol (2012) 9:147–55. doi: 10.1038/nrurol.2011.236
15. Dahl C, Gronbaek K, Guldberg P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin Chim Acta (2011) 412:831–6. doi: 10.1016/j.cca.2011.02.013
16. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324:930–5. doi: 10.1126/science.1170116
17. Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell (2011) 8:200–13. doi: 10.1016/j.stem.2011.01.008
18. Ko M, An J, Rao A. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr Opin Cell Biol (2015) 37:91–101. doi: 10.1016/j.ceb.2015.10.009
19. Mohr F, Dohner K, Buske C, Rawat VP. TET genes: new players in DNA demethylation and important determinants for stemness. Exp Hematol (2011) 39:272–81. doi: 10.1016/j.exphem.2010.12.004
20. Aravind L, Koonin EV. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol (2001) 2:research0007.1. doi: 10.1186/gb-2001-2-3-research0007
21. Loenarz C, Schofield CJ. Oxygenase catalyzed 5-methylcytosine hydroxylation. Chem Biol (2009) 16:580–3. doi: 10.1016/j.chembiol.2009.06.002
22. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333:1303–7. doi: 10.1126/science.1210944
23. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science (2011) 333:1300–3. doi: 10.1126/science.1210597
24. Wang J, Tang J, Lai M, Zhang H. 5-Hydroxymethylcytosine and disease. Mutat Res Rev Mutat Res (2014) 762:167–75. doi: 10.1016/j.mrrev.2014.09.003
25. Sun W, Zang L, Shu Q, Li X. From development to diseases: The role of 5hmC in brain. Genomics (2014) 104:347–51. doi: 10.1016/j.ygeno.2014.08.021
26. Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet (2000) 1:11–9. doi: 10.1038/35049533
27. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet (2002) 3:415–28. doi: 10.1038/nrg816
28. Chen K, Zhang J, Guo Z, Ma Q, Xu Z, Zhou Y, et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res (2016) 26:103–18. doi: 10.1038/cr.2015.150
29. Mendoza-Perez J, Gu J, Herrera LA, Tannir NM, Matin SF, Karam JA, et al. Genomic DNA Hypomethylation and Risk of Renal Cell Carcinoma: A Case-Control Study. Clin Cancer Res (2016) 22:2074–82. doi: 10.1158/1078-0432.CCR-15-0977
30. Ficz G, Gribben JG. Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics (2014) 104:352–7. doi: 10.1016/j.ygeno.2014.08.017
31. Thomson JP, Ottaviano R, Unterberger EB, Lempiainen H, Muller A, Terranova R, et al. Loss of Tet1-Associated 5-Hydroxymethylcytosine Is Concomitant with Aberrant Promoter Hypermethylation in Liver Cancer. Cancer Res (2016) 76:3097–108. doi: 10.1158/0008-5472.CAN-15-1910
32. Chen S, Zhou Q, Liu T, Zhang W, Zeng X-T, Guo Z. Prognostic value of downregulated 5-hydroxymethylcytosine expression in renal cell carcinoma: a 10 year follow-up retrospective study. J Cancer (2020) 11:1212–22. doi: 10.7150/jca.38283
33. Fan M, He X, Xu X. Restored expression levels of TET1 decrease the proliferation and migration of renal carcinoma cells. Mol Med Rep (2015) 12:4837–42. doi: 10.3892/mmr.2015.4058
34. Si Y, Liu J, Shen H, Zhang C, Wu Y, Huang Y, et al. Fisetin decreases TET1 activity and CCNY/CDK16 promoter 5hmC levels to inhibit the proliferation and invasion of renal cancer stem cell. J Cell Mol Med (2019) 23:1095–105. doi: 10.1111/jcmm.14010
35. Ge G, Peng D, Xu Z, Guan B, Xin Z, He Q, et al. Restoration of 5-hydroxymethylcytosine by ascorbate blocks kidney tumour growth. EMBO Rep (2018) 19:e45401. doi: 10.15252/embr.201745401
36. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
37. Shelar S, Shim EH, Brinkley GJ, Kundu A, Carobbio F, Poston T, et al. Biochemical and Epigenetic Insights into L-2-Hydroxyglutarate, a Potential Therapeutic Target in Renal Cancer. Clin Cancer Res (2018) 24:6433–46. doi: 10.1158/1078-0432.CCR-18-1727
38. Shenoy N, Bhagat TD, Cheville J, Lohse C, Bhattacharyya S, Tischer A, et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J Clin Invest (2019) 130:1612–25. doi: 10.1172/JCI98747
39. Shenoy N. Epigenetic dysregulation by aberrant metabolism in renal cell carcinoma can be reversed with Ascorbic acid. Mol Cell Oncol (2019) 6:1595309. doi: 10.1080/23723556.2019.1595309
40. Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, et al. L-2-Hydroxyglutarate: An Epigenetic Modifier and Putative Oncometabolite in Renal Cancer. Cancer Discov (2014) 4:1290–8. doi: 10.1158/2159-8290.CD-13-0696
41. Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet (2014) 15:127–50. doi: 10.1146/annurev-genom-090413-025405
42. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A (1974) 71:3971–5. doi: 10.1073/pnas.71.10.3971
43. Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev (2015) 29:2037–53. doi: 10.1101/gad.269415.115
44. Heck AM, Wilusz CJ. Small changes, big implications: The impact of m(6)A RNA methylation on gene expression in pluripotency and development. Biochim Biophys Acta Gene Regul Mech (2019) 1862:194402. doi: 10.1016/j.bbagrm.2019.07.003
45. Zhao W, Qi X, Liu L, Ma S, Liu J, Wu J. Epigenetic Regulation of m(6)A Modifications in Human Cancer. Mol Ther Nucleic Acids (2020) 19:405–12. doi: 10.1016/j.omtn.2019.11.022
46. Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The Critical Role of RNA m6A Methylation in Cancer. Cancer Res (2019) 79:1285–92. doi: 10.1158/0008-5472.CAN-18-2965
47. Selberg S, Blokhina D, Aatonen M, Koivisto P, Siltanen A, Mervaala E, et al. Discovery of Small Molecules that Activate RNA Methylation through Cooperative Binding to the METTL3-14-WTAP Complex Active Site. Cell Rep (2019) 26:3762–71.e5. doi: 10.1016/j.celrep.2019.02.100
48. Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy (2019) 15:1419–37. doi: 10.1080/15548627.2019.1586246
49. He L, Li J, Wang X, Ying Y, Xie H, Yan H, et al. The dual role of N6-methyladenosine modification of RNAs is involved in human cancers. J Cell Mol Med (2018) 22:4630–9. doi: 10.1111/jcmm.13804
50. Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y, et al. METTL3 Facilitates Oral Squamous Cell Carcinoma Tumorigenesis by Enhancing c-Myc Stability via YTHDF1-Mediated m6A Modification. Mol Ther - Nucleic Acids (2020) 20:1–12. doi: 10.1016/j.omtn.2020.01.033
51. Hesser CR, Karijolich J, Dominissini D, He C, Glaunsinger BA. N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog (2018) 14:e1006995. doi: 10.1371/journal.ppat.1006995
52. Berlivet S, Scutenaire J, Deragon JM, Bousquet-Antonelli C. Readers of the m(6)A epitranscriptomic code. Biochim Biophys Acta Gene Regul Mech (2019) 1862:329–42. doi: 10.1016/j.bbagrm.2018.12.008
53. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet (2014) 15:293–306. doi: 10.1038/nrg3724
54. Niu Y, Zhao X, Wu Y-S, Li M-M, Wang X-J, Yang Y-G. N6-methyl-adenosine (m6A) in RNA: An Old Modification with A Novel Epigenetic Function. Genomics Proteomics Bioinf (2013) 11:8–17. doi: 10.1016/j.gpb.2012.12.002
55. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol (2018) 11(1):48. doi: 10.1186/s13045-018-0590-8
56. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol (2014) 16:191–8. doi: 10.1038/ncb2902
57. Chen T, Hao Y-J, Zhang Y, Li M-M, Wang M, Han W, et al. m6A RNA Methylation Is Regulated by MicroRNAs and Promotes Reprogramming to Pluripotency. Cell Stem Cell (2015) 16:289–301. doi: 10.1016/j.stem.2015.01.016
58. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature (2015) 526:591–4. doi: 10.1038/nature15377
59. Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature (2017) 543:573–6. doi: 10.1038/nature21671
60. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med (2017) 23:1369–76. doi: 10.1038/nm.4416
61. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother (2019) 112:108613. doi: 10.1016/j.biopha.2019.108613
62. Wang S, Chai P, Jia R, Jia R. Novel insights on m(6)A RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer (2018) 17:101. doi: 10.1186/s12943-018-0847-4
63. Zhou J, Wang J, Hong B, Ma K, Xie H, Li L, et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging (2019) 11:1633–47. doi: 10.18632/aging.101856
64. Strick A, von Hagen F, Gundert L, Klumper N, Tolkach Y, Schmidt D, et al. The N(6) -methyladenosine (m(6) A) erasers alkylation repair homologue 5 (ALKBH5) and fat mass and obesity-associated protein (FTO) are prognostic biomarkers in patients with clear cell renal carcinoma. BJU Int (2020) 125:617–24. doi: 10.1111/bju.15019
65. Wen L, Yu Y, Lv H, He Y, Yang B. FTO mRNA expression in the lower quartile is associated with bad prognosis in clear cell renal cell carcinoma based on TCGA data mining. Ann Diagn Pathol (2019) 38:1–5. doi: 10.1016/j.anndiagpath.2018.10.009
66. Zheng Z, Mao S, Guo Y, Zhang W, Liu J, Li C, et al. N6methyladenosine RNA methylation regulators participate in malignant progression and have prognostic value in clear cell renal cell carcinoma. Oncol Rep (2020) 43:1591–605. doi: 10.3892/or.2020.7524
67. Green NH, Galvan DL, Badal SS, Chang BH, LeBleu VS, Long J, et al. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene (2019) 38:6211–25. doi: 10.1038/s41388-019-0869-4
68. Lin H, Huang B, Wang H, Liu X, Hong Y, Qiu S, et al. MTHFD2 Overexpression Predicts Poor Prognosis in Renal Cell Carcinoma and is Associated with Cell Proliferation and Vimentin-Modulated Migration and Invasion. Cell Physiol Biochem (2018) 51:991–1000. doi: 10.1159/000495402
Keywords: DNA methylation, DNA hydroxymethylation, RNA methylation, ten-eleven translocases, tumor
Citation: Feng C, Huang X, Li X and Mao J (2020) The Roles of Base Modifications in Kidney Cancer. Front. Oncol. 10:580018. doi: 10.3389/fonc.2020.580018
Received: 04 July 2020; Accepted: 19 October 2020;
Published: 13 November 2020.
Edited by:
Xiang Shu, Vanderbilt University Medical Center, United StatesReviewed by:
Alessandra Montecucco, Italian National Research Council, ItalyTanja Kunej, University of Ljubljana, Slovenia
Copyright © 2020 Feng, Huang, Li and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuekun Li, eHVla3VuX2xpQHpqdS5lZHUuY24=; Jianhua Mao, bWFvamg4OEB6anUuZWR1LmNu