 Takuma Uo
Takuma Uo Cynthia C. Sprenger
Cynthia C. Sprenger Stephen R. Plymate
Stephen R. Plymate- 1Department of Medicine, University of Washington, Seattle, WA, United States
- 2Geriatrics Research Education and Clinical Center, VA Puget Sound Health Care System, Seattle, WA, United States
Metabolic reprogramming is associated with re/activation and antagonism of androgen receptor (AR) signaling that drives prostate cancer (PCa) progression to castration resistance, respectively. In particular, AR signaling influences the fates of citrate that uniquely characterizes normal and malignant prostatic metabolism (i.e., mitochondrial export and extracellular secretion in normal prostate, mitochondrial retention and oxidation to support oxidative phenotype of primary PCa, and extra-mitochondrial interconversion into acetyl-CoA for fatty acid synthesis and epigenetics in the advanced PCa). The emergence of castration-resistant PCa (CRPC) involves reactivation of AR signaling, which is then further targeted by androgen synthesis inhibitors (abiraterone) and AR-ligand inhibitors (enzalutamide, apalutamide, and daroglutamide). However, based on AR dependency, two distinct metabolic and cellular adaptations contribute to development of resistance to these agents and progression to aggressive and lethal disease, with the tumor ultimately becoming highly glycolytic and with imaging by a tracer of tumor energetics, 18F-fluorodoxyglucose (18F-FDG). Another major resistance mechanism involves a lineage alteration into AR-indifferent carcinoma such a neuroendocrine which is diagnostically characterized by robust 18F-FDG uptake and loss of AR signaling. PCa is also characterized by metabolic alterations such as fatty acid and polyamine metabolism depending on AR signaling. In some cases, AR targeting induces rather than suppresses these alterations in cellular metabolism and energetics, which can be explored as therapeutic targets in lethal CRPC.
Introduction
Normal cells gain distinctive capabilities to overcome the restrictions in the tissue of origin to initiate primary tumor formation (1–3). The phenotypic traits in the original environment often determine the molecular processes that drive the progression to advanced and metastatic tumors (4, 5). This is true for reprogramming of cellular and energy metabolism during cancer progression (6–8).
“The Warburg effect (aerobic glycolysis)” observed by Otto Warburg nearly a century ago is the phenomenon that cancer cells preferentially convert most glucose to lactate even in the presence of oxygen by which mitochondrial oxidative phosphorylation can proceed to generate ATP more efficiently(9, 10). His original hypothesis also emphasized that dysfunction of mitochondria is the initiating factor for cancer formation (11, 12). While not maximizing ATP production/glucose, aerobic glycolysis permits cancer cells to efficiently convert glucose into the biomass (e.g., nucleotides, amino acids, and lipids) for cell growth and proliferation (13, 14). As opposed to Warburg’s notion, most-if not all-cancer cells rely on functional mitochondria for their survival (13, 15).
Prostate cancer (PCa) is unique from a metabolic perspective. Ironically, the normal prostatic epithelial cell is one of the best cell types that fit to the original Warburg’s theory: mitochondria must be dysfunctional to get higher rate of glycolysis. Instead, primary PCa does not exhibit the Warburg effect. Contrary to other cancer cells, malignant transformation involves the conversion from energy-inefficient (“glycolytic”) secretory epithelial cells to energy-efficient (“oxidative”) PCa cells (16–20) (Figure 1).
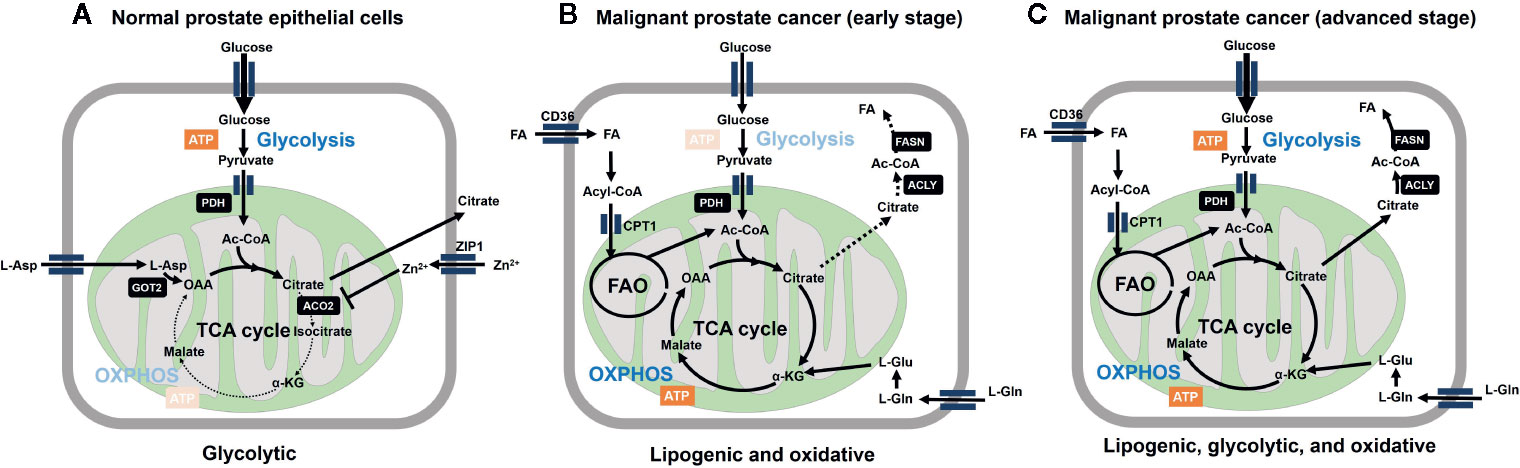
Figure 1 Metabolic reprogramming is involved in malignant transformation of prostatic cells. (A) Normal prostate epithelial cells express zinc transporter ZIP1 facilitating intracellular accumulation of zinc ion, which contributes to inhibition of m-aconitase (ACO2) at mitochondria. This inhibition results in truncation of tricarboxylic acid cycle (TCA) cycle and release of citrate to the extracellular space. Citrate production is supported by increasing the substrate pools for citrate synthase, acetyl-CoA (Ac-CoA) and oxaloacetic acid (OAA) at the mitochondria. OAA is supplied as the result of action of mitochondrial aspartate aminotransferase (GOT2) on L-aspartate. The level of mitochondrial acetyl-CoA is associated with increased expression of pyruvate dehydrogenase E1 component subunit alpha (PDHE1α) of pyruvate dehydrogenase complex (PDH). From bioenergetic point of view, normal prostatic cell is supported by aerobic glycolysis. (B) Marked decrease in zinc levels due to depletion of ZIP1 represents an essential early event in the development of PCa malignancy, which relieves m-aconitase to establish a complete TCA cycle. These metabolic alterations are functionally related to low citrate level and the general low avidity of 18F-FDG in primary PCa. Fatty acids (FA) are incorporated through CD36, followed by CPT1-mediated entry into mitochondria to serve as the substrate for fatty acid oxidation (FAO). L-Glutamine also serves as the precursor of TCA cycle intermediates after conversion into L-glutamate. ATP-citrate lyase (ACLY) cleaves citrate to produce acetyl-CoA to serve as the substrate for fatty acid synthase (FASN). (C) Further malignant transformation promotes glycolysis (through increased expression of glycolytic enzymes). While lipogenic trait is enhanced, multiple combinations of/all energy source pathways are theoretically available at this stage. Therefore, it is important to determine which metabolic pathway dominates for survival of given tumors for the future metabolism-based precision therapy.
Androgen receptor (AR) plays pivotal roles in both normal and malignant prostate cells. Indeed, AR transcriptional program supports PCa viability during the course from primary tumor formation to progression to metastasis. AR has the capabilities of regulating virtually all aspects of cellular metabolism (glucose, lipid, amino acid, nucleotides, etc.) (20–24). Conversely, pre-receptor control of “androgen” metabolism, which is dictated by tissue localization and abundance of steroidogenic enzymes and metabolism, ultimately determines activity of the holoreceptor for the transcriptional output (25, 26). Nevertheless, PCa exhibits specified metabolic and energetic phenotypes depending on the stage of disease progression (18, 19, 23). For example, while AR signaling persists, the transition from oxidative to glycolytic metabolism occurs during the progression to advanced PCa (27–29). Lipogenesis is continuously maintained by AR during the development of PCa (30–32). AR antagonism is highly effective in counteracting AR signaling thus altering associated metabolic programs, but tumors evolve by acquiring androgen-independent AR activation in adenocarcinoma or bypassing AR requirement through transdifferentiation to more aggressive and lethal AR-indifferent carcinomas (33). This cellular transformation results in drastic metabolic adaptation to promote aerobic glycolysis (29, 34, 35).
Understanding of the relationship between these distinctive metabolic features and AR signaling in PCa will lead to identification of metabolic vulnerabilities that offer the opportunity for diagnosis and therapy. In this review, we will characterize metabolic phenotypes of PCa in relation to AR signaling and review the current knowledge of metabolism-based imaging tools and therapeutic interventions to target cancer metabolism.
Androgen Action in Prostate Function and Metabolism: Zinc, Truncated Tricarboxylic Acid Cycle Cycle, Citrate Metabolism
Androgens are hormones required for development and maintenance of the male reproductive system. The functions of prostatic cells in both normal and malignant condition have been characterized by the relationship to the status and availability of androgen and its cognate receptor AR. Upon binding to androgen, AR which is otherwise sequestered in the cytoplasm translocates to nucleus and acts as sequence-specific dimerized transcription factors (36).
The unique metabolic processes in the prostate are well adapted to fulfill the major function as a secretory tissue to generate prostatic fluid comprised of high concentration of citrate along with zinc, lipids, and kallikrein enzymes including prostate-specific antigen (PSA) (37, 38) (Figure 1). Typically, citrate is either retained and oxidized in the mitochondria to generate energy as an essential intermediate in the citric acid cycle, or is exported into the cytoplasm where it is cleaved by ATP citrate lyase (ACLY) to generate acetyl-CoA, which is used for fatty acid (FA) synthesis (39).
The normal human prostate retains the capability of accumulating the highest levels of zinc in any soft tissue of the human body through expression of specific zinc transporters (ZIP1–4 for uptake and ZnT1–10 for release) (40). High levels of mitochondrial zinc inhibit mitochondrial aconitase, resulting in truncation of tricarboxylic acid cycle (TCA) cycle at the first step of citrate oxidation (17, 41). Androgen signaling enhances citrate production by increasing the substrate pools for citrate synthase, acetyl-CoA and oxaloacetic acid (OAA) at the mitochondria. The level of mitochondrial acetyl-CoA is associated with increased expression of pyruvate dehydrogenase E1 component subunit alpha (PDHE1α) (42). Aspartate uptake is through the excitatory amino acid transporter SLC1A1/EAAC1 (43). Followed by transamination processes to generate OAA at the mitochondria (44). Mammalian cells typically produce ~38 ATP/glucose through the combined actions of glycolysis and TCA cycle oxidation on glucose. On the other hand, the normal prostatic epithelia can generate only ~14 ATP/glucose due to truncation in TCA cycle resulting in the loss of ~24 ATP/glucose (19).
Marked decrease in zinc levels due to depletion of ZIP1 represents an essential early event in the development of PCa malignancy (45), which relieves mitochondrial aconitase to establish a complete TCA cycle (18, 19). These metabolic alterations are functionally related to low citrate level and the general low avidity of 18F-FDG in primary PCa (46, 47).
AR Drives PCa by Regulating Central Metabolism
Multi-omics studies (transcriptome, proteomics, cistrome, and metabolome) define the AR as a master regulator that orchestrates cellular metabolism to fuel proliferation and growth of PCa cells (20–22, 48, 49). Specifically, AR transcriptionally regulates multiple pathways of energy and biomass supply, including glycolysis, mitochondrial respiration, metabolism of FA (synthesis, ß-oxidation, and uptake), nucleotides, amino acids, and polyamines. Thus, drastic metabolic alterations are expected to inevitably follow AR inhibition and re/activation during the progression to lethal CRPC along with AR antagonism therapy.
Glucose Metabolism
AR determines bioenergetic traits through regulation of components in glycolytic pathway (GLUT1, HK1, HK2, and PFK2/PFKFB) and pyruvate flux into mitochondria (PDH, MPC2) (21, 42, 50). AR signaling increases expression of glucose-6-phosphate dehydrogenase (G6PD) which directs glucose-6-phosphate from glycolysis to the pentose phosphate pathway (PPP) for generation of NADPH and nucleotide precursors (51). The conversion from pyruvate to lactate is catalyzed by LDH proteins including AR-target LDHA (52, 53). Hyperpolarized 13C magnetic resonance spectroscopic imaging (MRSI) demonstrates that in vivo conversion [1-13C] lactate into [1-13C] pyruvate occurred more efficiently in PDX models of AR-driven CRPC than those of AR-negative PCa (54). Monocarboxylate transporter MCT4 is upregulated in CRPC and contributes to completion of successful aerobic glycolysis through secretion of lactate. Indeed, MCT4-targeting antisense oligonucleotides (ASO) provide significant tumor suppressive activity in cellular and xenograft models of CRPC (55). Overall, AR is capable of promoting both glycolysis and pyruvate oxidation, indicating AR’s predominant roles in both glycolytic and oxidative PCa tumors.
FA Metabolism
AR regulates FA metabolism by controlling expression of more than 20 enzymes involved in many aspects of lipid metabolism, including uptake, trafficking, synthesis, and degradation (32, 49).
AR and the master regulator of lipid homeostasis sterol regulatory-element binding protein (SREBP) regulate each other in a positive feedback system (32, 49, 56, 57). SREBP’s transcriptional targets include ELOV6 and SCD1 (58) while fatty acid synthase (FASN) and ACC (acetyl-CoA carboxylase) are co-targeted by both SREBP and AR (49, 59). Thus, AR activation accelerates FA synthesis, particularly as the form of mono-unsaturated and saturated FA (31, 32). Conversely, AR inhibition leads to marked reduction of de novo lipogenesis and permits incorporation of dietary FA enriched in polyunsaturated FA which are prone to lipid peroxidation when subjected to oxidizers such as arsenic trioxide (60, 61).
In addition to citrate oxidation, fatty acid oxidation (FAO) is the dominant energy producing pathway through decomposition of de novo or exogenous FA (62–65). Both FA synthesis and FAO have been recently shown to play key roles in cancer cell growth and proliferation (49, 63). This is an apparently contradictory situation where catabolism and anabolism of the same group of metabolites co-exist in the same cells. Also, FA synthesis and FAO have traditionally been considered incompatible due to the inhibitory effects of malonyl-CoA (the product of ACC1 which serves as the substrate for FASN) on carnitine palmitoyltransferase 1 (CPT1) in the carnitine shuttle (the rate limiting step for the transport of FA into the mitochondria) (66). Nevertheless, pharmacological or genetic inhibition of FASN resulted in decreased FAO as well as oxygen consumption, suggesting the existence of simultaneous FA synthesis and oxidation in the cells (67). Moreover, combined inhibition of FASN and FAO produced additive therapeutic effects in PCa, demonstrating that two pathways coexisting and feeding each other in some situations (65, 68). More excitingly, CPT1A-mediated FAO is reportedly to linked to epigenetics by supplying acetyl-CoA for histone acetylation (69).
On the other hand, there is growing body of evidence that PCa utilizes exogenous FA derived from diet or adipocytes (70–72). Blockade of this incorporation by CD36 inhibition is antitumorigenic (71). These reports emphasize that pharmacological intervention in FA metabolism has therapeutic benefit.
Amino Acids
AR regulates amino acid catabolism through expression of amino acid transporters (LAT1, LAT3, ASCT1, ASCT2) (43, 73–76). LATs and ASCTs are for bulky and small neutral amino acids, respectively. In particular, ASCT2 prefers the conditionally essential amino acid glutamine as the substrate. Glutamine undergoes glutaminolysis to generate TCA cycle intermediates via glutamate production as an alternative energy source, providing pharmacological glutamine starvation as a therapeutic strategy (74, 77–80).
One Carbon Metabolism Network
AR regulates one-carbon metabolism network consisting of the two folate cycle pathways (DHFR, GNMT, SARDH), and methionine cycle (MAT, AHCY) which interact with trans-sulfuration pathway (CBS, CTH) and polyamine synthesis (ODC1, AMD1) (81–83). The methionine cycle contributes to the formation of S-adenosyl-methionine (SAM), the universal methyl donor for protein and DNA methyltransferase reactions(84). Thus, this metabolism may contribute to AR-driven malignant progression by promoting DNA synthesis and changing DNA and histone methylation status (81). As discussed below, availability of SAM determines neuroendocrine PCa (NEPC) status which is AR independent (85).
Addiction to Altered Metabolism
Dependence of AR on reprogrammed metabolic characteristics occurs in FA and ornithine metabolism. AR signaling is blunted when genetic or pharmacological inhibition of the rate-limiting enzymes in the pathways, such as ODC1, FASN, and CPT1 (67, 68, 82).
Metabolic Plasticity in Relation to Anti-AR Therapy and the Resistance Mechanisms
Since 1950s, inhibition of AR activity has remained a mainstay in the treatment of advanced PCa (86–89). Although most patients with PCa initially respond to AR inhibition, they eventually develop castration-resistant PCa (CRPC) (36, 90, 91). The emergence of CRPC usually involves reactivation of AR signaling(92–97), which is then further targeted by as androgen synthesis inhibitors (abiraterone) and AR-ligand inhibitors (enzalutamide, apalutamide, and daroglutamide) (98, 99). Nevertheless, resistance to these agents and progression to lethal disease are essentially universal by developing adaptive resistance to these target therapies through two distinct groups of mechanisms based on AR dependency (100) (Figures 2, 3). Continued AR activation occurs by multiple mechanisms including increased AR expression in close association with enhanced intracrine or paracrine androgen synthesis (Figure 2, Group 1), AR gene mutations enabling promiscuous ligand interaction, and expression of constitutively active AR variants (AR-Vs)(Group 2) (100–103). AR antagonism can also promote lineage crisis and cellular plasticity to bypass AR blockade and generate neuroendocrine PCa (NEPC)(Group 1, 2→Group 3, Group 4→Group 3) (33, 104–106). Transformation into treatment-induced NEPC (t-NEPC) requires lineage plasticity in adeno-PCa to bypass AR blockade along with three major events: (i) The loss of AR expression. (ii) Alternative splicing of REST transcript by SRRM4 leading to the loss of REST activity that represses neuroendocrine gene expression. (iii) Activation of NE transcription factors e.g. ASCL1 and BRN2 that determine the commitment to a NE lineage (105–108). Among many other factors, EZH2 stands out to regulate NEPC-specific gene expression through epigenetic machinery (105). Importantly, N-MYC forms a transcription repressor complex with EZH2 to repress AR transcription program (109). It is noteworthy that AKT1-mediated phosphorylation drives a non-epigenetic mode of EZH2 action as AR coactivator to support androgen-independent AR activation during CRPC development and progression. Another emerging cell type is double negative PCa (DNPC)(Group 1, 2→Group 4), which is negative for both AR and neuroendocrine markers and may represent an intermediate phenotype between AR expressing adenocarcinoma and the neuroendocrine phenotype (110).
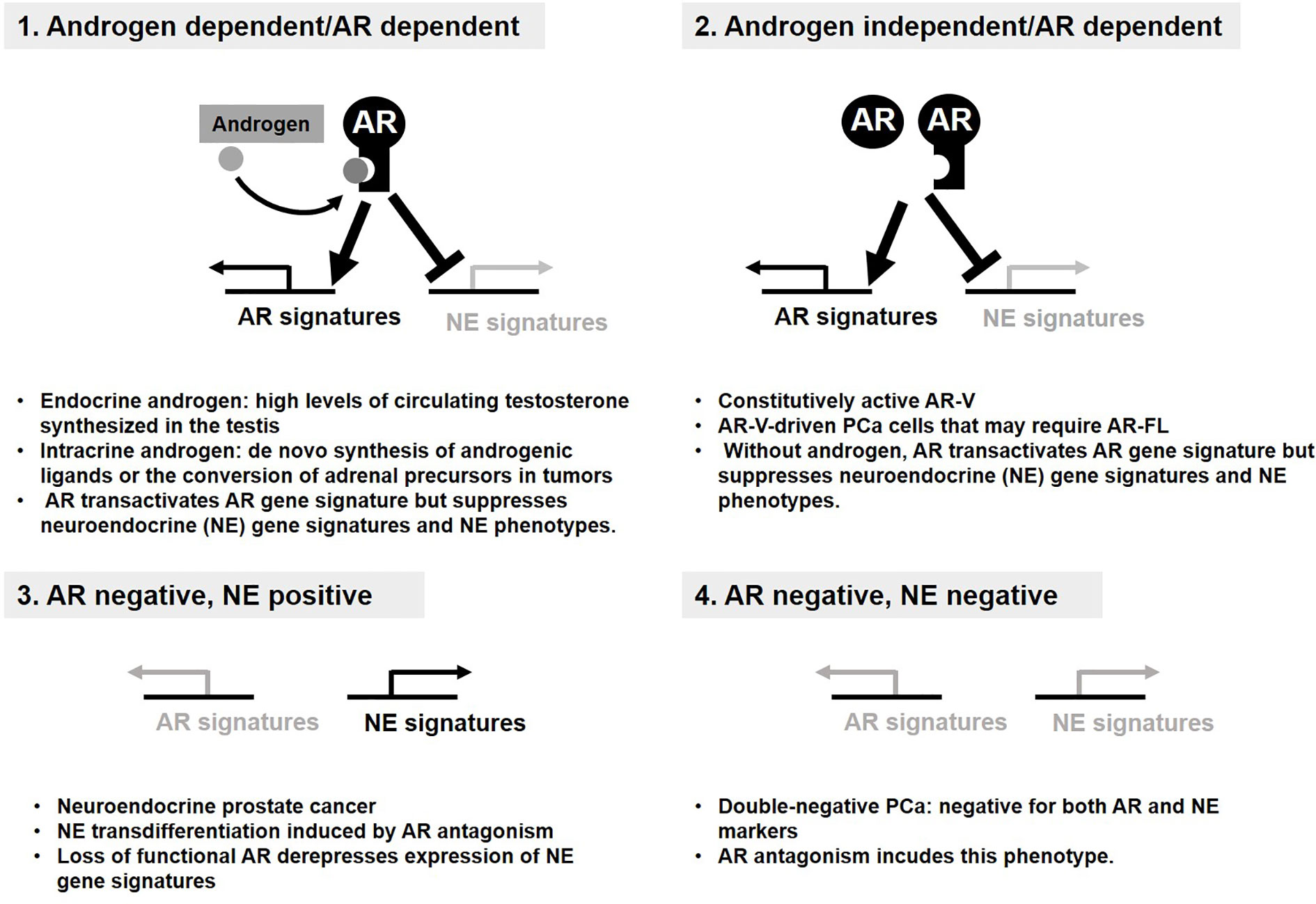
Figure 2 Androgen receptor (AR) status defines four distinct groups of prostate cancer (PCa). Four distinct groups of PCa display the resistance mechanism to anti-AR therapy. AR signaling supports survival and growth of PCa and suppresses transdifferentiation into neuroendocrine. (1, 2). Loss of AR signaling derepresses expression of NE gene signatures required for NE phenotypes (3). Double-negative PCa bypasses AR requirement without NE phenotype (4).
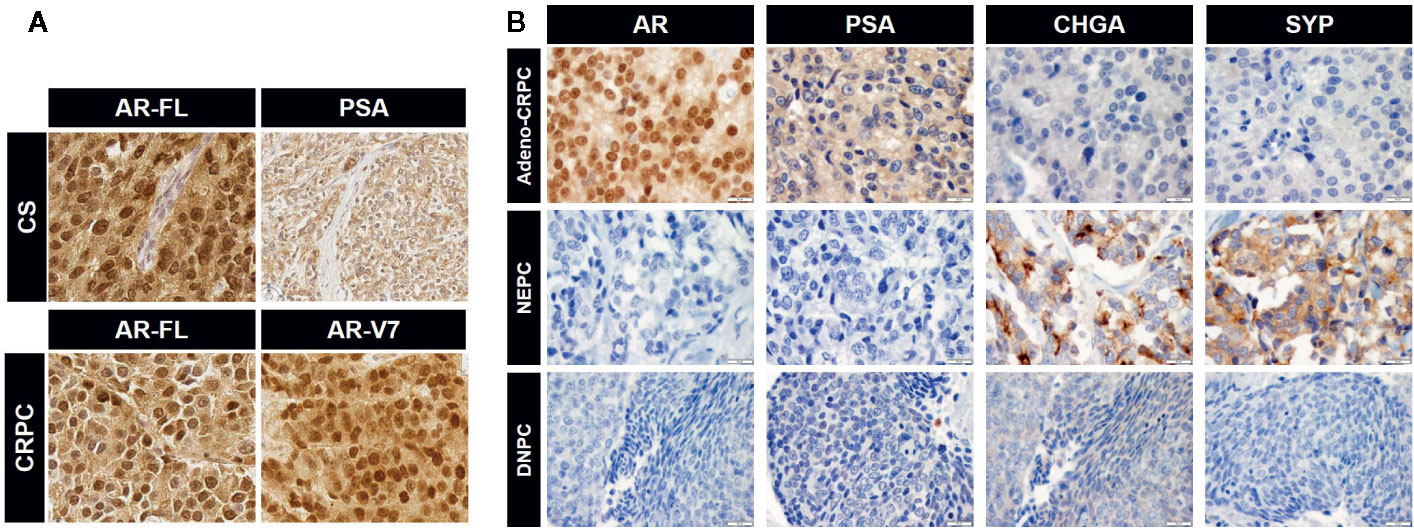
Figure 3 Immunohistochemical images for the expression of androgen receptor (AR), prostate-specific antigen (PSA), and neuroendocrine PCa (NEPC) markers. (A) Expression of full-length AR (AR-FL) and PSA in castration-sensitive (CS) PCa and AR-FL and AR-V7 in CRPC. Note uniform nuclear staining of AR-V7. CS and CRPC correspond to group (1) and (2) in Figure 2, respectively. (B) Expression of specific markers for each CRPC type. CRPC adenocarcinoma are positive for AR and its transcriptional target PSA but negative for NEPC markers chromogranin A (CHGA) and synaptophysin (SYP). NEPC is positive for NEPC markers and negative for AR and PSA. DNPC is negative for AR, PSA, and NEPC markers (image courtesy of Dr. Colm Morrissey at University of Washington). Representative images for the data in Figure 2 (2: Adeno-CRPC, 3: NEPC, 4: DNPC). Scale bar=20 µm.
Alteration in Pre-Receptor Control of Dihydroxytestosterone Metabolism
5α-Reduction of testosterone (T) in prostate results in the formation of the more potent ligand dihydroxytestosterone (DHT) to activate AR. Thus, ADT is the frontline treatment and directed toward disruption of T-DHT-AR axis by suppression of gonadal T by medical or surgical castration (26). Resistance to gonadal T depletion namely CRPC is associated with AR activity which is achieved by a gain-of-function in AR itself and/or sufficient intratumoral amounts of T and DHT to activate AR (25, 26, 101). Metastatic prostate tumor cells synthesize their own androgens through de novo steroidogenesis, which involves upregulation of enzymes required for stepwise synthesis from cholesterol to T and DHT (93, 111). Another strategy requires adrenal synthesis and supply of dehydroepiandrosterone (DHEA) and its sulfate (DHEA-S) which are converted to Δ4-androstenedione (AD) by 3β-hydroxysteroid dehydrogenase/isomerase (HSD3B1) in PCa (25, 26). AD is converted to DHT through canonical (AD→T→DHT) or alternative (“backdoor”) pathway involving the intermediate androstenedione to form DHT. Importantly, a gain-of-function mutation in HSD3B1 (N367T) leads to stabilization of the enzyme which confers two distinct survival benefits to PCa (112, 113). This variant supports CRPC status to bypass depletion of gonadal testosterone by facilitating the synthesis of AD thus flux to DHT from adrenal DHEA and DHEA-S (112). The resistance to anti-AR therapy is acquired by this variant which more efficiently converts the androgen synthesis inhibitor abiraterone into the precursor of potent AR agonist (113).
AR Dependent Mechanisms: Full-Length AR (AR-FL) and AR-V7 Specific Signaling?
As discussed above, AR is the master regulator of cellular metabolism. The questions remain as to whether AR-Vs are simply a constitutively active substitute for liganded-AR-FL to control cellular metabolism. Do AR-V7 and AR-FL differentially contribute to a selective adaptation during metabolic rewiring that occurs in CRPC progression? To answer this question, androgen-treated LNCaP and LNCaP engineered to co-express AR-V7 were used to extract AR-FL and AR-V7 signaling, respectively (114). AR-V7 specific metabolic signatures include reduced citrate level as a result of enhanced utilization rather than a failure to synthesize citrate. AR-V7 enhanced glycolytic flux more effectively than AR-FL with enhanced conversion of glutamine to citrate via reductive carboxylation (114). These findings suggest that AR-V alters flux of a subset of metabolites to provide growth advantage. As of yet, no such data has been generated to address the functional contribution of endogenous AR-Vs to bioenergetic phenotypes.
AR Indifferent CRPC: Drastic Metabolic Changes Are Associated With Cellular Lineage Alterations
MYC family proteins regulate virtually all genes involved in glycolysis not only by controlling their express levels but shifting alternative splicing toward glycolytic isoform PKM2 over PKM1 (115, 116). Moreover, MYC, increases mitochondrial export of acetyl groups as the form of citrate and the resulting acetyl-CoA contributes to histone acetylation by histone acetyltransferase GCN5 (117). Indeed, there exists the interplay between the epigenetic landscape and metabolism (118). For example, pyruvate generated from glycolysis is the main substrate for acetyl-CoA, a central metabolite coordinating the activity of the histone acetyltransferase (HAT) enzymes. Increased expression of the histone lysine demethylase KDM8 is observed in the context of treatment-induced NEPC and transactivated expression of EZH2 (119). Mechanistically, the KDM8-mediated PKM2 nuclear translocation results in the transcriptional activation of glycolytic program, including GLUT1, HK2, PKM2, LDHA, etc.) and downregulation of genes for pyruvate dehydrogenase complex (PDHA1 and PDHB1) to reduce the direction of pyruvate to mitochondria. As a proof of concept, inhibition of glycolysis lead to growth inhibition (119). Phosphoglycerate dehydrogenase (PHGDH) is the first enzyme branching from glycolysis in the serine biosynthesis which involved in one-carbon metabolism to supply S-adenosyl methionine (SAM) (120). SAM in turn serves as the substrate for DNA and protein methyltransferases. Cancer metabolism is linked to epigenetics in this scenario. Upregulation of PHGDH is common in NEPC thus facilitating methylation-related epigenetic modifiers such as EZH2 (105).
Positron Emission Tomography–Based Metabolic Phenotyping
In vivo metabolic phenotyping involves the steps for profiling and characterizing energetic phenotypes of tumors, which has a great diagnostic value for PCa patients. In this regard, 18F-FDG, 18F- or 11C-labeled acetate, and 18F- or 11C-labeled choline represent the three most studied positron emission tomography (PET) radiotracers in the PCa field (121, 122). Biochemical characteristics of tumors correlate well with uptake of each radiotracer (Figure 4). Acetate uptake is increased concomitantly with elevated FASN activity (123, 124). Upregulation of choline kinase (CK), which is associated with malignancy, promotes phosphorylation of choline to be incorporated in cellular membrane as the form of phosphatidylcholine (125, 126). While both acetate and choline uptake serve as a basis of powerful PET imaging, it has been well accepted that PCa displays less avidity to 18F-FDG (46, 47). However, largely depending on the disease phase, 84% of mCRPC patients have at least one 18F-FDG positive metastasis. Moreover, 85% of 18F-FDG positive metastasis displayed positivity for another tracer 18F-fluorodihydrotestosterone (18F-FDHT) used as indicator of AR(-FL) expression (29). On the other hand, prostate specific membrane antigen (PSMA) is “imageable” AR-target gene product (127). Thus, 68Ga-PSMA-PET imaging reflects relative changes in treatment-dependent AR activity thus providing high diagnostic values (128). The expression levels of glucose uptake–associated genes, including GLUTs and hexokinases to provide a genomic rationalization for the previously reported 18F-FDG avidity of PSMA-suppressed PC tumors such as NEPC and DNPC (35, 129). Non-invasive imaging tools have not been available for oxidative phosphorylation in tumors. Oxidative tumors can be monitored by the agent 4-[18F]fluorobenzyl triphenylphosphonium (18FBnTP) whose uptake is driven by mitochondrial membrane potential (ΔΨm) (130). Thus, combined use of these diagnostic tools will be powerful to characterize bioenergetic phenotypes of PCa tumors and determine treatment options.
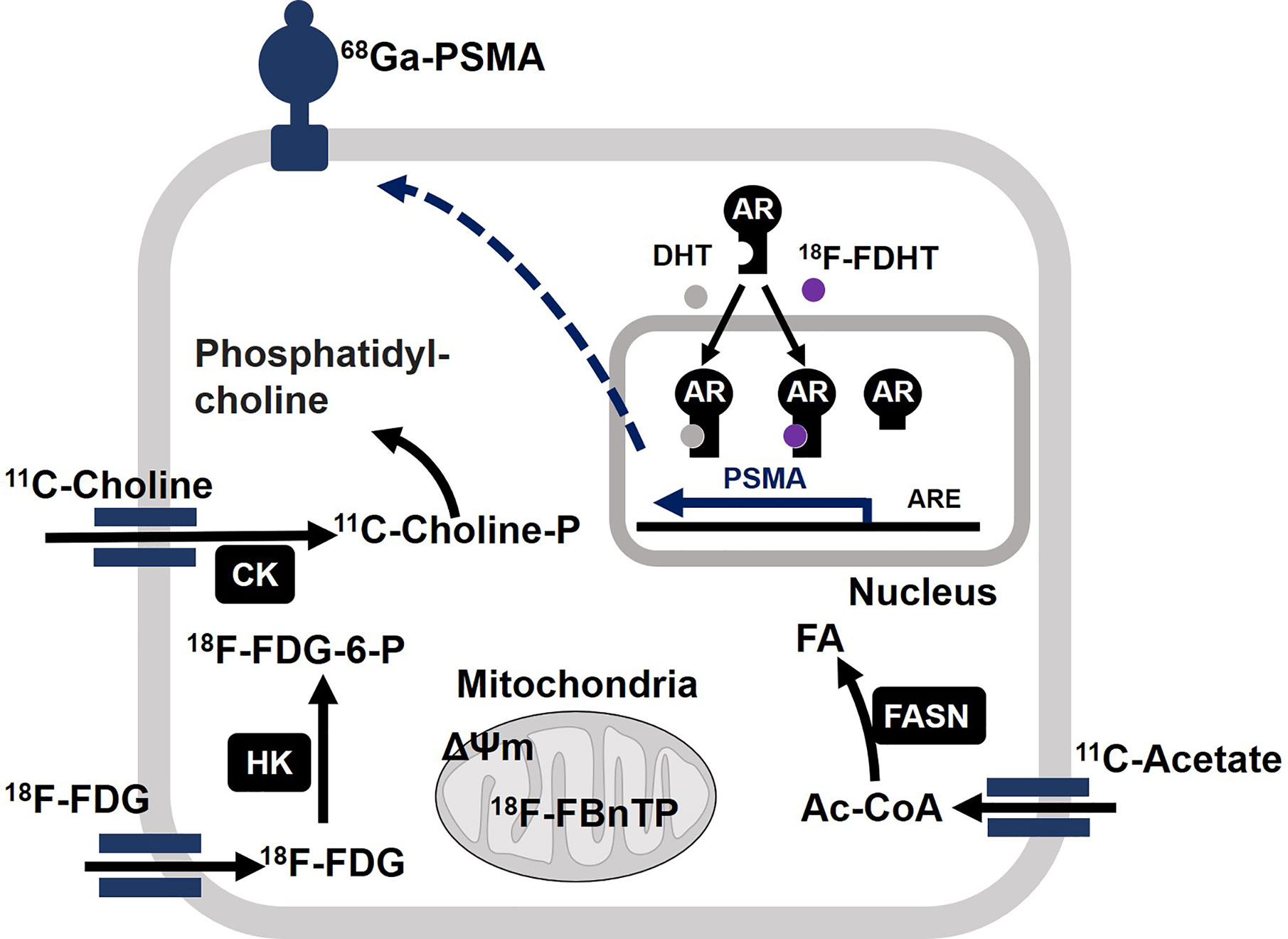
Figure 4 Molecular basis of actions of positron emission tomography (PET) radiotracers in prostate cancer (PCa). Acetate is converted to acetyl-CoA (Ac-CoA) which serves as a substrate for FASN to produce fatty acids (FA). After enzymatic modification by choline kinase (CK), 11C-choline is incorporated into cell membrane as the form of phosphatidylcholine. After incorporation into cell, 18F-FDG undergoes phosphorylation by hexokinase (HK) and accumulates as the form of 18F-FDG-6-P. Mitochondrial membrane potential (ΔΨm) drives accumulation of 4-[18F]fluorobenzyl triphenylphosphonium (18F-FBnTP) at mitochondria. 18F signal is indicative of respiration-competent functional mitochondria. Binding of ligand dihydrotestosterone (DHT) activates full-length AR as a transcriptional factor to upregulate target genes such as PSMA. Accordingly, the presence of full-length AR can be monitored by 18F-FDHT. It is noteworthy that constitutively active AR variant fails to bind to 18F-FDHT. Accordingly, 18F-FDHT negativity does not necessarily mean tumors are negative for any form of AR. 68Ga-labeled antagonistic ligand for PSMA can be used to monitor tumors with active AR signaling.
Tumor Metabolism in CRPC Is Observed Across Various Cancer Types?
As discussed above, PCa develops adaptive resistance to AR-targeting therapy through two distinct groups of mechanism based on AR dependency. In addition to alterations in AR structure of function, AR-dependent mechanism involves aberrant pre-receptor metabolism of steroids which is arguably unique to CRPC. AR-independent paths include transdifferentiation into NEPC and DNPC. Nevertheless, advanced CRPC, NEPC, and DNPC are ultimately addicted to aerobic glycolysis which is associated with high avidity of FDG in PET scan (29, 35). Ironically, Warburg effects occur in virtually all types of cancers and may represent the final form of tumor metabolism (13, 23, 131). Consistently, systems biology approach was used to analyze the expression of metabolic genes across 20 different cancer types and their impact on clinical outcome, which demonstrates that downregulation of mitochondrial genes is associated with the worst clinical outcome across all cancer types (132). Interdependence of AR and FASN drives AR-dependent CRPC progression (67), but overexpression of FASN is the rule rather than the exception in many types of cancers (133). Cancer cells appear to undergo a tissue-specific metabolic rewiring, which converges toward a common metabolic landscape. One may ask “What metabolic programs differentiate one cancer type from the others?”. A recent report from the Vander Heiden group specifically addressed this issue by testing whether tissue-of-origin dictates cancer dependence on specific metabolic pathways (134). Mouse models of pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung carcinoma (NSCLC) have the same genetic background with Kras mutation and TP53 deletion, While PDAC tumors have decreased branched-chain amino acids (BCAA) uptake, NSCLC tumors incorporate free BCAAs into tissue protein and use BCAAs as a nitrogen source while PDAC tumors have decreased BCAA uptake. Expression pattern of BCAA metabolizing enzymes in original tissues reflect these metabolic differences in tumors, arguing both tumor genetics and tissue context define cancer dependence on specific metabolic pathways (134). While TP53 and RB1 are commonly tumor suppressive in many cancer types (135), their combined inactivation promotes cell plasticity in PCa to undergo NEPC differentiation (136, 137). In this scenario, PCa-specific metabolic status might permit this lineage transition.
Therapeutic Interventions
Two biological events are emerging as hallmarks of cancer: reprogramming of energy metabolism and evading immune destruction (1). The latter is an active area of research as cancer immunotherapy. Metastatic PCa with CDK12 inactivating mutations (3-7% incidence) has durable responses to PD-1 blockade by checkpoint inhibitors (138, 139). As for targeted therapy in cancer metabolism, gain of function mutations in isocitrate dehydrogenases (IDH1 and IDH2) result in the production of the “oncometabolite” 2-hydroxyglutarate (140, 141). Targeting mutant IDH is attractive but limited in PCa: IDH mutations account for only 1-2% of PCa incidence, which is much lower than other tumors, e.g. glioma (~50%) (142, 143). For PCa, dysregulated FA metabolism, which is mechanistically linked to aberrant AR and/or SREBP signaling (49, 144), has multiple candidate factors for pharmacological inhibition, including SREBP (fatostatin) (145), acetyl-CoA carboxylase (ND-646, GS-0976) (146, 147), and SCD1 (Merck Frosst Cpd 3) (148). IPI-9119 (67) and TVB-2640 (80) are selective FASN inhibitors for potential clinical use. Treatment with IPI-9119 led to disruption of the interdependence between AR and FASN and extensive reduction in AR signaling (67). Energy disruptors aim to reduce intracellular ATP level by inhibiting glycolysis or disturbing mitochondrial mechanisms leading to oxidative phosphorylation (33, 149). Several options are available for pharmacological inhibition of glucose metabolism: glucose uptake (phloretin) (150) and glycolytic enzymes (3-bromopyruvate and Koningic acid for GAPDH) (151, 152). Complex I (NADH–quinone oxidoreductase) is the largest respiratory complex of the mitochondrial oxidative phosphorylation system (153). Complex I inhibition has been shown to be a potential clinical repressor of prostate growth based on early correlative and retrospective studies in men with PCa who had received metformin for treatment of their associated diabetes mellitus (154, 155). Thus, mitochondrial energy metabolism emerges as cancer therapy target (156). In addition to direct inhibition on oxidative phosphorylation (BAY87-2243 and IACS-010759 for complex I) (157, 158), the strategies can be developed to prevent entry of the precursors of TCA cycle intermediates into mitochondria. Glutamine utilization can be prevented by inhibiting glutamine uptake and metabolism (CB839 for glutaminase and V9302 for ASCT2) (159–161). CPT1 inhibition prevents the entry of FA into mitochondria and thus downstream FAO (65). On the other hand, MSDC-0160 inhibits pyruvate entry into mitochondria by mitochondrial pyruvate carrier (144).
Therapeutic targets in cancer metabolism in many cases exist even in the normal cells, which adds potential toxicity and non-specificity to drugs targeting metabolic pathways (162). It is necessary to define their specific action in the context of tumor initiation and progression. The successful application of metabolic inhibitors will lie in accurate metabolic phenotyping and stratification of tumors to predict which respond to the given drugs.
Discussion
We have described how PCa is unique from other cancers from the metabolic point of view. In addition, AR signaling persists in normal and malignant prostatic cells except for when AR antagonism triggers the transition to highly glycolytic AR-indifferent carcinoma. AR determines virtually all aspects of cellular metabolism while a selected phenotype is dominant depending on the stage of disease progression. Accordingly, the question remains as to what directs AR toward specified metabolic preference. The underlying mechanisms may include the presence of AR-Vs, differential actions of AR co-regulators, epigenetics, and tumor microenvironment. Understanding and targeting the selective AR-metabolome axis may provide the unique therapeutic opportunity for AR-driven CRPC which is resistant to current anti-AR therapy.
Except for targeting mutant IDHs, metabolic inhibitors are potentially active regardless of tumor genetic subtype and thus beneficial to the large majority of men with CRPC who are not currently candidates for precision medicine (e.g., DNA repair defects for PARP inhibitors or CDK12 loss for immunotherapy) (138, 163). Nevertheless, appropriate tumor imaging at spatial resolution (e.g., use of PET radiotracers) may facilitate select effective metabolic therapy by determining what bioenergetic phenotype dominates in tumors (glycolytic, lipogenic, or oxidative) (121, 122). For instance, FASN inhibition may be selected when 11C acetate uptake suggests tumors are lipogenic. High avidity to 18F-FDG is supported by expression of glycolytic gene signature in NEPC, providing a rationale to target glucose metabolism for therapy. Tumor plasticity adds another layer of complexity to PCa as it develops and spreads. Altered metabolic pathways may be dispensable or indispensable depending on the stage of tumor progression. This is true for de novo FA synthesis whose pharmacological inhibition is detrimental in some cases (antitumorigenic regardless of availability of exogenous lipids) but tolerable in others (e.g., rescued by lipids derived from diet and adipose tissues) (67, 70).
To develop effective metabolism-based target therapy (164), it is crucial to identify metabolic pathways that define the stage of tumor progression depending on AR and cellular lineage status. The success of future therapies may be enhanced by the combination of the prescribed metabolic inhibitors such as metformin and statins (155, 165).
Author Contributions
TU and SRP conceived and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
VA merit review award (I01BX003324) and DoD award (W81XWH-17-1-0323 W81XWH-20-1-0146, W81XWH-17-1-0484) to SRP. NCI P50 CA097186, and the Institute for Prostate Cancer Research at the University of Washington and Fred Hutchinson Cancer Research Center.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer (2017) 17(4):239–53. doi: 10.1038/nrc.2017.5
4. Sieber OM, Tomlinson SR, Tomlinson IP. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer (2005) 5(8):649–55. doi: 10.1038/nrc1674
5. Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence M, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature (2015) 518(7539):360–4. doi: 10.1038/nature14221
6. Mayers JR, Vander Heiden MG. Nature and Nurture: What Determines Tumor Metabolic Phenotypes? Cancer Res (2017) 77(12):3131–4. doi: 10.1158/0008-5472.CAN-17-0165
7. Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab (2012) 15(2):157–70. doi: 10.1016/j.cmet.2011.12.015
8. Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol (2013) 31(6):522–9. doi: 10.1038/nbt.2530
9. Warburg O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften (1924) 12(50):1131–7. doi: 10.1007/BF01504608
10. Flier J, Mueckler M, Usher P, Lodish H. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science (1987) 235(4795):1492–5. doi: 10.1126/science.3103217
11. Warburg O. On respiratory impairment in cancer cells. Science (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.267
12. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
13. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
14. Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science (2010) 329(5998):1492–9. doi: 10.1126/science.1188015
15. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell (2017) 168(4):657–69. doi: 10.1016/j.cell.2016.12.039
16. Costello LC, Franklin RB. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Mol Cancer (2006) 5(1):17. doi: 10.1186/1476-4598-5-17
17. Costello LC, Franklin RB. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch Biochem Biophys (2016) 611:100–12. doi: 10.1016/j.abb.2016.04.014
18. Costello LC, Franklin RB. Citrate metabolism of normal and malignant prostate epithelial cells. Urology (1997) 50(1):3–12. doi: 10.1016/S0090-4295(97)00124-6
19. Costello LC, Franklin RB. The intermediary metabolism of the prostate: a key to understanding the pathogenesis and progression of prostate malignancy. Oncology (2000) 59(4):269–82. doi: 10.1159/000012183
20. Bader DA, McGuire SE. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat Rev Urol (2020) 17(4):214–31. doi: 10.1038/s41585-020-0288-x
21. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J (2011) 30(13):2719–33. doi: 10.1038/emboj.2011.158
22. Barfeld SJ, Itkonen HM, Urbanucci A, Mills IG. Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocr Relat Cancer (2014) 21(4):T57–66. doi: 10.1530/ERC-13-0515
23. Giunchi F, Fiorentino M, Loda M. The Metabolic Landscape of Prostate Cancer. Eur Urol Oncol (2019) 2(1):28–36. doi: 10.1016/j.euo.2018.06.010
24. Centenera MM, Harris JM, Tilley WD, Butler LM. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol Endocrinol (2008) 22:2373–82. doi: 10.1210/me.2008-0017
25. Zhang A, Zhang J, Plymate S, Mostaghel EA. Classical and Non-Classical Roles for Pre-Receptor Control of DHT Metabolism in Prostate Cancer Progression. Horm Cancer (2016) 7(2):104–13. doi: 10.1007/s12672-016-0250-9
26. Auchus RJ, Sharifi N. Sex Hormones and Prostate Cancer. Annu Rev Med (2020) 71:33–45. doi: 10.1146/annurev-med-051418-060357
27. Gonzalez-Menendez P, Hevia D, Mayo JC, Sainz RM. The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int J Cancer (2018) 142(12):2414–24. doi: 10.1002/ijc.31165
28. Kelsey R. Prostate cancer: Glycolysis and AR expression as biomarkers. Nat Rev Urol (2018) 15(1):2. doi: 10.1038/nrurol.2017.205
29. Fox JJ, Gavane SC, Blanc-Autran E, Nehmeh S, Gonen M, Beattie B, et al. Positron Emission Tomography/Computed Tomography-Based Assessments of Androgen Receptor Expression and Glycolytic Activity as a Prognostic Biomarker for Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol (2018) 4(2):217–24. doi: 10.1001/jamaoncol.2017.3588
30. Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst (2009) 101(7):519–32. doi: 10.1093/jnci/djp030
31. Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology (1996) 137(10):4468–74. doi: 10.1210/endo.137.10.8828509
32. Swinnen JV, Heemers H, van de Sande T, de Schrijver E, Brusselmans K, Heyns W, et al. Androgens, lipogenesis and prostate cancer. J Steroid Biochem Mol Biol (2004) 92(4):273–9. doi: 10.1016/j.jsbmb.2004.10.013
33. Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res (2019) 25(23):6916–24. doi: 10.1158/1078-0432.CCR-19-1423
34. Spratt DE, Gavane S, Tarlinton L, Fareedy SB, Doran MG, Zelefsky MJ, et al. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate (2014) 74(11):1153–9. doi: 10.1002/pros.22831
35. Bakht MK, Lovnicki JM, Tubman J, Stringer KF, Chiaramonte J, Reynolds MR, et al. Differential Expression of Glucose Transporters and Hexokinases in Prostate Cancer with a Neuroendocrine Gene Signature: A Mechanistic Perspective for (18)F-FDG Imaging of PSMA-Suppressed Tumors. J Nucl Med (2020) 61(6):904–10. doi: 10.2967/jnumed.119.231068
36. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer (2001) 1(1):34–45. doi: 10.1038/35094009
37. Costello LC, Franklin RB. Prostatic fluid electrolyte composition for the screening of prostate cancer: a potential solution to a major problem. Prostate Cancer Prostatic Dis (2009) 12(1):17–24. doi: 10.1038/pcan.2008.19
38. Robinson BR JMM, Ro JY, Divatia M. Precision molecular pathology of prostate cancer. New York: SPRINGER (2019).
39. Mycielska ME, Patel A, Rizaner N, Mazurek MP, Keun H, Patel A, et al. Citrate transport and metabolism in mammalian cells: prostate epithelial cells and prostate cancer. Bioessays (2009) 31(1):10–20. doi: 10.1002/bies.080137
40. Kolenko V, Teper E, Kutikov A, Uzzo R. Zinc and zinc transporters in prostate carcinogenesis. Nat Rev Urol (2013) 10(4):219–26. doi: 10.1038/nrurol.2013.43
41. Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem (1997) 272(46):28875–81. doi: 10.1074/jbc.272.46.28875
42. Costello LC, Liu Y, Zou J, Franklin RB. The pyruvate dehydrogenase E1 alpha gene is testosterone and prolactin regulated in prostate epithelial cells. Endocr Res (2000) 26(1):23–39. doi: 10.1080/07435800009040143
43. Franklin RB, Zou J, Yu Z, Costello LC. EAAC1 is expressed in rat and human prostate epithelial cells; functions as a high-affinity L-aspartate transporter; and is regulated by prolactin and testosterone. BMC Biochem (2006) 7:10. doi: 10.1186/1471-2091-7-10
44. Franklin RB, Kukoyi BI, Akuffo V, Costello LC. Testosterone stimulation of mitochondrial aspartate aminotransferase levels and biosynthesis in rat ventral prostate. J Steroid Biochem (1987) 28(3):247–56. doi: 10.1016/0022-4731(87)91015-6
45. Franklin RB, Feng P, Milon B, Desouki MM, Singh KK, Kajdacsy-Balla A, et al. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol Cancer (2005) 4(1):32. doi: 10.1186/1476-4598-4-32
46. Jadvar H. Is There Use for FDG-PET in Prostate Cancer? Semin Nucl Med (2016) 46(6):502–6. doi: 10.1053/j.semnuclmed.2016.07.004
47. Sahin E, Elboga U, Kalender E, Basibuyuk M, Demir HD, Celen YZ. Clinical significance of incidental FDG uptake in the prostate gland detected by PET/CT. Int J Clin Exp Med (2015) 8(7):10577–85.
48. Awad D, Pulliam TL, Lin C, Wilkenfeld SR, Frigo DE. Delineation of the androgen-regulated signaling pathways in prostate cancer facilitates the development of novel therapeutic approaches. Curr Opin Pharmacol (2018) 41:1–11. doi: 10.1016/j.coph.2018.03.002
49. Butler LM, Centenera MM, Swinnen JV. Androgen control of lipid metabolism in prostate cancer: novel insights and future applications. Endocr Relat Cancer (2016) 23(5):R219–27. doi: 10.1530/ERC-15-0556
50. Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, Kim JW, et al. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J (2011) 433(1):225–33. doi: 10.1042/BJ20101104
51. Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, et al. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis (2014) 3:e103. doi: 10.1038/oncsis.2014.18
52. Audet-Walsh E, Dufour CR, Yee T, Zouanat FZ, Yan M, Kalloghlian G, et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev (2017) 31(12):1228–42. doi: 10.1101/gad.299958.117
53. Audet-Walsh E, Yee T, McGuirk S, Vernier M, Ouellet C, St-Pierre J, et al. Androgen-Dependent Repression of ERRgamma Reprograms Metabolism in Prostate Cancer. Cancer Res (2017) 77(2):378–89. doi: 10.1158/0008-5472.CAN-16-1204
54. Zacharias N, Lee J, Ramachandran S, Shanmugavelandy S, McHenry J, Dutta P, et al. Androgen Receptor Signaling in Castration-Resistant Prostate Cancer Alters Hyperpolarized Pyruvate to Lactate Conversion and Lactate Levels In Vivo. Mol Imaging Biol MIB Off Publ Acad Mol Imaging (2019) 21(1):86–94. doi: 10.1007/s11307-018-1199-6
55. Choi SY, Xue H, Wu R, Fazli L, Lin D, Collins CC, et al. The MCT4 Gene: A Novel, Potential Target for Therapy of Advanced Prostate Cancer. Clin Cancer Res (2016) 22(11):2721–33. doi: 10.1158/1078-0432.CCR-15-1624
56. Heemers HV, Verhoeven G, Swinnen JV. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol Endocrinol (2006) 20(10):2265–77. doi: 10.1210/me.2005-0479
57. Heemers H, Verrijdt G, Organe S, Claessens F, Heyns W, Verhoeven G, et al. Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J Biol Chem (2004) 279(29):30880–7. doi: 10.1074/jbc.M401615200
58. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer (2020) 122(1):4–22. doi: 10.1038/s41416-019-0650-z
59. Poulose N, Mills IG, Steele RE. The impact of transcription on metabolism in prostate and breast cancers. Endocr Relat Cancer (2018) 25(9):R435–R52. doi: 10.1530/ERC-18-0048
60. Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res (2010) 70(20):8117–26. doi: 10.1158/0008-5472.CAN-09-3871
61. Tousignant KD, Rockstroh A, Poad BLJ, Talebi A, Young RSE, Taherian Fard A, et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab (2020) 8(1):11. doi: 10.1186/s40170-020-00217-6
62. Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol (2014) 2(2):111–20.
63. Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis (2006) 9(3):230–4. doi: 10.1038/sj.pcan.4500879
64. Liu Y, Zuckier LS, Ghesani NV. Dominant uptake of fatty acid over glucose by prostate cells: a potential new diagnostic and therapeutic approach. Anticancer Res (2010) 30(2):369–74.
65. Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther (2014) 13(10):2361–71. doi: 10.1158/1535-7163.MCT-14-0183
66. Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest (2012) 122(6):1958–9. doi: 10.1172/JCI63967
67. Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci U S A (2019) 116(2):631–40. doi: 10.1073/pnas.1808834116
68. Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijon MA, et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget (2017) 8(34):56051–65. doi: 10.18632/oncotarget.17359
69. Joshi M, Stoykova GE, Salzmann-Sullivan M, Dzieciatkowska M, Liebman LN, Deep G, et al. CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions. Cells (2019) 8(10):1115. doi: 10.3390/cells8101115
70. Balaban S, Nassar ZD, Zhang AY, Hosseini-Beheshti E, Centenera MM, Schreuder M, et al. Extracellular Fatty Acids Are the Major Contributor to Lipid Synthesis in Prostate Cancer. Mol Cancer Res (2019) 17(4):949–62. doi: 10.1158/1541-7786.MCR-18-0347
71. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med (2019) 11(478):eaau5758. doi: 10.1126/scitranslmed.aau5758
72. Tousignant KD, Rockstroh A, Taherian Fard A, Lehman ML, Wang C, McPherson SJ, et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol Cancer Res (2019) 17(5):1166–79. doi: 10.1158/1541-7786.MCR-18-1147
73. Ono M, Oka S, Okudaira H, Nakanishi T, Mizokami A, Kobayashi M, et al. [(14)C]Fluciclovine (alias anti-[(14)C]FACBC) uptake and ASCT2 expression in castration-resistant prostate cancer cells. Nucl Med Biol (2015) 42(11):887–92. doi: 10.1016/j.nucmedbio.2015.07.005
74. Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst (2013) 105(19):1463–73. doi: 10.1093/jnci/djt241
75. Putluri N, Shojaie A, Vasu VT, Nalluri S, Vareed SK, Putluri V, et al. Metabolomic profiling reveals a role for androgen in activating amino acid metabolism and methylation in prostate cancer cells. PloS One (2011) 6(7):e21417. doi: 10.1371/journal.pone.0021417
76. White MA, Lin C, Rajapakshe K, Dong J, Shi Y, Tsouko E, et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol Cancer Res (2017) 15(8):1017–28. doi: 10.1158/1541-7786.MCR-16-0480
77. Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol (2015) 236(3):278–89. doi: 10.1002/path.4518
78. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
79. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med (2018) 24(2):194–202. doi: 10.1038/nm.4464
80. Broer A, Fairweather S, Broer S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front Pharmacol (2018) 9:785. doi: 10.3389/fphar.2018.00785
81. Corbin JM, Ruiz-Echevarria MJ. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int J Mol Sci (2016) 17(8):1208. doi: 10.3390/ijms17081208
82. Shukla-Dave A, Castillo-Martin M, Chen M, Lobo J, Gladoun N, Collazo-Lorduy A, et al. Ornithine Decarboxylase Is Sufficient for Prostate Tumorigenesis via Androgen Receptor Signaling. Am J Pathol (2016) 186(12):3131–45. doi: 10.1016/j.ajpath.2016.08.021
83. MacLean HE, Chiu WS, Notini AJ, Axell AM, Davey RA, McManus JF, et al. Impaired skeletal muscle development and function in male, but not female, genomic androgen receptor knockout mice. FASEB J (2008) 22(8):2676–89. doi: 10.1096/fj.08-105726
84. Sanderson SM, Gao X, Dai Z, Locasale JW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer (2019) 19(11):625–37. doi: 10.1038/s41568-019-0187-8
85. Huang J, Duran A, Reina-Campos M, Valencia T, Castilla EA, Muller TD, et al. Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell (2018) 33(4):770–84 e6. doi: 10.1016/j.ccell.2018.03.001
86. Huggins C, Hodges C. Studies on prostatic cancer - I The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res (1941) 1(4):293–7.
87. Sharifi N, Gulley JL, Dahut WL. An update on androgen deprivation therapy for prostate cancer. Endocr Relat Cancer (2010) 17(4):R305–15. doi: 10.1677/ERC-10-0187
88. Sharifi N. Steroid sidestep: evading androgen ablation by abiraterone. Clin Cancer Res (2015) 21(6):1240–2. doi: 10.1158/1078-0432.CCR-14-2899
89. Sharifi N. Prostate cancer: CYP17A1 inhibitor failure-lessons for future drug development. Nat Rev Urol (2015) 12(5):245–6. doi: 10.1038/nrurol.2015.66
90. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med (2011) 364:1995–2005. doi: 10.1056/NEJMoa1014618
91. Fizazi K, Scher H, Miller K, Basch E, Sternberg C, Cella D, et al. Effect of enzalutamide on time to first skeletal-related event, pain, and quality of life in men with castration-resistant prostate cancer: results from the randomised, phase 3 AFFIRM trial. Lancet Oncol (2014) 15:1147–56. doi: 10.1016/S1470-2045(14)70303-1
92. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med (2004) 10(1):33–9. doi: 10.1038/nm972
93. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res (2008) 68(11):4447–54. doi: 10.1158/0008-5472.CAN-08-0249
94. Mostaghel EA, Plymate SR, Montgomery B. Molecular pathways: targeting resistance in the androgen receptor for therapeutic benefit. Clin Cancer Res (2014) 20:791–8. doi: 10.1158/1078-0432.CCR-12-3601
95. Mostaghel E, Marck B, Plymate S, Vessella R, Balk S, Matsumoto A, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res (2011) 17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728
96. Taplin M, Antonarakis E, Ferrante K, Horgan K, Blumenstein B, Saad F, et al. Clinical factors associated with AR-V7 detection in ARMOR3-SV, a randomized trial of galeterone (Gal) vs enzalutamide (Enz) in men with AR-V7+metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol (2017) 35. doi: 10.1200/JCO.2017.35.15_suppl.5005
97. Attard G, Parker C, Eeles RA, Schröder F, Tomlins SA, Tannock I, et al. Prostate cancer. Lancet (2015) 387(10013):70–82. doi: 10.1016/S0140-6736(14)61947-4
98. Rice MA, Malhotra SV, Stoyanova T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front Oncol (2019) 9:801. doi: 10.3389/fonc.2019.00801
99. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer (2015) 15(12):701–11. doi: 10.1038/nrc4016
100. Nelson PS. Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. J Clin Oncol (2012) 30(6):644–6. doi: 10.1200/JCO.2011.39.1300
101. Uo T, Plymate SR, Sprenger CC. Allosteric alterations in the androgen receptor and activity in prostate cancer. Endocr Relat Cancer (2017) 24(9):R335–R48. doi: 10.1530/ERC-17-0108
102. Uo T, Plymate SR, Sprenger CC. The potential of AR-V7 as a therapeutic target. Expert Opin Ther Targets (2018) 22(3):201–16. doi: 10.1080/14728222.2018.1439016
103. Luo J, Attard G, Balk SP, Bevan C, Burnstein K, Cato L, et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur Urol (2018) 73(5):715–23. doi: 10.1016/j.eururo.2017.11.038
104. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res (2014) 20(11):2846–50. doi: 10.1158/1078-0432.CCR-13-3309
105. Davies A, Zoubeidi A, Selth LA. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr Relat Cancer (2020) 27(2):R35–50. doi: 10.1530/ERC-19-0420
106. Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol (2018) 15(5):271–86. doi: 10.1038/nrurol.2018.22
107. Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest (2019) 129(10):4492–505. doi: 10.1172/JCI128212
108. Zhang X, Coleman IM, Brown LG, True LD, Kollath L, Lucas JM, et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin Cancer Res (2015) 21(20):4698–708. doi: 10.1158/1078-0432.CCR-15-0157
109. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell (2016) 30(4):563–77. doi: 10.1016/j.ccell.2016.09.005
110. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell (2017) 32(4):474–89 e6. doi: 10.1016/j.ccell.2017.09.003
111. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res (2008) 68(15):6407–15. doi: 10.1158/0008-5472.CAN-07-5997
112. Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell (2013) 154(5):1074–84. doi: 10.1016/j.cell.2013.07.029
113. Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature (2015) 523(7560):347–51. doi: 10.1038/nature14406
114. Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget (2015) 6(31):31997–2012. doi: 10.18632/oncotarget.5585
115. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature (2010) 463(7279):364–8. doi: 10.1038/nature08697
116. Dang CV. MYC. metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med (2013) 3(8):a014217. doi: 10.1101/cshperspect.a014217
117. Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J (2006) 25(12):2723–34. doi: 10.1038/sj.emboj.7601152
118. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab (2012) 16(1):9–17. doi: 10.1016/j.cmet.2012.06.001
119. Wang HJ, Pochampalli M, Wang LY, Zou JX, Li PS, Hsu SC, et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene (2019) 38(1):17–32. doi: 10.1038/s41388-018-0414-x
120. Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, et al. Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell (2019) 35(3):385–400 e9. doi: 10.1016/j.ccell.2019.01.018
121. Jadvar H. Prostate cancer: PET with 18F-FDG, 18F- or 11C-acetate, and 18F- or 11C-choline. J Nucl Med (2011) 52(1):81–9. doi: 10.2967/jnumed.110.077941
122. Emonds KM, Swinnen JV, Lerut E, Koole M, Mortelmans L, Mottaghy FM. Evaluation of androgen-induced effects on the uptake of [18F]FDG, [11C]choline and [11C]acetate in an androgen-sensitive and androgen-independent prostate cancer xenograft model. EJNMMI Res (2013) 3(1):31. doi: 10.1186/2191-219X-3-31
123. Vavere AL, Kridel SJ, Wheeler FB, Lewis JS. 1-11C-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J Nucl Med (2008) 49(2):327–34. doi: 10.2967/jnumed.107.046672
124. Yoshimoto M, Waki A, Yonekura Y, Sadato N, Murata T, Omata N, et al. Characterization of acetate metabolism in tumor cells in relation to cell proliferation: acetate metabolism in tumor cells. Nucl Med Biol (2001) 28(2):117–22. doi: 10.1016/S0969-8051(00)00195-5
125. Jadvar H, Gurbuz A, Li X, Shahinian A, Conti PS. Choline autoradiography of human prostate cancer xenograft: effect of castration. Mol Imaging (2008) 7(3):147–52. doi: 10.2310/7290.2008.00018
126. Kotzerke J, Prang J, Neumaier B, Volkmer B, Guhlmann A, Kleinschmidt K, et al. Experience with carbon-11 choline positron emission tomography in prostate carcinoma. Eur J Nucl Med (2000) 27(9):1415–9. doi: 10.1007/s002590000309
127. Zha Z, Ploessl K, Choi SR, Wu Z, Zhu L, Kung HF. Synthesis and evaluation of a novel urea-based (68)Ga-complex for imaging PSMA binding in tumor. Nucl Med Biol (2018) 59:36–47. doi: 10.1016/j.nucmedbio.2017.12.007
128. Hope TA, Truillet C, Ehman EC, Afshar-Oromieh A, Aggarwal R, Ryan CJ, et al. 68Ga-PSMA-11 PET Imaging of Response to Androgen Receptor Inhibition: First Human Experience. J Nucl Med (2017) 58(1):81–4. doi: 10.2967/jnumed.116.181800
129. Meziou S, Ringuette Goulet C, Hovington H, Lefebvre V, Lavallée É, Bergeron M, et al. GLUT1 expression in high-risk prostate cancer: correlation with 18F-FDG-PET/CT and clinical outcome. Prostate Cancer Prostatic Dis (2020) 23(3):441–8. doi: 10.1038/s41391-020-0202-x
130. Momcilovic M, Jones A, Bailey ST, Waldmann CM, Li R, Lee JT, et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature (2019) 575(7782):380–4. doi: 10.1038/s41586-019-1715-0
131. Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res (2006) 66(18):8927–30. doi: 10.1158/0008-5472.CAN-06-1501
132. Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun (2016) 7:13041. doi: 10.1038/ncomms13041
133. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer (2016) 16(11):732–49. doi: 10.1038/nrc.2016.89
134. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science (2016) 353(6304):1161–5. doi: 10.1126/science.aaf5171
135. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell (2002) 2(2):103–12. doi: 10.1016/S1535-6108(02)00102-2
136. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (2017) 355(6320):84–8. doi: 10.1126/science.aah4307
137. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (2017) 355(6320):78–83. doi: 10.1126/science.aah4199
138. Manogue C, Cotogno P, Ledet E, Lewis B, Wyatt AW, Sartor O. Biomarkers for Programmed Death-1 Inhibition in Prostate Cancer. Oncologist (2019) 24(4):444–8. doi: 10.1634/theoncologist.2018-0546
139. Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell (2018) 173(7):1770–82 e14. doi: 10.1016/j.cell.2018.04.034
140. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19(1):17–30. doi: 10.1016/j.ccr.2010.12.014
141. Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest (2013) 123(9):3652–8. doi: 10.1172/JCI67228
142. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discovery (2013) 3(7):730–41. doi: 10.1158/2159-8290.CD-13-0083
143. Ghiam AF, Cairns RA, Thoms J, Dal Pra A, Ahmed O, Meng A, et al. IDH mutation status in prostate cancer. Oncogene (2012) 31(33):3826. doi: 10.1038/onc.2011.546
144. Bader DA, Hartig SM, Putluri V, Foley C, Hamilton MP, Smith EA, et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat Metab (2019) 1(1):70–85. doi: 10.1038/s42255-018-0002-y
145. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther (2014) 13(4):855–66. doi: 10.1158/1535-7163.MCT-13-0797
146. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med (2016) 22(10):1108–19. doi: 10.1038/nm.4181
147. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology (2018) 155(5):1463–73 e6. doi: 10.1053/j.gastro.2018.07.027
148. Ramtohul YK, Black C, Chan CC, Crane S, Guay J, Guiral S, et al. SAR and optimization of thiazole analogs as potent stearoyl-CoA desaturase inhibitors. Bioorg Med Chem Lett (2010) 20(5):1593–7. doi: 10.1016/j.bmcl.2010.01.083
149. Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta (2011) 1807(6):552–61. doi: 10.1016/j.bbabio.2010.10.012
150. Gonzalez-Menendez P, Hevia D, Rodriguez-Garcia A, Mayo JC, Sainz RM. Regulation of GLUT Transporters by Flavonoids in Androgen-Sensitive and -Insensitive Prostate Cancer Cells. Endocrinology (2014) 155(9):3238–50. doi: 10.1210/en.2014-1260
151. Valenti D, Vacca RA, de Bari L. 3-Bromopyruvate induces rapid human prostate cancer cell death by affecting cell energy metabolism, GSH pool and the glyoxalase system. J Bioenerg Biomembr (2015) 47(6):493–506. doi: 10.1007/s10863-015-9631-y
152. Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab (2017) 26(4):648–59 e8. doi: 10.1016/j.cmet.2017.08.017
153. Urra FA, Munoz F, Lovy A, Cardenas C. The Mitochondrial Complex(I)ty of Cancer. Front Oncol (2017) 7:118. doi: 10.3389/fonc.2017.00118
154. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (2005) 330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7
155. Whitburn J, Edwards CM, Sooriakumaran P. Metformin and Prostate Cancer: a New Role for an Old Drug. Curr Urol Rep (2017) 18(6):46. doi: 10.1007/s11934-017-0693-8
156. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol (2015) 11(1):9–15. doi: 10.1038/nchembio.1712
157. Ellinghaus P, Heisler I, Unterschemmann K, Haerter M, Beck H, Greschat S, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med (2013) 2(5):611–24. doi: 10.1002/cam4.112
158. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24(7):1036–46. doi: 10.1038/s41591-018-0052-4
159. Zacharias NM, McCullough C, Shanmugavelandy S, Lee J, Lee Y, Dutta P, et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci Rep (2017) 7(1):16159. doi: 10.1038/s41598-017-16327-z
160. Truini A, Alama A, Dal Bello MG, Coco S, Vanni I, Rijavec E, et al. Clinical Applications of Circulating Tumor Cells in Lung Cancer Patients by CellSearch System. Front Oncol (2014) 4:242. doi: 10.3389/fonc.2014.00242
161. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870
162. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/sciadv.1600200
163. Mateo J, Boysen G, Barbieri CE, Bryant HE, Castro E, Nelson PS, et al. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur Urol (2017) 71(3):417–25. doi: 10.1016/j.eururo.2016.08.037
164. Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol (2017) 24(9):1161–80. doi: 10.1016/j.chembiol.2017.08.028
Keywords: androgen receptor, castration-resistant prostate cancer, metabolic reprogramming 18F-FDG, neuroendocrine, aerobic glycolysis, fatty acid metabolism, mitochondria
Citation: Uo T, Sprenger CC and Plymate SR (2020) Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 10:580617. doi: 10.3389/fonc.2020.580617
Received: 06 July 2020; Accepted: 25 September 2020;
Published: 09 October 2020.
Edited by:
Isabel R. Schlaepfer, University of Colorado, United StatesReviewed by:
Jone Stanley, Texas A&M University, United StatesAnca Maria Cimpean, Victor Babes University of Medicine and Pharmacy, Romania
Copyright © 2020 Uo, Sprenger and Plymate. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takuma Uo, dHVvQHV3LmVkdQ==; Stephen R. Plymate, c3BseW1hdGVAdXcuZWR1