Abstract
Tumor cells rewire metabolism to meet their increased nutritional demands, allowing the maintenance of tumor survival, proliferation, and expansion. Enhancement of glycolysis and glutaminolysis is identified in most, if not all cancers, including multiple myeloma (MM), which interacts with a hypoxic, acidic, and nutritionally deficient tumor microenvironment (TME). In this review, we discuss the metabolic changes including generation, depletion or accumulation of metabolites and signaling pathways, as well as their relationship with the TME in MM cells. Moreover, we describe the crosstalk among metabolism, TME, and changing function of immune cells during cancer progression. The overlapping metabolic phenotype between MM and immune cells is discussed. In this sense, targeting metabolism of MM cells is a promising therapeutic approach. We propose that it is important to define the metabolic signatures that may regulate the function of immune cells in TME in order to improve the response to immunotherapy.
Introduction
Multiple myeloma (MM) is a hematologic malignant proliferative disease characterized by the unrestricted proliferation of plasma cells in the bone marrow with overproduction of monoclonal immunoglobulin or light chain proteins (1). Although the overall survival of MM patients has significantly improved in the recent years, this disease is still incurable (2). Treatment options are limited especially for refractory and recurrent patients who have been treated with proteasome inhibitors, immunomodulatory drugs, as well as monoclonal antibodies. Immunotherapies, including oncolytic vaccines, checkpoint inhibitors, and adoptive cellular immunotherapy, offer a potentially effective treatment to these patients. However, the therapeutic efficacy is limited to a small number of population. Thus, it is urgent to look for alternative therapies.
Metabolic reprogramming is considered a hallmark of cancers and metabolism is altered in most, if not all cancer cells, regardless of the type of cancers, to meet the needs of energy and biosynthesis for rapid cell proliferation, and to adapt to the tumor microenvironment (TME). Glycolysis and glutaminolysis enhancement are two of the most common but vital modalities in TME (3, 4). Recent studies have revealed that metabolic reprogramming may affect the TME and, more importantly, could impact the function of immune cells. Therefore, some researchers suggested that the treatment response of MM might depend, at least in part, on the function of immune cells and tumor cell metabolic status in the TME (5).
In this review, we discuss the metabolic reprogramming in MM, which is associated with a hypoxic, acidic, and nutritionally deficient TME, and how these changes impede the function of immune cells. Metabolic changes of glucose and glutamine are described in detail. We suggest that integration of targeting tumor metabolism with immunotherapy could be a rational approach to modulate the TME and improve therapeutic efficacy of MM.
Metabolic Changes in MM Cells
In contrast to the reliance on oxidative phosphorylation (OXPHOS) to obtain energy in normal cells, most cancer cells adapted to their microenvironment rely heavily on aerobic glycolysis, converting glucose into lactic acid, for rapid production of adenosine triphosphate (ATP) to provide competitive advantages to cancer cells, thus meeting the requirements of rapid division and growth. This phenomenon is known as the Warburg effect (6), which is a hallmark of cancers, and is utilized in the 18F-fluorodeoxyglucose PET (18F- FDG–PET) as a sensitive diagnostic and prognostic tool in clinic (7, 8). MM is also reported to be dependent on glycolysis due to an elevated glycolytic gene profile, as well as its susceptibility to glycolysis inhibitors, such as inhibitors of glucose transporter (GLUT) and key glycolytic enzyme (9). The GLUT family comprises of 14 GLUT subtypes (10), among which GLUT1 overexpression is associated with poor clinical outcomes in various cancers. Researchers have found that GLUT1 up-regulation enables MM cells to elevate glucose uptake and GLUT1-specific inhibition can selectively induce death in MM cells with high GLUT1 level (11). However, other study has demonstrated that MM cells rely on GLUT4 for fundamental glucose uptake, maintenance of growth, survival and Mcl-1 expression (12). After glucose is transported into the cells, it is transformed into lactate and produces ATP through a multi-step metabolism depending on several key enzymes, including hexokinase 2 (HK2), phosphofructokinase (PFK), pyruvate kinase M2 (PKM2) and lactate dehydrogenase A (LDHA) that are highly expressed in MM (13) (Figure 1). Aerobic glycolysis enhancement also activates the pentose phosphate bypass pathway (PPP) and leads to increased production of reduced nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH), both benefit tumor cells against oxidative damage (14). Since oxidative stress is one of the important mechanisms of using bortezomib, drug resistance may be accompanied by increased antioxidant capacity. GP Soriano et al. have proposed that the generation of NADPH makes MM cells more tolerant to proteasome inhibitors (15). Lactate, as a glycolysis product, is transported by monocarboxylate transporters (MCTs) MCT1 and MCT4. Several studies have indicated that lactate can be used as a fuel for OXPHOS (16, 17). Fujiwara et al. found that MCT1 expression, which preferentially promotes lactate import, is up-regulated in MM cells under aerobic conditions, while MCT4, which favors lactate export, is upregulated under hypoxic conditions (18). It has been suggested that lactate secreted from hypoxic cancer cells may be used by normoxic cancer cells as an energy source since it can be converted to pyruvate, and then enter the tricarboxylic acid (TCA) cycle, establishing a metabolic symbiosis between hypoxic and normoxic cancer cells, which is important for the fast-growth and progression of tumors (19). Sevim et al. have proved that MM cells are capable of enhancing mitochondrial OXPHOS under ritonavir treatment (20). Subsequently, Christopher et al. have reported that increased OXPHOS in MM cells is associated with CD38-driven mitochondrial transfer (21).
Figure 1
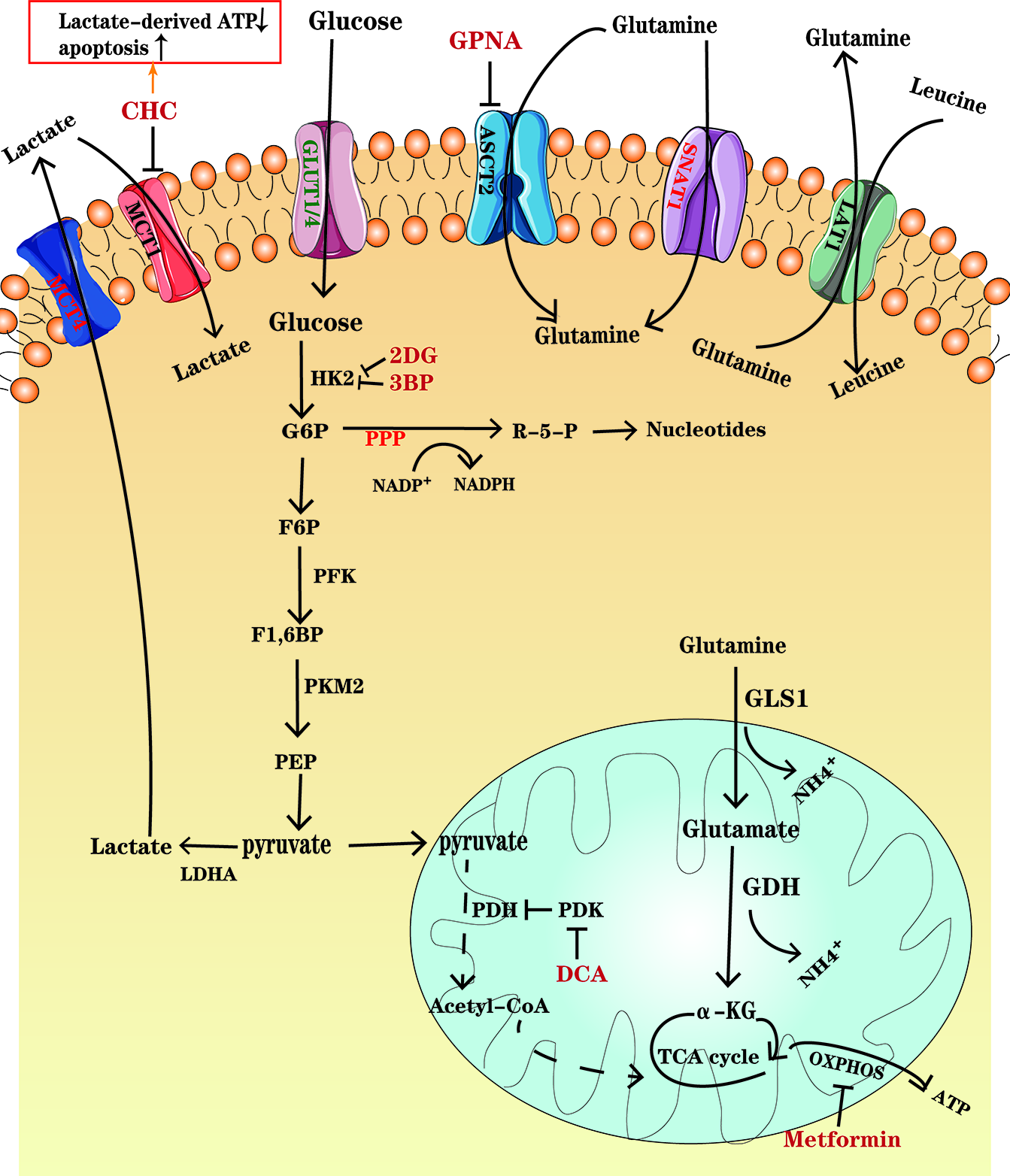
An overview of glucose and glutamine metabolism in MM cells. Glucose enters MM cells through GLUT1 and GLUT4 and is transformed into lactate depending on several key enzymes, including HK2, PFK, PMK2, and LDHA. Lactate is transported out of the cells through MCT4. Some cancer cells in normoxic areas may exploit lactate secreted from hypoxic cancer cells as an energy source and import it into cells by MCT1. PDK, the PDH inhibitor, is overexpressed in MM ells, which results in a decrease in pyruvate entering TCA cycle. Glutamine, as the major substrate for TCA cycle intermediates, enters the MM cells through transporters ASCT2 and SNAT1, in which ASCT2 is the major type. Glutamine can be exported out of the cell through antiporters LAT1 in exchange for other amino acids such as leucine. Glutamine is converted to glutamate by the enzyme GLS and further to a-KG by GDH, which fuel the TCA cycle. Drugs, such as CHC, GPNA, 2DG and so on, have been designed to inhibit the important enzymes and transporters and thus to disturb metabolism in MM cells. Further details are found in the main body of text.
Studies using myeloma cell lines have also shown the significance of glutamine in plasma cell metabolism. The growth of MM cells is limited by glutamine depletion (22). Glutamine, which is an important nitrogen donor for synthesis of amino acids and nucleotides, and the major substrate for TCA cycle intermediates, is indispensable to vigorous cell proliferation (23). Bortezomib-resistant cell lines have shown enhanced mitochondrial function fueled by glutamine rather than glucose. Therefore, interfering with glutamine metabolism has great potential in the treatment and overcoming drug resistance in MM (24). c-Myc is an important factor, which contributes to the tumorigenic phenotype of myeloma cells and increases glutamine transporters and glutaminase (GLS) expression to favor glutaminolysis (25). In glutaminolysis, glutamine, transported into the cells through transporters such as SLC1A5 (also known as ASCT2) and SNAT1 (26), is converted to glutamate by the enzyme GLS and further to α-ketoglutarate (α-KG) by glutamate dehydrogenase (GDH), which fuels the TCA cycle (27, 28) (Figure 1). A study has demonstrated that in the development of monoclonal immunoglobulin disease, CD138+ cells enhance the expression of glutamine transporters ASCT2, LAT1 and SNAT1. However, only ASCT2 inhibition in human myeloma cell lines (HMCLs) causes a marked decrease in glutamine uptake and cell growth (29). It has also been shown that HMCLs express GLS1 and are sensitive to its inhibition, whereas they negligibly express glutamine synthetase (GS) and GLS2 (29), which demonstrates that MM cells mainly depend on extracellular glutamine uptake.
In summary, myeloma cells appear to be more dependent on glycolysis and glutaminolysis than normal plasma cells. Therefore, targeting glycolysis and glutaminolysis could be used for the treatment of the disease. However, the compensatory enhancement of mitochondrial OXPHOS when glycolysis is inhibited in MM should also be noted.
Metabolic Changes in Immune Cells
While it is well known that cancer cells undergo metabolic reprogramming, as described above, there is a growing recognition that such metabolic alterations also occur in immune cells, which affects the function of immune cells and contribute to tumor immune escape (30–32).
The increasing emergence of evidence implies that polarized macrophages exhibit different metabolic patterns. M1 macrophages induce increased anabolic metabolism, such as glycolysis, PPP and fatty acid biosynthesis (FAS), whereas M2 macrophages induce OXPHOS. Specifically, aerobic glycolysis is promoted by toll like receptor (TLR) -induced signaling, which stabilizes hypoxia-inducible factor (HIF)-1α and boosts mammalian target of rapamycin (mTOR) activity (33). Similarly, T cells use distinct metabolic pathways in the process of activation and differentiation. Regarded as quiescent population, naïve and memory T cells mainly rely on OXPHOS and fatty acid β-oxidation (FAO) to produce ATP and show reduced rates of nutrient uptake and biosynthesis (34, 35). However, after activation, T-effector cells rapidly switch towards glycolysis and glutaminolysis to produce ATP and metabolic resources rapidly and meet the demand for daughter cell generation and suppress FAO (36). This change is orchestrated by T cell receptor (TCR) and CD28, which activate the phosphatidylinositol 3’-kinase (PI3K) -AKT-mTOR pathway and the transcription factors HIF-1α and c-myc, thus contributing to the up-regulation of metabolic enzymes, glucose and amino acid transporters (37). Likewise, activated NK cells also boost glycolysis (38). PI3K is required for multiple key aspects of NK cell biology, including maturation, homing, priming, and functioning (39).
Interestingly, some types of anti-tumor immune cells share many metabolic needs with MM cells, which leads to an energy inter-dependence that may cause metabolic competition between them. In general, cancer cells have better access to nutrients than immune cells, inducing hypoxic and acidic areas with nutrient depletion and lactate accumulation, thus promoting the development and spread of tumors and suppressing immune surveillance (40). For example, the lack of glucose and glutamine impairs TCR signaling, and is harmful for glycolysis and anti-tumor functions of T-effector cells. However, regulatory T (Treg) cells, preferring FAO to glycolysis, are able to survive under these conditions and act as immunosuppressive cells. Actually, the activation of AMP-activated protein kinase (AMPK), a sensor of nutrient stress, correlates with Treg cells expansion (41). Although the specific mechanism of metabolic switch in immune cells during MM development has not been thoroughly studied, this competition has led to the hypothesis that controlling the metabolism of MM cells may inhibit their growth by not only killing them directly, but also restoring the function of immune cells indirectly.
Autologous stem cell transplant (ASCT) and other novel anti-MM therapies have emerged in recent years. Such treatments have changed the composition and metabolic profile of MM cells and immune cells, particularly T cells. Compared with healthy donors, immune cells in relapsed/refractory MM (RRMM) patients increase FAO and mitochondrial respiration (42). This might result from MM cells creating a microenvironment of chronic inflammation in the bone marrow, particularly the increased production of IL-6, which drives the metabolic alteration of immune cells (43, 44). Anti-MM therapies and ASCT might disrupt the composition of immune cell populations and promote metabolic rewiring. In the TME, there are a significant reduction of CD4+ naïve T cells, an increase in CD8+ memory T cells and an increase in CD4+ T cells that overexpressed PD1, that might perturb the fitness in immunotherapies, particularly chimeric antigen receptor (CAR)-T therapy. Understanding these changes might guide ASCT and other immune therapies for MM. A study suggested that storing up T cells before ASCT rather than at relapse may offer a more effective CAR-T therapy (42).
Molecular Signaling Pathways and Transcription Factors Leading to Metabolic Reprogramming in MM
As explained above, MM and immune cells show complex metamorphosis in metabolism in the course of tumor progression. Below, we discuss a few crucial molecular signaling pathways and transcription factors that regulate metabolism in MM (Figure 2).
Figure 2
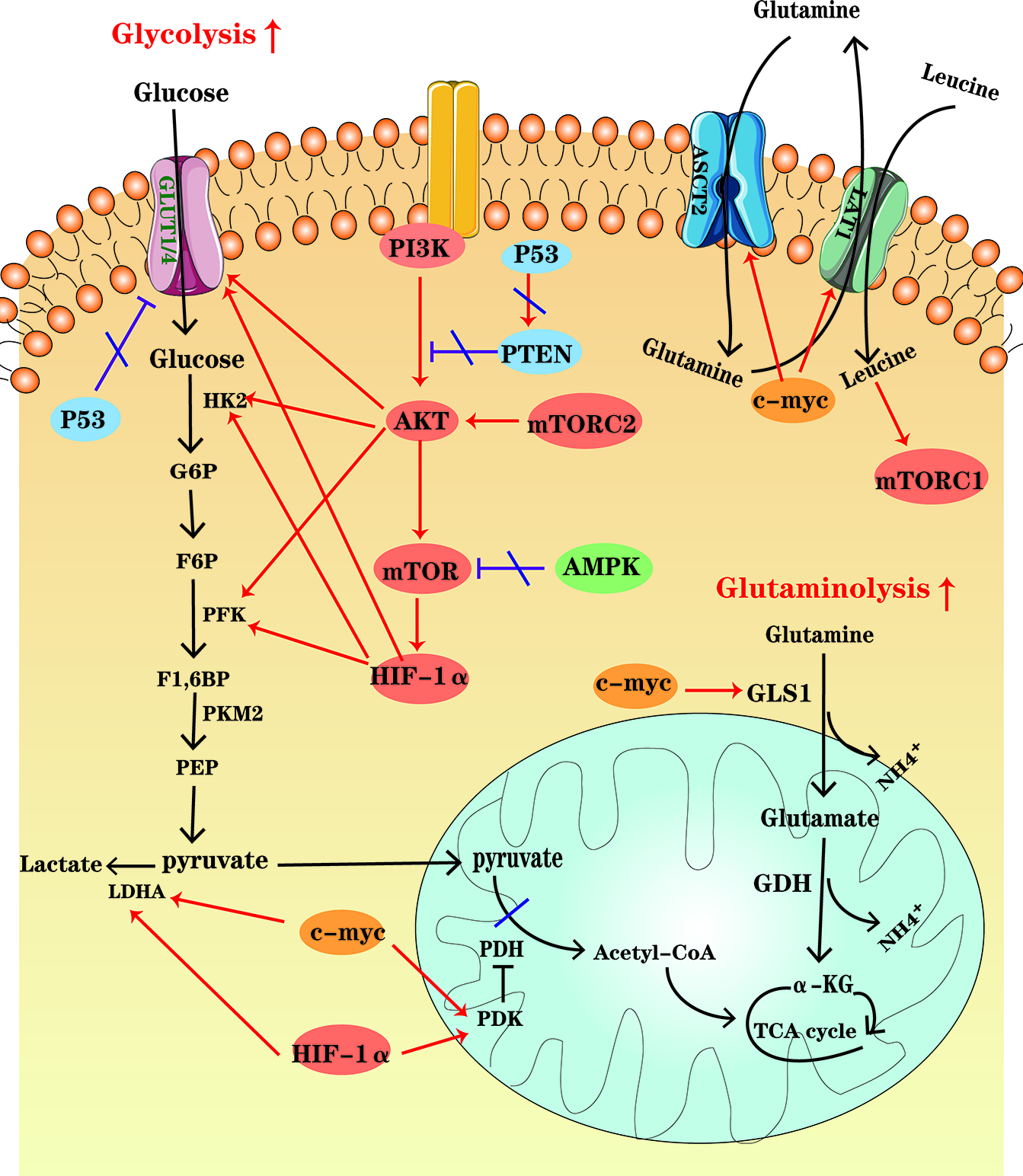
An overview of cellular metabolic pathways in MM cells. In MM cells, PI3K-AKT-mTOR pathway up-regulation, the high HIF-1 expression and the increased c-myc activity enhance glycolysis and glutaminolysis, which foster expression of key enzymes and transporters. At the same time, AMPK down-regulation, as well as P53 deficiency contribute to the metabolic reprogramming of MM cells in the glycolytic direction.
PI3K-AKT Pathway
The PI3K-Akt signaling pathway is activated by a variety of cellular stimuli, to regulate transcription, translation, proliferation, growth, survival and other basic cell functions. It has been found that PI3K-Akt signaling pathway is upregulated in MM, which is activated by some key cytokines associated with MM pathogenesis, such as interleukin (IL)-6 (45, 46) and stromal-derived factor (SDF)-1 (47, 48). Once it is activated, Akt can promote glycolysis by activating several glycolytic enzymes, including HK and PFK, and upregulating GLUTs (49). Akt is also a major trigger of mTOR activation. mTOR, the serine/threonine kinase, consists of mTORC1 and mTORC2 complexes. mTORC1 can drive the expression of several glycolytic enzymes such as PFK by increasing HIF-1α translation, promoting a shift in glucose metabolism from OXPHOS to glycolysis. The most important role of mTORC2 is likely to foster Akt phosphorylation and activation (50).
AMPK Pathway
Expressed in essentially all eukaryotic cells, AMPK is a sensor of cellular energy status. When the ratios of AMP : ATP and ADP : ATP increase, it indicates cellular energy stress and positively regulates signal transduction pathways that generate ATP, FAO and autophagy, and to negatively regulates ATP-consuming biosynthesis processes, including gluconeogenesis, FAS, and protein synthesis (51). Moreover, contributing to direct phosphorylation and activation of tuberous sclerosis complex 2 (TSC2), AMPK activation results in suppression of the mTOR signaling pathway (50). Tumor cells seem to downregulate AMPK under selective pressure, thereby limiting its inhibitory effect on cell growth and proliferation (52). Studies have shown that up-regulation of the AMPK signaling pathway may be beneficial for MM treatment. For instance, metformin can inhibit mitochondrial complex I activity, and thus cause an increase in AMP : ATP and ADP : ATP ratios, which activate AMPK indirectly and inhibit mTOR signaling, decreasing IL-6R expression (46).
Transcription Factors: HIF-1α, c-MYC, and P53
Composed of an inducibly expressed HIF-1α subunit and a constitutively expressed HIF-1β subunit, HIF-1 is a transcription factor known as a major regulator of hypoxia, which is a common condition in TME. Previous studies have shown that hypoxia is a characteristic of the specific bone marrow niche of MM and regions of hypoxia develop during MM progression (53, 54). Immunohistochemical staining has shown that HIF-1α is highly expressed in MM bone marrow. HIF-1α is an important regulator of cellular metabolism (55). HIF-1α induces the expression of glycolytic genes, including GLUT1, HK2, PFKFB3, LDHA, and pyruvate dehydrogenase kinase (PDK), and TCA cycle suppressors (56, 57). Inhibition of HIF-1α enables drug resistant MM cells to restore sensitivity (58, 59).
Members of the MYC family are important oncogenes involved in the development of malignant cells, and c-MYC activity is enhanced in MM (60). Actually, MYC regulates all genes involved in glycolysis and most genes in glutaminolysis (61). For instance, combining with HIF, c-MYC induces the expression of glucose transporters and enzymes such as LDHA to enhance glycolysis and PDK1 to inhibit pyruvate dehydrogenase (PDH) activity, thereby impairing mitochondrial function. Besides, c-MYC plays a significant role in inducing the major transporter and enzyme expression for cancer cell glutamine metabolism, including ASCT2 and GLS1 (4, 61).
The tumor suppressor P53 is altered in approximately 50% of human cancers (62). The aberrant TP53 gene, resulted from the deletion or mutation of the TP53 gene (TP53mut), is one of the key biomarkers of poor prognosis of MM (63). P53 represses glycolysis and thus favors OXPHOS by downregulating SLC2A1/4, which encodes the glucose transporters GLUT1 and GLUT4, and upregulating PTEN, a tumor suppressor gene, which inhibits PI3K-Akt pathway. In other words, P53 defect contributes to the metabolic rewiring of cancer cells in a more glycolytic direction (64).
The Impact of Cellular Metabolic Changes on the TME and Immunosurveillance
The growth of MM requires a large amount of oxygen and nutrients, and produces a lot of lactate. This metabolic shift shapes the TME towards a hypoxic, acidic, and nutritionally deficient one, which supports cancer proliferation and metastasis (65, 66). However such TME is extremely unfavorable for immune cells to exert their antitumor effects (5, 31, 67) (Figure 3).
Figure 3
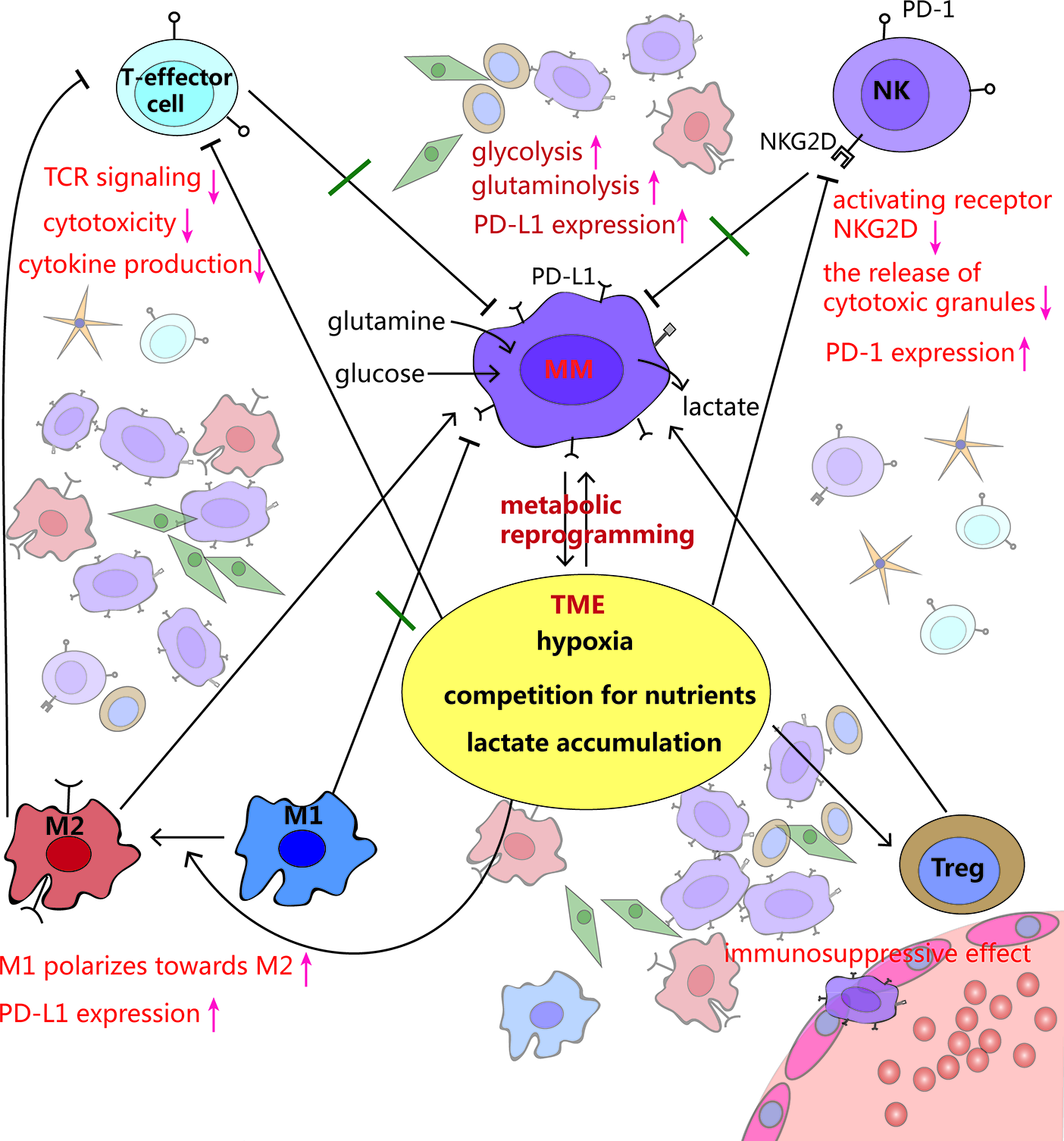
Crosstalk among MM cells, immune cells and tumor microenvironment (TME). MM cells rewire metabolism, such as increased glycolysis and glutaminolysis, to adapt to the hypoxic TME. Meanwhile, this makes the TME more anoxic, acidic and less nutritious, which inhibits T effector cells and NK cells, stimulates polarization from pro-inflammatory M1 macrophages toward a cancer-promoting M2 phenotype, as well as promotes Tregs development, leading to immune evasion.
Hypoxia
Since tumor aerobic glycolysis consumes a large amount of oxygen, the TME is often anoxic. Studies have indicated that overwhelming anti-tumor immunity is mostly due to hypoxia in the TME (68, 69).
Ikeda et al. have demonstrated that under low oxygen conditions, miR-210 is activated in MM cells (70). Remarkably, Noman et al. have shown that high level of miR-210, regulated by hypoxia, significantly blunts the susceptibility of tumor cell to cytotoxic T cell-mediated lysis by silencing of PTPN1, HOXA1, and TP53I11 in melanoma and lung cancer (71). Among them, PTPN1 and TP53I11 were found dramatically downregulated in MM (72).
NK cells have unique recognition function and cytotoxic function, that play crucial roles in immune monitoring and fighting against cancer cells (73). The receptors NKG2D and DNAM-1 are required for NK cell-mediated killing by identifying ligands RAE-1 and PVR expressed on MM cells, respectively (74, 75). However, the expression of these receptors decreases on NK cells derived from MM patients, resulting in impairment of NK cell function (76, 77). Several studies have suggested that hypoxia decreases NKG2D expression on NK cell surface, partially by hypoxic-tumor-derived micro-vesicles expressing immune modulatory factor TGF-β (78, 79). Similarly, NKG2D ligand-bearing micro-vesicles interfere with the function of NK cells, which kills cancer cells in a NKG2D-dependent manner (78).
Hypoxia, via HIF-1α, directly upregulates PD-L1 expression in various tumor cells by directly binding to the HRE in PD-L1 gene promoter, causing immunosuppressive TME (51, 80). The expression of PD-L1 on plasma cells (PCs) has been reported to be higher in MM patients than those of MGUS patients. Compared to PCs from healthy donors, PD-L1 expression on cells from minimal residual disease (MRD) positive MM patients is also upregulated (81, 82). Additionally, NK cells, derived from patients with MM, express PD-1 whereas normal NK cells do not. PD-1 inhibits NK cell cytotoxicity through the engagement of PD-L1/PD-1 pathway (83).
Lactate Accumulation
As a result of enhanced glycolysis, high concentration of lactate exerts its immunosuppressive function by suppressing lymphocyte proliferation, cytokine production and cytotoxic activity (36, 84). Studies have reported that tumor cell–derived lactate, which lowers the pH of TME to values below 6-6.5, is able to render T lymphocytes anergic due to reduced cytokine secretion including IFN-γ, IL-2, and TNF-α, decreased expression of TCR and IL-2 receptor CD25, as well as impairment of STAT5 and ERK activation after TCR binding. Buffering the pH to a physiologic level can reverse T cell anergy (66, 85).
Macrophages have great plasticity and exhibit different phenotypes when stimulated by the environment. The number of macrophages increased in the BMM of MM patients and support MM cell proliferation and survival through contact- dependent and –independent pro-proliferative molecule STAT3 activation (86). Mediated by G protein-coupled receptors (GPCRs), that sense the tumor acidic environment, macrophages express inducible cyclic AMP early repressor (ICER), which inhibits TLR-dependent NF-κB signaling, and thus prevents macrophages from polarizing toward a pro-inflammatory phenotype (87). Recently, Chen et al. (88) found that lactic acid can be linked to histones in the macrophage genome in the form of epigenetic modification of lactylation, regulating the switches of relevant genes, and promoting the transformation of macrophages from the pro-inflammatory and anticancer M1 type to the anti-inflammatory and cancer-promoting M2 type. Expressed lactate-induced ligands PD-L1, these M2-like tumor-associated macrophages (TAMs) also can blunt effector T cell function (89).
Targeting Metabolic Vulnerabilities in MM
As explained in the previous sections of this review, the metabolic pattern of MM cells is different from that of normal PCs, due to its eagerness for nutrients, especially glucose and glutamine. Importantly, the metabolic competition between MM and immune cells, together with the TME may exert an unfavorable effect on the functions of anti-tumor immune cells. Therefore, targeting cell metabolism by making use of the obvious distinctions between MM and normal cells, or integrating MM cell metabolism manipulation with immunotherapy seems to be an attractive anticancer strategy. However, the overlapping metabolic requirements between MM and immune cells should be taken into consideration.
Glucose metabolism is the focus of cancer metabolism research, including MM. Therefore, drugs have been designed to target glycolysis for cancer treatment. Most of them are proposed to inhibit key glycolytic enzymes (Figure 1).
HK2, the first interesting enzyme that catalyze glycolysis, is highly expressed in MM cells and is the most important target of 3-bromopyruvate (3BP), the small alkylating compound. Entering cells through MCTs, 3BP utilizes the distinct differences in MCT expression between tumor and normal cells, making cancer cells susceptible to it and decreasing the harm to normal cells with a proper concentration (90). In a few cancer researches, 3BP has been shown to be strikingly effective because of its multiple targets, enabling it to inhibit both glycolysis and mitochondrial OXPHOS (91). In addition to disruption of ATP production, 3BP can cause a remarkable decrease of reduced GSH level, as well as induce cell apoptosis, that were confirmed on MM cells (92).
2-Deoxyglucose (2DG), a glucose analog, is phosphorylated by HK2 and thus form a non-metabolizable substance, 2-DG-6-phosphate after entering cancer cells, which interferes with glycolysis (13). However, 2DG alone has not been reported to eradicate cancers in animals quickly. It has also been shown to decrease the secretion of cytokines and compromise the effector functions of T cells (93). In contrast, 2DG used to inhibit glycolysis has been demonstrated to favor the generation of memory T cells, whose number within tumors relates to a better overall survival (94–96). However, it has not been fully studied in MM. According to the current evidence, 3BP may has a better therapeutic effect compared to 2DG in MM.
Similar to HK2, PKM2 is another important enzyme of glycolysis. Mitosis gene A (NIMA)-related kinase 2 (NEK2), transcriptionally modulated by c-MYC in MM cells, can promote aerobic glycolysis by regulating PKM splicing and increasing the PKM2/PKM1 ratio. Myeloma patients with NEK2 and PKM2 overexpression have reduced event-free and overall survival (97). It has recently been revealed that P5091,a nonreversible USP7 inhibitor, can deplete NEK2 protein in vitro and in vivo, resulting in suppression of NEK2 downstream target, thus overcoming bortezomib resistance (98).
As a fuel for OXPHOS, lactate is incorporate into MM cells by MCT1. Inhibition of MCT1 by its inhibitor, such as α-cyano-4-hydroxycinnamic acid (CHC), or by gene-silencing technique, has proved to reduce lactate incorporation and lactate-derived ATP production significantly, as well as induce MM cell apoptosis. Additionally, the combination of CHC with dichloroacetate (DCA), a PDK inhibitor, can enhance MM cell death. DCA has also additive cytotoxicity when used in combination with bortezomib (16, 99). However, it promotes the formation of Treg cell (100).
Previous studies have demonstrated that inhibition of GLUT4 leads to apoptosis and cell arrest in MM cells. However, a portion of MM cells survive with medication targeting glycolysis, likely through the compensatory enhancement of mitochondrial OXPHOS (20). Metformin, universally described as an antihyperglycemic drug, can inhibit the activity of mitochondrial complex I (Figure 1), activate AMPK pathway, as well as downregulate mTOR (101), which induce MM cell autophagy and cell cycle arrest in G0/G1, rather than cell apoptosis (102). Combination of the GLUT4 inhibitor ritonavir with the complex I inhibitor metformin, which increases dependence on glycolytic ATP production, has been shown to overcome ritonavir resistance and effectively elicit cell apoptosis in both in vivo and vitro studies of MM (20). Additionally, metformin enhances cytotoxic T cell function through phosphorylation of PD-L1 at S195, which induces PD-L1 glycosylation and degradation in a breast cancer model (103).
Glutaminolysis enhancement is another metabolic change observed in MM, which is important for maintenance of energy production and redox control (28). A subset of cancer cells are resistance to treatment of glycometabolism inhibition, which may result from compensatory increased glutamine consumption, suggesting that inhibition of both glutaminolysis and glycolysis might be a feasible therapy (40).
Glutamine transporter ASCT2 can be inhibited by l-γ-glutamyl-p-nitroanilide (GPNA), thus decreasing glutamine uptake and suppressing cancer cell proliferation (104). As mentioned before, GS expression in MM cells is absent, whereas GLS expression is elevated. It has been shown that GLS inhibitor CB-839 in combination with proteasome inhibitors carfilzomib exerts a synergistic cytotoxic effect (105). Recently, Powell et al. (106) found that the compound JHU083, which blocks glutamine metabolism can disrupt tumor metabolism, paralyze the “Warburg effect” of tumors, reverse hypoxia, polyacid, and nutrient deficiency in the TME, and relieve the immunosuppression ability of tumor microenvironment. In addition, this small molecule can also reprogram the metabolism of T cells, directly activating T cells, increasing their longevity and promoting memory T cells formation. The researchers have named this treatment as “metabolic checkpoint” inhibitors. Since Powell et al. have studied only four cancer cell lines, colon cancer MC38, lymphoma el-4, colon cancer CT26, and melanoma B16, till date, MM treatment remains to be explored.
Conclusion
MM is an extremely heterogeneous and complex disease and is incurable in spite of great advances in therapeutic strategies. Numerous studies on metabolic reprogramming have revealed various targets in the process of glucose and glutamine metabolism. Metabolic reprogramming in MM creates a hypoxic, acidic, and nutritionally deficient TME that inhibits normal cell growth and immune cell function. The overlapping metabolic phenotype between MM and immune cells must be identified. Targeting metabolic pathways and promoting the function of immune cells could be a novel strategy for the cure of MM.
Funding
This work was supported by the National Natural Science Foundation of China (81400156).
Statements
Author contributions
SW and D-HY formulated the topic of the review. SW, HK, and JK drafted the manuscript. D-HY edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; F1,6BP, fructose-1,6-biphosphate; PEP, phosphoenolpyruvate; TGF-β, transforming growth factor beta; PD-1, programmed cell death-1; PD-L1, programmed cell death-ligand 1.
References
1
TamuraH. Immunopathogenesis and immunotherapy of multiple myeloma. Int J Hematol (2018) 107(3):278–85. doi: 10.1007/s12185-018-2405-7
2
D’AgostinoMBertaminiLOlivaSBoccadoroMGayF. Pursuing a Curative Approach in Multiple Myeloma: A Review of New Therapeutic Strategies. Cancers (Basel) (2019) 11(12). doi: 10.3390/cancers11122015
3
HanahanDWeinbergRA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
4
BiswasSK. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity (2015) 43(3):435–49. doi: 10.1016/j.immuni.2015.09.001
5
WegielBVuerichMDaneshmandiSSethP. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front Oncol (2018) 8:284. doi: 10.3389/fonc.2018.00284
6
Vander HeidenMGCantleyLCThompsonCB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
7
CavoMTerposENanniCMoreauPLentzschSZweegmanSet al. Role of (18)F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: a consensus statement by the International Myeloma Working Group. Lancet Oncol (2017) 18(4):e206–17. doi: 10.1016/S1470-2045(17)30189-4
8
TakahashiMESMosciCSouzaEMBrunettoSQEtchebehereESantosAOet al. Proposal for a Quantitative 18F-FDG PET/CT Metabolic Parameter to Assess the Intensity of Bone Involvement in Multiple Myeloma. Sci Rep Uk (2019) 9(1). doi: 10.1038/s41598-019-52740-2
9
D’SouzaLBhattacharyaD. Plasma cells:You are what you eat. Immunol Rev (2019) 288(1):161–77. doi: 10.1111/imr.12732
10
MuecklerMThorensB. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med (2013) 34(2-3):121–38. doi: 10.1016/j.mam.2012.07.001
11
MatsumotoTJimiSMigitaKTakamatsuYHaraS. Inhibition of glucose transporter 1 induces apoptosis and sensitizes multiple myeloma cells to conventional chemotherapeutic agents. Leukemia Res (2016) 41:103–10. doi: 10.1016/j.leukres.2015.12.008
12
McBrayerSKChengJCSinghalSKrettNLRosenSTShanmugamM. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood (2012) 119(20):4686–97. doi: 10.1182/blood-2011-09-377846
13
ElACDe VeirmanKMaesKDe BruyneEMenuE. Metabolic Features of Multiple Myeloma. Int J Mol Sci (2018) 19(4):1200. doi: 10.3390/ijms19041200
14
StuaniLSabatierMSarryJ. Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Biol (2019) 17(1). doi: 10.1186/s12915-019-0670-4
15
SorianoGPBesseLLiNKrausMBesseAMeeuwenoordNet al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia (2016) 30(11):2198–207. doi: 10.1038/leu.2016.102
16
FujiwaraSWadaNKawanoYOkunoYKikukawaYEndoSet al. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp Hematol Oncol (2015) 4(1):12. doi: 10.1186/s40164-015-0008-z
17
SonveauxPVégranFSchroederTWerginMCVerraxJRabbaniZNet al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest (2008) 118(12):3930–42. doi: 10.1172/JCI36843
18
FujiwaraSWadaNKawanoYOkunoYKikukawaYEndoSet al. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp Hematol Oncol (2015) 4:12. doi: 10.1186/s40164-015-0008-z
19
de la Cruz-LópezKGCastro-MuñozLJReyes-HernándezDOGarcía-CarrancáAManzo-MerinoJ. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front Oncol (2019) 9:1143. doi: 10.3389/fonc.2019.01143
20
Dalva-AydemirSBajpaiRMartinezMAdekolaKUAKandelaIWeiCet al. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin Cancer Res (2015) 21(5):1161–71. doi: 10.1158/1078-0432.CCR-14-1088
21
MarleinCRPiddockREMistryJJZaitsevaLHellmichCHortonRHet al. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res (2019) 79(9):2285–97. doi: 10.1158/0008-5472.CAN-18-0773
22
GiulianiNChiuMBolzoniMAccardiFBianchiMGToscaniDet al. The potential of inhibiting glutamine uptake as a therapeutic target for multiple myeloma. Expert Opin Ther Targets (2017) 21(3):231–4. doi: 10.1080/14728222.2017.1279148
23
YangCKoBHensleyCTJiangLWastiATKimJet al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell (2014) 56(3):414–24. doi: 10.1016/j.molcel.2014.09.025
24
ZaalEABerkersCR. The Influence of Metabolism on Drug Response in Cancer. Front Oncol (2018) 8:500. doi: 10.3389/fonc.2018.00500
25
HirscheyMDDeBerardinisRJDiehlAMEDrewJEFrezzaCGreenMFet al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol (2015) 35:S129–50. doi: 10.1016/j.semcancer.2015.10.002
26
WiseDRThompsonCB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
27
JinLAlesiGNKangS. Glutaminolysis as a target for cancer therapy. Oncogene (2016) 35(28):3619–25. doi: 10.1038/onc.2015.447
28
AltmanBJStineZEDangCV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
29
BolzoniMChiuMAccardiFVescoviniRAiroldiIStortiPet al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood (2016) 128(5):667–79. doi: 10.1182/blood-2016-01-690743
30
RennerKSingerKKoehlGEGeisslerEKPeterKSiskaPJet al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol (2017) 8:248. doi: 10.3389/fimmu.2017.00248
31
ChangCQiu JOSullivanDBuckMDNoguchiTCurtisJDet al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
32
ShiRTangYQMiaoH. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm (2020) 1(1):47–68. doi: 10.1002/mco2.6
33
HoPLiuP. Metabolic communication in tumors: a new layer of immunoregulation for immune evasion. J Immunother Cancer (2016) 4(1). doi: 10.1186/s40425-016-0109-1
34
ZhangLRomeroP. Metabolic Control of CD8+ T Cell Fate Decisions and Antitumor Immunity. Trends Mol Med (2018) 24(1):30–48. doi: 10.1016/j.molmed.2017.11.005
35
MichalekRDRathmellJC. The metabolic life and times of a T-cell. Immunol Rev (2010) 236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x
36
YinZBaiLLiWZengTTianHCuiJ. Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy. J Exp Clin Canc Res (2019) 38(1). doi: 10.1186/s13046-019-1409-3
37
SiskaPJRathmellJC. T cell metabolic fitness in antitumor immunity. Trends Immunol (2015) 36(4):257–64. doi: 10.1016/j.it.2015.02.007
38
GardinerCMFinlayDK. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front Immunol (2017) 8:367. doi: 10.3389/fimmu.2017.00367
39
MaceEM. Phosphoinositide-3-Kinase Signaling in Human Natural Killer Cells: New Insights from Primary Immunodeficiency. Front Immunol (2018) 9:445. doi: 10.3389/fimmu.2018.00445
40
AfonsoJSantosLLLongatto-FilhoABaltazarF. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat Rev Urol (2020) 17(2):77–106. doi: 10.1038/s41585-019-0263-6
41
AllisonKECoomberBLBridleBW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology (2017) 152(2):175–84. doi: 10.1111/imm.12777
42
CookeREQuinnKMQuachHHarrisonSPrinceHMKoldejRet al. Conventional Treatment for Multiple Myeloma Drives Premature Aging Phenotypes and Metabolic Dysfunction in T Cells. Front Immunol (2020) 11:2153. doi: 10.3389/fimmu.2020.02153
43
HarmerDFalankCReaganMR. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front Endocrinol (2019) 9:788. doi: 10.3389/fendo.2018.00788
44
KlarquistJChitrakarAPennockNKilgoreABlainTZhengCet al. Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci Immunol (2018) 3(27). doi: 10.1126/sciimmunol.aas9822
45
HuJHuW. Targeting signaling pathways in multiple myeloma: Pathogenesis and implication for treatments. Cancer Lett (2018) 414:214–21. doi: 10.1016/j.canlet.2017.11.020
46
MishraAKDingliD. Metformin inhibits IL-6 signaling by decreasing IL-6R expression on multiple myeloma cells. Leukemia (2019) 33(11):2695–709. doi: 10.1038/s41375-019-0470-4
47
PiddockRBowlesKRushworthS. The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma. Cancers (2017) 9(12):29. doi: 10.3390/cancers9040029
48
BouyssouJMCGhobrialIMRoccaroAM. Targeting SDF-1 in multiple myeloma tumor microenvironment. Cancer Lett (2016) 380(1):315–8. doi: 10.1016/j.canlet.2015.11.028
49
DemelHFeuereckerBPiontekGSeidlCBlechertBPickhardAet al. Effects of topoisomerase inhibitors that induce DNA damage response on glucose metabolism and PI3K/Akt/mTOR signaling in multiple myeloma cells. Am J Cancer Res (2015) 5(5):1649.
50
SaxtonRASabatiniDM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
51
Riera-DomingoCAudigeAGranjaSChengWCHoPCBaltazarFet al. Immunity, Hypoxia, and Metabolism-the Menage a Trois of Cancer: Implications for Immunotherapy. Physiol Rev (2020) 100(1):1–102. doi: 10.1152/physrev.00018.2019
52
HardieDG. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin Cancer Res (2015) 21(17):3836–40. doi: 10.1158/1078-0432.CCR-14-3300
53
AzabAKHuJQuangPAzabFPitsillidesCAwwadRet al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood (2012) 119(24):5782–94. doi: 10.1182/blood-2011-09-380410
54
ImaiTMuzBYehCHYaoJZhangRAzabAKet al. Direct measurement of hypoxia in a xenograft multiple myeloma model by optical-resolution photoacoustic microscopy. Cancer Biol Ther (2017) 18(2):101–5. doi: 10.1080/15384047.2016.1276137
55
HuJHandisidesDRVan ValckenborghEDe RaeveHMenuEVandeBIet al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood (2010) 116(9):1524–7. doi: 10.1182/blood-2010-02-269126
56
SemenzaGL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123(9):3664–71. doi: 10.1172/JCI67230
57
HayN. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer (2016) 16(10):635–49. doi: 10.1038/nrc.2016.77
58
MaisoPHuynhDMoschettaMSaccoAAljawaiYMishimaYet al. Metabolic Signature Identifies Novel Targets for Drug Resistance in Multiple Myeloma. Cancer Res (2015) 75(10):2071–82. doi: 10.1158/0008-5472.CAN-14-3400
59
RizzieriDPaulBKangY. Metabolic alterations and the potential for targeting metabolic pathways in the treatment of multiple myeloma. J Cancer Metastasis Treat (2019) 2019. doi: 10.20517/2394-4722.2019.05
60
HolienTVåtsveenTKHellaHWaageASundanA. Addiction to c-MYC in multiple myeloma. Blood (2012) 120(12):2450–3. doi: 10.1182/blood-2011-08-371567
61
StineZEWaltonZEAltmanBJHsiehALDangCV. MYC, Metabolism, and Cancer. Cancer Discov (2015) 5(10):1024–39. doi: 10.1158/2159-8290.CD-15-0507
62
FlyntEBishtKSridharanVOrtizMTowficFThakurtaA. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells-Basel (2020) 9(2):287. doi: 10.3390/cells9020287
63
PetrackovaAMinarikJSedlarikovaLLibigerovaTHamplovaAKrhovskaPet al. Diagnostic deep-targeted next-generation sequencing assessment ofTP53 gene mutations in multiple myeloma from the whole bone marrow. Brit J Haematol (2020) 189:e122–e5. doi: 10.1111/bjh.16547
64
LacroixMRiscalRArenaGLinaresLKLe CamL. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol Metab (2020) 33:2–22. doi: 10.1016/j.molmet.2019.10.002
65
ScharpingNDelgoffeG. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines (2016) 4(4):46. doi: 10.3390/vaccines4040046
66
CassimSPouyssegurJ. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int J Mol Sci (2020) 21(1):157. doi: 10.3390/ijms21010157
67
Overacre-DelgoffeAEVignaliDAA. Treg Fragility: A Prerequisite for Effective Antitumor Immunity? Cancer Immunol Res (2018) 6(8):882–7. doi: 10.1158/2326-6066.CIR-18-0066
68
NomanMZHasmimMLequeuxAXiaoMDuhemCChouaibSet al. Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells-Basel (2019) 8(9):1083. doi: 10.3390/cells8091083
69
ChouaibSMessaiYCouveSEscudierBHasmimMNomanMZ. Hypoxia Promotes Tumor Growth in Linking Angiogenesis to Immune Escape. Front Immunol (2012) 3:21. doi: 10.3389/fimmu.2012.00021
70
IkedaSKitadateAAbeFSaitohHMichishitaYHatanoYet al. Hypoxia-inducible microRNA-210 regulates the DIMT1-IRF4 oncogenic axis in multiple myeloma. Cancer Sci (2017) 108(4):641–52. doi: 10.1111/cas.13183
71
NomanMZBuartSRomeroPKetariSJanjiBMariBet al. Hypoxia-Inducible miR-210 Regulates the Susceptibility of Tumor Cells to Lysis by Cytotoxic T Cells. Cancer Res (2012) 72(18):4629–41. doi: 10.1158/0008-5472.CAN-12-1383
72
JankerLMayerRLBileckAKreutzDMaderJCUtpatelKet al. Metabolic, Anti-apoptotic and Immune Evasion Strategies of Primary Human Myeloma Cells Indicate Adaptations to Hypoxia. Mol Cell Proteomics (2019) 18(5):936–53. doi: 10.1074/mcp.RA119.001390
73
ArtisDSpitsH. The biology of innate lymphoid cells. Nature (2015) 517(7534):293–301. doi: 10.1038/nature14189
74
AntonangeliFSorianiARicciBPonzettaABenigniGMorroneSet al. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. Oncoimmunology (2016) 5(10):e1218105. doi: 10.1080/2162402X.2016.1218105
75
El-SherbinyYMMeadeJLHolmesTDMcGonagleDMackieSLMorganAWet al. The Requirement for DNAM-1, NKG2D, and NKp46 in the Natural Killer Cell-Mediated Killing of Myeloma Cells. Cancer Res (2007) 67(18):8444–9. doi: 10.1158/0008-5472.CAN-06-4230
76
PaulBKangSZhengZKangY. The challenges of checkpoint inhibition in the treatment of multiple myeloma. Cell Immunol (2018) 334:87–98. doi: 10.1016/j.cellimm.2018.10.003
77
GuillereyCNakamuraKVuckovicSHillGRSmythMJ. Immune responses in multiple myeloma: role of the natural immune surveillance and potential of immunotherapies. Cell Mol Life Sci (2016) 73(8):1569–89. doi: 10.1007/s00018-016-2135-z
78
NomanMZJanjiBBerchemGChouaibS. miR-210 and hypoxic microvesicles: Two critical components of hypoxia involved in the regulation of killer cells function. Cancer Lett (2016) 380(1):257–62. doi: 10.1016/j.canlet.2015.10.026
79
DuanSGuoWXuZHeYLiangCMoYet al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer (2019) 18(1). doi: 10.1186/s12943-019-0956-8
80
BarsoumIBSmallwoodCASiemensDRGrahamCH. A Mechanism of Hypoxia-Mediated Escape from Adaptive Immunity in Cancer Cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
81
PaivaBAzpilikuetaAPuigNOcioEMSharmaROyajobiBOet al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia (2015) 29(10):2110–3. doi: 10.1038/leu.2015.79
82
JelinekTPaivaBHajekR. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front Immunol (2018) 9:2431. doi: 10.3389/fimmu.2018.02431
83
BensonDMBakanCEMishraAHofmeisterCCEfeberaYBecknellBet al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti–PD-1 antibody. Blood (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
84
FischerKHoffmannPVoelklSMeidenbauerNAmmerJEdingerMet al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
85
CalcinottoAFilipazziPGrioniMIeroMDe MilitoARicupitoAet al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72(11):2746–56. doi: 10.1158/0008-5472.CAN-11-1272
86
De BeuleNDe VeirmanKMaesKDe BruyneEMenuEBreckpotKet al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J Pathol (2017) 241(4):534–46. doi: 10.1002/path.4860
87
BohnTRappSLutherNKleinMBruehlTKojimaNet al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol (2018) 19(12):1319–29. doi: 10.1038/s41590-018-0226-8
88
ZhangDTangZHuangHZhouGCuiCWengYet al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
89
SethPCsizmadiaEHedblomAVuerichMXieHLiMet al. Deletion of lactate dehydrogenase-A in myeloid cells triggers antitumor immunity. Cancer Res (2017) 77(13):3632–43. doi: 10.1158/0008-5472.CAN-16-2938
90
LisPDylągMNiedźwieckaKKoYPedersenPGoffeauAet al. The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate. Molecules (2016) 21(12):1730. doi: 10.3390/molecules21121730
91
FanTSunGSunXZhaoLZhongRPengY. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers (2019) 11(3):317. doi: 10.3390/cancers11030317
92
NiedźwieckaKDylągMAugustyniakDMajkowska-SkrobekGCal-BąkowskaMKoYHet al. Glutathione may have implications in the design of 3-bromopyruvate treatment protocols for both fungal and algal infections as well as multiple myeloma. Oncotarget (2016) 7(40):65614. doi: 10.18632/oncotarget.11592
93
RennerKGeiselhöringerAFanteMBrussCFärberSSchönhammerGet al. Metabolic plasticity of human T cells: Preserved cytokine production under glucose deprivation or mitochondrial restriction, but 2-deoxy-glucose affects effector functions. Eur J Immunol (2015) 45(9):2504–16. doi: 10.1002/eji.201545473
94
SukumarMLiuJJiYSubramanianMCromptonJGYuZet al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
95
PanYKupperTS. Metabolic Reprogramming and Longevity of Tissue-Resident Memory T Cells. Front Immunol (2018) 9:1347. doi: 10.3389/fimmu.2018.01347
96
JamesonSCMasopustD. Understanding Subset Diversity in T Cell Memory. Immunity (2018) 48(2):214–26. doi: 10.1016/j.immuni.2018.02.010
97
GuZXiaJXuHFrechITricotGZhanF. NEK2 Promotes Aerobic Glycolysis in Multiple Myeloma Through Regulating Splicing of Pyruvate Kinase. J Hematol Oncol (2017) 10(1). doi: 10.1186/s13045-017-0392-4
98
Franqui-MachinRHaoMBaiHGuZZhanXHabelhahHet al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J Clin Invest (2018) 128(7):2877–93. doi: 10.1172/JCI98765
99
FujiwaraSKawanoYYukiHOkunoYNosakaKMitsuyaHet al. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Brit J Cancer (2013) 108(1):170–8. doi: 10.1038/bjc.2012.527
100
KouidhiSBen AyedFBenammar ElgaaiedA. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front Immunol (2018) 9:353. doi: 10.3389/fimmu.2018.00353
101
AmaralISilvaCCorreia-BrancoAMartelF. Effect of metformin on estrogen and progesterone receptor-positive (MCF-7) and triple-negative (MDA-MB-231) breast cancer cells. BioMed Pharmacother (2018) 102:94–101. doi: 10.1016/j.biopha.2018.03.008
102
WangYXuWYanZZhaoWMiJLiJet al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Canc Res (2018) 37(1). doi: 10.1186/s13046-018-0731-5
103
ChaJYangWXiaWWeiYChanLLimSet al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol Cell (2018) 71(4):606–20. doi: 10.1016/j.molcel.2018.07.030
104
ChenLCuiH. Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach. Int J Mol Sci (2015) 16(9):22830–55. doi: 10.3390/ijms160922830
105
ThompsonRMDytfeldDReyesLRobinsonRMSmithBManevichYet al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget (2017) 8(22):35863–76. doi: 10.18632/oncotarget.16262
106
LeoneRDZhaoLEnglertJMSunIOhMSunIet al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (2019) v2588:366. doi: 10.1126/science.aav2588
Summary
Keywords
metabolic reprogramming, multiple myeloma, signaling pathways, tumor microenvironment, immune cell dysfunction
Citation
Wu S, Kuang H, Ke J, Pi M and Yang D-H (2021) Metabolic Reprogramming Induces Immune Cell Dysfunction in the Tumor Microenvironment of Multiple Myeloma. Front. Oncol. 10:591342. doi: 10.3389/fonc.2020.591342
Received
04 August 2020
Accepted
30 November 2020
Published
15 January 2021
Volume
10 - 2020
Edited by
Rentian Feng, University of Pittsburgh, United States
Reviewed by
Yogesh Chawla, Mayo Clinic, United States; Peter J. Siska, University Medical Center Regensburg, Germany
Updates
Copyright
© 2021 Wu, Kuang, Ke, Pi and Yang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaojie Wu, Wshj510515@163.com; Dong-Hua Yang, Yangd1@stjohns.edu
†These authors have contributed equally to this work
This article was submitted to Cancer Molecular Targets and Therapeutics, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.