Abstract
The human TP53 locus, located on the short arm of chromosome 17, encodes a tumour suppressor protein which functions as a tetrameric transcription factor capable of regulating the expression of a plethora of target genes involved in cell cycle arrest, apoptosis, DNA repair, autophagy, and metabolism regulation. TP53 is the most commonly mutated gene in human cancer cells and TP53 germ-line mutations are responsible for the cancer-prone Li-Fraumeni syndrome. When mutated, the TP53 gene generally presents missense mutations, which can be distributed throughout the coding sequence, although they are found most frequently in the central DNA binding domain of the protein. TP53 mutations represent an important prognostic and predictive marker in cancer. The presence of a TP53 mutation does not necessarily imply a complete P53 inactivation; in fact, mutant P53 proteins are classified based on the effects on P53 protein function. Different models have been used to explore these never-ending facets of TP53 mutations, generating abundant experimental data on their functional impact. Here, we briefly review the studies analysing the consequences of TP53 mutations on P53 protein function and their possible implications for clinical outcome. The focus shall be on Chronic Lymphocytic Leukemia (CLL), which also has generated considerable discussion on the role of TP53 mutations for therapy decisions.
Introduction
The human tumour suppressor gene TP53, located at 17p13.1 locus, encodes a 393 amino acid-long protein, which was discovered in the 80s of last Century within a complex containing the viral SV40 large T antigen (1–3). Initially misclassified as an oncogene, because of the isolation of mutant cDNA clones capable of inducing cell transformation, the wild type (WT) TP53 gene was eventually classified as tumour suppressor, upon the definite demonstration of its capacity of inhibiting the growth and the oncogenic transformation of cells in culture (4). Concurrently, somatic TP53 mutations were identified in tumours (5, 6) and germ-line TP53 mutations were described in the Li-Fraumeni syndrome (LFS), the well-known hereditary cancer predisposition disorder (7).
The P53 protein consists of different functional domains including mainly a N-terminal transactivation domain (residues 1–61, TAD), a central DNA binding domain (residues 94–290, DBD), an oligomerization domain (residues 325–356, OD) and a C-terminal domain that regulates the DNA binding (residues 357–393, CTD) (Figure 1A) (10). While the TAD domain interacts with components of the transcription machinery, the OD and the DBD domains are necessary for the formation of the P53 tetramer which interacts with specific DNA target sequences, called P53 response elements (P53 REs) that are comprised of two degenerate decameric sequences [Pu (Purine)-Pu-Pu-C-A/T-AT-G-Py (Pyrimidine)-Py-Py] separated by a variable spacer (11). To complicate the scenario further, different isoforms of P53, resulting from the usage of alternative promoters and splicing sites, or alternative initiation sites of translation, have been recently described (12, 13).
Figure 1
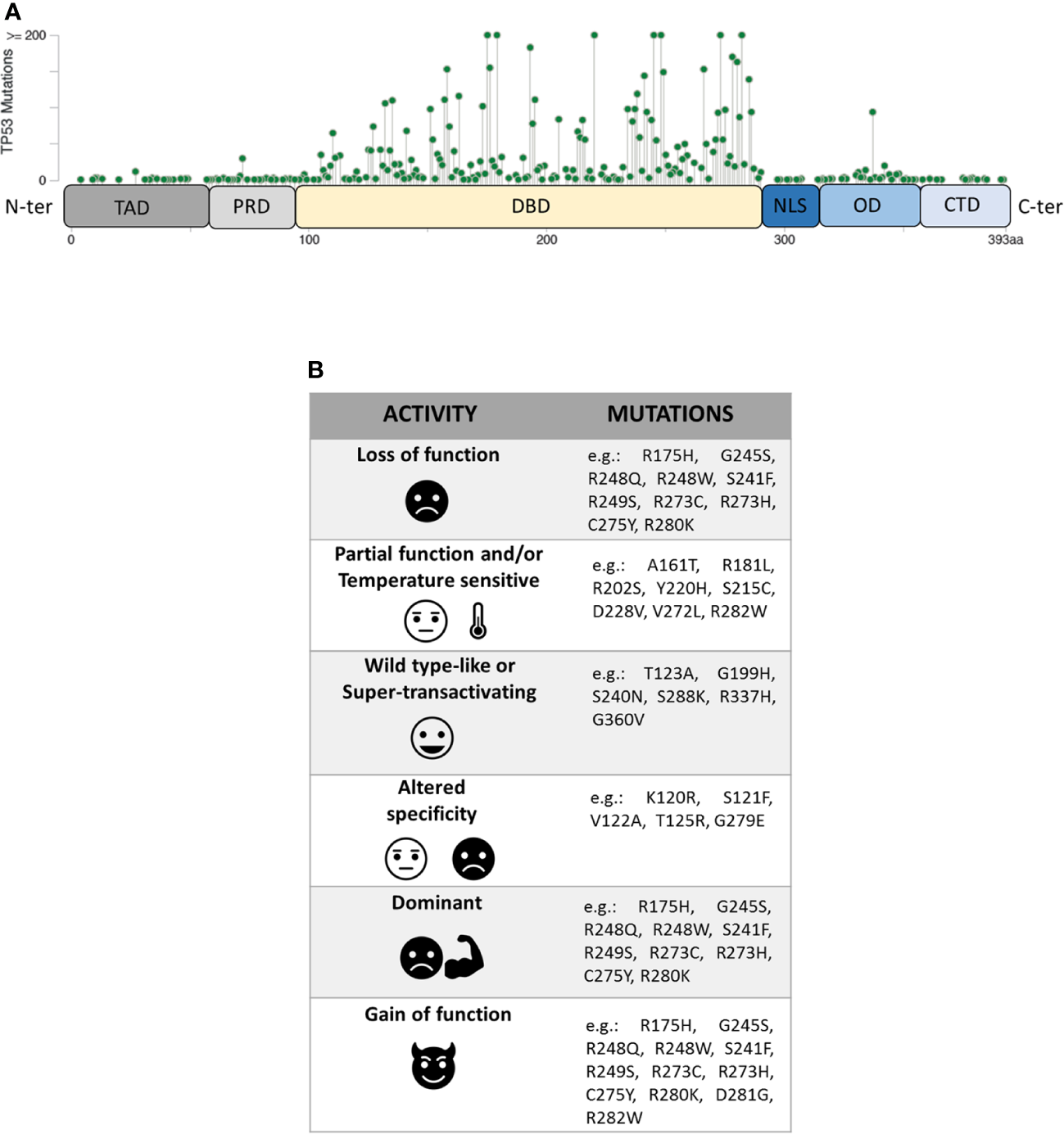
(A) Domain organization of P53 protein (TAD, transactivation domain; PRD, proline-rich domain; DBD, DNA-binding domain; NLS, nuclear localization signal; OD, oligomerization domain; CTD, C-terminal domain) and distribution of TP53 missense mutations from the cBioPortal online tool (TCGA) (8, 9). Missense mutations location (green signs) throughout the P53 protein is shown according to the frequency and the position of the amino acid hit. (B) Heterogeneity of mutants P53 with examples of the corresponding TP53 mutations.
P53 is a transcription factor (TF) which can be induced by endogenous and exogenous stresses (e.g. oncogenes and UV radiation); these stresses activate signals, which operate via post-translational modifications on P53 protein (e.g. phosphorylations, acetylations), and cause release of P53 from the mortal embrace with MDM2 protein, eventually leading to P53 activation (14). P53 protein can then enter the nucleus where it induces the expression of a plethora of target genes (15). However, increasing observations are reporting certain “non-transcriptional functions” of P53, that can contribute to tumour suppression activity (16).
TP53 Mutations Heterogeneity
Unlike other tumour suppressors, the TP53 gene is mostly altered by missense mutations, mainly involving the portion coding for the DBD domain of the P53 protein. Within this region, eight amino-acid substitutions (i.e. R175H, G245S, R248Q, R248W, R249S, R273H, R273S, and R282W), called hotspot mutations, characterize ~27% of all mutant P53 proteins identified in human cancers. However, the spectrum of missense mutations is extremely broad (Figure 1A) (8, 9, 17, 18) and varies in the different tumour types.
Mutant P53 proteins have been primarily categorized as “contact” or “structural”, depending on whether substituted amino-acid was directly involved in the interaction with DNA (e.g. R273H) or caused a global effect on the protein structure (e.g. R175H) (19, 20). Over the last 25 years, a large amount of experimental data on the functional impact of different amino-acid substitutions has been generated and different models have been used to explore the never-ending facets of the corresponding mutant proteins. One of these models is the yeast S. cerevisiae, exploiting the fact that P53 can act as TF also in this organism by binding P53 REs located upstream a reporter gene (21, 22). The assay, originated as a Functional Analysis of Separate Alleles in Yeast to study the TP53 status in somatic tumour and blood cells, has been unceasingly modified and upgraded in order to explore additional functions of P53 mutants (23–26). In 2003 Ishioka’s group tested the functionality of more than 2,000 different P53 single amino-acid substitutions expressed in yeast (27). Their work, along with that of others (23–26), led to a re-classification of the TP53 mutations based on their effects on P53 function generating the following categories: i) loss of function (LOF), ii) partial function and/or temperature sensitive (PF, TS), iii) WT-like or super-transactivating (WT-L, ST), iv) with altered specificity (AS) (i.e. active or partially active on some targets but inactive on others), and v) dominant-negative (DN), based on the ability to inhibit WT protein in a heterozygous condition. However, mutant P53 proteins might be classified also as gain of function (GOF) related to the acquisition of novel oncogenic activities, not shared with the WT protein (Figure 1B). This latter property is mainly based on the ability of the mutant P53 to interact with other TFs or with chromatin-modifying complexes, altering the cellular transcriptional profile (28–32). Therefore, mutant P53 proteins appear to constitute a functional rainbow (Figure 1B) (33).
Recently, two seminal papers confirmed the relevance of the heterogeneity of mutants P53 in terms of Relative Fitness Score (RFS) in in vitro cultures (34, 35). RFS has been used as an indicator of the functional impact of TP53 mutations in terms of selective growth; specifically, a high RFS indicates a higher fitness of the TP53 variant with preferential expansion within mixed cultured cells in competition with other TP53 variants, whereas a low RFS pinpoints preferential depletion in the same experimental condition. Moreover, Kotler et al. (34) showed that the loss of the anti-proliferative function of WT P53 largely correlates with the occurrence of cancer-associated TP53 mutations, and that selective GOF properties may further favour specific mutants P53 in vivo. An enhanced cellular fitness was also confirmed in association with the loss of WT P53 function or the DN effect associated to specific TP53 mutations (35).
Impact of TP53 Mutations Heterogeneity: From Mice to LFS Clinical Condition
A clear evidence of the different impact of TP53 mutations at the organism level came from the studies with Trp53 knock-in mice. In mice, the introduction of the R172H mutation (corresponding to the human R175H hotspot) at the germ-line level generates a tumor phenotype similar to that observed in Trp53 null mice, but with a much higher rate of metastasis (36). The GOF activity of R172H mutation was demonstrated to be associated with the functional inactivation of P63 and P73 TFs (37). In contrast, the R172P mutation in mice (corresponding to the human R175P, PF mutation) caused a delayed tumorigenesis rate with absence of chromosomal instability (38). These findings have been paralleled by observations made in patients affected by LFS, showing that the age of first tumor onset and the spectrum of observed tumors are dependent on the type of TP53 mutation (39).
The associations of the genotype with clinical outcome was explored in carriers of TP53 germ-line mutations also using a functional classification of the mutant P53 based on the quantification of their transactivation potential and DN effect in a yeast reporter assay. The analyses revealed that P53 proteins severely deficient in transactivation capability were more frequently associated with more severe cancer proneness syndromes (e.g. LFS) (40), whereas a further classification of these alleles, based on DN effects, did not distinguish clinical subclasses (41).
Impact of TP53 Mutations Heterogeneity in Somatic Cancers
Although the majority of the studies on the prognostic and predictive role of TP53 status in human cancers distinguish between patients harboring WT versus mutant proteins, some evidence favors a categorization of TP53 mutations since different mutant P53 proteins can have different biologic effects. Poeta et al. (42) proposed the distinction between “disruptive” and “non-disruptive” TP53 mutations; while disruptive mutations likely lead to a complete loss of activity of the P53 protein, non-disruptive mutants can encode proteins which retain some of the original functions. This classification was used to stratify patients with TP53 mutations in head and neck squamous cell carcinoma (42), advanced Non-Small Cell Lung Cancer (43), breast and ovarian tumors (44, 45) and esophageal squamous cell carcinoma (46). However, the association between the type of TP53 mutations and prognosis was significantly variable in the different cancers.
Recently, Dutta et al. (47) analyzed data from 1,537 patients with Acute Myeloid Leukemia (AML) in order to determine a correlation between TP53 mutations and clinical outcome. TP53 mutations have been classified according to (i) their impact on protein structure (disruptive versus non-disruptive), (ii) an evolutionary action score that takes into account the evolutionary sensitivity to sequence variation and amino-acid conservation (48) and (iii) the RFS (34). Only the RFS was capable of distinguishing among AML patients with a significantly different overall survival and event-free survival. All these observations complicate the scenario and the definition of the events which are drivers of the disease pathogenesis.
Impact of TP53 Mutations Heterogeneity: Restoration of WT P53 Functionality
Mutant P53 protein has been considered a promising target for the development of new anticancer strategies and, in the last two decades, several molecules have been developed with the aim of reinstating the WT function or eliminating the mutant P53 accumulated in cancer cells (49). Upon re-acquisition of its original WT properties, the P53 protein should become transactivation competent (50), and trigger an efficient apoptotic response following treatments to which the tumor cells are normally refractory. PRIMA-1 and its methylated derivative PRIMA-1Met (also named APR246) are the most widely studied molecules on which phase I/II clinical trials are in progress (51). These molecules were found capable of reconstituting the specific DNA binding capacities to different mutant proteins (e.g. R273H, R175H) and of inducing significant apoptosis in cancer cells carrying a mutant P53 protein (52). Another molecule, RITA, which interacts with P53 and inhibits its binding to MDM2, induces a P53-dependent gene transcription and cell death (53). Beside these, many others molecules, which target the interaction of WT P53 with negative regulators (e.g. Nutlins) or with the mutant P53 (e.g. CP31398), have been investigated, some of them being currently tested in clinical trials (53, 54).
A different approach is based on the potential inhibition of the GOF activities, obtained by promoting mutant P53 protein degradation. Since mutant P53 is stabilized by the heat shock protein HSP90, usually over-expressed in cancer cells (55), several HSP90 inhibitors, such as 17-AAG or Ganetespib, have been tested as anticancer molecules and their ability to trigger mutant P53 degradation has been demonstrated (56). Also Histone Deacetylase inhibitors (HDAC), such as SAHA, can induce the degradation of the mutant P53, restraining tumor growth in vivo (56, 57). Lastly, a role of autophagy to trigger mutant, but not WT, P53 deprivation has been shown in different cancer cells (58–60), identifying the modulation of autophagy as an emerging strategy for cancer therapy (61, 62).
Not Just a Question of TP53 Mutations
P53 total inactivation in human cancer cells is frequently caused by the alterations of both alleles, comprising the allelic loss due to deletion of the short arm of the chromosome 17 [del(17p)], and the concomitant mutation of the other allele. It is of note that Donehower et al. (63) performing a comprehensive assessment of the P53 pathway involvement in 32 cancers from The Cancer Genome Atlas, demonstrated the loss of the second allele in 91% of the cases with TP53 mutations. In addition, in heterozygous murine tumours carrying the hotspot GOF allele R248Q, the loss of the remaining WT TP53 allele was a necessary prerequisite for the stabilization of the mutant P53 and for the GOF properties to become evident in vivo (64). These observations suggest that a given TP53 mutation must operate in a specific cellular context to show its biological consequences (65).
Clinical Impact of TP53 Alterations: The Example of Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia (CLL) is the most common leukemia in the Western countries, characterized by the clonal expansion of CD5+ B cells in peripheral blood, lymph-nodes and bone marrow. CLL clinical course is highly heterogeneous (66), ranging from decades of survival with no need for treatment, to a rapid disease progression with the requirement for an early treatment (67). Such a scenario likely reflects the cellular and molecular heterogeneity of the disease. CLL cases present specific karyotype aberrations, the most frequent being 13q- (~55%), 11q- (~15%) 17p- (~8%) and +12 (~15%), which correlate with a different disease course and outcome (68). In addition, gene mutations (e.g. TP53, SF3B1, BIRC3, and NOTCH1) have been reported (69), which, again, may influence the disease course and outcome. B cell receptors (BCR) features expressed by the leukemic cells also dictate the subsequent patient fate as demonstrated by the fact that patients with somatically mutated IGHV genes in the leukemic cells have a better clinical course and outcome than the patients in whom such genes are not somatically mutated; it is generally assumed that stimulation of the leukemic cells by self or exogenous antigens may promote clonal expansion (70). This notion is supported by the observation that inhibitors of the BCR-dependent signal transducing pathway are efficient treatments for CLL (70). In addition, different CLL patients that share the same BCR have similar clinical courses (71). Finally, patients with complex karyotypes, detected by chromosome G-banding, may have a dire prognosis, even in the era of new drugs (72).
P53 dysfunction has certainly a role in the clinical evolution of CLL (73). The incidence of TP53 mutations is low at diagnosis (<10% of patients), although it rises in cases with progressive disease and reaches approximately 40% in refractory CLL (73–77). Furthermore, there is evidence that CLL patients with TP53 dysfunction [measured as del(17p) and/or TP53 mutations] progress more rapidly to stages requiring treatment. Together, these considerations indicate that TP53 alterations facilitate clonal expansion and disease progression irrespective of the impact they may have on therapy (78). The presence of a P53 dysfunction has a definite negative impact on the effect of chemo-immunotherapy, whereas such impact appears to be less pronounced in patients treated with BCR inhibitors (e.g. Ibrutinib or Idelalisib) or with apoptosis inducers (e.g. Venetoclax). Because of this, TP53 mutational screening for all patients before therapy start is recommended by the European Research Initiative on CLL group (ERIC) to avoid treatment protocols that are ineffective in patients with P53 dysfunction (79).
Detection of a del(17p) or of a TP53 mutation is generally assumed to be a sufficient indication for a P53 dysfunction. CLL patients with del(17p) carry a TP53 mutation in 80% to 90% of the cases, and ~60% of patients with TP53 mutations also harbor del(17p), as detected by FISH. Even in the absence of del(17p), the presence of a TP53 mutation appears to be more frequent in patients with a poor prognosis and a higher genetic complexity (80, 81). Moreover, CLL sub-clones carrying specific TP53 mutations can be positively selected upon treatment, ultimately becoming the prevalent expansion of an initially minor mutant component (69, 82–84).
Dealing With Complexity in CLL
The identification of molecular biomarkers together with certain clinical features of the disease may dictate the choice of treatment in CLL (85). Since a P53 dysfunction is the strongest predictor of chemo-refractoriness, the assessment of TP53 status is the first, and possibly most important, decisional node in the first-line treatment algorithm. Indeed, the presence of P53 dysfunctions prevents the use of chemo-immunotherapy in favour of BCR inhibitors or Venetoclax (78). However, although such drugs have improved the poor efficacy of chemo-immunotherapy in patients with del(17p) and/or TP53 mutations (86), all these treatments still pose some challenges in these patients. Furthermore, the real influence of a gene dosage effect [e.g. presence of del(17p) versus presence of both deletion and a TP53 mutation] in patients treated with the new drugs has still to be clarified (78).
Although genomic technologies are changing the practice of onco-haematology, with improved detection of driver lesions, genomic data, generated through different technologies, each with its own sensitivity, are often considered not only interchangeable [i.e. equivalence between the presence of del(17p) and of a TP53 mutation (TP53mut)], but are also subjected to oversimplification [i.e. equivalence between the presence of one TP53 alteration (mutation or deletion) and of both alterations)]. Even though a binary simplification (P53 dysfunction versus no P53 dysfunction) can be considered clinically usable, the actual situation is potentially more complicated than estimated (87) and a more realistic situation diverging from a simple binary scenario (noDel/noMut versus Del and/or Mut) could be conceived and proposed for the clinical use in the future (Figure 2). Furthermore, the abundance of the single TP53 alteration within the leukemic clone [i.e. Variant Allele Frequency (VAF) for a TP53 mutation and percentage of del(17p) positive cells] may represent a factor of relevance. For example, while all identified TP53 mutations were clonal with the Sanger sequencing method (VAF>10%), both clonal (VAF>10%) and sub-clonal (VAF<10%, as small as 0.3%) TP53 mutations can be detected with the introduction of Next Generation Sequencing technologies. Nevertheless, this information has not entered into clinical practice yet, although it may contribute to provide information on the effective P53 function in the leukemic clone and also on its potential prospective evolution. The last update of the guidelines released by ERIC still consider that clinical decisions should be taken based on the presence of a clonal TP53 mutation.
Figure 2
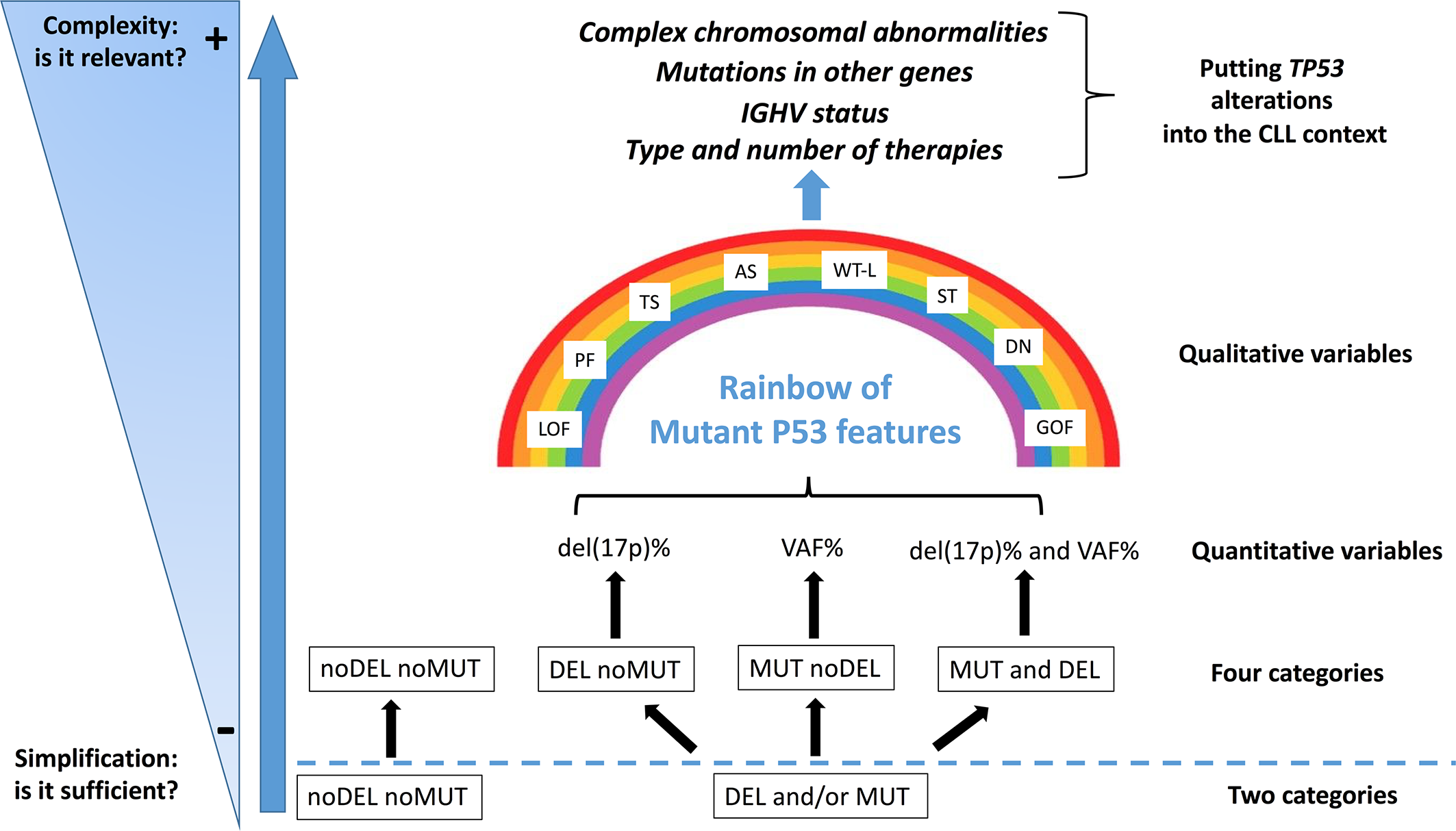
Del(17p) (DEL), detected by FISH, and/or TP53 mutation (s) (MUT), detected by DNA sequencing, as indicators of a P53 dysfunction. The present classification may represent an oversimplification of the conditions leading to an impaired P53 function, both in terms of quantity [del(17p) % or VAF, Variant Allele Frequency %] and quality (i.e. P53 mutant protein functionality: LOF, loss of function; PF, partial function; AS, altered specificity; TS, temperature sensitive; WT-L, wild-type like; ST, super-transactivating; GOF, gain of function; DN, dominant negative). Moreover, other molecular features (e.g. mutated IGHV versus un-mutated IGHV; SF3B1, BIRC3 and NOTCH1 mutations; 13q and 11q partial deletions) and the eventual previous patient management might influence the highly heterogeneous clinical course of the disease.
The percentage of del(17p) positive cell may also represents an important variable as it appears that, among patients with del(17p), those with a higher percentage of cells carrying the deletion have shorter survivals (88). Another layer of complexity is related to the fact that not all mutant P53 proteins appear to have the same functional consequences, as previously described. Although these issues, which are related to the effective P53 function in a leukemic clone, have emerged as real biological and clinical problems, they have not been so far deeply investigated in CLL (65). Finally, it should be stressed that TP53 alterations, although important, are not the sole alterations and should be considered together with other cytogenetic abnormalities which may occur concomitantly in the single patients and may affect per se the clinical course of CLL (Figure 2).
Concluding Remarks
In conclusion, is the binary scenario compatible with the underlying complexity in CLL? While a simplified vision is important for deciding clinical strategies, new studies appear necessary for assessing whether further levels of complexity in CLL classification, can lead to a more precise patient stratification. In this context, it is likely that future studies will define whether patients with del(17p) and a TP53 mutation might have a different clinical course from those who have only a TP53 mutation or only del(17p). Furthermore, a patient with a partial function TP53 mutation might show a different clinical course from those harbouring a complete loss of function TP53 mutation, as suggested by our present observations and studies in other experimental systems (33, 89, 90). All of these aspects regarding P53 dysfunction may affect therapy and consequently deserve an evaluation, possibly more extended than that currently used.
Funding
This work was supported by: Associazione Italiana Ricerca sul Cancro (AIRC) Grant 5 x mille n.9980, (to MF, FM, and AN); AIRC I.G. n.14326 (to MF), n.15426 (to FF), and n. 5506 (to GF); AIRC and Fondazione CaRiCal co-financed Multi-Unit Regional Grant 2014 n.16695 (to FM); Italian Ministry of Health 5x1000 funds 2013 (to GF) 2014 (to GC, SZ, and AI), 2015 (to FF and GF) and 2016 (to FF, GC, and GF); Current Research 2016 to GF; and 2016 (to FF and GC); Compagnia S. Paolo Turin Italy project 2017.0526 (to GF) Italian Ministry of Health: Alleanza Contro il Cancro (Hematology network).
Statements
Author contributions
All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Lane DP . Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature (1979) 278:261–3. doi: 10.1038/278261a0
2
Linzer DI Levine AJ . Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell (1979) 17:43–52. doi: 10.1016/0092-8674(79)90293-9
3
Kress M May E Cassingena R May P . Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol (1979) 31:472–83. doi: 10.1128/JVI.31.2.472-483.1979
4
Finlay CA Hinds PW Levine AJ . The p53 proto-oncogene can act as a suppressor of transformation. Cell (1989) 57:1083–93. doi: 10.1016/0092-8674(89)90045-7
5
Baker SJ Fearon ER Nigro JM Hamilton SR Preisinger AC Jessup JM et al . Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science (1989) 244:217–21. doi: 10.1126/science.2649981
6
Hollstein M Sidransky D Vogelstein B Harris CC . p53 mutations in human cancers. Science (1991) 253:49–53. doi: 10.1126/science.1905840
7
Malkin D Li FP Strong LC Fraumeni JF Nelson CE Kim DH et al . Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science (1990) 250:1233–8. doi: 10.1126/science.1978757
8
Cerami E Gao J Dogrusoz U Gross BE Sumer SO Aksoy BA et al . The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
9
Gao J Aksoy BA Dogrusoz U Dresdner G Gross B Sumer SO et al . Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:pl1. doi: 10.1126/scisignal.2004088
10
Joerger AC Fersht AR . The Tumor Suppressor p53: From Structures to Drug Discovery. Cold Spring Harbor Perspect Biol (2010) 2:a000919–a000919. doi: 10.1101/cshperspect.a000919
11
El-Deiry WS Kern SE Pietenpol JA Kinzler KW Vogelstein B . Definition of a consensus binding site for p53. Nat Genet (1992) 1:45–9. doi: 10.1038/ng0492-45
12
Wei J Zaika E Zaika A . p53 Family: Role of Protein Isoforms in Human Cancer. J Nucleic Acids (2012) 2012:1–19. doi: 10.1155/2012/687359
13
Anbarasan T Bourdon J-C . The Emerging Landscape of p53 Isoforms in Physiology, Cancer and Degenerative Diseases. IJMS (2019) 20:6257. doi: 10.3390/ijms20246257
14
Liu Y Tavana O Gu W . p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol (2019) 11:564–77. doi: 10.1093/jmcb/mjz060
15
Fischer M . Census and evaluation of p53 target genes. Oncogene (2017) 36:3943–56. doi: 10.1038/onc.2016.502
16
Ho T Tan BX Lane D . How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. IJMS (2019) 21:13. doi: 10.3390/ijms21010013
17
Leroy B Fournier JL Ishioka C Monti P Inga A Fronza G et al . The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res (2013) 41:D962–9. doi: 10.1093/nar/gks1033
18
Leroy B Ballinger ML Baran-Marszak F Bond GL Braithwaite A Concin N et al . Recommended Guidelines for Validation, Quality Control, and Reporting of TP53 Variants in Clinical Practice. Cancer Res (2017) 77(6):1250–60. doi: 10.1158/0008-5472.CAN-16-2179
19
Cho Y Gorina S Jeffrey PD Pavletich NP . Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (1994) 265:346–55. doi: 10.1126/science.8023157
20
Kitayner M Rozenberg H Rohs R Suad O Rabinovich D Honig B et al . Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat Struct Mol Biol (2010) 17:423–9. doi: 10.1038/nsmb.1800
21
Ishioka C Frebourg T Yan Y-X Vidal M Friend SH Schmidt S et al . Screening patients for heterozygous p53 mutations using a functional assay in yeast. Nat Genet (1993) 5:124–9. doi: 10.1038/ng1093-124
22
Flaman JM Frebourg T Moreau V Charbonnier F Martin C Chappuis P et al . A simple p53 functional assay for screening cell lines, blood, and tumors. Proc Natl Acad Sci (1995) 92:3963–7. doi: 10.1073/pnas.92.9.3963
23
Campomenosi P Monti P Aprile A Abbondandolo A Frebourg T Gold B et al . p53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene (2001) 20:3573–9. doi: 10.1038/sj.onc.1204468
24
Resnick MA Inga A . Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc Natl Acad Sci USA (2003) 100:9934–9. doi: 10.1073/pnas.1633803100
25
Šmardová J Šmarda J Koptíková J . Functional analysis of p53 tumor suppressor in yeast. Differentiation (2005) 73:261–77. doi: 10.1111/j.1432-0436.2005.00028.x
26
Monti P Bosco B Gomes S Saraiva L Fronza G Inga A . Yeast As a Chassis for Developing Functional Assays to Study Human P53. J Vis Exp (2019) 59071. doi: 10.3791/59071
27
Kato S Han S-Y Liu W Otsuka K Shibata H Kanamaru R et al . Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA (2003) 100:8424–9. doi: 10.1073/pnas.1431692100
28
Bisio A Ciribilli Y Fronza G Inga A Monti P . TP53 mutants in the tower of babel of cancer progression. Hum Mutat (2014) 35:689–701. doi: 10.1002/humu.22514
29
Di Como CJ Gaiddon C Prives C . p73 Function Is Inhibited by Tumor-Derived p53 Mutants in Mammalian Cells. Mol Cell Biol (1999) 19:1438–49. doi: 10.1128/MCB.19.2.1438
30
Strano S Fontemaggi G Costanzo A Rizzo MG Monti O Baccarini A et al . Physical Interaction with Human Tumor-derived p53 Mutants Inhibits p63 Activities. J Biol Chem (2002) 277:18817–26. doi: 10.1074/jbc.M201405200
31
Adorno M Cordenonsi M Montagner M Dupont S Wong C Hann B et al . A Mutant-p53/Smad Complex Opposes p63 to Empower TGFβ-Induced Metastasis. Cell (2009) 137:87–98. doi: 10.1016/j.cell.2009.01.039
32
Weissmueller S Manchado E Saborowski M Morris JP Wagenblast E Davis CA et al . Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell (2014) 157:382–94. doi: 10.1016/j.cell.2014.01.066
33
Sabapathy K Lane DP . Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol (2018) 15:13–30. doi: 10.1038/nrclinonc.2017.151
34
Kotler E Shani O Goldfeld G Lotan-Pompan M Tarcic O Gershoni A et al . Segal E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol Cell (2018) 71:178–190.e8. doi: 10.1016/j.molcel.2018.06.012
35
Giacomelli AO Yang X Lintner RE McFarland JM Duby M Kim J et al . Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet (2018) 50:1381–7. doi: 10.1038/s41588-018-0204-y
36
Liu G McDonnell TJ Montes de Oca Luna R Kapoor M Mims B El-Naggar AK et al . High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci (2000) 97:4174–9. doi: 10.1073/pnas.97.8.4174
37
Lang GA Iwakuma T Suh Y-A Liu G Rao VA Parant JM et al . Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell (2004) 119:861–72. doi: 10.1016/j.cell.2004.11.006
38
Liu G Parant JM Lang G Chau P Chavez-Reyes A El-Naggar AK et al . Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet (2004) 36:63–8. doi: 10.1038/ng1282
39
Xu J Qian J Hu Y Wang J Zhou X Chen H et al . Heterogeneity of Li-Fraumeni Syndrome links to unequal gain-of-function effects of p53 mutations. Sci Rep (2015) 4:4223. doi: 10.1038/srep04223
40
Monti P Ciribilli Y Jordan J Menichini P Umbach DM Resnick MA et al . Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin Cancer Res (2007) 13:3789–95. doi: 10.1158/1078-0432.CCR-06-2545
41
Monti P Perfumo C Bisio A Ciribilli Y Menichini P Russo D et al . Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol Cancer Res (2011) 9:271–9. doi: 10.1158/1541-7786.MCR-10-0496
42
Poeta ML Manola J Goldwasser MA Forastiere A Benoit N Califano JA et al . TP53 Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2007) 357:2552–61. doi: 10.1056/NEJMoa073770
43
Molina-Vila MA Bertran-Alamillo J Gasco A Mayo-de-las-Casas C Sanchez-Ronco M Pujantell-Pastor L et al . Nondisruptive p53 Mutations Are Associated with Shorter Survival in Patients with Advanced Non-Small Cell Lung Cancer. Clin Cancer Res (2014) 20:4647–59. doi: 10.1158/1078-0432.CCR-13-2391
44
Dobes P Podhorec J Coufal O Jureckova A Petrakova K Vojtesek B et al . Influence of mutation type on prognostic and predictive values of TP53 status in primary breast cancer patients. Oncol Rep (2014) 32:1695–702. doi: 10.3892/or.2014.3346
45
Bueno AM Molina MA Fielding A Bertran-Alamillo J Dougherty BA Lai Z et al . Disruptive mutations in TP53 associate with survival benefit in a PARPi trial in ovarian cancer. Ann Oncol (2017) 28:v626. doi: 10.1093/annonc/mdx440.036
46
Huang M Jin J Zhang F Wu Y Xu C Ying L et al . Non-disruptive mutation in TP53 DNA-binding domain is a beneficial factor of esophageal squamous cell carcinoma. Ann Transl Med (2020) 8:316–6. doi: 10.21037/atm.2020.02.142
47
Dutta S Pregartner G Rücker FG Heitzer E Zebisch A Bullinger L et al . Functional Classification of TP53 Mutations in Acute Myeloid Leukemia. Cancers (2020) 12:637. doi: 10.3390/cancers12030637
48
Neskey DM Osman AA Ow TJ Katsonis P McDonald T Hicks SC et al . Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res (2015) 75:1527–36. doi: 10.1158/0008-5472.CAN-14-2735
49
Muller PAJ Vousden KH . Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell (2014) 25:304–17. doi: 10.1016/j.ccr.2014.01.021
50
Bykov VJN Wiman KG . Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett (2014) 588:2622–7. doi: 10.1016/j.febslet.2014.04.017
51
Lehmann S Bykov VJN Ali D Andrén O Cherif H Tidefelt U et al . Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol (2012) 30:3633–9. doi: 10.1200/JCO.2011.40.7783
52
Bykov VJN Issaeva N Shilov A Hultcrantz M Pugacheva E Chumakov P et al . Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8:282–8. doi: 10.1038/nm0302-282
53
Selivanova G . Wild type p53 reactivation: from lab bench to clinic. FEBS Lett (2014) 588:2628–38. doi: 10.1016/j.febslet.2014.03.049
54
Duffy MJ Synnott NC McGowan PM Crown J O’Connor D Gallagher WM . p53 as a target for the treatment of cancer. Cancer Treat Rev (2014) 40:1153–60. doi: 10.1016/j.ctrv.2014.10.004
55
Trepel J Mollapour M Giaccone G Neckers L . Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer (2010) 10:537–49. doi: 10.1038/nrc2887
56
Li D Marchenko ND Schulz R Fischer V Velasco-Hernandez T Talos F et al . Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res (2011) 9:577–88. doi: 10.1158/1541-7786.MCR-10-0534
57
Alexandrova EM Yallowitz AR Li D Xu S Schulz R Proia DA et al . Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature (2015) 523:352–6. doi: 10.1038/nature14430
58
Foggetti G Ottaggio L Russo D Mazzitelli C Monti P Degan P et al . Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci Rep (2019) 39:BSR20181345. doi: 10.1042/BSR20181345
59
Rodriguez OC Choudhury S Kolukula V Vietsch EE Catania J Preet A et al . Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle (2012) 11:4436–46. doi: 10.4161/cc.22778
60
Garufi A Pucci D D’Orazi V Cirone M Bossi G Avantaggiati ML et al . Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis (2014) 5:e1271. doi: 10.1038/cddis.2014.217
61
Cordani M Butera G Pacchiana R Donadelli M . Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim Biophys Acta Rev Cancer (2017) 1867:19–28. doi: 10.1016/j.bbcan.2016.11.003
62
Cirone M Gilardini Montani MS Granato M Garufi A Faggioni A D’Orazi G . Autophagy manipulation as a strategy for efficient anticancer therapies: possible consequences. J Exp Clin Cancer Res (2019) 38:262. doi: 10.1186/s13046-019-1275-z
63
Donehower LA Soussi T Korkut A Liu Y Schultz A Cardenas M et al . Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep (2019) 28:1370–84.e5. doi: 10.1016/j.celrep.2019.07.001
64
Alexandrova EM Mirza SA Xu S Schulz-Heddergott R Marchenko ND Moll UM . p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis (2017) 8:e2661–1. doi: 10.1038/cddis.2017.80
65
Kastenhuber ER Lowe SW . Putting p53 in Context. Cell (2017) 170:1062–78. doi: 10.1016/j.cell.2017.08.028
66
Chiorazzi N Rai KR Ferrarini M . Chronic Lymphocytic Leukemia. N Engl J Med (2005) 352:804–15. doi: 10.1056/NEJMra041720
67
Gentile M Shanafelt TD Reda G Mauro FR Zirlik K Ciolli S et al . Validation of a biological score to predict response in chronic lymphocytic leukemia patients treated front-line with bendamustine and rituximab. Leukemia (2018) 32:1869–73. doi: 10.1038/s41375-018-0100-6
68
Döhner H Stilgenbauer S Benner A Leupolt E Kröber A Bullinger L et al . Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med (2000) 343:1910–6. doi: 10.1056/NEJM200012283432602
69
Nadeu F Delgado J Royo C Baumann T Stankovic T Pinyol M et al . Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood (2016) 127:2122–30. doi: 10.1182/blood-2015-07-659144
70
Hallek M . Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am J Hematol (2019) 94:1266–87. doi: 10.1002/ajh.25595
71
ten Hacken E Gounari M Ghia P Burger JA . The importance of B cell receptor isotypes and stereotypes in chronic lymphocytic leukemia. Leukemia (2019) 33:287–98. doi: 10.1038/s41375-018-0303-x
72
Jondreville L Krzisch D Chapiro E Nguyen-Khac F . The complex karyotype and chronic lymphocytic leukemia: prognostic value and diagnostic recommendations. Am J Hematol (2020) ajh.25956. doi: 10.1002/ajh.25956
73
Buccheri V Barreto WG Fogliatto LM Capra M Marchiani M Rocha V . Prognostic and therapeutic stratification in CLL: focus on 17p deletion and p53 mutation. Ann Hematol (2018) 97:2269–78. doi: 10.1007/s00277-018-3503-6
74
Rossi D Cerri M Deambrogi C Sozzi E Cresta S Rasi S et al . The Prognostic Value of TP53 Mutations in Chronic Lymphocytic Leukemia Is Independent of Del17p13: Implications for Overall Survival and Chemorefractoriness. Clin Cancer Res (2009) 15:995–1004. doi: 10.1158/1078-0432.CCR-08-1630
75
Zenz T for the European Research Initiative on CLL (ERIC) Vollmer D Trbusek M Smardova J Benner A et al . TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia (2010) 24:2072–9. doi: 10.1038/leu.2010.208
76
Pospisilova S on behalf of the European Research Initiative on CLL (ERIC) Gonzalez D Malcikova J Trbusek M Rossi D et al . ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia (2012) 26:1458–61. doi: 10.1038/leu.2012.25
77
Stilgenbauer S Schnaiter A Paschka P Zenz T Rossi M Döhner K et al . Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood (2014) 123:3247–54. doi: 10.1182/blood-2014-01-546150
78
Morabito F Gentile M Monti P Recchia AG Menichini P Skafi M et al . TP53 dysfunction in chronic lymphocytic leukemia: clinical relevance in the era of B-cell receptors and BCL-2 inhibitors. Expert Opin Invest Drugs (2020) 29(8):869–80. doi: 10.1080/13543784.2020.1783239
79
Malcikova J on behalf of the European Research Initiative on Chronic Lymphocytic Leukemia (ERIC) — TP53 network Tausch E Rossi D Sutton LA Soussi T et al . ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia—update on methodological approaches and results interpretation. Leukemia (2018) 32:1070–80. doi: 10.1038/s41375-017-0007-7
80
Zenz T Krober A Scherer K Habe S Buhler A Benner A et al . Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood (2008) 112:3322–9. doi: 10.1182/blood-2008-04-154070
81
Sutton L-A Rosenquist R . The complex interplay between cell-intrinsic and cell-extrinsic factors driving the evolution of chronic lymphocytic leukemia. Semin Cancer Biol (2015) 34:22–35. doi: 10.1016/j.semcancer.2015.04.009
82
Malcikova J Smardova J Rocnova L Tichy B Kuglik P Vranova V et al . Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood (2009) 114:5307–14. doi: 10.1182/blood-2009-07-234708
83
Rossi D Khiabanian H Spina V Ciardullo C Bruscaggin A Fama R et al . Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood (2014) 123:2139–47. doi: 10.1182/blood-2013-11-539726
84
Puente XS Beà S Valdés-Mas R Villamor N Gutiérrez-Abril J Martín-Subero JI et al . Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature (2015) 526:519–24. doi: 10.1038/nature14666
85
Moia R Patriarca A Schipani M Ferri V Favini C Sagiraju S et al . Precision Medicine Management of Chronic Lymphocytic Leukemia. Cancers (2020) 12:642. doi: 10.3390/cancers12030642
86
Burger JA . Treatment of Chronic Lymphocytic Leukemia. N Engl J Med (2020) 383:460–73. doi: 10.1056/NEJMra1908213
87
Chin-Yee B Sadikovic B Chin-Yee IH . Genomic data in prognostic models-what is lost in translation? The case of deletion 17p and mutant TP53 in chronic lymphocytic leukaemia. Br J Haematol (2020) 188:652–60. doi: 10.1111/bjh.15827
88
Yuan Y Zhu H Wu J Xia Y Liang J Wu W et al . The percentage of cells with 17p deletion and the size of 17p deletion subclones show prognostic significance in chronic lymphocytic leukemia. Genes Chromosomes Cancer (2019) 58:43–51. doi: 10.1002/gcc.22692
89
Cutrona G Matis S Colombo M Massucco C Baio G Valdora F et al . Effects of miRNA-15 and miRNA-16 expression replacement in chronic lymphocytic leukemia: implication for therapy. Leukemia (2017) 31:1894–904. doi: 10.1038/leu.2016.394
90
Cutrona G Tripodo C Matis S Recchia AG Massucco C Fabbi M et al . Microenvironmental regulation of the IL-23R/IL-23 axis overrides chronic lymphocytic leukemia indolence. Sci Transl Med (2018) 10(428):eaal1571. doi: 10.1126/scitranslmed.aal1571
Summary
Keywords
TP53 mutations, chronic lymphocytic leukemia, clinical impact, P53 protein function, reactivation of P53
Citation
Monti P, Menichini P, Speciale A, Cutrona G, Fais F, Taiana E, Neri A, Bomben R, Gentile M, Gattei V, Ferrarini M, Morabito F and Fronza G (2020) Heterogeneity of TP53 Mutations and P53 Protein Residual Function in Cancer: Does It Matter?. Front. Oncol. 10:593383. doi: 10.3389/fonc.2020.593383
Received
10 August 2020
Accepted
29 September 2020
Published
28 October 2020
Volume
10 - 2020
Edited by
Jerson L. Silva, Federal University of Rio de Janeiro, Brazil
Reviewed by
João Agostinho Machado-Neto, University of São Paulo, Brazil; Alvaro Galli, Italian National Research Council, Italy
Updates
Copyright
© 2020 Monti, Menichini, Speciale, Cutrona, Fais, Taiana, Neri, Bomben, Gentile, Gattei, Ferrarini, Morabito and Fronza.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilberto Fronza, gilberto.fronza@hsanmartino.it
This article was submitted to Molecular and Cellular Oncology, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.