 Ramiro Vázquez1,2†
Ramiro Vázquez1,2† Maria E. Riveiro1†
Maria E. Riveiro1† Caroline Berenguer-Daizé3
Caroline Berenguer-Daizé3 Anthony O’Kane4Julie Gormley4Olivier Touzelet4Keyvan Rezai5Mohamed Bekradda1L’Houcine Ouafik3,6*
Anthony O’Kane4Julie Gormley4Olivier Touzelet4Keyvan Rezai5Mohamed Bekradda1L’Houcine Ouafik3,6*- 1Preclinical Department, Early Drug Development Group (E2DG), Boulogne-Billancourt, France
- 2Center for Genomic Science of IIT@SEMM, Fondazione Istituto Italiano di Tecnologia (IIT), Milan, Italy
- 3Aix Marseille University, CNRS, INP, Institute of NeuroPhysiopathology, Marseille, France
- 4Discovery and Scientific Affairs Department, Fusion Antibodies plc., Belfast, United Kingdom
- 5Department of Radio-Pharmacology, Institute Curie-René Huguenin Hospital, Saint-Cloud, France
- 6APHM, CHU Nord, Service de Transfert d’Oncologie Biologique, Marseille, France
The development, maintenance and metastasis of solid tumors are highly dependent on the formation of blood and lymphatic vessels from pre-existing ones through a series of processes that are respectively known as angiogenesis and lymphangiogenesis. Both are mediated by specific growth-stimulating molecules, such as the vascular endothelial growth factor (VEGF) and adrenomedullin (AM), secreted by diverse cell types which involve not only the cancerogenic ones, but also those constituting the tumor stroma (i.e., macrophages, pericytes, fibroblasts, and endothelial cells). In this sense, anti-angiogenic therapy represents a clinically-validated strategy in oncology. Current therapeutic approaches are mainly based on VEGF-targeting agents, which, unfortunately, are usually limited by toxicity and/or tumor-acquired resistance. AM is a ubiquitous peptide hormone mainly secreted in the endothelium with an important involvement in blood vessel development and cardiovascular homeostasis. In this review, we will introduce the state-of-the-art in terms of AM physiology, while putting a special focus on its pro-tumorigenic role, and discuss its potential as a therapeutic target in oncology. A large amount of research has evidenced AM overexpression in a vast majority of solid tumors and a correlation between AM levels and disease stage, progression and/or vascular density has been observed. The analysis presented here indicates that the involvement of AM in the pathogenesis of cancer arises from: 1) direct promotion of cell proliferation and survival; 2) increased vascularization and the subsequent supply of nutrients and oxygen to the tumor; 3) and/or alteration of the cell phenotype into a more aggressive one. Furthermore, we have performed a deep scrutiny of the pathophysiological prominence of each of the AM receptors (AM1 and AM2) in different cancers, highlighting their differential locations and functions, as well as regulatory mechanisms. From the therapeutic point of view, we summarize here an exhaustive series of preclinical studies showing a reduction of tumor angiogenesis, metastasis and growth following treatment with AM-neutralizing antibodies, AM receptor antagonists, or AM receptor interference. Anti-AM therapy is a promising strategy to be explored in oncology, not only as an anti-angiogenic alternative in the context of acquired resistance to VEGF treatment, but also as a potential anti-metastatic approach.
Introduction
The term cancer comprises different types of pathologies characterized by uncontrolled proliferation of cells that, with the exception of those of hematological or lymphatic origin, give place to malignant tumor masses. Primary tumors grow supported by new vascularization resulting from pre-existing capillaries in a sequence of events that are collectively known as angiogenesis. This process is triggered in response to spontaneous or induced tissue hypoxia, a common phenomenon in solid tumors. These new vessels are also used by cancer cells to spread to other sites within the body after acquiring invasive potential, thereby causing metastasis and, without intervention, death. Surgery and radiotherapy constitute the first approaches in the treatment of localized tumors while systemic agents (chemotherapy, hormone and biological therapies) are the choice to confront the metastatic setting.
In this context, the discovery of tumor angiogenesis opened a new path in fighting cancer. Hypoxia-inducible factor-1α (HIF-1α) is the master switch of the cell machinery required to face O2-lacking periods in physiological and pathological conditions. One of its target genes is that which encodes for the vascular endothelial growth factor (VEGF), the best characterized angiogenic promoter, involved in the modulation of vessel permeability and remodeling, and endothelial cell survival, proliferation and migration (1). The current angiogenesis-targeting approaches approved in clinical practice are: 1) VEGF-blocking monoclonal antibodies (bevacizumab/Avastin®); 2) decoy receptors, ‘VEGF-trap’ (aflibercept/Zaltrap®); 3) tyrosine kinase inhibitors (sunitinib/Sutent®, sorafenib/Nexavar®, axitinib/Inlyta®); and 4) monoclonal antibodies targeting VEGF receptors (ramucirumab/Cyramza®) (2). These agents are being used in the treatment of breast, colorectal, hepatocellular, gastric, and lung among other cancers (2), increasing the effectiveness of conventional chemotherapy. However, a significant number of preclinical and clinical observations have shown that the process of angiogenesis is far from being clearly understood. Furthermore, this approach is not effective in all cancers and often has only limited impact on patient’s overall survival, which, added to the occurrence of frequent drug toxicity and the development of resistance, support the necessity to explore novel strategies aiming to influence alternative factors involved in tumor angiogenesis. In this regard, additional approaches are being tested in preclinical and clinical trials including: angiopoietins (Ang), epidermal growth factor (EGF), fibroblast growth factors (FGF1 and FGF2), hepatocyte growth factor (HGF), platelet-derived growth factor C (PDGF-C), or agents targeting angiogenesis indirectly by inhibiting oncogenic pathways (e.g., HER2, PI3k/AkT/mTOR, and mutated EGF receptor) or hormone signaling (3).
Adrenomedullin –onwards AM– is a regulatory peptide whose involvement in tumor progression and metastasis has become more evident in recent years. The whole literature supports the idea of AM as a survival factor for tumor cells, which can be produced either by the malignant cells themselves or by those located in adjacent/surrounding stroma. In general, AM expression is upregulated by hypoxia, and the excessive production of this peptide is associated with poorer prognosis in cancer patients (4, 5). Moreover, recent reports indicate that AM could be a master regulator upstream of the VEGF pathway and even induce HIF-1α expression, therefore attracting much interest as a therapeutic strategy (6, 7).
Here, we review the physiological and pathological processes mediated by AM, analyzing the advantages that the employment of anti-AM therapy may offer in oncology.
AM in Physiological Context
AM, Calcitonin-Like Peptides, and Their Physiological Roles
The isolation and characterization of AM was reported for the first time by Kitamura et al. in 1993 from human pheochromocytomas of adrenomedullary origin, identifying it as a potent vasodilator (8). The AM gene encodes for a 185-residue preprohormone composed of two bioactive peptides: AM and proadrenomedullin N-terminal 20-residue peptide (PAMP) (9, 10). The preprohormone is firstly processed into proadrenomedullin, from which 20 amino acids at the N-terminus later form PAMP. The remaining AM precursor is successively converted into a C-terminal glycine extended intermediate of 53 amino acids (AM-gly) with scant activity –≈5% of AM effect–, and further into the 52-amino acid biologically-active form by α-amidation of the tyrosine at the carboxy terminal by a peptidylglycine α-amidating monooxygenase (PAM; EC, 1.14.17.3 Figure 1A) (11). Together with the amidated C-terminus, an intramolecular disulfide bond in the N-terminus giving rise to a ring of six or seven residues represent the distinctive structural feature of the calcitonin-like peptide family, which, besides AM, includes calcitonin, the calcitonin gene-related peptide (CGRP), amylin (AMY), and intermedin (or AM2). It is interesting to note their limited sequence homology as evidenced in Figure 1B. Also, despite structural likeness and the fact they share some biological activities, their main physiological roles are diverse (12).
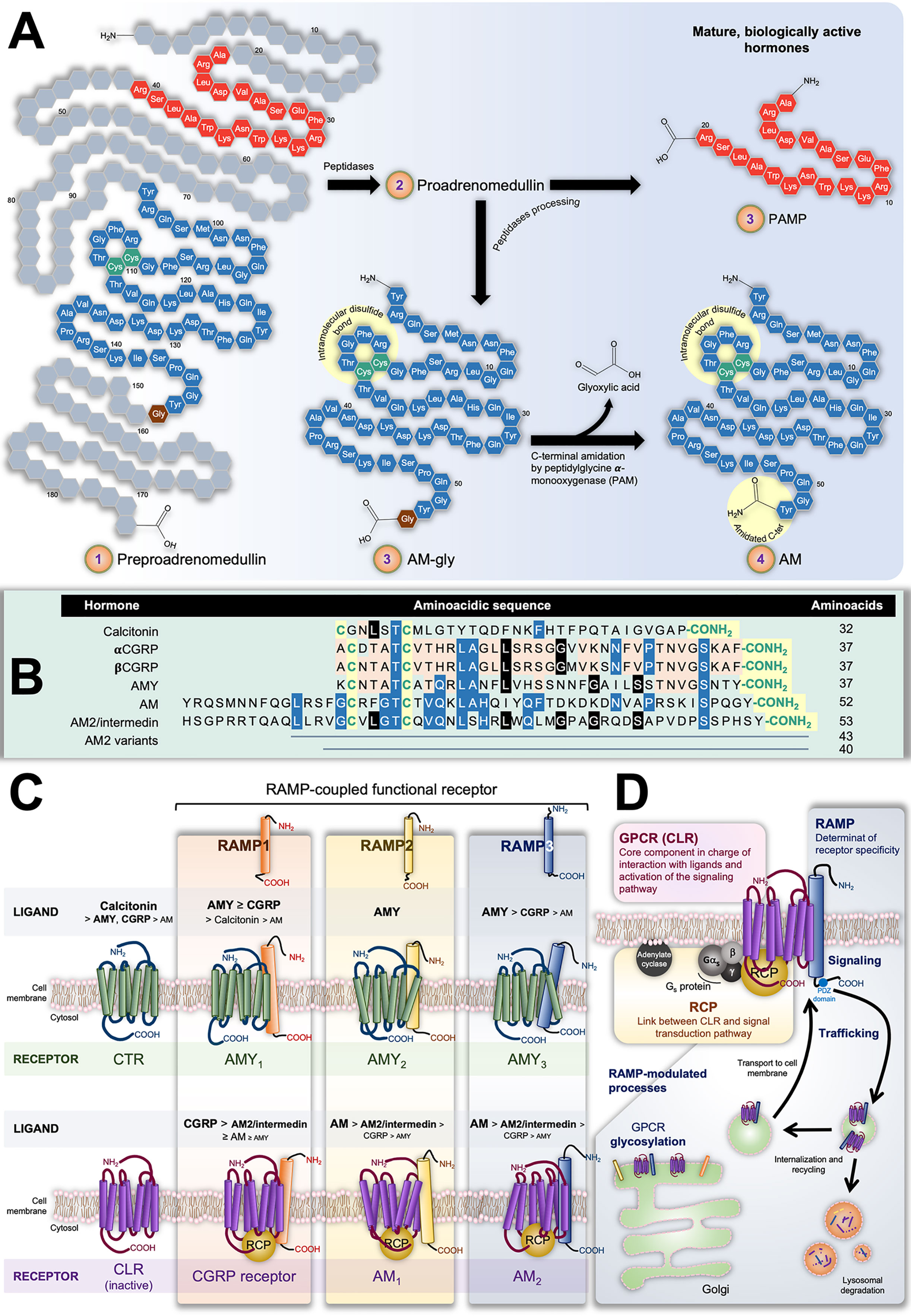
Figure 1 (A) Scheme representing preproadrenumedullin processing to give rise to the mature biologically active peptides adrenomedullin (AM) and proadrenomedullin N-terminal 20 peptide (PAMP), a potent angiogenic agent. Common structural features of the calcitonin-like peptide family (i.e., disulfide bond and amidated C-terminal) are highlighted by yellow circles. (B) Sequences of human components of this family. As in (A), conserved characteristic structures are highlighted in yellow. Different colors indicate position-shared amino acids amongst the AM (blue), AM2/Intermedin variants (black), calcitonin gene-related peptide CGRP), and amylin (AMY) (orange) peptides. (C) Receptors with their decreasing affinity for different ligands. The interaction of calcitonin receptor (CTR) and calcitonin-receptor like receptor (CLR) with receptor-activity modifying proteins (RAMPs) have been very well studied in terms of the ability of the former group to alter the GPCR specificity by acting on their ligand affinity. The CTR binds calcitonin whilst presenting low affinity for AMY and CGRP. In contrast, it forms three AMY-high-affinity and calcitonin-low-affinity receptors (AMY1, AMY2, and AMY3) in the presence of RAMP1, RAMP2, or RAMP3, respectively. On the other hand, CLR itself is not able to bind any known ligand. Nonetheless, the CLR/RAMP1 dimer results in a high-affinity CGRP receptor, while its combination with RAMP2 or RAMP3 respectively produces the AM1 and AM2 subtypes of the AM receptor. Both CLR/RAMP1, AM1 and AM2 also show moderate affinity for AM2/intermedin. (D) Constituents of the AM2 and their role in the tripartite complex.
Calcitonin is a 32-amino acid peptide encoded by the CALCA gene secreted by parafollicular cells –also known as C cells– in the thyroid gland. It plays a key function in the physiological homeostasis of serum Ca2+ inducing its absorption by bone tissue, and hence, hypocalcemia. CGRP results as an alternative splicing of the CALCA gene mRNA transcript (αCGRP) in the nervous system (13, 14), or as the direct (only mature) product of the CALCB gene (βCGRP) (15). CGRP –both isoforms– is widely distributed along the gastrointestinal tract and the central and peripheral nervous systems (16, 17). It is synthesized and released by sensory nerves and has much prominence in afferent neurotransmission. It also induces potent and long-lasting dilation of microvasculature of the nervous system and is involved in nociceptive signaling in migraines (17, 18). It likewise induces hypocalcemia with moderate potency (19). AMY, also referred to as islet amyloid polypeptide (IAPP), possesses a significant level of homology with CGRP, sharing 16 of 37 amino acids (20). AMY was originally identified in the pancreas, where it is co-synthesized, co-packaged, and co-released with insulin from islet β cells (21, 22). Nevertheless, further studies also localized this hormone in areas of the central nervous systems implicated in metabolic control, such as the hypothalamus (23). AMY deposits in type II diabetes are associated with the illness progression (24). In all, AMY is involved in the regulation of energy metabolism by mainly activating and modulating the satiating effect and stomach emptying through the central nervous systems (23, 25, 26), and antagonizing anabolic activity of insulin (4, 27). It also has a potent hypocalcemic activity (28).
AM displays several biological effects, in part due to its almost ubiquitous tissue expression. Apart from malignant and normal adrenal medullary cells, Kitamura et al. detected significant immunoreactive amounts of AM in the lung and kidney (8). Subsequent investigations reported AM expression in neurons and glial cells, but also in blood vessels (endothelial and smooth muscle cells), cardiomyocytes, macrophages, retinal epithelium adipose tissue, and different cancer cells. In fact, it is thought that all body tissues are able to secrete this peptide (29–31). In all cases, AM is not accumulated in vesicles, but its gene expression, synthesis, and subsequent secretion occur immediately in response to a vast series of biochemical (e.g., hormones and LPS) and physical (e.g., hypoxia and shear stress) cell type-dependent stimuli. In the context of this wide distribution of AM-secreting tissues, adrenal production is relative. Indeed, although present in lower concentrations (<1.5 fmol/mg), the total amount of AM in lung and kidney is higher than that produced by the adrenal medulla (≈150 fmol/mg) (8). Plasma concentration of total AM in humans (the sum of the less active, AM-gly and the mature active C-term amidated AM) varies from 2 to 20 fmol/ml (8, 32–35), being significantly increased in patients suffering from hypertension, chronic renal failure, heart failure, obesity, arteriosclerosis, and/or sepsis (30–37). However, it is worth noting that AM-gly is the main endogenous form of immunoreactive blood-circulating AM (38). In fact, the elevated concentration of total AM observed in hypertensive patients would be caused by AM-gly since plasmatic mature AM shows comparable levels between hypertensive and normotensive subjects (38). Kitamura et al. propound three non-mutually exclusive hypotheses to explain such paradoxical findings. 1) Most of the mature AM would act in situ by binding cell membrane receptors and thus a little fraction would be released to blood. 2) Alternatively, the scarce activity of AM-gly would not only facilitate its diffusion into peripheral vessels, but also extend its half-life. 3) Since the mature AM is thought to derive from AM-gly, both the location and activity of the PAM would therefore have a key role in regulating the AM/AM-gly ratio (38). In this sense, authors suggest that the misalignment in terms of PAM and AM expression amongst different tissues could explain the relatively high plasmatic levels of AM-gly. However, their hypothesis is based on the distribution of α-amidilating enzymes in rat (39), which could differ in humans as observed in the case of the lung (40).
AM acts as a circulating hormone but it also elicits multiple biological activities in a paracrine and/or autocrine manner. Its effects are of importance for cardiovascular homeostasis, growth and development of cardiovascular tissues, modulation of the lymphatic flow, regulation of body fluids and diabetes mellitus (8–10, 41–48). Systemic AM administration has been demonstrated to reduce arterial pressure, decrease peripheral vascular resistance, and increase heart rate and cardiac output (8, 49). Moreover, AM and PAMP act as potent angiogenic agents, being necessary for the maintenance of functional membrane microvasculature integrity (45, 47), and regulate lymphatic edema drainage promoting a faster healing of epithelial wounds (50, 51).
In the context of body fluid volume and renal function, AM exerts a tight control of the hypothalamic-pituitary-adrenal axis at all levels (33, 52). AM and its receptors are abundantly expressed in the central nervous system and its cellular components (53). It plays an important role in the regulation of specific blood-brain barrier properties (54), it also increases preganglionic sympathetic discharges (55) and exerts several neuroprotective actions against ischemic damage (56). Furthermore, relatively recent studies suggest that AM may be involved in the neuroendocrine response to stress and nociception (57).
In the digestive system, AM immunoreactivity is widely distributed in the mucosal and glandular epithelia of the stomach, esophagus, intestine, gallbladder, bile duct, and acini of the pancreas and salivary glands (58). It also regulates insulin secretion, directly acting on pancreatic cells (10), and is a potent inhibitor of basal gastrin-stimulated HCl secretion (59). Moreover, it has emerged as a novel and promising therapy for digestive pathologies related with inflammation such as gastric ulcers (60) and inflammatory bowel diseases (61). This is related to the local and systemic anti-inflammatory actions that AM is able to exert (62, 63). For example, it has been demonstrated that AM inhibits the secretion of pro-inflammatory cytokines into the medium by peripheral blood monocytes (64) and plays a role in the evolution of Th1/Th2 cytokine balance, decreasing pro-inflammatory cytokine levels (IL-6, IL-10, TNF-α, IFN-γ) (65–67). In addition to the regulatory role on immune cells, AM also decreases endothelial permeability, thus reducing the formation of inflammatory exudates (64). Likewise, it has been found in all epithelial surfaces that separate the external and internal environment and in all body secretions (68). This wide distribution suggests the possibility that AM has an immunity-related function. In this sense, it has been proven that AM displays potent antimicrobial action against Gram-positive and Gram-negative bacteria (69).
Despite other members of this family, AM possesses limited effect in bone tissue and therefore in calcemic regulation (12).
In 2004, two different groups independently reported the identification of an AM-high-homology peptide in humans. Takei et al., who had previously isolated this AM analog from pufferfish (70), called it AM2 (71); while Roh et al. employed the term intermedin (72). AM2/intermedin is a 53-amino acid peptide resulting from the cleavage of a (148-amino acid) pre-prohormone (72, 73). Rho et al. also reported other two further alternative cuts able to generate 40- or 47-amino acid versions of the hormone (72). In mice, it is highly expressed in submaxillary gland, stomach, pancreas, intestines, kidney, lung, mesentery, thymus, spleen, ovary (but not in testis), and the immune system (71). Both Takei et al. and Rho et al. emphasize the relatively significant levels of the AM2/intermedin in the pituitary gland, which would regulate hormone secretion in an autocrine and/or paracrine manner (71, 72). Like AM, AM2/intermedin possesses a strong hypotensive effect in normal and in spontaneous hypertensive rats (72) as well as cardioprotective effects against myocardial ischemia/reperfusion injury in rats (73).
Particularities of the AM (and Calcitonin-Like Peptide) Receptors
At this point, it is clear that AM and the other hormones of this family exert a broad variety of physiological effects by acting on diverse tissues and systems. What is striking is the fact that all these processes arise from their interaction with only two closely related proteins: calcitonin and calcitonin-like receptors (CTR and CLR, respectively). These are G protein-coupled receptors (GPCR) belonging to the B1 subfamily, which are linked with Gs and thereby signal through adenylate cyclase/cAMP (74). In principle, such apparent promiscuity could partially explain the superposition of biological activities displayed by some of these peptides. Nevertheless, the ligand affinity and thus pharmacological behavior of these GPCRs results from their additional heterodimerization with one of the three accessory receptor activity-modifying proteins (RAMP1, RAMP2 or RAMP3).
The three RAMPs share about 30% of homology in their ≈160-amino acid sequence that gives place to a common structure including a large extracellular N-terminal domain, a single transmembrane domain, and a very short cytoplasmic C-terminal tail (C-tail) (75). RAMPs are well conserved amongst mammals. Indeed, mouse and human amino acid sequence identity for RAMP1, RAMP2, and RAMP3 are 70%, 68%, and 84%, respectively (76). In the endoplasmic reticulum, either, RAMP1, RAMP2, or RAMP3 can pair with CLR or CTR acting as chaperones that confer ligand specificity and binding affinity. Simultaneously, that assembly allows the transport of the receptor complex to the plasma membrane. It has been shown that only RAMP3 bears a PSD-95/Discs large/ZO-1 homology (PDZ) domain in the C-tail which allows its interaction with different factors responsible for further endocytic receptor trafficking and recycling (77). Although RAMPs are ubiquitous throughout the body, there are differences in their tissue distribution, and the abundance of each isoform depends on the tissue type (78–80).
The pharmacology of the CTR –mediator of calcitonin effects– switches differentially in presence of RAMP1, RAMP2 or RAMP3, giving rise to the AMY heterodimeric receptors AMY1, AMY2, and AMY3, respectively. Analogously, the CGRP receptor results from dimerization of CLR with RAMP1, while AM receptors 1 (AM1) and 2 (AM2) correspondingly emerge from the assembly of CLR with RAMP2 or RAMP3. In parallel, AM2/intermedin exerts it effects through the CGRP receptor, AM1 and AM2, presenting the highest potency when binding the latter (72, 81). Figure 1C illustrates the CTR/RAMP- and CLR/RAMP-composed receptors and their respective affinity for the different ligands.
The modulating effect of RAMPs on the ligand affinity of CTR and CLR could be due to an allosteric change in the conformation of the GPCR, leading to the exposure of different binding epitopes; or to a direct contribution together with the GPCR with epitopes that interact with the hormones (82–85). As mentioned above, RAMPs do not only give rise to different AM-, CGRP-, and AM2/intermedin-preferring GPCRs, but also are involved in their trafficking from Golgi to the cell membrane, posttranslational modifications like glycosylation, signaling as well as recycling (82, 86, 87). Furthermore, it is worth mentioning that, although best studied and characterized, the modulating action of RAMPs is not exclusive to CTR and CLR. In this sense, RAMPs also operate as molecular chaperones and allosteric modulators of several GPCRs and their signaling pathways, including glucagon, growth hormone releasing hormone (GHRH), parathyroid hormone and chemokine receptors among others (82, 88, 89).
In the case of the AM and the CGRP receptors, full functionality requires a third element: the receptor-complement protein (RCP) (90, 91). This cytosolic component, bound through ionic interactions to the cell membrane, couples CLR with the Gs-mediated signaling transduction pathway (92, 93). Different reports point out that RCP is not necessary for AM and CGRP receptors to recognize their respective ligands; however, they are not able to activate adenylate cyclase in its absence (91–93).
In all, Figure 1D illustrates the complex structure of AM receptors formed by the three aforementioned proteins: CLR acting as the ligand-recognizing component whose affinity and membrane location depends on the RAMP chaperone member, and the RCP as the transduction-coupling constituent.
Nowadays, it is well accepted that AM, as well as the remaining calcitonin-like peptides, exert their physiological and pathological activities through the complex receptors accordingly mentioned above and shown in Figure 1C. Nevertheless, as Hay et al. deeply reviewed and clearly explained, there have been some misunderstandings regarding the apparent capacity of two other particular proteins to act as AM receptors: RDC1 and GPR182 (also known as L1-R/G10D and, mistakenly, AMDR) (94). In the middle 1990s, both proteins were proposed as mediators of AM pharmacological effects (95, 96). Although further studies have not confirmed this (86, 97), the suggestion that RDC1 and GPR182 may be AM receptors persist to this day. Currently, RDC1 and GPR182 are not accepted to be genuine AM or CGRP receptors (94). RDC1 is considered an atypical chemokine receptor, also known as C-X-C motif receptor 7 (CXCR7) (98), while GPR182 has remained as an orphan GPCR so far (99).
Involvement of AM Receptors in Mediating Physiological and Pathological Processes
AM2 and AM1 present comparable affinity for AM (81), thereby making difficult the distinction of their individual involvement in the aforementioned AM-mediated physiological activities. In these circumstances, their tripartite structure and the fact that CLR and RAMPs seldom migrate to the cell surface separately make the RAMP2- and RAMP3-focussed studies a good approach to differentiate the roles of AM1 and AM2. Naturally, when drawing conclusions, it must be always considered that RAMPs do not interact exclusively with CLR (and CTR).
Several studies indicate that murine AM (AM-/-), CLR (Calcrl-/-) or RAMP2 (Ramp2-/-) knockout embryos die at mid-gestation owing to severe edema as a consequence of altered angiogenesis and lymphatic vasculature (42, 45, 48, 100–104). Although there were divergences in terms of whether hemorrhage is present or not amongst the different models, which ultimately might be attributed to the employment of different mouse strains, the fact that the three knockout types resulted in a non-viable common phenotype not only suggests the importance of AM for embryonic development, but also evidences the necessity of the canonical AM1-mediated signaling in endothelial cells (105). In fact, this was confirmed by Kechele et al. achieving the survival of a Ramp2-/- fetus by engineering mice with endothelial-specific expression of Ramp2 under the control of the VE-cadherin promoter (104).
Nonetheless, it raised the question of whether AM1 requirement was circumscribed to uterine growth. Concerning this, Koyama et al. further made use of interesting murine models enabling the study of AM-/– and Ramp2-/–derived phenotypes in adulthood (47). Specifically, they developed endothelial cell-exclusive Ramp2 and AM knockout mice (E-RAMP2-/- and E-AM-/-, respectively) as well as (tamoxifen) drug-inducible E-RAMP2-/- mice (DI-E-RAMP2-/-), for Ramp2 deletion induction in adults. Contrary to conventional Ramp2-/- mice, most (≈95%) E-RAMP2-/- and E-AM-/- animals died during the peri-natal period, and not in utero. The surviving ones developed systemic and interstitial edema, vascular abnormalities and vasculitis throughout age. These effects were comparable in the DI-E-RAMP2-/- model but less severe in E-AM-/- mice, possibly given the fact that AM is produced by a wide range of tissues (47). Of note, Ramp2+/- and DI-E-RAMP2-/- animals presented a normal lymphatic system (48). In particular, RAMP2+/- mice showed substantially reduced fertility and other endocrinological alterations, including basal and maternal hyperprolactinemia, enlarged pituitary gland, accelerated mammary gland development, and altered skeletal properties among others (45, 101, 106). Nevertheless, these phenotypes were not reproducible in the Calcrl+/- genotype, implying they are not directly linked to AM/AM1 but respond to CLR-independent physiological roles of RAMP2 in other systems (106).
Tam et al. carried out a series of experiments in RAMP2-overexpressing mice demonstrating a prominent physiological role of AM1 in mediating the vasodilatory effects of AM in vascular smooth muscle cells (107). Likewise, their analysis indicates that AM would bind the CGRP receptor at high nanomolar-range concentrations once AM1 has been blocked. Interestingly, Pawlak et al. more recently drew a fully opposing conclusion. In a study employing Calrl+/-, Ramp2+/-, Ramp1-/-, Ramp3-/,- and Ramp1-/-/Ramp3-/- double-knockout mice, they demonstrated that the AM hypotensive effect would be mainly mediated by its action on CGRP receptor and secondarily on AM1 (108). These discrepancies must be clarified in future investigations.
On the other hand, Ramp3-/- mice presented normal fertility and angiogenesis in both embryos and as adults with no obvious phenotypic defects (48, 101). Postsurgical lymphoedema drainage was significantly delayed in these animals, pointing out AM2 as principal AM mediator in the regulation of lymphatic functionality (48). In addition, Dackor et al. observed that this genotype failed in gaining body weight in adulthood (101); however, this was not further confirmed by other authors (48). Interestingly, Dackor et al. also reported that Ramp3-/-, Ramp2+/-, and wild type genotypes showed comparable blood pressure. Noteworthy, this was not due to any compensatory up-regulation of either Ramp2 or Ramp3 gene expression, supporting the lack of functional redundancy of AM1 and AM2 in vivo (45, 47, 109, 110). In contrast, Ichikawa-Shindo et al., showed that Ramp2+/- mice displayed a slight but significant increase in blood pressure with respect to wild type animals accompanied by elevated compensatory levels of AM (45). Pawlak et al. also found elevated blood pressure in Ramp3-/- mice with respect to wild type ones, particularly in males (108). In line with this, Barrick et al., have observed that, in the RenTgMK transgene mice model of angiotensin II-induced chronic hypertension, renal damage, cardiac hypertrophy and cardiac apoptosis were substantially exacerbated in males over females when animals presented the Ramp3-/- genotype (109). The mechanistic reason that would explain these findings is not clear yet, but other RAMP3-regulated processes also seem to be influenced by gender (111). In fact, both AM and Ramp3 may be induced by estrogens, RAMP3 even being associated with the origin of menopausal obesity, although independently of AM-signaling (111, 112). On the other hand, Zhao et al. have not observed estrogen-induced AM secretion in human endometrial primary cells (113).
Regardless of the apparent sexual dimorphism, Ramp3 expression is consistently up-regulated in rodent models of cardiac hypertrophy, hypertension and heart failure, leading to the hypothesis that increased AM signaling through AM2 may have a cardioprotective aim. In this sense, Cueille et al. also reported increased RAMP3 and (RAMP1) in atria and ventricles from rats in a non-ischemic model of chronic cardiac insufficiency due to pressure overload caused by aortic banding for six months (114). No variations were observed in CLR and AM at that time. In a further independent study, on the other hand, the left ventricle of rats with aortocaval shunt-induced cardiac hypertrophy presented up-regulated gene expressions of AM, Calcrl, Ramp2, and Ramp3 compared with controls five weeks after the surgery (115). In a similar work, Oie et al. found that basal Ramp2 mRNA in rat ventricular cardiomyocytes and non-cardiomyocytes cells is significantly higher than Ramp3 mRNA transcript. However, this pattern was drastically inverted one week after a congestive heart failure induced by left coronary artery ligation (116).
LPS-induced septic shock in mice resulted in a strong decrease of Calrl and Ramp2 mRNA expression in lungs, accompanied by a substantial rise in the levels of RAMP3 in pulmonary tissue and the principal organs in immune system. This is thought to be a body response to dampen the inflammatory process (36, 110).
All these findings collectively suggest that AM1 is essential for angiogenesis, vascular homeostasis and embryonic development while AM2 is meaningfully involved in lymphatic system physiology and would be induced, under certain physiological or pathological conditions, to adjust the CLR signaling. It is also quite plausible that the particular PDZ domain-bearing C-tail of RAMP3, enabling its interaction with molecules involved in trafficking of GPCRs, could be of crucial importance in the precise and dynamic regulation of AM2 availability in the cell membrane.
The signal transduction pathways activated by AM in order to regulate processes such as vasodilation, cell survival, proliferation, migration, and vascular cord-like structure formation vary between species, organs, tissues, and cells. However, the main signaling pathways whereby AM exerts its actions involve cAMP, Akt, mitogen activated protein kinase (MAPK)-extracellular signal regulated protein kinase (ERK), and the tyrosine phosphorylation of focal adhesion kinase (P125FAK) (117, 118).
AM action in Malignant Context
Roles of AM in Cancer
Since its discovery from human pheochromocytoma extracts, cumulative clinical evidence has revealed the association between AM and a variety of tumor types, showing that it is expressed by malignant cells, endothelial cells, pro-angiogenic cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), immature monocytic cells including TIE2+ monocytes, VEGFR1+ hemangiocytes, and CD11b+ myeloid cells within the tumor microenvironment (119–122).
Elevated circulating AM has been reported in the plasma of patients with lung and gastrointestinal cancers (123), as well as in untreated Cushing’s disease due to pituitary ACTH-producing adenoma, as compared with normal subjects (124). The increase in the former case seems related to ACTH production and immediately normalizes after surgical excision of the tumor. AM overexpression could be considered as a compensatory response to the elevated circulating cortisol. However, it is worth remarking that it was observed that AM concentration was about double on the side where the adenoma was localized, suggesting it may be directly produced by the tumor (124).
Patients with intraocular or orbital tumors presented significantly elevated AM mRNA levels in the malignant tissues in comparison with those having proliferative vitreoretinopathy, proliferative diabetic retinopathy, preretinal macular fibrosis, and acute retinal necrosis; indicating that AM may play a specific role in the pathogenesis of these cancers (125).
AM expression has been also described in human malignant pleural mesothelioma (MPM) and melanoma biopsies (119, 126). In colorectal cancer, the AM mRNA transcript level has been suggested as a useful marker for predicting high risk for relapse and cancer-related death in patients who undergo curative resection (127). Moreover, it has been found that plasma AM levels positively correlated with malignancy (120). Notably, increased expression levels of AM have been observed in samples harboring a mutation in KRAS. Instead, with the exception of scattered positive staining at the base of the glands in neuroendocrine cells, normal colon tissues were negative for AM expression (121, 122).
High expression of AM was found in human glioma samples, especially in the most aggressive form, namely, glioblastoma, whereas it was low in anaplastic astrocytoma and barely detectable in the low-grade astrocytoma and oligodendroglioma (128, 129).
Increased in AM mRNA expression was also reported in samples from those prostate cancer patients presenting the worse prognoses, as indicated by high Gleason’s scores, while practically absent in tissues samples collected from benign pathologies (130). Similarly, different works indicate that elevated AM expression associates with higher incidence of metastasis, larger residual size of tumors after cytoreduction, and shorter disease-free and overall survival time in epithelial ovarian cancer patients (5, 131), and correlates with tumor grading and metastasis in osteosarcoma (4) and hepatocellular carcinoma (23, 132).
Analogously, elevated tissue AM mRNA is a distinguishing feature of clear-cell renal carcinomas compared with other kidney tumors, and it is associated with an increased risk of relapse after curative nephrectomy due to this type of carcinoma (133, 134). Actually, Michelsen et al. reported significantly increased plasma AM concentrations in patients with renal malignant disease compared with healthy controls (133). Nevertheless, as mentioned above, renal failure is associated with elevated blood levels of AM. Therefore, impaired function of the affected kidney may be partially responsible for AM up-regulation. For this reason, plasma AM may not be suited as a tumor marker for renal cancer (80, 134).
Quantification of plasmatic AM concentrations showed no substantial difference between breast cancer patients and healthy women (135). Nonetheless, a significant positive correlation between tumor diameter and plasma AM levels was observed, suggesting not only that the breast malignancies were actually the source of the circulating AM, but also that tumors require a critical size until a noteworthy increase of secreted AM is detectable in blood (120, 135).
The AM gene has been found markedly overexpressed in patients with pancreatic cancer when compared with controls with benign/cystic pancreatic diseases or pancreatitis (136, 137). Likewise, the expression of AM was higher in subjects with pancreatic cancer and diabetes mellitus as compared with those not suffering from the former. These increased levels were detected in malignant tissue, at both RNA and protein levels, and as circulating AM (137).
Preclinical research does not only support these findings, but also has made great efforts in order to explain them. In this regard, it is worth remarking upon the work carried out by Prof. Ouafik’s group showing a correlation between AM expression and disease stage, progression or vascular density in the context of human glioblastoma (128), prostate (130, 138, 139), colon (121), MPM (126), pheochromocytomas (140), lung (141), and renal (134) cancers. In summary, these as well as studies from other laboratories indicate that AM itself does not cause cancer but can contribute to its pathogenesis in three main ways: 1) directly stimulating cell growth and inhibiting apoptosis (142–144); 2) inducing tumor angiogenesis and lymphangiogenesis, thereby supplying nutrients and oxygen to the tumor while boosting metastasis (45, 103, 126, 139, 145); 3) and/or changing the phenotype of cells, leading them to exhibit a more aggressive behavior (126, 138).
With respect to the first mechanism, several studies have demonstrated that AM exposure enhances cell growth and/or invasion in in vitro and/or in vivo models of breast (146), colon (121), prostate (139), renal (134), and glioblastoma (128) tumors, amongst others. The intrinsic processes behind these effects are not completely clear yet. Nevertheless, AM has proven to be able to inhibit apoptosis of tumor and endothelial cells by down-regulating pro-apoptotic factors such as fragmented PARP, Bax, and activated caspases (143, 144, 147).
In addition, AM protects malignant cells from hypoxia-induced cell death by up-regulation of Bcl-2 in an autocrine/paracrine manner in endometrial carcinoma and osteosarcoma cells (144, 148). In the former, the mechanism involves activation of the MEK/ERK1/2 signaling pathway (148). Similarly, AM was able to significantly increase proliferation and invasiveness of androgen-independent prostate cancer cell lines through stimulation of cAMP and the activation of the CRAF/MEK/ERK/MAPK pathway (139).
Furthermore, intraperitoneal injections of AM stimulated the growth of prostate androgen-dependent cell-derived tumors in castrated animals, suggesting that the peptide might be involved in tumor resurgence after testosterone ablation as in the case of patients treated with androgen-deprivation therapy (138).
Greillier et al. also reported increased activation of the CRAF/MEK/ERK/MAPK pathway in the AM-induced proliferation and invasiveness of the MPM-derived MSTO-211H and H2452 cell lines (126). In the case of hepatocellular carcinoma, the autocrine proliferative effect of AM in hypoxic conditions would be mediated by the PI3K signaling pathway (132). Likewise, the AM-activated PI3K/Akt pathway has been proved to protect endothelial cells from the apoptosis induced by hyperosmotic mannitol therapy for brain edema (149).
More interestingly, AM could also indirectly modulate MYC activity by regulating MYC-associated factor X (Max). This is based on the results of Shichiri et al. in quiescent rat endothelial cells, in which AM stimulated the expression of Max without affecting MYC, and so prevented apoptosis triggered by serum deprivation (150). AM-induced MYC/Max perturbation may result in favor of anti-apoptotic and proliferative effects of MYC.
On the other hand, tamoxifen is an anti-estrogen employed in the treatment of hormone-dependent breast cancer. A major concern of this therapy is its proliferative-inducing effect in endometrium, which significantly increases the risk of malignization with the long-term intake (151). The mitogenic and anti-apoptotic properties of AM point to this peptide as the principal intermediary of tamoxifen’s adverse endometrial effects through non-canonical estrogen receptor-mediated mechanisms (113, 144, 152). Partial agonistic effects of tamoxifen on the estrogen receptor would be able to stimulate AM transcription via an activator protein-1 (AP-1)-directed pathway rather than estrogen signaling response elements in a cell type specific manner (113).
AM activity also appears to extend beyond the malignant cells, being able to act in the tumor environment. There, it may reduce the effectiveness of the immune system to destroy cancer cells by decreasing the expression of pro-inflammatory cytokines and inhibiting the activation of the alternative complement pathway by binding to complement factor H (153, 154).
Infiltrations of myelomonocytic cells are common in the stroma of different tumors, such as pancreatic ductal adenocarcinoma (PDAC) and melanoma, and are related to poor prognosis (119, 120, 154). In their models of PDAC, Xu et al. demonstrated that AM promotes myelomonocytic cell migration and invasion and a pro-tumor phenotype through activation of the MAPK, PI3K/Akt, and NOS signaling pathways, as well as the expression and activity of the matrix metalloproteinase-2 (120). Comparable results were obtained by Chen et al. reporting not only that AM is involved in macrophage polarization –acquisition of a pro-tumor behavior–, but also that AM produced by TAMs is a central factor in macrophage-induced angiogenesis and melanoma growth (119).
More recently, CAFs have also been reported to play a key role in secreting and mediating AM pro-metastatic and angiogenic effects in melanoma (155), pancreatic (156), and breast (157) tumors.
The best-studied activity of AM in the tumor environment is the induction of neovascularization. In this regard, some works have shown that AM did not induce any direct mitogenic effect in the B16/F10 (melanoma), S180 (sarcoma), MDA-MB-231 (breast), A549 (lung), and pancreatic cancer cell lines; instead, when injected in mice, it would induce their growth as tumor masses by providing a suitable blood flow (119, 136, 141, 146, 158, 159).
Decreased O2 partial pressure and the resulting increase of HIF-1α have been implicated as one of the underlying pathways causing AM overexpression in human tumors (160). Hypoxia-stimulated AM expression has been observed in a broad variety of tumor cells, including endometrial (144), osteosarcoma (148), colorectal (121, 122), renal (161), hepatic (132), prostate and promyelocytic leukemia (162), pancreatic (136, 158), and so on. Actually, since tamoxifen has proven to induce hypoxia in xenografts, this has been proposed to be an alternative mechanism by which this anti-estrogen may induce AM in endometrium (144, 163). Thus, given the fact that localized hypoxia is an intrinsic hallmark of most solid tumors, it is not surprising that plasma AM is elevated in distinct common cancer types (133). It was described that AM expression correlates with both VEGF and HIF-1α in colorectal cancer (127). Furthermore, It was observed that AM could be an upstream modulator of the HIF-1α/VEGF pathway, suggesting that AM might induce angiogenesis through VEGF expression as consequence of JNK and AP-1 activation in epithelial ovarian cancer (6, 7). Analogously, several in vitro and in vivo studies have demonstrated the involvement of hypoxia, HIF-1α, AM, and AM receptors in HCC development, and how anti-AM therapeutic approaches would be of great value to deal with this disease (164–166).
On the other hand, it must be considered that the major sources of AM are not only malignant cells themselves, but also the vascular endothelium. This was clearly demonstrated in a work by Iimuri et al. in which S180 sarcoma tumorigenic cells presented more difficulty in developing tumors in AM-heterozygous knockout (AM+/-) mice –expressing ≈50% of circulating AM– compared to wild type ones (159).
AM-producing tumors are characterized by an increased vascular density (163) that correlates with the probability of metastasis occurring (167). Indeed, once trapped in the lymphatic capillaries, the angiogenic potential of AM-overexpressing cancer cells would facilitate neovascularization and hence macroscopic metastatic growth (146). In line with this, the analysis of AM expression and the clinicopathologic features of breast cancer patients showed that axillary lymph node metastasis markedly correlated with the AM protein levels in tumors (135). In colorectal cancer, AM was one of the most selectively upregulated genes in cells with a mutant KRAS under hypoxic conditions (122). Silencing of K-ras drastically reduced AM expression levels in hypoxia compared to control, indicating that AM is a key target for the K-ras oncogenic action in poorly oxygenated tumors. In addition, knockdown of AM suppressed tumor growth and impaired angiogenesis in colon tumor xenografts (122). Studies in HUVEC indicate that AM-induced pro-angiogenic effects would be mediated by PI3K/Akt, ERK, and P125FAK (118).
Besides modifying the nature of TAMs –inducing a M2 phenotype–, AM expression has proven to be associated with more aggressive forms of glioma (128), prostate (130, 138), ovarian (5, 131) and breast cancers, generally correlated with increased metastatic capacity (5, 146). In prostate cancer, AM promotes the appearance of a neuroendocrine-like phenotype –usually presented in the clinic as an aggressive and drug-resistant form– in an androgen-regulated manner in the testosterone-dependent LNCaP cell line (138). These cells normally express both AM as well as AM1 and AM2. However, AM production is significantly increased upon the removal of androgen both in culture and in vivo (when injected in immune-suppressed mice), suggesting that paracrine/autocrine AM secretion represents a survival mechanism that prostate malignant cells employ to regulate neuron-like differentiation when facing androgen deprivation conditions. Such a phenotypic effect would be mediated by AM-induced nuclear translocation of cGMP-dependent protein kinase (PKG), and subsequent regulation of gene expression.
In vitro studies with colon cancer cells have shown the capacity of AM to promote a more invasive phenotype (122).
In summary, Figure 2 illustrates all the above-mentioned AM autocrine/paracrine pro-tumor actions on malignant cells and/or different components of the tumor stroma.
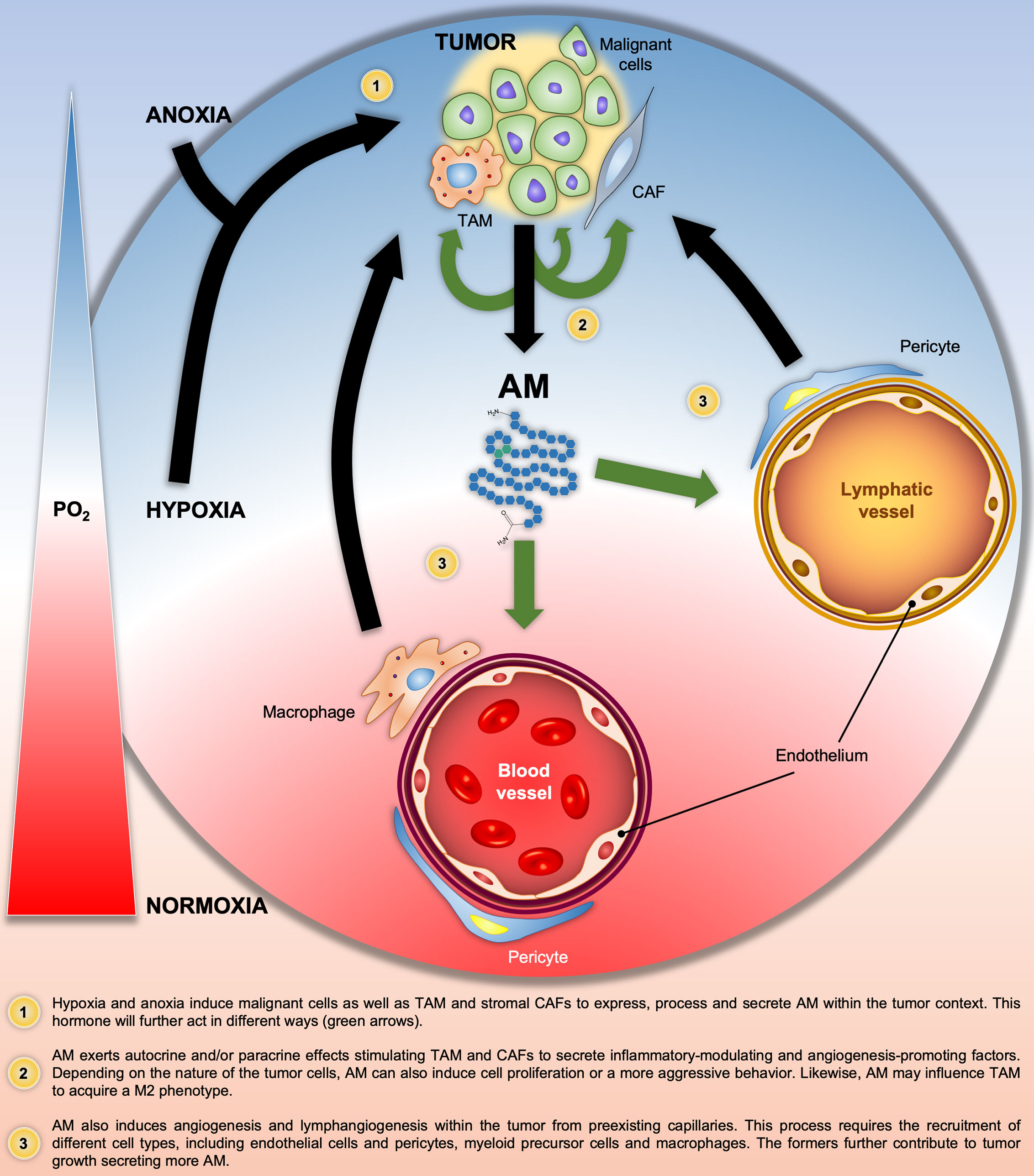
Figure 2 Graphical summary of the Adrenomedullin (AM)-related pathologic processes in solid tumors. Black and green arrows indicate the progression of the pathologic processes and the AM target cells, respectively. PO2, O2 partial pressure.
AM1 and AM2 in Cancer
Cancer cells employ circulating AM as well as that produced by themselves to promote tumor growth through the aforementioned mechanisms. In this section, we will review the role of key mediators of AM’s pro-tumor activity: AM1 and AM2. The findings discussed below are summarized in Table 1.
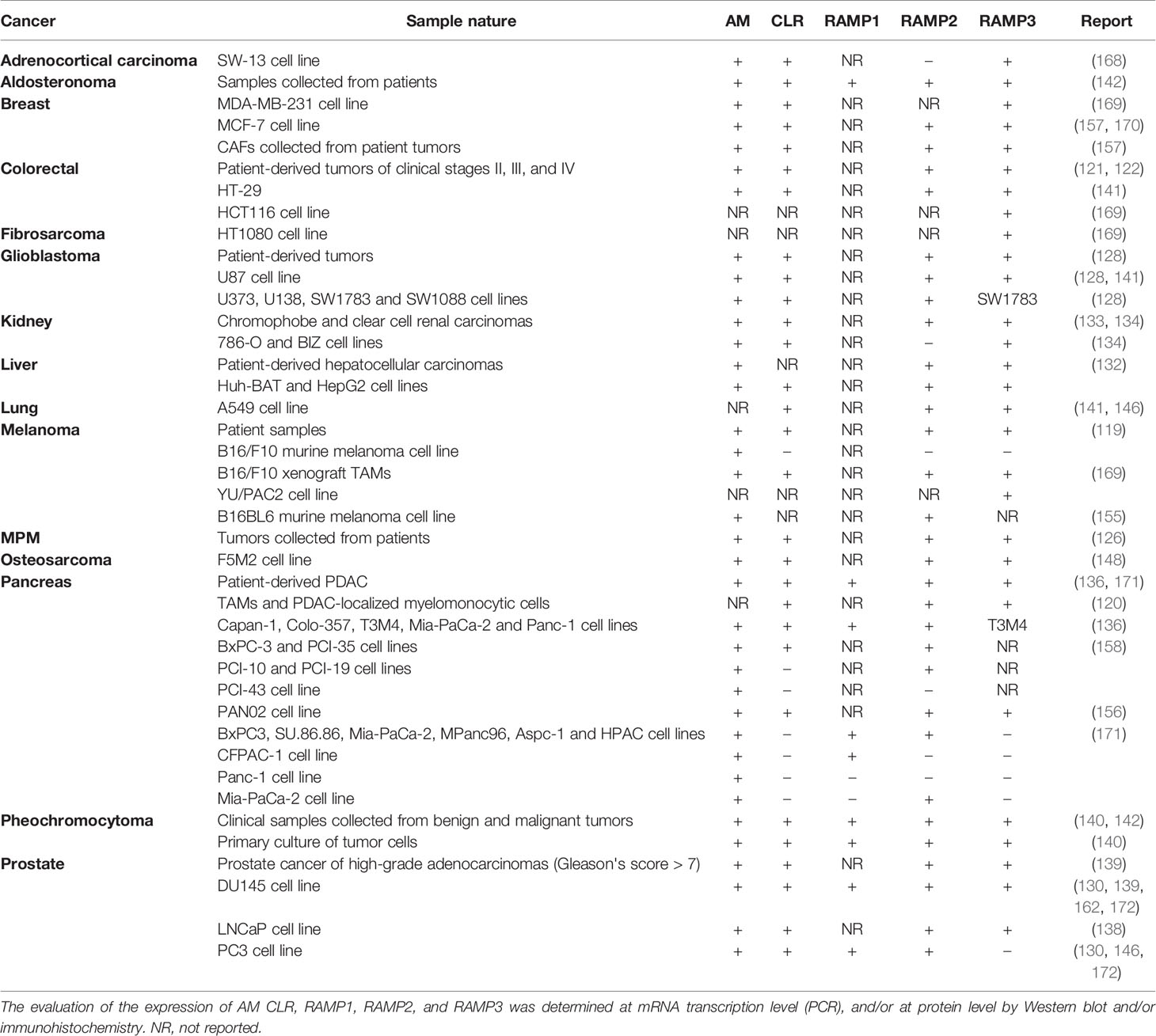
Table 1 Confirmed expression of AM, CLR, RAMP1, RAMP2, and RAMP3 in different tumors.
Berenguer et al. observed that both AM1 and AM2 were responsible for mediating AM-induction of a neuroendocrine phenotype in the androgen-sensitive LNCaP cell line. Moreover, they described that, while the levels of CLR, RAMP2 and RAMP3 were not regulated by androgen status, levels of AM mRNA and immunoreactive AM increased 4- to 7-fold after androgen withdrawal in vitro and in LNCaP xenografts in animals after castration (138). Mazzocchi et al. did not find evidence of significant differences in the level of expression of either Calcrl or Ramp1 and Ramp2 mRNAs between prostate hyperplasia and cancer specimens. Nevertheless, they observed a clear higher expression of the Ramp3 gene in the former group. Unfortunately, these authors did not specify the type and androgen dependency status of the cancers (172). Furthermore, these results were partially mirrored in in vitro studies with DU145 and PC3 androgen-independent prostate cancer cell lines. In this regard, Calcrl, Ramp1 and Ramp2 mRNA transcripts were detected in both cell types. In contrast, Ramp3 expression was restricted to the DU145 cells, indicating that they resemble prostate cancer epithelial cells more closely than the PC3 cell line. Of note, AM showed proliferation-inducing and anti-apoptotic activities only in DU145 cells (130, 139, 172), pointing at AM2 as the mediator of such effects. This finding was supported by the fact that the AM antagonist CGRP8-37 was more effective than AM (22-52) inhibiting DU145 proliferation. It is well known the higher selectivity of CGRP8-37 for AM2 with respect to AM1 (172).
In clinical samples of renal cell carcinoma, RAMP3 has shown to be predominantly expressed in inflammatory cells associated with the tumor whereas RAMP2 was mainly localized in the malignant cells (134). On the other hand –and as part of the same study–, although authors suggest that the in vitro cell proliferation, invasion, and migration activated by exogenous AM would be mediated by both AM1 and AM2, RAMP2 was barely detected in the renal cancer cell lines employed (786-O and BIZ) (134), indicating that they may not be representative models of that cancer.
Chen et al. observed that in vivo tumor growth is significantly attenuated by AM antagonists in the murine B16/F10 melanoma cell line, even when components of the AM receptors were not detected (119). In contrast, TAMs were found to express not only AM, but also CRLR, RAMP2, and RAMP3. These cells are thought to be the main producers of hypoxia-induced AM within the tumor masses and so the mediators of angiogenesis. Moreover, the peptide will act in a paracrine and autocrine way inducing the acquisition of an M2 phenotype.
In the clinic, most human melanoma tissues were positive for CRLR, RAMP2, and RAMP3 –apart from AM–, while their expression levels were much lower in control healthy samples (119). Nevertheless, it would be of great value to determine the distribution of these proteins amongst different tumor cell types.
Ishikawa et al. obtained similar results in a series of five pancreatic cancer cell lines. In this respect, although all of them presented AM, only two (BxPC-3 and PCI-35) showed CLR and RAMP2 proteins. PCI-10 and PCI-19 cells showed RAMP2, and the PCI-43 cell line neither CLR nor RAMP2 (158). Interestingly, although anti-AM therapy had no effect in the in vitro proliferation of none the cell lines, it substantially abrogated PCI-43 cell-derived tumors in vivo, clearly implying that AM is acting on other components of the tumor environment.
A subsequent independent series of experiments performed by Keleg et al. in five other pancreatic cancer cell lines (Capan-1, Colo-357, T3M4, Mia-PaCa-2, and Panc-1) were partially in agreement with the previous results (136). The five assessed cell lines expressed AM, CLR, RAMP1, and RAMP2, whereas RAMP3 was detected in only one of them. Studies with patient samples showed increases in median Calcrl mRNA expression in PDAC in comparison to normal pancreatic tissues. In contrast, they observed >2- and >7-fold reduction in the median transcripts of Ramp1 and Ramp3, respectively, in PDAC tissues compared to normal pancreas samples while no differences were detected in the expression of Ramp2 between healthy and malignant tissues. Immunohistochemical analysis of CLR and RAMP1, RAMP2 and RAMP3 demonstrated moderate to strong staining in islets in the normal pancreas. In PDAC tissue, however, CLR colocalized with RAMP1 and RAMP2, and they were prominently expressed in malignant cells while RAMP3 was not detected. More recently, Xu et al. reported CLR, RAMP2, and RAMP3 in the pro-tumorigenic myelocytic cells associated with PDAC (120), which is in line with the capacity of AM to recruit different cell types reported by Kaafarani et al. (141).
Considering the AM pro-tumor effect, as well as RAMP1, RAMP2 and RAMP3 expression levels, it could be said that the results published by Ramachandran et al. in pancreatic cancer are partially in agreement with the previous ones. More specifically, none of the assayed cell lines (BxPC3, MiaPaCa-2, CFPAC-1, HPAC, MPanc96, Panc-1, Aspc-1, and SU.86.86) presented Ramp3 or CLR expression. However, there are discrepancies in terms of Ramp1 and Ramp2 expression in BxPC-3, Panc-1, and Mia-PaCa-2 cells (171). Instead, authors attributed the AM-mediated pro-tumoral activity to the GPR182 receptor. They observed that by silencing GPR182 the basal growth was reduced as well as the AM-stimulated growth and invasive capacity of the malignant cells when compared with control shRNA (171). As in the case of the first report proposing GPR182 as an AM receptor, these findings have not been yet confirmed. Indeed, as concluded some paragraphs above, most of the cumulative evidence suggests that AM-induced tumor development in pancreatic cancer is mediated by AM1 and AM2. It is also noteworthy that they evaluated RCP expression, finding it in all the cell lines except CFPAC-1.
In all, these findings indicate that AM1 would be more relevant than AM2 in the pro-tumor action of AM in PDAC. Likewise, in vitro and in vivo evidence strongly suggest that AM induced tumor growth by mainly acting on tumor stroma components (120, 136, 158), proposing that the focus should not be on the malignant cells per se but in the peripheral stroma. In line with this, very recent experiments carried out by Dai et al. inoculating PAN02 tumor cells in the spleen of Ramp3-/- and DI-E-RAMP2-/- mice not only demonstrated the active involvement of CAFs in the tumorigenic and metastatic capacity of these pancreatic cells, but also the different roles played by AM1 and AM2 (156). In this sense, when injected in DI-E-RAMP2-/- animals, despite the tumor masses being significantly reduced and accompanied by defective angiogenesis, metastases to the liver were notably increased. In them, CAFs presented elevated RAMP3 and podoplanin (PDPN), a marker of lymphatic endothelial cells and lymphangiogenesis associated with a poor prognosis in various types of cancers. On the other hand, in Ramp3-/- mice, although tumor growth and angiogenesis were not affected (with respect to Ramp3+/+ animals), tumors presented a less aggressive phenotype as indicated by the marked reduction in liver metastases as well as the number of PDPN-positive CAFs. In summary, in pancreatic cancer AM1 would be essential in promoting tumor growth and angiogenesis while antagonizing an AM2-mediated pro-metastatic effect (156).
This group had previously observed an increased propensity to lung metastasis in DI-E-RAMP2-/- mice by generating B16BL6 melanoma cell line-derived tumors (155). Most tellingly, endothelial cell-specific RAMP2 overexpression in E-RAMP2 Tg mice strongly reduced lung metastasis and promoted survival. The authors hypothesized that the irregularly shaped, tortuous, and hyperpermeable vessels resulting from deficient angiogenesis (consequent of the DI-E-RAMP2-/- phenotype) would be suitable substrate for the formation of pre-metastatic niches in distant organs (155). The sum of these findings suggests that a physiologically active AM/AM1 signaling system may be crucial to reduce the likelihood of metastasis in at least pancreatic and melanoma tumors.
Benyahia et al. more recently also demonstrated that CAFs extracted from invasive human breast adenocarcinomas are more competent than normal fibroblasts in enhancing MCF-7 cell growth by induction of stable vascularization when injected into immunocompromised mice (157). The authors demonstrated that AM is one of the CAF-derived factors responsible for endothelial cell-like and pericyte recruitment.
CLR, RAMP2 and RAMP3 have been also detected in glioblastoma tumors –both patient-collected samples and in immortalized malignant cell lines (128)– and in MPM (126). Indeed, expression levels in the former have shown to be much higher than in normal adjacent (pleural) tissue (126). Analogously, CLR, RAMP2, and RAMP3 immunostaining was scarcely detectable in the colonic epithelia of the crypts in normal tissue whereas the epithelial compartment in the well-differentiated adenocarcinoma samples showed strong staining for all these proteins (121). Evidence suggests that AM would act in a paracrine and autocrine manner in colorectal tumors; however, it is not yet clear the individual roles of AM1 and AM2 in the physio-pathology of the disease (121, 141). This is also applicable in hepatocellular carcinoma cell lines (132).
In vitro studies determined that the human SW-13 adrenocortical carcinoma-derived cell line only expressed Calcrl and Ramp3, while normal adrenocortical cells presented Ramp1, Ramp2 and Ramp3 mRNA transcripts, suggesting that the AM proliferative effects in the malignant cells are mediated by AM2 stimulation (168).
Clinical aldosteronoma samples were shown to express CLR, RAMP1, RAMP2, and RAMP3 mRNA, the levels of the two former being much higher than those of RAMP1 (142).
Studies with clinical samples and in primary culture of tumor cells have shown that, although benign pheochromocytomas secrete more AM than malignant ones, both share comparable expression levels of AM and (and CGRP) receptor components (CLR and RAMPs), RAMP1 being markedly abundant (up to 12-fold) with respect to RAMP2 and RAMP3 (140). Nevertheless, Thouënnon et al. suggest that RDC1 could be the predominant receptor for the autocrine effect of AM in this type of tumor basing their hypothesis on three pillars: 1) the elevated rate of expression of RDC1 over AM receptors components; 2) the fact that, among all genes examined in benign and malignant pheochromocytomas, RDC1 was the only one that exhibited a significant differential expression between the two tumor subtypes, suggesting that it could be involved in malignant transformation; and 3) the assumption that AM is not pharmacologically active on the CGRP receptor. Considering the aforementioned strongly increased levels of RAMP1 over RAMP2 and RAMP3, authors presume that the competition for CLR would leave scarce possibility for the assembly of AM1 and AM2. If this is combined with the third pillar, there would not be a place for AM action in these cells. However, at this point it may be of particular interest to reconsider the, already mentioned, articles of Tam et al. (107) and Pawlak et al. (108) describing AM cardiovascular effects mediated through the CLR/RAMP1 complex. Considering that Thouënnon et al. do not give any proof of RDC1-activated signaling specifically in response to AM, the CGRP receptor seems more plausible as the AM target in pheochromocytoma. This is indeed supported by results obtained in the RDC1-downmodulated rat pheochromocytoma-derived PC12 cell lines suggesting that, although RDC1 may be involved in cell survival in serum deprivation conditions, it does not appear to be implicated in the AM-induced proliferative effect (140).
In line with the idea of alternative receptors, Zudaire et al. have reported that, in the tumor context, AM is able to induce mast cells to secrete different inflammation and angiogenesis mediators such as histamine and β-hexosaminidase by way of a process that would not be mediated by AM classical receptors, but, instead, by its interaction with other cell membrane proteins through electric charges. This effect is of particular pharmacological interest given the fact that it would escape from the action of AM1 or AM2 antagonists (160).
In all, except in the work of Ramachandran et al., AM pro-tumor effects always correlate with the occurrence of the necessary subunits to assemble functional AM1 and/or AM2, whether directly in cancer cells or in diverse stroma cell components (e.g., TAMs, CAFs, endothelium, etc.) (173). Likewise, there appears to be a differential predominance by one type of receptor depending on the type of cancer, which is of much importance in the eventual development of therapeutic approaches. In this sense, specific anti-AM2 agents would be more efficient in prostate cancers than in PDACs.
AM-Targeting Agents and Potential Employment in Oncology
Several strategies have been proposed to inhibit AM-mediated processes with potential application in oncology, including impediment of the AM-receptor interaction by antagonistic ligands, direct targeting of AM by specific blocking antibodies, and even regulation of the AM mRNA transcript. All these potential therapeutic approaches are discussed in the following paragraphs and summarized in Figure 3.
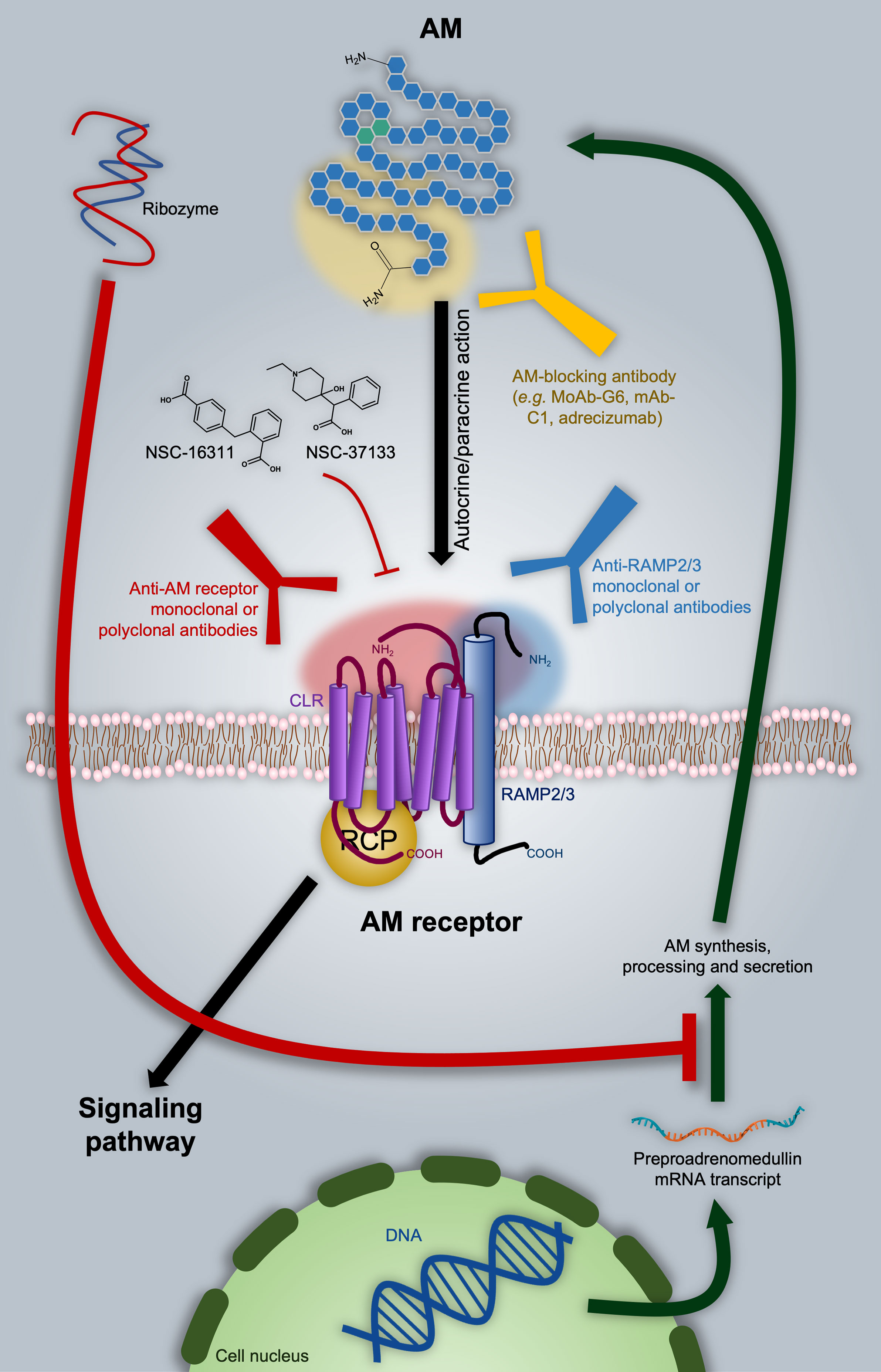
Figure 3 Adrenomedullin (AM)-based therapeutic approaches in oncology. As discussed in the text, one of the options is directly targeting AM with blocking antibodies, as in the case of the MoAb-G6, mAb-C1, and adrecizumab. A ribozyme has proven to be effective in diminishing AM expression. Nevertheless, it still presents significant disadvantages from the pharmacokinetic point of view. AM-receptor antagonists are represented by NSC-16311 and NSC-37133. These compounds have more favorable pharmacokinetic features with respect to peptides like AM (22–52)CONH2 (not shown). The binding of AM to its receptors can be also impeded by targeting the former with specific antibodies. In this respect, we introduce the possibility of employing anti-RAMP2 or anti-RAMP3 antibodies in order to get selective inhibition of AM1 or AM2 respectively. Potential epitope domains on AM and AM receptor components are indicated as shaded areas with the respective color of the targeting antibody.
The first AM antagonists arose as result of structure-activity relationship (SAR) studies indicating that both the ring formed between amino acids 16 and 21 and the C-terminal amide are essential for the full demonstration of the AM hypotensive effect (174). On this basis, different laboratories undertook the search for truncated, pharmacologically inactive versions of AM able to antagonize it by competing for receptors. These investigations gave place to different types of derivatives: 1) AM (16-52)CONH2, AM (13-52)CONH2 (174), and AM (15-52)CONH2 (175) peptides with hypotensive and vasodilatory activity comparable to that shown by AM; 2) AM (22-52)CONH2 –also known as AM antagonist (AMA) and weakly selective for AM1 over AM2–, AM (1-52)COOH (176, 177), and AM (37-52)CONH2 (178), showing significantly lower receptor binding and pharmacological response; 3) AM (16-21), AM (16-31), AM (1-25) (179), and AM (11-26) (176), as inducers of increased systemic arterial pressure in rat; and 4) AM (33-52)CONH2 and AM (1-10)COOH, not able to bind AM receptors (174).
Together with the CGRP derivative CGPR (8-37), some molecules belonging to group 2 have been commonly employed as AM antagonists (172, 180), shown to reduce AM-mediated endothelial cell proliferation, migration, angiogenesis, tumor growth, and vascularization (5, 121, 157–159). However, whatever the pharmacological use the aforementioned peptides may have, their short half-life, low potency, and lack of selectivity circumscribe them only into the research field (175, 177, 180, 181).
It is worth mentioning that, at the moment at which most of those ligands were identified, the structures of the AM receptors were still unknown. Indeed, in many cases the evaluation of their antagonistic potency was determined by measuring their capacity to displace 125I-AM from the cell surface (174). Much effort has been made since then in order to get a molecular comprehension of the interaction of AM with its receptors for the further design and development of more selective and potent antagonists. In point of fact, nowadays it is accepted that while the C-terminal half of the AM binds the complex extracellular domain (ECD) formed by the N-terminals of CLR and RAMPs, the AM N-terminal portion –more precisely, residues 16-30– interacts with the CLR 7-transmembrane domain of the receptor, so stabilizing the conformation that will induce cytoplasmic signaling (83, 84, 182). In this context, Moad, and Pioszak identified the AM (37-52)CONH2 fragment as the minimal structure required to interact with the binding region of the ECD complex (178).
Robinson et al. carried out a series of studies with a number of peptide chimeras formed with AM, intermedin/AM2 and CGRP fragments, identifying several molecules with increased affinity compared to AM (22-52)CONH2 for AM receptors. Interestingly, the peptide called ‘C7’ [AM (22-36)-AM2 (23-30)-AM (37-52)] presented significantly higher selectivity for AM2 with respect to AM1 and the CGRP receptor, which is of great value from the pharmacological point of view in discriminating the individual role of each receptor in AM activity (183). In addition, these, and other authors, have demonstrated that selectivity is strongly established by the interaction of residue 74 in RAMPs with a series of a few amino acids in the C-terminal of AM (181, 183). More recently, Booe et al. identified that AM residues at position 45 and 50 are crucial in determining the affinity of AM for its receptors, as demonstrated by the significantly increased antagonistic capacity of AM (37-52)CONH2 after A45W and Q50W substitutions (84). Complementing this finding, Fischer et al. proved that the conformation of the ring 16-21 in AM is not only determined by the amino acid at position 22 (Thr), but also has a strong influence on the selectivity within the receptor system (182).
Unfortunately, so far, there are no further reports regarding the potential application in oncology of AM antagonists derived from these more recent studies. On the other hand, even if they resulted in more potent and selective molecules, the issue regarding their high metabolization rate would still not be solved.
The development of efficient, druggable small molecules is an alternative strategy to overcome the limiting poor selectivity and potency as well as short half-life often presented by peptide ligands. In this sense, Martínez et al. developed an elegant antibody-mediated method to screen for potential AM-antagonists in chemical libraries (180). Amongst the compounds identified, the piperidine derivative of phenylacetic acid, 16311 (also known as NSC-16311), elevated blood pressure when administered to normotensive (anesthetized) rats. On the other hand, the benzoic acid derivative, 37133 (also referred as NSC-37133), had no hemodynamic effects (180). In a later publication, the same group reported that both NSC-16311 and NSC-37133 as well as several other analogs exerted anti-proliferative effects in the human breast cancer cell line T47D, probably related to their AM-antagonistic behavior. In line with this, the AM-antagonistic action of NSC-16311 was further employed by Portal-Núñez et al. to demonstrate the AM contribution to the carcinogenic effect of tobacco combustion products in pulmonary tissue (184). More recently, Siclari et al. proved that NSC-16311 and NSC-37133 may be effective against breast cancer bone metastases due to their selective blockade of the osteolytic and pro-tumor-invasive actions of AM (146).
Taking advantage of the screening assay developed by Martínez et al. and complementing it by 3D quantitative SAR (3D-QSAR) and cell signaling studies, Roldós et al. further identified critical functional groups to be considered for the design of this type of ligands (185). Specifically, an aromatic ring, a hydrogen bond donor and a free carboxylic group seems to be essential to get AM-negative modulators to be eventually assessed as antiangiogenic and anti-cancer agents (185). However, it is notable that to date there are no clinical trials involving small-molecule AM antagonists.
In contrary, different selective small-molecule CGRP antagonists achieved clinical status for the treatment of migraine, including BIBN4096BS/olcegepant (186) (NCT02194777, NCT02194322, etc.), MK-0974/telcagepant (187) (NCT01294709, NCT00432237, etc.), MK-3207 (188) (NCT00712725, NCT00548353), and the very recently Food and Drug Administration (FDA)-approved MK-1602/ubrogepant (Ubrelvy®) (189) (NCT04179474, NCT02867709, etc.). This indicates the potential for developing selective antagonists for other CLR/RAMP heterodimers by exploiting key residue differences amongst RAMPs. In line with the aforementioned relevance of residue 74 in determining the selective interaction of RAMPs with their respective ligands, the tryptophan located in this position is crucial in mediating the binding of these compounds to RAMP1 (188). Instead, RAMP2 and RAMP3 share a glutamic acid residue at this position, indicating that such a class of anti-CGRP compounds may not be suitable scaffolds for the development of AM antagonists with oncological value. Even more so when taking into account that they were abandoned after some trial participants showed signs of hepatotoxicity (81)
On the other hand, the list of anti-CGRP agents with therapeutic action in migraine is not limited only to antagonists of the CLR/RAMP1 receptor, but also contains the commercially available, CGRP-neutralizing antibody fremanezumab (Ajovy®) (190) (NCT03308968, NCT04041284, etc.). Both mono- and polyclonal AM-blocking antibodies have been preclinically employed to inhibit AM pro-tumor and/or pro-angiogenic activity in glioblastoma (128), melanoma (119), lung (170), prostate (138, 139), ovarian, breast, and colon (121, 170) cancer models. Just to give examples, MoAb-G6 (170) and [anti-AM-(46–52)] mAb-C1 (191) are some of the monoclonal antibodies tested in these studies. Of note, no sign of toxicity was observed with this therapy (139). Nevertheless, the most relevant AM-blocking antibody is adrecizumab, currently under evaluation in cardiogenic and septic shock patients (NCT03083171, NCT04252937, etc.) (192). Results from these trials will be of great value given the fact that they will provide information on eventual side effects. Another question raised by the application of this approach concerns the impact competition with the AM-gly fraction –the most abundant circulating form of the hormone– may have on the bioavailability of the AM-neutralizing antibodies. To our knowledge, there are no reports in this respect.
Around 30% of all pharmaceutical products currently employed exert their therapeutic actions by modulating GPCRs (45, 75). The vast majority is represented by small molecules, then there is a minimal proportion composed by peptides, and, interestingly, the list is completed with only two FDA-approved monoclonal antibodies: mogamulizumab/Poteligeo®, employed in the treatment of mycosis fungoides or Sézary syndrome by targeting the CC chemokine receptor 4 (CCK4); and AMG 334/erenumab/Aimovig®, targeting CGRP receptor and prescribed for migraine prophylaxis (193–195). The approval of these two monoclonal antibodies gives promise to the potential of the currently available AM1-, AM2-, and CLR-targeting antibodies with proved tumor growth-inhibitory activity in preclinical models of colon, glioblastoma, PDAC, lung and breast cancer (120, 141, 157, 196). The anti-tumor effects of these antibodies are a direct consequence of their anti-angiogenic capacity. Indeed, treated tumor have been reported to appear pale or translucent, with diminished vasculature, permeability and recruitment of pericytes and macrophages and other myeloid cells (120, 141, 157, 196). Most tellingly, it must be noted that Kaafarani et al. did not observe signs of toxicity associated with long-term therapy (60 days). Histological analysis revealed no abnormalities of physiological vasculature in heart, liver, kidney, lung, and spleen. This could be partially explained by the specificity of the antibodies. In addition, it may mean that tumor angiogenic cells overexpressing AM1 and AM2 would become more susceptible to AM signaling blockade than the quiescent vasculature-associated cell components (141).
Over the years, several process-intrinsic challenges have limited the interest of the pharmaceutical industry in developing monoclonal antibodies against membrane proteins (195). However, the theory suggests that the complex structure of AM (and CGPR) receptors may compensate such restrictions, simultaneously offering encouraging advantages. A key factor in the development of such agents is to get antigens as close to the native conformation as possible, which is not a simple task for those proteins constantly moving immersed in a liquid film of phospholipids, as in the case of GPCRs. In this regard, it is thought that the mutual stabilization of the proteins (CLR and RAMPs) within the heterodimeric structure of AM1 and AM2 could allow them to remain longer in a physiologically relevant conformation. Moreover, the bipartite ECD creates a larger antigenic surface that increases the range of epitopes available to the antibodies, and so the chances of obtaining an appropriate therapeutic candidate (195). In contrast, the individual RAMP accessory proteins are more conventional single-pass receptors, and taken in isolation, represent more attractive targets, as recombinant expression of the extracellular domain is much less technically challenging. The RAMP components themselves represent individual therapeutic targets since they constitute strategic tools to define the pharmacological selectivity. In this sense, although its deficiency affects tumor vasculature and growth, RAMP2 does not appear to be a logical candidate for targeting given its role in preventing metastasis. In contrast, RAMP3 blockade in the context of a healthy AM/AM1 signaling is expected to considerably suppress tumor metastatic capacity (155, 156). Moreover, even when partial, RAMP2 deletion resulted in abnormalities in various vascular endothelial cells, which suggests attention must be paid regarding side effects when considering RAMP2 as the therapeutic target (155). On the other hand, Ramp3-/- mice did not show major phenotypic alterations (48, 110). Therefore, the incorporation of anti-RAMP3 agents in current therapeutic protocols in oncology may represent a promising anti-metastatic approach.
Lastly, a less conventional approach was proposed by Taylor and Samson, which consisted in the design of a catalytically active ribozyme that specifically recognizes and cleaves the preproadrenomedullin mRNA transcript (197). This molecule showed a good enough half-life to observe physiological variations both in vitro, decreasing AM content in cultured vascular smooth muscle cells, and in vivo, where reduction of AM resulted in exaggerated water drinking by rats after overnight water restriction or central administration of angiotensin II. Nevertheless, as an RNA-based molecule, stability, and route of administration are still drawbacks to consider in this strategy.
Discussion
AM and VEGF share common features in that they are both hypoxically regulated, increase vascular permeability and are key pro-angiogenic factors. Indeed, their deficiency causes dysfunctional vascularization, even leading to embryonic lethality in mice when absolute (198, 199). It has been observed that both molecules can interact in an alternate complementary fashion in angiogenesis-involving processes. VE-cadherin is a protein component of endothelial cell-to-cell adherent junctions with a key role in the maintenance of vascular integrity, neovessel assembly and remodeling of pericyte-endothelial cell association. Dr. Ouafik and collaborators have demonstrated that AM blockade, using anti-AM or anti-AM receptor antibodies, increases endothelial cell permeability by inhibiting cell-cell contacts predominantly through disruption of the VE-cadherin/β-catenin/Akt signaling pathway (200). At a molecular level, AM blockade induces phosphorylation of VE-cadherin at the critical residue Tyr731, preventing binding of its cytoplasmic tail with β-catenin and thus disrupting vasculature structure. In contrast, the activation of the VEGF receptor 2 increases the phosphorylation of VE-cadherin VE at Tyr731 inducing loss of cell polarity and lumen formation, consequently allowing VEGF to stimulate endothelial cell proliferation (201). They also interact at the cell signaling level since VEGF activates a number of different intracellular signaling pathways, including ERK’s and PI3K/Akt to ensure endothelial cell survival and proliferation (202), which can be inhibited by AM blockade leading to endothelial cell death by apoptosis (personal communication of Prof. Ouafik’s group).
The general outcome of anti-angiogenesis therapy is still unsatisfactory, principally due to low efficacy and the development of tumor-acquired resistance and adverse effects. Although the causes are not clear, several hypotheses have been posed in order to explain the discrepancy observed in terms of the effectiveness of anti-VEGF treatments amongst (in-mice) preclinical studies and the clinic. One reason may be in the administration of higher doses in animals, that in general are younger and relatively healthier than patients, and for which therapy-related toxic reactions are often omitted (203).
Also, attention must be paid to the fact that anti-VEGF agents do not act directly on tumor cells, but on their supply of nutrients and O2. In this sense, residual hypoxia-resistant cells –usually associated with the cancer stem cell-like subpopulation– would act as a remnant source of VEGF and other factors that combined, would overcome the VEGF signaling blockade (203).
AM meets all the requirements to alternatively bypass VEGF inhibition, including a direct mitogen activity on malignant cells. Indeed, Gao et al. have demonstrated not only that AM expression was significantly (4-fold) increased in sunitinib-resistant renal carcinoma cells, but also that the treatment with AM (22-52)CONH2 markedly reduced the growth of tumors derived from those cells (204). In all, evidence points to AM as an alternative target in tumors resistant to anti-VEGF agents. In this respect, AM also offers a broader front of action within the milieu of solid tumors, being able to act as modulator of endothelial cell interactions with other endothelial cells and/or pericytes, recruiter of different cell types into hypoxic areas and stimulator of cancer cell proliferation. On the other hand, the mitogenic activity of VEGF is mainly restricted to endothelial cells.
However, such multitasking role carries increased risk of adverse effects. In this regard, hypertension and its associated secondary events (i.e., myocardial infarction and stroke) may be the main complication in anti-AM therapy, which is indeed an adverse effect observed in patients undergoing VEGF-inhibiting treatments. In general, however, anti-VEGF agents are well tolerated, especially when compared with chemotherapy; although, undesirable effects can limit their administration in some cases. These include venous and arterial thromboembolism, proteinuria, hemorrhage, gastrointestinal perforation and impaired wound healing (203, 205, 206). Actually, the former has been already reported in anti-AM therapy preclinical models. Mice-based studies indicate that the major adverse effects related to AM-inhibitory treatments may involve development of secondary lymphoedema (207, 208) and altered wound healing capacity (50). Both are of importance for those patients who undergo surgery and/or radiotherapy or might present a genetic susceptibility to the risk of developing secondary lymphoedema (208). However, they are manageable and usually pose a significantly lower risk than the tumors being treated.
One aspect to consider in any therapy is the capacity of identifying prognostic factors. Although in anti-VEGF regimens these types of molecules have remained elusive, VEGF itself has been postulated as general biomarker of angiogenic activity and tumor progression in cancer patients (129, 209). In this respect, increased circulating values of VEGF generally correlate with worse outcome (206, 209). In the case of anti-AM therapy, VEGF should be thought, at least, as the starting prognostic parameter to be evaluated.
We are of the opinion that there is mounting evidence of the potential of anti-AM strategies in oncology, representing more than just an anti-angiogenesis therapy, to merit further investigation. The efforts should be onwards focused on the discussion and development of efficient therapeutic approaches, taking also into account the feasibility of combination therapy with conventional chemotherapy. For example, Li et al. observed a significant potentiation of the tumor growth-inhibitory effect of cisplatin in AM-deficient liver cancer cells (210).
Considering the already described involvement of AM in different physiological activities, the extrapolation of a bevacizumab/Avastin®-like approach (directly targeting the ligand) may, in principle, not be a first option as a therapeutic strategy for AM. Another point to take into account in this respect is the fact that the AM-gly is the major circulating form of this protein. Thus, a potential antibody should be able to discern the AM mature variant in order to avoid the bioavailability-reducing competition with the inactive one, highlighting the AM amidated C-terminal as the antigenic epitope of choice.
As pointed out above, current clinical trials with adrecizumab will be more than interesting in addressing the feasibility of this strategy in oncology. Nevertheless, the blockade of AM receptors might represent a more suitable alternative. Moreover, the recent FDA approval of the CGRP receptor-targeting erenumab/Aimovig® antibody encourages the application of the anti-AM receptors strategy in oncology. One of the key challenges regarding this approach is the ability to selectively target individual components.
Collectively, data discussed in the previous sections suggests that both AM1 and AM2 possess crucial roles within the tumor structure regulating diverse functional aspects of the cell components. In this context, RAMP2 and RAMP3 themselves represent target candidates. However, AM2 blockade could be presented as more advantageous by decreasing the chances of metastasis and having less impact on the physiological angiogenic processes. Furthermore, although it can be said it is a general feature of the AM system, the AM2 signaling pathway in particular appears to be in a physiological dormant state ready to assemble and operate in response to diverse pathological conditions. This could explain the low incidence of adverse effects observed when targeting this receptor in preclinical models.
In the light of the current evidence, AM inhibition is a promising first-in-class therapeutic strategy in oncology and its potential needs to be further explored for the development of selective pharmacological options in cancer patients.
Author Contributions
RV: As primary author, conceived the original idea of the article, drafting and writing the paper, and made the figures. MR: primary author conceived the original idea of the article, and drafting and writing most of the paper. CB-D, AO’K, JG, OT, and KR: Participated in revisions to scientific content of specific sections of the manuscript. MB: Participated in the critical revision, editing, and writing of the manuscript. Provided revisions to scientific content of the manuscript. L’HO: Conceived the original idea and provided critical revision of the manuscript as well as the final approval of the version to publish. All authors contributed to the article and approved the submitted version.
Conflict of Interest
Authors AO’K, JG, and OT are employed by the company Fusion Antibodies. RV, MR, and MB are stockholders of Early Drug Development Group.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is dedicated to the memory of Iva Ricchi, Tata.
Abbreviations
ACTH, Adrenocorticotropic hormone; AM, adrenomedullin; AM1, adrenomedullin receptor type 1; AM2, adrenomedullin receptor type 2; AM2, adrenomedullin 2 (intermedin); CAF, tumor-associated fibroblast. AMY, amylin; cAMP, cyclic-adenosine monophosphate; CGRP, calcitonin gene-related peptide; cGMP, cyclic guanosine monophosphate; CLR, calcitonin-receptor like receptor; CTR, calcitonin receptor; ECD, extracellular domain; ERK (1/2), extracellular signal regulated protein kinase (1/2); GPCR, G protein-coupled receptor; HIF-1α, hypoxia-inducible factor-1α; LPS, Lipopolysaccharides; MAPK, mitogen activated protein kinase; MPM, malignant pleural mesothelioma; PAM, peptidylglycine α-amidating monooxygenase; PAMP, Proadrenomedullin N-terminal 20 peptide; PDAC, pancreatic ductal adenocarcinoma; PDPN, podoplanin; PDZ, PSD-95/Discslarge/ZO-1; PKG, cGMP-dependent protein kinase or protein kinase G; RAMP, receptor-activity modifying protein; RCP, receptor-complement protein; SAR, structure-activity relationship; TAM, tumor-associated macrophage; VEGF, vascular endothelial growth factor.
References
1. Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci (2001) 224:201–7. doi: 10.1016/S0165-6147(00)01676-X
2. Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol (2019) 121:27. doi: 10.1186/s13045-019-0718-5
3. Fallah A, Sadeghinia A, Kahroba H, Samadi A, Heidari HR, Bradaran B, et al. Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. BioMed Pharmacother (2019) 110:775–85. doi: 10.1016/j.biopha.2018.12.022
4. Dai X, Ma W, He XJ, Jha RK. Elevated expression of adrenomedullin is correlated with prognosis and disease severity in osteosarcoma. Med Oncol (2013) 301:347. doi: 10.1007/s12032-012-0347-0
5. Deng B, Zhang S, Miao Y, Han Z, Zhang X, Wen F, et al. Adrenomedullin expression in epithelial ovarian cancers and promotes HO8910 cell migration associated with upregulating integrin alpha5beta1 and phosphorylating FAK and paxillin. J Exp Clin Cancer Res (2012) 31:19. doi: 10.1186/1756-9966-31-19
6. Chen Q, Chen P, Pang X, Hu Y, Zhang Y. Adrenomedullin Up-regulates the Expression of Vascular Endothelial Growth Factor in Epithelial Ovarian Carcinoma Cells via JNK/AP-1 Pathway. Int J Gynecol Cancer (2015) 256:953–60. doi: 10.1097/IGC.0000000000000465
7. Zhang Y, Xu Y, Ma J, Pang X, Dong M. Adrenomedullin promotes angiogenesis in epithelial ovarian cancer through upregulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor. Sci Rep (2017) 7:40524. doi: 10.1038/srep40524
8. Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H, et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun (1993) 1922:553–60. doi: 10.1006/bbrc.1993.1451
9. Eto T. A review of the biological properties and clinical implications of adrenomedullin and proadrenomedullin N-terminal 20 peptide (PAMP), hypotensive and vasodilating peptides. Peptides (2001) 2211:1693–711. doi: 10.1016/S0196-9781(01)00513-7
10. Zudaire E, Cuttitta F, Martinez A. Regulation of pancreatic physiology by adrenomedullin and its binding protein. Regul Pept (2003) 1121-3:121–30. doi: 10.1016/S0167-0115(03)00030-2
11. Kitamura K, Sakata J, Kangawa K, Kojima M, Matsuo H, Eto T. Cloning and characterization of cDNA encoding a precursor for human adrenomedullin. Biochem Biophys Res Commun (1993) 1942:720–5. doi: 10.1006/bbrc.1993.1881
12. Naot D, Musson DS, Cornish J. The Activity of Peptides of the Calcitonin Family in Bone. Physiol Rev (2019) 991:781–805. doi: 10.1152/physrev.00066.2017
13. Amara SG, Jonas V, Rosenfeld MG, Ong ES, Evans RM. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature (1982) 2985871:240–4. doi: 10.1038/298240a0
14. Rosenfeld MG, Mermod JJ, Amara SG, Swanson LW, Sawchenko PE, Rivier J, et al. Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature (1983) 3045922:129–35. doi: 10.1038/304129a0
15. Hoppener JW, Steenbergh PH, Zandberg J, Geurts van Kessel AH, Baylin SB, Nelkin BD, et al. The second human calcitonin/CGRP gene is located on chromosome 11. Hum Genet (1985) 703:259–63. doi: 10.1007/BF00273453
16. Glowka TR, Steinebach A, Stein K, Schwandt T, Lysson M, Holzmann B, et al. The novel CGRP receptor antagonist BIBN4096BS alleviates a postoperative intestinal inflammation and prevents postoperative ileus. Neurogastroenterol Motil (2015) 277:1038–49. doi: 10.1111/nmo.12584
17. Edvinsson L. Role of CGRP in Migraine. Handb Exp Pharmacol (2019) 255:121–30. doi: 10.1007/164_2018_201
18. Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol (1990) 282:183–7. doi: 10.1002/ana.410280213
19. Roos BA, Fischer JA, Pignat W, Alander CB, Raisz LG. Evaluation of the in vivo and in vitro calcium-regulating actions of noncalcitonin peptides produced via calcitonin gene expression. Endocrinology (1986) 1181:46–51. doi: 10.1210/endo-118-1-46
21. Hartter E, Svoboda T, Ludvik B, Schuller M, Lell B, Kuenburg E, et al. Basal and stimulated plasma levels of pancreatic amylin indicate its co-secretion with insulin in humans. Diabetologia (1991) 341:52–4. doi: 10.1007/BF00404025
22. Cooper GJ. Amylin compared with calcitonin gene-related peptide: structure, biology, and relevance to metabolic disease. Endocr Rev (1994) 152:163–201. doi: 10.1210/edrv-15-2-163
23. Li Z, Kelly L, Heiman M, Greengard P, Friedman JM. Hypothalamic Amylin Acts in Concert with Leptin to Regulate Food Intake. Cell Metab (2015) 226:1059–67. doi: 10.1016/j.cmet.2015.10.012
24. Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes (1999) 482:241–53. doi: 10.2337/diabetes.48.2.241
25. Boyle CN, Lutz TA, Le Foll C. Amylin - Its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol Metab (2018) 8:203–10. doi: 10.1016/j.molmet.2017.11.009
26. Asthana S, Mallick B, Alexandrescu AT, Jha S. IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim Biophys Acta Biomembr (2018) 6:S0005-2736(18)30061-0. doi: 10.1016/j.bbamem.2018.02.020
27. Castle AL, Kuo CH, Han DH, Ivy JL. Amylin-mediated inhibition of insulin-stimulated glucose transport in skeletal muscle. Am J Physiol (1998) 2753:E531–6. doi: 10.1152/ajpendo.1998.275.3.E531
28. Datta HK, Zaidi M, Wimalawansa SJ, Ghatei MA, Beacham JL, Bloom SR, et al. In vivo and in vitro effects of amylin and amylin-amide on calcium metabolism in the rat and rabbit. Biochem Biophys Res Commun (1989) 1622:876–81. doi: 10.1016/0006-291X(89)92391-7
29. Minamino N, Kikumoto K, Isumi Y. Regulation of adrenomedullin expression and release. Microsc Res Tech (2002) 571:28–39. doi: 10.1002/jemt.10048
30. Nomura I, Kato J, Tokashiki M, Kitamura K. Increased plasma levels of the mature and intermediate forms of adrenomedullin in obesity. Regul Pept (2009) 1581-3:127–31. doi: 10.1016/j.regpep.2009.08.003
31. Kawano S, Kawagoe Y, Kuwasako K, Shimamoto S, Igarashi K, Tokashiki M, et al. Gender-related alterations in plasma adrenomedullin level and its correlation with body weight gain. Endocr Connect (2015) 41:43–9. doi: 10.1530/EC-14-0131
32. Ishimitsu T, Nishikimi T, Saito Y, Kitamura K, Eto T, Kangawa K, et al. Plasma levels of adrenomedullin, a newly identified hypotensive peptide, in patients with hypertension and renal failure. J Clin Invest (1994) 945:2158–61. doi: 10.1172/JCI117573
33. Jougasaki M, Wei CM, Aarhus LL, Heublein DM, Sandberg SM, Burnett JC Jr. Renal localization and actions of adrenomedullin: a natriuretic peptide. Am J Physiol (1995) 268(4 Pt 2):F657–63. doi: 10.1152/ajprenal.1995.268.4.F657
34. Suzuki Y, Horio T, Hayashi T, Nonogi H, Kitamura K, Eto T, et al. Plasma adrenomedullin concentration is increased in patients with peripheral arterial occlusive disease associated with vascular inflammation. Regul Pept (2004) 1181-2:99–104. doi: 10.1016/j.regpep.2003.11.001
35. Kato J, Kitamura K, Eto T. Plasma adrenomedullin level and development of hypertension. J Hum Hypertens (2006) 208:566–70. doi: 10.1038/sj.jhh.1002033
36. Ono Y, Okano I, Kojima M, Okada K, Kangawa K. Decreased gene expression of adrenomedullin receptor in mouse lungs during sepsis. Biochem Biophys Res Commun (2000) 2711:197–202. doi: 10.1006/bbrc.2000.2606
37. Voors AA, Kremer D, Geven C, Ter Maaten JM, Struck J, Bergmann A, et al. Adrenomedullin in heart failure: pathophysiology and therapeutic application. Eur J Heart Fail (2019) 212:163–71. doi: 10.1002/ejhf.1366
38. Kitamura K, Kato J, Kawamoto M, Tanaka M, Chino N, Kangawa K, et al. The intermediate form of glycine-extended adrenomedullin is the major circulating molecular form in human plasma. Biochem Biophys Res Commun (1998) 2442:551–5. doi: 10.1006/bbrc.1998.8310
39. Sakata J, Mizuno K, Matsuo H. Tissue distribution and characterization of peptide C-terminal alpha-amidating activity in rat. Biochem Biophys Res Commun (1986) 1401:230–6. doi: 10.1016/0006-291X(86)91080-6
40. Saldise L, Martinez A, Montuenga LM, Treston A, Springall DR, Polak JM, et al. Distribution of peptidyl-glycine alpha-amidating mono-oxygenase (PAM) enzymes in normal human lung and in lung epithelial tumors. J Histochem Cytochem (1996) 441:3–12. doi: 10.1177/44.1.8543779
41. Samson WK. Adrenomedullin and the control of fluid and electrolyte homeostasis. Annu Rev Physiol (1999) 61:363–89. doi: 10.1146/annurev.physiol.61.1.363
42. Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci USA (2001) 982:615–9. doi: 10.1073/pnas.98.2.615
43. Wang C, Dobrzynski E, Chao J, Chao L. Adrenomedullin gene delivery attenuates renal damage and cardiac hypertrophy in Goldblatt hypertensive rats. Am J Physiol Renal Physiol (2001) 2806:F964–71. doi: 10.1152/ajprenal.2001.280.6.F964
44. Nicholls MG. Hemodynamic and hormonal actions of adrenomedullin. Braz J Med Biol Res (2004) 378:1247–53. doi: 10.1590/S0100-879X2004000800016
45. Ichikawa-Shindo Y, Sakurai T, Kamiyoshi A, Kawate H, Iinuma N, Yoshizawa T, et al. The GPCR modulator protein RAMP2 is essential for angiogenesis and vascular integrity. J Clin Invest (2008) 1181:29–39. doi: 10.1172/JCI33022
46. Cam-Etoz B, Isbil-Buyukcoskun N, Ozluk K. Cardiovascular effects of the intracerebroventricular injection of adrenomedullin: roles of the peripheral vasopressin and central cholinergic systems. Braz J Med Biol Res (2012) 453:250–5. doi: 10.1590/S0100-879X2012007500027
47. Koyama T, Ochoa-Callejero L, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Iinuma N, et al. Vascular endothelial adrenomedullin-RAMP2 system is essential for vascular integrity and organ homeostasis. Circulation (2013) 1277:842–53. doi: 10.1161/CIRCULATIONAHA.112.000756
48. Yamauchi A, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Kawate H, Igarashi K, et al. Functional differentiation of RAMP2 and RAMP3 in their regulation of the vascular system. J Mol Cell Cardiol (2014) 77:73–85. doi: 10.1016/j.yjmcc.2014.09.017
49. Kataoka Y, Miyazaki S, Yasuda S, Nagaya N, Noguchi T, Yamada N, et al. The first clinical pilot study of intravenous adrenomedullin administration in patients with acute myocardial infarction. J Cardiovasc Pharmacol (2010) 564:413–9. doi: 10.1097/FJC.0b013e3181f15b45
50. Harada K, Yamahara K, Ohnishi S, Otani K, Kanoh H, Ishibashi-Ueda H, et al. Sustained-release adrenomedullin ointment accelerates wound healing of pressure ulcers. Regul Pept (2011) 1681-3:21–6. doi: 10.1016/j.regpep.2011.02.014
51. Garcia-Honduvilla N, Cifuentes A, Bellon JM, Bujan J, Martinez A. The angiogenesis promoter, proadrenomedullin N-terminal 20 peptide (PAMP), improves healing in both normoxic and ischemic wounds either alone or in combination with autologous stem/progenitor cells. Histol Histopathol (2013) 281:115–25. doi: 10.14670/HH-28.115
52. Nishikimi T. Adrenomedullin in the kidney-renal physiological and pathophysiological roles. Curr Med Chem (2007) 1415:1689–99. doi: 10.2174/092986707780830943
53. Serrano J, Alonso D, Fernandez AP, Encinas JM, Lopez JC, Castro-Blanco S, et al. Adrenomedullin in the central nervous system. Microsc Res Tech (2002) 572:76–90. doi: 10.1002/jemt.10053
54. Kis B, Abraham CS, Deli MA, Kobayashi H, Niwa M, Yamashita H, et al. Adrenomedullin, an autocrine mediator of blood-brain barrier function. Hypertens Res (2003) 26 Suppl:S61–70. doi: 10.1291/hypres.26.S61
55. Saita M, Shimokawa A, Kunitake T, Kato K, Hanamori T, Kitamura K, et al. Central actions of adrenomedullin on cardiovascular parameters and sympathetic outflow in conscious rats. Am J Physiol (1998) 2744:R979–84. doi: 10.1152/ajpregu.1998.274.4.R979
56. Miyashita K, Itoh H, Arai H, Suganami T, Sawada N, Fukunaga Y, et al. The neuroprotective and vasculo-neuro-regenerative roles of adrenomedullin in ischemic brain and its therapeutic potential. Endocrinology (2006) 1474:1642–53. doi: 10.1210/en.2005-1038
57. Fernandez AP, Serrano J, Tessarollo L, Cuttitta F, Martinez A. Lack of adrenomedullin in the mouse brain results in behavioral changes, anxiety, and lower survival under stress conditions. Proc Natl Acad Sci USA (2008) 10534:12581–6. doi: 10.1073/pnas.0803174105
58. Marutsuka K, Hatakeyama K, Sato Y, Yamashita A, Sumiyoshi A, Asada Y. Immunohistological localization and possible functions of adrenomedullin. Hypertens Res (2003) 26 Suppl:S33–40. doi: 10.1291/hypres.26.S33
59. Rossowski WJ, Cheng BL, Jiang NY, Coy DH. Examination of somatostatin involvement in the inhibitory action of GIP, GLP-1, amylin and adrenomedullin on gastric acid release using a new SRIF antagonist analogue. Br J Pharmacol (1998) 1255:1081–7. doi: 10.1038/sj.bjp.0702160
60. Fukuda K, Tsukada H, Onomura M, Saito T, Kodama M, Nakamura H, et al. Effect of adrenomedullin on ion transport and muscle contraction in rat distal colon. Peptides (1998) 196:1043–7. doi: 10.1016/S0196-9781(98)00043-6
61. Ashizuka S, Kita T, Inatsu H, Kitamura K. Adrenomedullin: a novel therapy for intractable ulcerative colitis. Inflammation Bowel Dis (2013) 192:E26–7. doi: 10.1002/ibd.22891
62. Rulle S, Ah Kioon MD, Asensio C, Mussard J, Ea HK, Boissier MC, et al. Adrenomedullin, a neuropeptide with immunoregulatory properties induces semi-mature tolerogenic dendritic cells. Immunology (2012) 1362:252–64. doi: 10.1111/j.1365-2567.2012.03577.x
63. Talero E, Di Paola R, Mazzon E, Esposito E, Motilva V, Cuzzocrea S. Anti-inflammatory effects of adrenomedullin on acute lung injury induced by Carrageenan in mice. Mediators Inflamm (2012) 2012:717851. doi: 10.1155/2012/717851
64. Hippenstiel S, Witzenrath M, Schmeck B, Hocke A, Krisp M, Krull M, et al. Adrenomedullin reduces endothelial hyperpermeability. Circ Res (2002) 917:618–25. doi: 10.1161/01.RES.0000036603.61868.F9
65. Ashizuka S, Ishikawa N, Kato J, Yamaga J, Inatsu H, Eto T, et al. Effect of adrenomedullin administration on acetic acid-induced colitis in rats. Peptides (2005) 2612:2610–5. doi: 10.1016/j.peptides.2005.05.007
66. Talero E, Sanchez-Fidalgo S, de la Lastra CA, Illanes M, Calvo JR, Motilva V. Acute and chronic responses associated with adrenomedullin administration in experimental colitis. Peptides (2008) 2911:2001–12. doi: 10.1016/j.peptides.2008.07.013
67. Ashizuka S, Inagaki-Ohara K, Kuwasako K, Kato J, Inatsu H, Kitamura K. Adrenomedullin treatment reduces intestinal inflammation and maintains epithelial barrier function in mice administered dextran sulphate sodium. Microbiol Immunol (2009) 5310:573–81. doi: 10.1111/j.1348-0421.2009.00159.x
68. Martinez A, Elsasser TH, Muro-Cacho C, Moody TW, Miller MJ, Macri CJ, et al. Expression of adrenomedullin and its receptor in normal and malignant human skin: a potential pluripotent role in the integument. Endocrinology (1997) 13812:5597–604. doi: 10.1210/endo.138.12.5622
69. Allaker RP, Zihni C, Kapas S. An investigation into the antimicrobial effects of adrenomedullin on members of the skin, oral, respiratory tract and gut microflora. FEMS Immunol Med Microbiol (1999) 234:289–93. doi: 10.1111/j.1574-695X.1999.tb01250.x
70. Ogoshi M, Inoue K, Takei Y. Identification of a novel adrenomedullin gene family in teleost fish. Biochem Biophys Res Commun (2003) 3114:1072–7. doi: 10.1016/j.bbrc.2003.10.111
71. Takei Y, Inoue K, Ogoshi M, Kawahara T, Bannai H, Miyano S. Identification of novel adrenomedullin in mammals: a potent cardiovascular and renal regulator. FEBS Lett (2004) 5561-3:53–8. doi: 10.1016/S0014-5793(03)01368-1
72. Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem (2004) 2798:7264–74. doi: 10.1074/jbc.M305332200
73. Yang JH, Jia YX, Pan CS, Zhao J, Ouyang M, Yang J, et al. Effects of intermedin1-53 on cardiac function and ischemia/reperfusion injury in isolated rat hearts. Biochem Biophys Res Commun (2005) 3273:713–9. doi: 10.1016/j.bbrc.2004.12.071
74. Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol (2001) 212. REVIEWS3013. doi: 10.1186/gb-2001-2-12-reviews3013
75. Hay DL, Pioszak AA. Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annu Rev Pharmacol Toxicol (2016) 56:469–87. doi: 10.1146/annurev-pharmtox-010715-103120
76. Husmann K, Sexton PM, Fischer JA, Born W. Mouse receptor-activity-modifying proteins 1, -2 and -3: amino acid sequence, expression and function. Mol Cell Endocrinol (2000) 1621-2:35–43. doi: 10.1016/S0303-7207(00)00212-4
77. Bomberger JM, Parameswaran N, Hall CS, Aiyar N, Spielman WS. Novel function for receptor activity-modifying proteins (RAMPs) in post-endocytic receptor trafficking. J Biol Chem (2005) 28010:9297–307. doi: 10.1074/jbc.M413786200
78. Oliver KR, Kane SA, Salvatore CA, Mallee JJ, Kinsey AM, Koblan KS, et al. Cloning, characterization and central nervous system distribution of receptor activity modifying proteins in the rat. Eur J Neurosci (2001) 144:618–28. doi: 10.1046/j.0953-816x.2001.01688.x
79. Parameswaran N, Spielman WS. RAMPs: The past, present and future. Trends Biochem Sci (2006) 3111:631–8. doi: 10.1016/j.tibs.2006.09.006
80. Wunder F, Rebmann A, Geerts A, Kalthof B. Pharmacological and kinetic characterization of adrenomedullin 1 and calcitonin gene-related peptide 1 receptor reporter cell lines. Mol Pharmacol (2008) 734:1235–43. doi: 10.1124/mol.107.042283
81. Hay DL, Garelja ML, Poyner DR, Walker CS. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br J Pharmacol (2018) 1751:3–17. doi: 10.1111/bph.14075
82. Wootten DL, Simms J, Hay DL, Christopoulos A, Sexton PM. Receptor activity modifying proteins and their potential as drug targets. Prog Mol Biol Transl Sci (2010) 91:53–79. doi: 10.1016/S1877-1173(10)91003-X
83. Booe JM, Walker CS, Barwell J, Kuteyi G, Simms J, Jamaluddin MA, et al. Structural Basis for Receptor Activity-Modifying Protein-Dependent Selective Peptide Recognition by a G Protein-Coupled Receptor. Mol Cell (2015) 586:1040–52. doi: 10.1016/j.molcel.2015.04.018
84. Booe JM, Warner ML, Roehrkasse AM, Hay DL, Pioszak AA. Probing the Mechanism of Receptor Activity-Modifying Protein Modulation of GPCR Ligand Selectivity through Rational Design of Potent Adrenomedullin and Calcitonin Gene-Related Peptide Antagonists. Mol Pharmacol (2018) 934:355–67. doi: 10.1124/mol.117.110916
85. Watkins HA, Chakravarthy M, Abhayawardana RS, Gingell JJ, Garelja M, Pardamwar M, et al. Receptor Activity-modifying Proteins 2 and 3 Generate Adrenomedullin Receptor Subtypes with Distinct Molecular Properties. J Biol Chem (2016) 29122:11657–75. doi: 10.1074/jbc.M115.688218
86. McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature (1998) 3936683:333–9. doi: 10.1038/30666
87. Nag K, Sultana N, Kato A, Dranik A, Nakamura N, Kutsuzawa K, et al. Ligand-induced internalization, recycling, and resensitization of adrenomedullin receptors depend not on CLR or RAMP alone but on the receptor complex as a whole. Gen Comp Endocrinol (2015) 212:156–62. doi: 10.1016/j.ygcen.2014.04.029
88. Weston C, Lu J, Li N, Barkan K, Richards GO, Roberts DJ, et al. Modulation of Glucagon Receptor Pharmacology by Receptor Activity-modifying Protein-2 (RAMP2). J Biol Chem (2015) 29038:23009–22. doi: 10.1074/jbc.M114.624601
89. Mackie DI, Nielsen NR, Harris M, Singh S, Davis RB, Dy D, et al. RAMP3 determines rapid recycling of atypical chemokine receptor-3 for guided angiogenesis. Proc Natl Acad Sci USA (2019) 11648:24093–9. doi: 10.1073/pnas.1905561116
90. Egea SC, Dickerson IM. Direct interactions between calcitonin-like receptor (CLR) and CGRP-receptor component protein (RCP) regulate CGRP receptor signaling. Endocrinology (2012) 1534:1850–60. doi: 10.1210/en.2011-1459
91. Dickerson IM. Role of CGRP-receptor component protein (RCP) in CLR/RAMP function. Curr Protein Pept Sci (2013) 145:407–15. doi: 10.2174/13892037113149990057
92. Evans BN, Rosenblatt MI, Mnayer LO, Oliver KR, Dickerson IM. CGRP-RCP, a novel protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin receptors. J Biol Chem (2000) 27540:31438–43. doi: 10.1074/jbc.M005604200
93. Prado MA, Evans-Bain B, Oliver KR, Dickerson IM. The role of the CGRP-receptor component protein (RCP) in adrenomedullin receptor signal transduction. Peptides (2001) 2211:1773–81. doi: 10.1016/S0196-9781(01)00517-4
94. Hay DL, Walker CS, Poyner DR. Adrenomedullin and calcitonin gene-related peptide receptors in endocrine-related cancers: opportunities and challenges. Endocr Relat Cancer (2011) 181:C1–14. doi: 10.1677/ERC-10-0244
95. Kapas S, Catt KJ, Clark AJ. Cloning and expression of cDNA encoding a rat adrenomedullin receptor. J Biol Chem (1995) 27043:25344–7. doi: 10.1074/jbc.270.43.25344
96. Kapas S, Clark AJ. Identification of an orphan receptor gene as a type 1 calcitonin gene-related peptide receptor. Biochem Biophys Res Commun (1995) 2173:832–8. doi: 10.1006/bbrc.1995.2847
97. Kennedy SP, Sun D, Oleynek JJ, Hoth CF, Kong J, Hill RJ. Expression of the rat adrenomedullin receptor or a putative human adrenomedullin receptor does not correlate with adrenomedullin binding or functional response. Biochem Biophys Res Commun (1998) 2443:832–7. doi: 10.1006/bbrc.1998.8349
98. Yun HJ, Ryu H, Choi YS, Song IC, Jo DY, Kim S, et al. C-X-C motif receptor 7 in gastrointestinal cancer. Oncol Lett (2015) 103:1227–32. doi: 10.3892/ol.2015.3407
99. Schmid CD, Schledzewski K, Mogler C, Waldburger N, Kalna V, Marx A, et al. GPR182 is a novel marker for sinusoidal endothelial differentiation with distinct GPCR signaling activity in vitro. Biochem Biophys Res Commun (2018) 4971:32–8. doi: 10.1016/j.bbrc.2018.01.185
100. Shimosawa T, Shibagaki Y, Ishibashi K, Kitamura K, Kangawa K, Kato S, et al. Adrenomedullin, an endogenous peptide, counteracts cardiovascular damage. Circulation (2002) 1051:106–11. doi: 10.1161/hc0102.101399
101. Dackor RT, Fritz-Six K, Dunworth WP, Gibbons CL, Smithies O, Caron KM. Hydrops fetalis, cardiovascular defects, and embryonic lethality in mice lacking the calcitonin receptor-like receptor gene. Mol Cell Biol (2006) 267:2511–8. doi: 10.1128/MCB.26.7.2511-2518.2006
102. Imai Y, Shiindo T, Maemura K, Kurihara Y, Nagai R, Kurihara H. Evidence for the physiological and pathological roles of adrenomedullin from genetic engineering in mice. Ann N Y Acad Sci (2001) 947:26–33; discussion -4. doi: 10.1111/j.1749-6632.2001.tb03927.x
103. Fritz-Six KL, Dunworth WP, Li M, Caron KM. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J Clin Invest (2008) 1181:40–50. doi: 10.1172/JCI33302
104. Kechele DO, Dunworth WP, Trincot CE, Wetzel-Strong SE, Li M, Ma H, et al. Endothelial Restoration of Receptor Activity-Modifying Protein 2 Is Sufficient to Rescue Lethality, but Survivors Develop Dilated Cardiomyopathy. Hypertension (2016) 683:667–77. doi: 10.1161/HYPERTENSIONAHA.116.07191
105. Kadmiel M, Fritz-Six KL, Caron KM. Understanding RAMPs through genetically engineered mouse models. Adv Exp Med Biol (2012) 744:49–60. doi: 10.1007/978-1-4614-2364-5_5
106. Kadmiel M, Fritz-Six K, Pacharne S, Richards GO, Li M, Skerry TM, et al. Research resource: Haploinsufficiency of receptor activity-modifying protein-2 (RAMP2) causes reduced fertility, hyperprolactinemia, skeletal abnormalities, and endocrine dysfunction in mice. Mol Endocrinol (2011) 257:1244–53. doi: 10.1210/me.2010-0400
107. Tam CW, Husmann K, Clark NC, Clark JE, Lazar Z, Ittner LM, et al. Enhanced vascular responses to adrenomedullin in mice overexpressing receptor-activity-modifying protein 2. Circ Res (2006) 982:262–70. doi: 10.1161/01.RES.0000200737.63865.58
108. Pawlak JB, Wetzel-Strong SE, Dunn MK, Caron KM. Cardiovascular effects of exogenous adrenomedullin and CGRP in Ramp and Calcrl deficient mice. Peptides (2017) 88:1–7. doi: 10.1016/j.peptides.2016.12.002
109. Barrick CJ, Lenhart PM, Dackor RT, Nagle E, Caron KM. Loss of receptor activity-modifying protein 3 exacerbates cardiac hypertrophy and transition to heart failure in a sex-dependent manner. J Mol Cell Cardiol (2012) 521:165–74. doi: 10.1016/j.yjmcc.2011.10.021
110. Dackor R, Caron K. Mice heterozygous for adrenomedullin exhibit a more extreme inflammatory response to endotoxin-induced septic shock. Peptides (2007) 2811:2164–70. doi: 10.1016/j.peptides.2007.08.012
111. Liu T, Kamiyoshi A, Tanaka M, Iida S, Sakurai T, Ichikawa-Shindo Y, et al. RAMP3 deficiency enhances postmenopausal obesity and metabolic disorders. Peptides (2018) 110:10–8. doi: 10.1016/j.peptides.2018.10.006
112. Watanabe H, Takahashi E, Kobayashi M, Goto M, Krust A, Chambon P, et al. The estrogen-responsive adrenomedullin and receptor-modifying protein 3 gene identified by DNA microarray analysis are directly regulated by estrogen receptor. J Mol Endocrinol (2006) 361:81–9. doi: 10.1677/jme.1.01825
113. Zhao Y, Hague S, Manek S, Zhang L, Bicknell R, Rees MC. PCR display identifies tamoxifen induction of the novel angiogenic factor adrenomedullin by a non estrogenic mechanism in the human endometrium. Oncogene (1998) 163:409–15. doi: 10.1038/sj.onc.1201768
114. Cueille C, Pidoux E, de Vernejoul MC, Ventura-Clapier R, Garel JM. Increased myocardial expression of RAMP1 and RAMP3 in rats with chronic heart failure. Biochem Biophys Res Commun (2002) 2942:340–6. doi: 10.1016/S0006-291X(02)00487-4
115. Yoshihara F, Nishikimi T, Okano I, Hino J, Horio T, Tokudome T, et al. Upregulation of intracardiac adrenomedullin and its receptor system in rats with volume overload-induced cardiac hypertrophy. Regul Pept (2005) 1271-3:239–44. doi: 10.1016/j.regpep.2004.12.017
116. Oie E, Vinge LE, Andersen GO, Yndestad A, Krobert KA, Sandberg C, et al. RAMP2 and RAMP3 mRNA levels are increased in failing rat cardiomyocytes and associated with increased responsiveness to adrenomedullin. J Mol Cell Cardiol (2005) 381:145–51. doi: 10.1016/j.yjmcc.2004.10.009
117. Nishimatsu H, Suzuki E, Nagata D, Moriyama N, Satonaka H, Walsh K, et al. Adrenomedullin induces endothelium-dependent vasorelaxation via the phosphatidylinositol 3-kinase/Akt-dependent pathway in rat aorta. Circ Res (2001) 891:63–70. doi: 10.1161/hh1301.092498
118. Kim W, Moon SO, Sung MJ, Kim SH, Lee S, So JN, et al. Angiogenic role of adrenomedullin through activation of Akt, mitogen-activated protein kinase, and focal adhesion kinase in endothelial cells. FASEB J (2003) 1713:1937–9. doi: 10.1096/fj.02-1209fje
119. Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res (2011) 1723:7230–9. doi: 10.1158/1078-0432.CCR-11-1354
120. Xu M, Qi F, Zhang S, Ma X, Wang S, Wang C, et al. Adrenomedullin promotes the growth of pancreatic ductal adenocarcinoma through recruitment of myelomonocytic cells. Oncotarget (2016) 734:55043–56. doi: 10.18632/oncotarget.10393
121. Nouguerede E, Berenguer C, Garcia S, Bennani B, Delfino C, Nanni I, et al. Expression of adrenomedullin in human colorectal tumors and its role in cell growth and invasion in vitro and in xenograft growth in vivo. Cancer Med (2013) 22:196–207. doi: 10.1002/cam4.51
122. Wang L, Gala M, Yamamoto M, Pino MS, Kikuchi H, Shue DS, et al. Adrenomedullin is a therapeutic target in colorectal cancer. Int J Cancer (2014) 1349:2041–50. doi: 10.1002/ijc.28542
123. Ehlenz K, Koch B, Preuss P, Simon B, Koop I, Lang RE. High levels of circulating adrenomedullin in severe illness: correlation with C-reactive protein and evidence against the adrenal medulla as site of origin. Exp Clin Endocrinol Diabetes (1997) 1053:156–62. doi: 10.1055/s-0029-1211745
124. Letizia C, Di Iorio R, De Toma G, Marinoni E, Cerci S, Celi M, et al. Circulating adrenomedullin is increased in patients with corticotropin-dependent Cushing’s syndrome due to pituitary adenoma. Metabolism (2000) 496:760–3. doi: 10.1053/meta.2000.6241
125. Udono T, Totsune K, Takahashi K, Abe T, Sato M, Shibahara S, et al. Increased expression of adrenomedullin mRNA in the tissues of intraocular and orbital tumors. Am J Ophthalmol (2000) 1294:555–6. doi: 10.1016/S0002-9394(99)00442-0
126. Greillier L, Tounsi A, Berenguer-Daize C, Dussault N, Delfino C, Benyahia Z, et al. Functional Analysis of the Adrenomedullin Pathway in Malignant Pleural Mesothelioma. J Thorac Oncol (2016) 111:94–107. doi: 10.1016/j.jtho.2015.09.004
127. Uemura M, Yamamoto H, Takemasa I, Mimori K, Mizushima T, Ikeda M, et al. Hypoxia-inducible adrenomedullin in colorectal cancer. Anticancer Res (2011) 312:507–14.
128. Ouafik L, Sauze S, Boudouresque F, Chinot O, Delfino C, Fina F, et al. Neutralization of adrenomedullin inhibits the growth of human glioblastoma cell lines in vitro and suppresses tumor xenograft growth in vivo. Am J Pathol (2002) 1604:1279–92. doi: 10.1016/S0002-9440(10)62555-2
129. Takahashi K, Satoh F, Hara E, Murakami O, Kumabe T, Tominaga T, et al. Production and secretion of adrenomedullin by cultured choroid plexus carcinoma cells. J Neurochem (1997) 682:726–31. doi: 10.1046/j.1471-4159.1997.68020726.x
130. Rocchi P, Boudouresque F, Zamora AJ, Muracciole X, Lechevallier E, Martin PM, et al. Expression of adrenomedullin and peptide amidation activity in human prostate cancer and in human prostate cancer cell lines. Cancer Res (2001) 613:1196–206.
131. Hata K, Takebayashi Y, Akiba S, Fujiwaki R, Iida K, Nakayama K, et al. Expression of the adrenomedullin gene in epithelial ovarian cancer. Mol Hum Reprod (2000) 610:867–72. doi: 10.1093/molehr/6.10.867
132. Park SC, Yoon JH, Lee JH, Yu SJ, Myung SJ, Kim W, et al. Hypoxia-inducible adrenomedullin accelerates hepatocellular carcinoma cell growth. Cancer Lett (2008) 2712:314–22. doi: 10.1016/j.canlet.2008.06.019
133. Michelsen J, Thiesson H, Walter S, Ottosen PD, Skott O, Jensen BL. Tissue expression and plasma levels of adrenomedullin in renal cancer patients. Clin Sci (Lond) (2006) 1111:61–70. doi: 10.1042/CS20060030
134. Deville JL, Bartoli C, Berenguer C, Fernandez-Sauze S, Kaafarani I, Delfino C, et al. Expression and role of adrenomedullin in renal tumors and value of its mRNA levels as prognostic factor in clear-cell renal carcinoma. Int J Cancer (2009) 12510:2307–15. doi: 10.1002/ijc.24568
135. Oehler MK, Fischer DC, Orlowska-Volk M, Herrle F, Kieback DG, Rees MC, et al. Tissue and plasma expression of the angiogenic peptide adrenomedullin in breast cancer. Br J Cancer (2003) 8910:1927–33. doi: 10.1038/sj.bjc.6601397
136. Keleg S, Kayed H, Jiang X, Penzel R, Giese T, Buchler MW, et al. Adrenomedullin is induced by hypoxia and enhances pancreatic cancer cell invasion. Int J Cancer (2007) 1211:21–32. doi: 10.1002/ijc.22596
137. Aggarwal G, Ramachandran V, Javeed N, Arumugam T, Dutta S, Klee GG, et al. Adrenomedullin is up-regulated in patients with pancreatic cancer and causes insulin resistance in beta cells and mice. Gastroenterology (2012) 1436:1510–7. doi: 10.1053/j.gastro.2012.08.044
138. Berenguer C, Boudouresque F, Dussert C, Daniel L, Muracciole X, Grino M, et al. Adrenomedullin, an autocrine/paracrine factor induced by androgen withdrawal, stimulates ‘neuroendocrine phenotype’ in LNCaP prostate tumor cells. Oncogene (2008) 274:506–18. doi: 10.1038/sj.onc.1210656
139. Berenguer-Daize C, Boudouresque F, Bastide C, Tounsi A, Benyahia Z, Acunzo J, et al. Adrenomedullin blockade suppresses growth of human hormone-independent prostate tumor xenograft in mice. Clin Cancer Res (2013) 1922:6138–50. doi: 10.1158/1078-0432.CCR-13-0691
140. Thouennon E, Pierre A, Tanguy Y, Guillemot J, Manecka DL, Guerin M, et al. Expression of trophic amidated peptides and their receptors in benign and malignant pheochromocytomas: high expression of adrenomedullin RDC1 receptor and implication in tumoral cell survival. Endocr Relat Cancer (2010) 173:637–51. doi: 10.1677/ERC-10-0109
141. Kaafarani I, Fernandez-Sauze S, Berenguer C, Chinot O, Delfino C, Dussert C, et al. Targeting adrenomedullin receptors with systemic delivery of neutralizing antibodies inhibits tumor angiogenesis and suppresses growth of human tumor xenografts in mice. FASEB J (2009) 2310:3424–35. doi: 10.1096/fj.08-127852
142. Forneris M, Gottardo L, Albertin G, Malendowicz LK, Nussdorfer GG. Expression and function of adrenomedullin and its receptors in Conn’s adenoma cells. Int J Mol Med (2001) 86:675–9. doi: 10.3892/ijmm.8.6.675
143. Martinez A, Vos M, Guedez L, Kaur G, Chen Z, Garayoa M, et al. The effects of adrenomedullin overexpression in breast tumor cells. J Natl Cancer Inst (2002) 9416:1226–37. doi: 10.1093/jnci/94.16.1226
144. Oehler MK, Norbury C, Hague S, Rees MC, Bicknell R. Adrenomedullin inhibits hypoxic cell death by upregulation of Bcl-2 in endometrial cancer cells: a possible promotion mechanism for tumour growth. Oncogene (2001) 2023:2937–45. doi: 10.1038/sj.onc.1204422
145. Hague S, Zhang L, Oehler MK, Manek S, MacKenzie IZ, Bicknell R, et al. Expression of the hypoxically regulated angiogenic factor adrenomedullin correlates with uterine leiomyoma vascular density. Clin Cancer Res (2000) 67:2808–14.
146. Siclari VA, Mohammad KS, Tompkins DR, Davis H, McKenna CR, Peng X, et al. Tumor-expressed adrenomedullin accelerates breast cancer bone metastasis. Breast Cancer Res (2014) 166:458. doi: 10.1186/s13058-014-0458-y
147. Abasolo I, Montuenga LM, Calvo A. Adrenomedullin prevents apoptosis in prostate cancer cells. Regul Pept (2006) 1331-3:115–22. doi: 10.1016/j.regpep.2005.09.026
148. Wu XY, Hao CP, Ling M, Guo CH, Ma W. Hypoxia-induced apoptosis is blocked by adrenomedullin via upregulation of Bcl-2 in human osteosarcoma cells. Oncol Rep (2015) 342:787–94. doi: 10.3892/or.2015.4011
149. Kim W, Moon SO, Sung MJ, Kim SH, Lee S, Kim HJ, et al. Protective effect of adrenomedullin in mannitol-induced apoptosis. Apoptosis (2002) 76:527–36. doi: 10.1023/A:1020695110648
150. Shichiri M, Kato H, Doi M, Marumo F, Hirata Y. Induction of max by adrenomedullin and calcitonin gene-related peptide antagonizes endothelial apoptosis. Mol Endocrinol (1999) 138:1353–63. doi: 10.1210/mend.13.8.0324
151. Carthew P, Edwards RE, Nolan BM, Martin EA, Heydon RT, White IN, et al. Tamoxifen induces endometrial and vaginal cancer in rats in the absence of endometrial hyperplasia. Carcinogenesis (2000) 214:793–7. doi: 10.1093/carcin/21.4.793
152. Withers DJ, Coppock HA, Seufferlein T, Smith DM, Bloom SR, Rozengurt E. Adrenomedullin stimulates DNA synthesis and cell proliferation via elevation of cAMP in Swiss 3T3 cells. FEBS Lett (1996) 3781:83–7. doi: 10.1016/0014-5793(95)01427-6
153. Zudaire E, Portal-Nunez S, Cuttitta F. The central role of adrenomedullin in host defense. J Leukoc Biol (2006) 802:237–44. doi: 10.1189/jlb.0206123
154. Larrayoz IM, Martinez-Herrero S, Garcia-Sanmartin J, Ochoa-Callejero L, Martinez A. Adrenomedullin and tumour microenvironment. J Transl Med (2014) 12:339. doi: 10.1186/s12967-014-0339-2
155. Tanaka M, Koyama T, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Kawate H, et al. The endothelial adrenomedullin-RAMP2 system regulates vascular integrity and suppresses tumour metastasis. Cardiovasc Res (2016) 1114:398–409. doi: 10.1093/cvr/cvw166
156. Dai K, Tanaka M, Kamiyoshi A, Sakurai T, Ichikawa-Shindo Y, Kawate H, et al. Deficiency of the adrenomedullin-RAMP3 system suppresses metastasis through the modification of cancer-associated fibroblasts. Oncogene (2020) 399:1914–30. doi: 10.1038/s41388-019-1112-z
157. Benyahia Z, Dussault N, Cayol M, Sigaud R, Berenguer-Daize C, Delfino C, et al. Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget (2017) 89:15744–62. doi: 10.18632/oncotarget.14999
158. Ishikawa T, Chen J, Wang J, Okada F, Sugiyama T, Kobayashi T, et al. Adrenomedullin antagonist suppresses in vivo growth of human pancreatic cancer cells in SCID mice by suppressing angiogenesis. Oncogene (2003) 228:1238–42. doi: 10.1038/sj.onc.1206207
159. Iimuro S, Shindo T, Moriyama N, Amaki T, Niu P, Takeda N, et al. Angiogenic effects of adrenomedullin in ischemia and tumor growth. Circ Res (2004) 954:415–23. doi: 10.1161/01.RES.0000138018.61065.d1
160. Zudaire E, Martinez A, Garayoa M, Pio R, Kaur G, Woolhiser MR, et al. Adrenomedullin is a cross-talk molecule that regulates tumor and mast cell function during human carcinogenesis. Am J Pathol (2006) 1681:280–91. doi: 10.2353/ajpath.2006.050291
161. Fujita Y, Mimata H, Nasu N, Nomura T, Nomura Y, Nakagawa M. Involvement of adrenomedullin induced by hypoxia in angiogenesis in human renal cell carcinoma. Int J Urol (2002) 96:285–95. doi: 10.1046/j.1442-2042.2002.00469.x
162. Garayoa M, Martinez A, Lee S, Pio R, An WG, Neckers L, et al. Hypoxia-inducible factor-1 (HIF-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: a possible promotion mechanism of carcinogenesis. Mol Endocrinol (2000) 146:848–62. doi: 10.1210/mend.14.6.0473
163. Oehler MK, Hague S, Rees MC, Bicknell R. Adrenomedullin promotes formation of xenografted endometrial tumors by stimulation of autocrine growth and angiogenesis. Oncogene (2002) 2118:2815–21. doi: 10.1038/sj.onc.1205374
164. Li F, Yang R, Zhang X, Liu A, Zhao Y, Guo Y. Silencing of hypoxiainducible adrenomedullin using RNA interference attenuates hepatocellular carcinoma cell growth in vivo. Mol Med Rep (2014) 103:1295–302. doi: 10.3892/mmr.2014.2320
165. Park SJ, Kim JG, Son TG, Yi JM, Kim ND, Yang K, et al. The histone demethylase JMJD1A regulates adrenomedullin-mediated cell proliferation in hepatocellular carcinoma under hypoxia. Biochem Biophys Res Commun (2013) 4344:722–7. doi: 10.1016/j.bbrc.2013.03.091
166. Drimal J, Faberova V, Schmidtova L, Bednarikova M, Drimal J Jr., Drimal D. The ACAT inhibitor VULM1457 significantly reduced production and secretion of adrenomedullin (AM) and down-regulated AM receptors on human hepatoblastic cells. Gen Physiol Biophys (2005) 244:397–409.
167. Fidler IJ, Kripke ML. The challenge of targeting metastasis. Cancer Metastasis Rev (2015) 344:635–41. doi: 10.1007/s10555-015-9586-9
168. Albertin G, Rucinski M, Carraro G, Forneris M, Andreis P, Malendowicz LK, et al. Adrenomedullin and vascular endothelium growth factor genes are overexpressed in the regenerating rat adrenal cortex, and AM and VEGF reciprocally enhance their mRNA expression in cultured rat adrenocortical cells. Int J Mol Med (2005) 163:431–5. doi: 10.3892/ijmm.16.3.431
169. Brekhman V, Lugassie J, Zaffryar-Eilot S, Sabo E, Kessler O, Smith V, et al. Receptor activity modifying protein-3 mediates the protumorigenic activity of lysyl oxidase-like protein-2. FASEB J (2011) 251:55–65. doi: 10.1096/fj.10-162677
170. Miller MJ, Martinez A, Unsworth EJ, Thiele CJ, Moody TW, Elsasser T, et al. Adrenomedullin expression in human tumor cell lines. Its potential role as an autocrine growth factor. J Biol Chem (1996) 27138:23345–51. doi: 10.1074/jbc.271.38.23345
171. Ramachandran V, Arumugam T, Hwang RF, Greenson JK, Simeone DM, Logsdon CD. Adrenomedullin is expressed in pancreatic cancer and stimulates cell proliferation and invasion in an autocrine manner via the adrenomedullin receptor, ADMR. Cancer Res (2007) 676:2666–75. doi: 10.1158/0008-5472.CAN-06-3362
172. Mazzocchi G, Malendowicz LK, Ziolkowska A, Spinazzi R, Rebuffat P, Aragona F, et al. Adrenomedullin (AM) and AM receptor type 2 expression is up-regulated in prostate carcinomas (PC), and AM stimulates in vitro growth of a PC-derived cell line by enhancing proliferation and decreasing apoptosis rates. Int J Oncol (2004) 256:1781–7. doi: 10.3892/ijo.25.6.1781
173. Albertin G, Sorato E, Oselladore B, Mascarin A, Tortorella C, Guidolin D. Involvement of vascular endothelial growth factor signaling in CLR/RAMP1 and CLR/RAMP2-mediated pro-angiogenic effect of intermedin on human vascular endothelial cells. Int J Mol Med (2010) 262:289–94. doi: 10.3892/ijmm_00000464
174. Eguchi S, Hirata Y, Iwasaki H, Sato K, Watanabe TX, Inui T, et al. Structure-activity relationship of adrenomedullin, a novel vasodilatory peptide, in cultured rat vascular smooth muscle cells. Endocrinology (1994) 1356:2454–8. doi: 10.1210/endo.135.6.7988431
175. Santiago JA, Garrison EA, Ventura VL, Coy DH, Bitar K, Murphy WA, et al. Synthetic human adrenomedullin and adrenomedullin 15-52 have potent short-lived vasodilator activity in the hindlimb vascular bed of the cat. Life Sci (1994) 555:PL85–90. doi: 10.1016/0024-3205(94)00652-0
176. Kitamura K, Matsui E, Kato J, Katoh F, Kita T, Tsuji T, et al. Adrenomedullin 11-26: a novel endogenous hypertensive peptide isolated from bovine adrenal medulla. Peptides (2001) 2211:1713–8. doi: 10.1016/S0196-9781(01)00529-0
177. Hay DL, Howitt SG, Conner AC, Schindler M, Smith DM, Poyner DR. CL/RAMP2 and CL/RAMP3 produce pharmacologically distinct adrenomedullin receptors: a comparison of effects of adrenomedullin22-52, CGRP8-37 and BIBN4096BS. Br J Pharmacol (2003) 1403:477–86. doi: 10.1038/sj.bjp.0705472
178. Moad HE, Pioszak AA, Selective CGRP. and adrenomedullin peptide binding by tethered RAMP-calcitonin receptor-like receptor extracellular domain fusion proteins. Protein Sci (2013) 2212:1775–85. doi: 10.1002/pro.2377
179. Champion HC, Friedman DE, Lambert DG, Murphy WA, Coy DH, Kadowitz PJ. Adrenomedullin 16-31 has pressor activity in the rat but not the cat. Peptides (1997) 181:133–6. doi: 10.1016/S0196-9781(96)00251-3
180. Martinez A, Julian M, Bregonzio C, Notari L, Moody TW, Cuttitta F. Identification of vasoactive nonpeptidic positive and negative modulators of adrenomedullin using a neutralizing antibody-based screening strategy. Endocrinology (2004) 1458:3858–65. doi: 10.1210/en.2003-1251
181. Qi T, Christopoulos G, Bailey RJ, Christopoulos A, Sexton PM, Hay DL. Identification of N-terminal receptor activity-modifying protein residues important for calcitonin gene-related peptide, adrenomedullin, and amylin receptor function. Mol Pharmacol (2008) 744:1059–71. doi: 10.1124/mol.108.047142
182. Fischer JP, Els-Heindl S, Schonauer R, Bierer D, Kobberling J, Riedl B, et al. The Impact of Adrenomedullin Thr22 on Selectivity within the Calcitonin Receptor-like Receptor/Receptor Activity-Modifying Protein System. ChemMedChem (2018) 1317:1797–805. doi: 10.1002/cmdc.201800329
183. Robinson SD, Aitken JF, Bailey RJ, Poyner DR, Hay DL. Novel peptide antagonists of adrenomedullin and calcitonin gene-related peptide receptors: identification, pharmacological characterization, and interactions with position 74 in receptor activity-modifying protein 1/3. J Pharmacol Exp Ther (2009) 3312:513–21. doi: 10.1124/jpet.109.156448
184. Portal-Nunez S, Shankavaram UT, Rao M, Datrice N, Atay S, Aparicio M, et al. Aryl hydrocarbon receptor-induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. Cancer Res (2012) 7222:5790–800. doi: 10.1158/0008-5472.CAN-12-0818
185. Roldos V, Martin-Santamaria S, Julian M, Martinez A, Choulier L, Altschuh D, et al. Small-molecule negative modulators of adrenomedullin: design, synthesis, and 3D-QSAR study. ChemMedChem (2008) 39:1345–55. doi: 10.1002/cmdc.200800066
186. Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med (2004) 35011:1104–10. doi: 10.1056/NEJMoa030505
187. Salvatore CA, Hershey JC, Corcoran HA, Fay JF, Johnston VK, Moore EL, et al. Pharmacological characterization of MK-0974 [N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxamide], a potent and orally active calcitonin gene-related peptide receptor antagonist for the treatment of migraine. J Pharmacol Exp Ther (2008) 3242:416–21. doi: 10.1124/jpet.107.130344
188. Salvatore CA, Moore EL, Calamari A, Cook JJ, Michener MS, O’Malley S, et al. Pharmacological properties of MK-3207, a potent and orally active calcitonin gene-related peptide receptor antagonist. J Pharmacol Exp Ther (2010) 3331:152–60. doi: 10.1124/jpet.109.163816
189. Dodick DW, Lipton RB, Ailani J, Halker Singh RB, Shewale AR, Zhao S, et al. Ubrogepant, an Acute Treatment for Migraine, Improved Patient-Reported Functional Disability and Satisfaction in 2 Single-Attack Phase 3 Randomized Trials, ACHIEVE I and II. Headache (2020) 604:686–700. doi: 10.1111/head.13766
190. Silberstein SD, Dodick DW, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, et al. Fremanezumab for the Preventive Treatment of Chronic Migraine. N Engl J Med (2017) 37722:2113–22. doi: 10.1056/NEJMoa1709038
191. Isumi Y, Minamino N, Katafuchi T, Yoshioka M, Tsuji T, Kangawa K, et al. Adrenomedullin production in fibroblasts: its possible function as a growth regulator of Swiss 3T3 cells. Endocrinology (1998) 1395:2552–63. doi: 10.1210/endo.139.5.6004
192. Blet A, Deniau B, Geven C, Sadoune M, Caillard A, Kounde PR, et al. Adrecizumab, a non-neutralizing anti-adrenomedullin antibody, improves haemodynamics and attenuates myocardial oxidative stress in septic rats. Intensive Care Med Exp (2019) 71:25. doi: 10.1186/s40635-019-0255-0
193. Shi L, Lehto SG, Zhu DX, Sun H, Zhang J, Smith BP, et al. Pharmacologic Characterization of AMG 334, a Potent and Selective Human Monoclonal Antibody against the Calcitonin Gene-Related Peptide Receptor. J Pharmacol Exp Ther (2016) 3561:223–31. doi: 10.1124/jpet.115.227793
194. Goadsby PJ, Reuter U, Hallstrom Y, Broessner G, Bonner JH, Zhang F, et al. A Controlled Trial of Erenumab for Episodic Migraine. N Engl J Med (2017) 37722:2123–32. doi: 10.1056/NEJMoa1705848
195. Dolgin E. First GPCR-directed antibody passes approval milestone. Nat Rev Drug Discovery (2018) 177:457–9. doi: 10.1038/nrd.2018.103
196. Fernandez-Sauze S, Delfino C, Mabrouk K, Dussert C, Chinot O, Martin PM, et al. Effects of adrenomedullin on endothelial cells in the multistep process of angiogenesis: involvement of CRLR/RAMP2 and CRLR/RAMP3 receptors. Int J Cancer (2004) 1086:797–804. doi: 10.1002/ijc.11663
197. Taylor MM, Samson WK. Ribozyme compromise of adrenomedullin mRNA reveals a physiological role in the regulation of water intake. Am J Physiol Regul Integr Comp Physiol (2002) 2826:R1739–45. doi: 10.1152/ajpregu.00696.2001
198. Dackor R, Fritz-Six K, Smithies O, Caron K. Receptor activity-modifying proteins 2 and 3 have distinct physiological functions from embryogenesis to old age. J Biol Chem (2007) 28225:18094–9. doi: 10.1074/jbc.M703544200
199. Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol (1999) 2092:254–67. doi: 10.1006/dbio.1999.9253
200. Khalfaoui-Bendriss G, Dussault N, Fernandez-Sauze S, Berenguer-Daize C, Sigaud R, Delfino C, et al. Adrenomedullin blockade induces regression of tumor neovessels through interference with vascular endothelial-cadherin signalling. Oncotarget (2015) 610:7536–53. doi: 10.18632/oncotarget.3167
201. Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun (2013) 4:1672. doi: 10.1038/ncomms2683
202. Fujio Y, Walsh K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J Biol Chem (1999) 27423:16349–54. doi: 10.1074/jbc.274.23.16349
203. Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res (2012) 728:1909–14. doi: 10.1158/0008-5472.CAN-11-3406
204. Gao Y, Li J, Qiao N, Meng Q, Zhang M, Wang X, et al. Adrenomedullin blockade suppresses sunitinib-resistant renal cell carcinoma growth by targeting the ERK/MAPK pathway. Oncotarget (2016) 739:63374–87. doi: 10.18632/oncotarget.11463
205. Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer (2007) 9612:1788–95. doi: 10.1038/sj.bjc.6603813
206. Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med (2012) 210. doi: 10.1101/cshperspect.a006577
207. Jin D, Harada K, Ohnishi S, Yamahara K, Kangawa K, Nagaya N. Adrenomedullin induces lymphangiogenesis and ameliorates secondary lymphoedema. Cardiovasc Res (2008) 803:339–45. doi: 10.1093/cvr/cvn228
208. Nikitenko LL, Shimosawa T, Henderson S, Makinen T, Shimosawa H, Qureshi U, et al. Adrenomedullin haploinsufficiency predisposes to secondary lymphedema. J Invest Dermatol (2013) 1337:1768–76. doi: 10.1038/jid.2013.47
209. Poon RT, Fan ST, Wong J. Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol (2001) 194:1207–25. doi: 10.1200/JCO.2001.19.4.1207
Keywords: adrenomedullin, angiogenesis, cancer, metastasis, AM1 and AM2, RAMP1-3
Citation: Vázquez R, Riveiro ME, Berenguer-Daizé C, O’Kane A, Gormley J, Touzelet O, Rezai K, Bekradda M and Ouafik L’H (2021) Targeting Adrenomedullin in Oncology: A Feasible Strategy With Potential as Much More Than an Alternative Anti-Angiogenic Therapy. Front. Oncol. 10:589218. doi: 10.3389/fonc.2020.589218
Received: 30 July 2020; Accepted: 02 November 2020;
Published: 06 January 2021.
Edited by:
Marco Rossi, University of Catanzaro, ItalyReviewed by:
Kevin Xueying Sun, Harbin Medical University, ChinaAlfredo Berruti, University of Brescia, Italy
Copyright © 2021 Vázquez, Riveiro, Berenguer-Daizé, O’Kane, Gormley, Touzelet, Rezai, Bekradda and Ouafik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: L’Houcine Ouafik, bGhvdWNpbmUub3VhZmlrQHVuaXYtYW11LmZy
†These authors have contributed equally to this work