 Luigi Ombrato
Luigi Ombrato Marco Montagner
Marco Montagner- 1Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
- 2Department of Molecular Medicine, School of Medicine and Surgery, University of Padua, Padua, Italy
Metastases are a major cause of cancer-related death and despite the fact that they have been focus of intense research over the last two decades, effective therapies for patients with distant secondary lesions are still very limited. In addition, in some tumor types metastases can grow years after the patients have been declared clinically cured, indicating that disseminated cancer cells (DCCs) persist undetected for years, even decades in a quiescent state. Clinical and experimental data highlight the importance of the immune system in shaping the fitness and behaviour of DCCs. Here, we review mechanisms of survival, quiescence and outgrowth of DCCs with a special focus on immune-regulation and we highlight the latest cutting-edge techniques for modelling the biology of DCCs in vitro and for studying the metastatic niche in vivo. We believe that a wide dissemination of those techniques will boost scientific findings towards new therapies to defeat metastatic relapses in cancer patients.
Clinical Problem
According to a recent analysis, the proportion of cancer deaths with metastases as contributing cause, ranged from 9.3% for CNS cancers to 90.4% and 80.2% for ovarian and colon cancer, respectively (1, 2). Metastases can be detected in concomitance with the primary tumor (synchronous) or at a later stage (metachronous). Although most tumors cover the same steps of metastatic dissemination (i.e., extravasation, dissemination through blood or lymphatics, intravazation, and establishment in the metastatic niche), the time required to form overt lesions significantly differs according to the tissue of origin and cancer subtypes. While breast, prostate, renal cell cancers, as well as sarcomas and melanomas show long latency and the time required to develop metachronous metastasis might reach 15 years, 85% of relapses from colon cancer are detected within 3 years (medium latency), and lung cancers often spread at distant sites within a few weeks (short latency) (1, 3–5). When the time required for a DCC to form an overt metastasis after the removal of the primary tumor is long (arbitrarily usually set as 5 years), latency is often referred to as “dormancy”. Importantly, different metastatic latencies might underlie different mechanisms in the acquisition of aggressive traits, and at the same time significantly impact on our capacity to intervene, as the time preceding the metastatic onset offers a therapeutic window so far underexploited. Thus, it is a priority to understand the biology of DCCs, cell intrinsic and extrinsic determinants of their death, survival and growth at the secondary site.
One factor that profoundly affects relapse of DCCs is the cell of origin and its genetic landscape, as exemplified by breast cancers. More than half of breast tumors positive for estrogen receptor (ER) relapse after 5 years of diagnosis and mastectomy, with a progressive increase in recurrence risk from 5 to 20 years in patients treated with adjuvant endocrine therapy (6, 7). This is in contrast with data from patients with ER negative breast cancers, where relapses mostly occur within the first two years (7). Interestingly, while averaging over a long time is required for meta-analysis of different case series, analysing events at shorter intervals in homogeneous case series allows the identification of a multi-peaks pattern of breast cancer recurrence (8, 9). This observation leads to a fundamental question: why do dormant DCCs (DDCCs) reawaken in cured patients with no apparent clinical condition? Beyond stochastic local perturbations, paraphysiological signals involved in exit from dormancy have yet to be identified, but candidates are, for example, hormones or factors related to lifestyle, such as diet (10, 11).
Interestingly, a recent report provided experimental evidence in support of a longstanding clinical observation, i.e., the effect of systemic inflammation on reawakening of DDCCs (8, 12). The paramount role of inflammation and immune surveillance on the behaviour of DDCCs has been unequivocally demonstrated by inadvertently transplanted malignant tumors (13–16). Demand for transplant organs far exceeds available donors, thus, occasionally, donors with a history of cancer were accepted provided that they were disease-free long enough to be considered cured (>10 years). In several cases, recipient patients developed metastases after transplantation of heart, kidney, lungs or liver. Most frequently transplanted tumors were renal cell cancer, cutaneous malignant melanoma, malignant glioblastoma (which is usually considered non-metastatic). Several concepts can be drawn from these reports: i) the presence of a malignant cancer was unknown for some donors, supporting the concept of an early dissemination, ii) the presence of DDCCs in organs that are not considered sites of secondary tumors, indicating that dissemination is not a prerogative of few organs, iii) immune system has a central role in controlling outgrowth of DDCCs, as when the organ was removed and immunosuppression discontinued, malignant cells were rejected by the host (host versus graft). This is supported by the empirical evidence that more metastatic lesions are observed in immunocompromized experimental mouse models compared to wild-type strains (12). The importance of the immune control of DDCCs is further reinforced by the clinical evidence showing discrete peaks of recurrence in patients after resection of the primary tumor, likely as a consequence of systemic inflammation (2, 12, 17). Importantly, perioperative resolution of the inflammatory status prevents outgrowth of otherwise DDCCs (12, 17).
These clinical evidences highlight the role of immunity in the control of DCCs survival and growth and strongly support a better understanding of the dynamic and complex immune tumor microenvironment (TME) at the metastatic site at a single cell level. However, this has been difficult to achieve so far due to the lack of tools to study local interactions between DCCs and their neighbouring cells. In this review we will first clarify key definitions in the dormancy phenotype and then summarise the current knowledge on the non-immune as well as immune-related mechanisms of dormancy. At the end of the review we will highlight recent technological advances that might greatly push forward our knowledge of the molecular mechanisms associated with dormancy.
Definitions
Dormancy can be used to describe two very distinct phenomena: primary tumor dormancy and metastatic dormancy. The former indicates the time required by an evolving cancer cell to overcome oncogene-induced senescence or apoptosis, metabolic adaptation, evade immune clearance and induce neoangiogenesis, thus forming a detectable tumor mass (18). Metastatic dormancy, instead, indicates the time required by a DCC to overtake the attrition due to seeding in a hostile environment and develop an overt lesion. Although in some cases, determinants of dormancy might be shared among primary tumor and metastases [such as ERK/p38 ratio and fibronectin (19, 20)], they are likely to be distinct processes.
Another distinction often used is between “cellular dormancy”, i.e., cells undergoing reversible G0/G1 cell cycle arrest, and “tumor mass dormancy”, indicating small clusters of cells where proliferation is balanced by death induced by lack of nutrients (angiogenic dormancy) and/or by immune clearance (immune dormancy). Although useful to rationalize the dormant phenotype, this sharp distinction has little experimental support and likely the two conditions coexist, with DDCCs dynamically fluctuating between the two states during their history. For example, a small, but detectable, proportion of early DCCs (eDCCs) from experimental breast cancers are found positive for proliferation markers (21), despite they are often referred to as “non-proliferative”. Moreover, Aguirre-Ghiso and colleagues showed, with an elegant genetically-encoded fluorescent marker dilution assay, that post-hypoxic DCCs, which are much less proliferative than post-normoxic DCCs, still undergo considerable proliferation over two weeks (22). This dynamic heterogeneity is not unique to DDCCs, as a significant number of Ki67-negative cells are found even in DCCs from aggressive cell lines, such as MDA-MB-231 (23). The development of longitudinal assays that keep track of the proliferative history of DCCs will help to understand if cellular and tumor mass dormancy are static or dynamic entities and which of them contributes to aggressive lesions.
Mechanisms of Survival, Quiescence, and Reawakening of DDCCs
The fate of disseminated cells is driven by a combination of cell intrinsic, extrinsic and stochastic events (1). Cell intrinsic programs involve oncogenes and tumor suppressors, membrane proteins (integrins, receptors etc…), intracellular components (such as cytoskeletal proteins and mechanotransducers), signaling pathways and sensors that integrate genetic and microenvironmental inputs and translate them into cellular processes. Cell extrinsic programs include triggers from the niche, such as stromal cells, tissue architecture, biophysical and biochemical cues, as reviewed in (24, 25). Intrinsic and extrinsic signals do not act on their own, rather they are nodes and connectors of a complex and dynamic network where extrinsic signals from TME (organ specific or shared) funnel into key intrinsic signaling hubs.
Immune-Independent Mechanisms
P-ERK/P-p38 ratio is perhaps the most widely validated dormancy hub so far (25, 26). While activated ERK drives exit from dormancy and growth, P-p38 promotes growth arrest via several mechanisms, such as MSK1, DEC2, NDRG1, NR2F1, and ultimately p21 and p27 induction (26, 27). Several signals converge on p38, such as TGFβ2 (28), BMP7 (29) as well as the metastasis suppressors MKK4 and Nm23 (30–32). Another determinant of dormancy/growth signaling is the PI3K/Akt/mTOR axis, whose activation drives survival and exit from dormancy (33–37). Different integrin dimers, often in conjunction with Src, have been consistently linked with survival of DDCCs and/or metastatic outbreak (20, 24, 38–45). Several signaling pathways have also been linked so far with quiescence and metastatic fitness: TGFβ and BMP pathways (23, 28, 29, 46–48), canonical and non-canonical Wnt pathway (21, 49–51), YAP/TAZ (41, 42), Notch (49, 52), JAK/STAT (53, 54). Recently, ER stress response and autophagy have been convincingly linked with survival of DDCCs in vitro and in vivo (55–58).
Several fibrous and non-fibrous ECM proteins have been shown to be key determinants of metastatic fitness (40): collagen I (39, 59), fibronectin (20, 38, 60), periostin (23, 50), tenascin C (49), thrombospondin (23, 41). Beside ECM proteins, hypoxia present at the primary site primes breast cancer DCCs for dormancy upon seeding to secondary organs (22).
Stromal cells provide organ-specific niches that regulate both quiescence and reactivation. Bone is probably the most characterised niche, since it is the preferred metastatic target of prostate and breast cancer (61). Osteoblasts release the growth arrest specific 6 (GAS6) ligand that binds to the Axl subfamily of receptors inducing dormancy (47, 62–65). Importantly, DDCCs can hijack endogenous signals regulating hematopoietic stem cells’ (HSC) reversible quiescence. For example, the chemokine CXCL12 from bone endothelial cells and mesenchymal progenitors induces dormancy in DCCs and HSC (1, 66–69). On the contrary, in preclinical models of bone metastasis, RANK-stimulated osteoclasts are reported to mobilize DDCCs and trigger proliferation (43, 70–72). Lung is another common homing site for DDCCs and interaction of breast DDCCs with type I pneumocytes is key for the activation of a dormant gene program in DCCs (20). In this context, cellular protrusions are required to gather survival signals from the microenvironment (20, 73, 74). Importantly, stromal derived BMP2 and TGFβ2 keep DDCCs in a latent state (28, 46), while collagen-rich fibrotic lung transforms DDCCs into aggressive metastatic cells (39), a similar mechanism was observed in fibrotic liver (75). Lastly, the perivascular niche regulates DCCs behaviour and chemoresistance in multiple organs (23, 45, 76).
Immune-Related Mechanisms
Immune cells are known to play a key role in shaping the TME in primary tumor and metastasis (77–79) and several evidences show that their recruitment at distant sites anticipates cancer cells colonization (59, 80–86). Moreover, extracellular vesicles (EVs) from the primary tumor have been shown to induce a premetastatic niche at the metastatic site [reviewed in (87, 88)]. Notably, the protein content of exosomes is critical to their function and it defines where cancer cells metastasise (89) and also influences response to chemotherapy of DCCs (90).
Because of their acknowledged tumor modulatory function immune cells have not surprisingly become a valid therapeutic target. Immunotherapy has finally proven its efficacy in treating patients and promises to further change the standard of care for cancer treatment in the coming years (91–94). However, a complete resolution of the TME as well as the understanding of this local crosstalk is far to be achieved, possibly limiting the efficacy of current immunotherapeutic options to a small number of patients. This local crosstalk has been shown to also occur via EVs. Immune cell derived exosomes have been initially shown to function as immunomodulators by carrying molecules able to induce a T-cell response (95, 96). However, metastatic cells can release exosomes expressing PD-L1 on their surface and are therefore able to suppress cytotoxic T cells (97–99). These mechanisms might influence a positive response to immunotherapy. Moreover, a further boost in immunotherapy might come from a better understanding of the immune diversity in the TME and the way immune cells locally interact, as this can help to predict therapeutic responsiveness (91, 100). Since immune cells are important in limiting metastatic outgrowth and keeping DCCs in an indolent state (5, 26, 101, 102), it is tempting to foresee a role of immunotherapy in targeting dormant DCCs (103). However, this possibility is currently restrained by a limited understanding of how immune cells interact with DCCs. In the next sections we will summarise the current knowledge on the role of immune cells specifically in metastatic dormancy.
Innate Immunity and Dormancy
Macrophages have long been known to play a role in cancer (77) and the intriguing finding that they polarise their status to support cancer growth paved the way for studies on immune cell pro-tumorigenic functions. For example, macrophages support tumor growth by several means, among them a direct inhibition of tumor suppressive immune cells (104, 105). Macrophages are able to directly promote breast DCCs survival in the lung via a VCAM1-α4 integrin binding (44). Interestingly, the aberrant expression of VCAM1 in bone-disseminated breast DCCs promotes the recruitment of monocytic osteocytic progenitors and subsequent transition from indolent growth to overt bone metastasis (43). Macrophages also sustain early dissemination and metastasis in the HER2+ model of breast cancer (106) and have been shown to interact with residual tumor cells and promote tumor recurrence in a HER2-driven breast cancer (107).
Neutrophils represent another abundant component of innate immunity whose contribution in cancer has only started to be elucidated in the last few years (108). Neutrophils have been shown to boost lung metastasis from breast cancer (81, 109–111) and to reawaken DDCCs (41, 107). Their ability to reawaken DDCCs in the lung following LPS exposure is strongly dependent on the release of neutrophils extracellular traps (NETs) (41). Notably, the metastatic outgrowth of DDCCs induced by LPS-mediated inflammation is rescued following neutrophil depletion, but not when depleting macrophages with anti-CCL2 (107), indicating a unique role for neutrophils in this context.
Another innate immune population, the NK cells, has been associated with the clearance of DCCs. NK cells play a key role in immune surveillance during metastatic dissemination (112). Indeed, the expression of NK cell-activating ligands on cancer cells is critical for their clearance (113) and the upregulation of NK cell-activating receptors render cancer cells more susceptible to NK cell-mediated killing (114). Moreover, neutrophils-mediated NK-cell depletion promotes outgrowth of disseminated carcinoma cells (115). By using a “latency competent model” of breast and lung carcinoma, Massague and colleagues showed that while NK cells clear most of the disseminated cells at first instance, some cancer cells stochastically enter quiescence and downregulate ligands for NK cells to evade the immune surveillance. Importantly, these quiescent DCCs keep their tumorigenic potential and can re-enter cell cycle to metastases when NK surveillance is released (116).
Adaptive Immune System and Dormancy
Cancer immunoediting has been recognized as a process by which the immune system controls cancer growth, with a primary role of adaptive immunity (117, 118). Schreiber and Smyth laboratories made important contributions to show how T cells maintain cancer cell quiescence and how the depletion of CD4+ and CD8+ cells, but not NK cells, allows primary tumor growth in a carcinogen-induced model of sarcoma (119, 120). A key role for a subpopulation of T cells, the tissue-resident memory CD8+ cells, in maintaining a durable immune-cancer equilibrium, has also been shown in skin melanoma (121).
Importantly, T-cells also control cancer outgrowth when DCCs colonize secondary sites. Persistent endoplasmic reticulum (ER) stress plays a role in maintaining pancreatic DCCs quiescence and protecting them from a CD8+ T-cell-mediated response. Indeed, the combination of ER stress relief and T-cell depletion allows liver metastasis formation (57). Another study supports a pre-eminent role for CD8+ T-cells, but not CD4+ cells, in the immunosurveillance of DCCs in a model of spontaneous melanoma (122). CD8+ T-cells have also been shown to induce a state of dormancy in murine B cell lymphoma via the production of INFy (123), while in fibrosarcoma, either CD8+ T-cell or NK cell depletion lead to spontaneous metastasis in immune-competent mice. An orchestrated response involving different immune subpopulations has been suggested in a model of chemotherapy-induced dormancy in ER negative breast cancer. Here, the signaling activation of the IRF7/IFN-β/IFNAR axis following chemotherapy is associated with a reduction in granulocytes and expansion of T and B lymphocytes and dendritic cells (124).
In Vivo Mouse Models to Study Immune-DDCCs Crosstalk
In the past decades several mouse models have been generated, each of them with strength and limitations in modeling the metastatic cascade [reviewed in (125)]. Experimental metastatic spread can be achieved i) by spontaneous dissemination of cells after formation of a primary tumor (either by cancer cell transplant or genetically-induced) or ii) by injecting cancer cells in the bloodstream (either allografts of mouse cells, or xenografts of human cells). The former have the advantage to mimic all the stages of metastatic colonization, the latter are more rapid and allow genetic manipulation of the injected cells, a prerequisite for some labeling techniques described later. Transgenic mouse models of dormant/indolent metastatic mammary cancers are worth mentioning, because they provide a good opportunity to study DDCCs in an in vivo immunocompetent animal. So far, three transgenic breast cancer models with spreading of indolent BCCs (breast cancer cells)have been reported: MMTV-Her2 (126), MMTV-PyMT (127), MMTV-Wnt1 (128). MMTV-Her2 and MMTV-PyMT models were used to support the early dissemination hypothesis, whereby dormant BCCs could be retrieved from the lung before the detection of the primary tumor lesion (127). Similarly, mammary cancers originating from the MMTV-Wnt1 transgene spread asymptomatic cells to lung and lymph nodes (128). Importantly, disseminated cells can be reawakened from dormancy following systemic triggers like bone marrow transplant or surgery. More recently, MMTV-Her2 and MMTV-PyMT mice have been used to describe the role of progesterone receptor, Her2 and partial-EMT into early dissemination (21, 126). The main limitation of the aforementioned models is that murine and human immune systems have important differences that undermined clinical translation of several preclinical findings. Moreover, the use of human cancer cells requires the use of immunodecifient or immunocompromised mice, which obviously fail to capture DDCCs-immune TME crosstalk.
Development of “humanized mice” started thirty years ago with the aim of studying human diseases in mice with a human immune system. Humanized mice are immunocompromised mice transplanted with human peripheral blood mononuclear cells (PBMCs), hematopoietic stem cells (HSCs) or human fetal tissues (thymus and liver) (129). Engraftment of patient-derived xenograft, or PDX (tumor fragments or single-cell suspension from tumor resections), are also employed to reconstitute the TME in mouse models. So far these models have been exploited mainly as preclinical testing platforms, as treatment responses in PDX have been correlated to those observed in patients in several cancer types (130), but they hold great potential to uncover previously overlooked human-specific aspects of immune-DDCCs crosstalk.
Technological Advances in Studying the Tumor Microenvironment
Advances in biomaterial technologies, including 3D bioprinting, are fundamental to model TME in vitro (131–136). The use of complex multi-cultures to mimic and perturb metastatic dormancy in vitro has been rapidly expanding, as reviewed in (24). More recently, the advances in microfluidic technologies are also boosting the development of cancer-on-chip models to better recapitulate multiple parameters of the TME complexity in vitro (137, 138) (Figure 1).
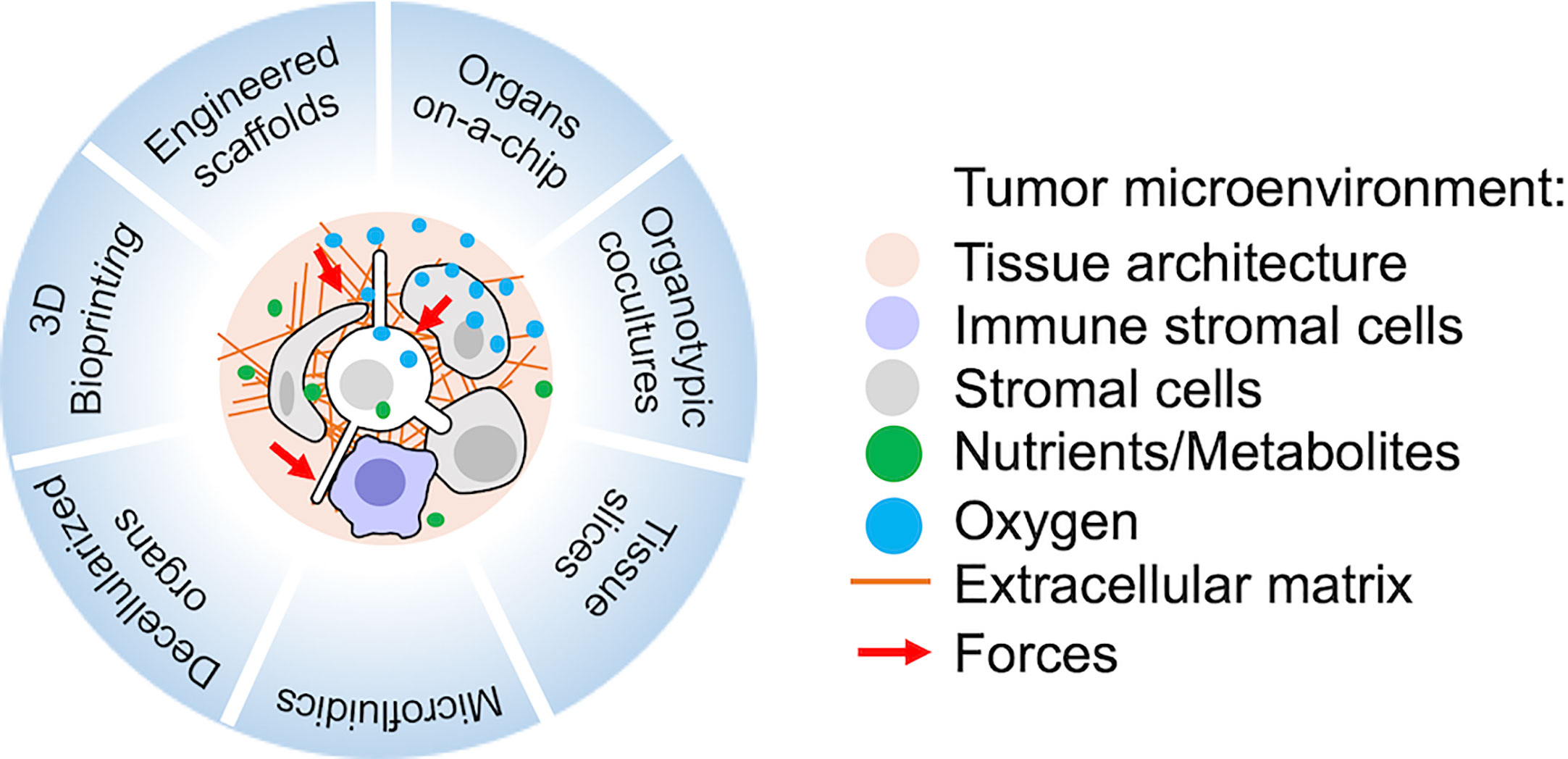
Figure 1 Recent technologies suitable for the study of metastatic tumor microenvironment (TME) in vitro or ex vivo. Relevant reviews with references to the original works and protocols are provided in the main text.
Biomaterial Technologies and 3D Bioprinting
The availability of new biomaterials also improved the studies of metastatic and immune cell interactions in vivo. Advancements in biomaterials allow to mimic the natural architecture of human tissues with scaffolds of tunable properties, either of natural (for example Matrigel) or synthetic origin (such as PCL, PLGA or PEG, polycaprolactone, polylactic-co-glycolic acid, polyethylene glycol, respectively) (132, 134). 3D scaffolds have been used to study the effects of ECM components and physical tissue properties as well as to dissect interactions between disseminated cells and stromal cells, such as fibroblasts, endothelial cells and macrophages (24, 134). Heterotopically implanted 3D hydrogels have been used to recreate artificial metastatic niches in vivo. Interestingly, following implantation they were infiltrated by immune cells and able to attract DCCs (139–141). Moreover, these systems can be easily manipulated to release cytokines and attract specific immune populations (140), representing a powerful tool to study how metastatic cells interact with host cells in vivo at a molecular level. However, they do not reflect the actual metastatic site composition whose replication remains difficult. This challenge has been partially overcome for leukemic tumors, thanks to the ability of HSC and progenitor cells to engraft and re-create a bone marrow environment. Humanized bone marrow environments have been used to study cellular interactions with human HSCs as well as malignant leukemic cells (142, 143). Interestingly, the engineered human bone-marrow niche can recapitulate main features of the pre-metastatic niche and attract DCCs, allowing to study the progression of the metastatic cascade (144). Importantly, 3D scaffolds have been successfully used as platforms for drug screening.
Three-dimensional bioprinting represents the most sophisticated strategy to achieve spatial control of matrix properties, integration of perfusable vascular networks and precise cancer-stroma cellular interaction (135). With 3D bioprinting, tissue spheroids, microcarriers, cell clusters, pellets, biomaterials and/or decellularized ECM can be deposited as bioinks under the control of computer designed patterns (135). With this technology, cancer models for several tissues have been generated, suggesting that metastatic niches as well could be designed in the near future.
Decellularized Organs and Precision Cut Tissue Slices
Hundreds of ECM proteins and carbohydrates are known to date, and their combination is key to the specificity of any cellular niche. Thus, reconstituting the exact ECM composition in vitro is almost impossible. For these reasons, several groups developed protocols to remove the cellular components of cell/ECM constructs, leaving decellularized ECM (dECM) that can be used for advanced in vitro model systems. dECM can be derived from native tissues or from tissues/organs generated in vitro. Decellularization protocols include chemical, physical or enzymatical approaches (or combinations) and the method clearly affects how much the resulting dECM resembles the native ECM of origin (145, 146). Decellularized tissues have been used to mimic breast cancer colonization of lungs (147) and adipose tissue (148). Importantly, dECM can be derived from patients, this allowed Pinto and colleagues to study crosstalk between colon cancer cells and macrophages within dECM from healthy and compromised tissues (149).
Long term ex vivo cultures of precision cut tissue slices and decellularized organs could be repurposed to study TME-DDCCs crosstalk in several secondary organs, such as lung, liver and brain (146, 150–153). Moreover, the use of intravital imaging technology combined with skin-fold chambers or optical windows has also allowed researchers to examine complex events in vivo, particularly in the context of primary tumors (154–156), but also in studying metastasis in organs such as bone, brain, liver and lung (157–159). However, the study of the metastatic TME in vivo remains technically difficult, particularly at an early stage of the disease, when small tumor nodules need to be spatially located in the metastatic tissue. This challenge is even bigger when dormant cell clusters or single DCCs need to be visualised and their neighbouring cells identified.
Microfluidic Systems and Organs-on-a-Chip
Another significant technological improvement for the design of metastatic niche in vitro is the development of microfluidic scaffolds. These platforms allow modelling of barriers and interfaces of tissues as well as a tight control of forces, perfusion and strains. Interfaces can be based on synthetic materials, hydrogels or self-assembled (160, 161). Organs-on-a-chip employ a combination of all the above techniques to generate organotypic models with geometrically defined multicellular composition, mechanical/electrical/biochemical stimulation and controlled liquid flow (160, 161). Organs-on-a-chip have been generated for lungs, heart, kidney, liver, muscle, while chips recapitulating immune responses have been underexplored so far, with the notable exceptions reviewed in ref. (162).
Laser-Capture Microdissection
The introduction of laser-capture microdissection technology has been largely used to study TME over the last 15 years (163, 164). The possibility to laser-cut a piece of tumor from a tissue section and specifically isolate cells from the TME by fluorescence activated cell sorting (FACS) enormously contributed to our knowledge in the field. However, despite being a powerful methodology this has some major limitations, mostly due the quality of the isolated material from a fixed tissue. Moreover, this approach could be very complicated to adopt when the spatial location of small metastatic nodules is required. Techniques involving the labeling of stromal cells within the niche could overcome these limitations. Once labeled, these cells can be isolated as live cells by flow cytometry, allowing their functional characterization ex vivo. Nowadays, the possibility to couple in vivo labeling techniques with state-of-art single cell analysis could enormously extend what we know about the role of the TME in the coming years.
In Vivo Labeling of Metastatic Niche
In this paragraph, we will discuss in detail some recently developed in vivo labeling methods. These systems have potential to be optimized in the context of dormancy and may finally reveal the “dormant niche” in vivo.
The techniques most commonly used to identify and isolate cells from tissues, including the most recent ones we describe in this section, imply using Fluorescence-Activated Cell Sorting (FACS) during the procedure. FACS has indeed proved to be a key asset to study the TME and the use of specific cellular markers has been critical to characterise different cell populations within the TME. In Supplementary Table 1, we provide a list of markers that might be useful to identify the cellular populations in the TME (this list has to be considered as a simplified guide to roughly discriminate the most abundant cellular components and need to be refined according to specific experimental needs and continuously revised as new findings emerge). However, advances in new technologies, and particularly the advent of the single cell RNA sequencing (scRNA-seq), keep revealing how the expression of markers initially thought to be exclusive of one lineage population are actually shared among different cell populations. Moreover, high heterogeneity and plasticity have been observed within the same cellular components in the TME, for example in tumor-associated macrophages and cancer-associated fibroblasts among the others (165–170). All this complexity makes it difficult to distinctively isolate some cellular sub-populations. The possibility to couple unbiased niche labeling methods with scRNA-seq could help to define more precise combinations of markers to identify specific subpopulations.
The generation of a genetic mouse model expressing a photoactivatable GFP (171) coupled with the two-photon microscopy technology allowed photoactivation of specific regions of inguinal lymph nodes with a technique called NICHE-seq (Figure 2) (172). Labeled cells were isolated by FACS and analysed by single cell RNA-sequencing. The same approach has shown potential to photoactivate regions surrounding melanoma cells (172). The main limitation is represented by the physical accessibility of the tissue to imaging and photoactivation, and by the requirement to precisely locate the cancer cell within an entire tissue, that can be particularly challenging in the case of isolated DDCCs.
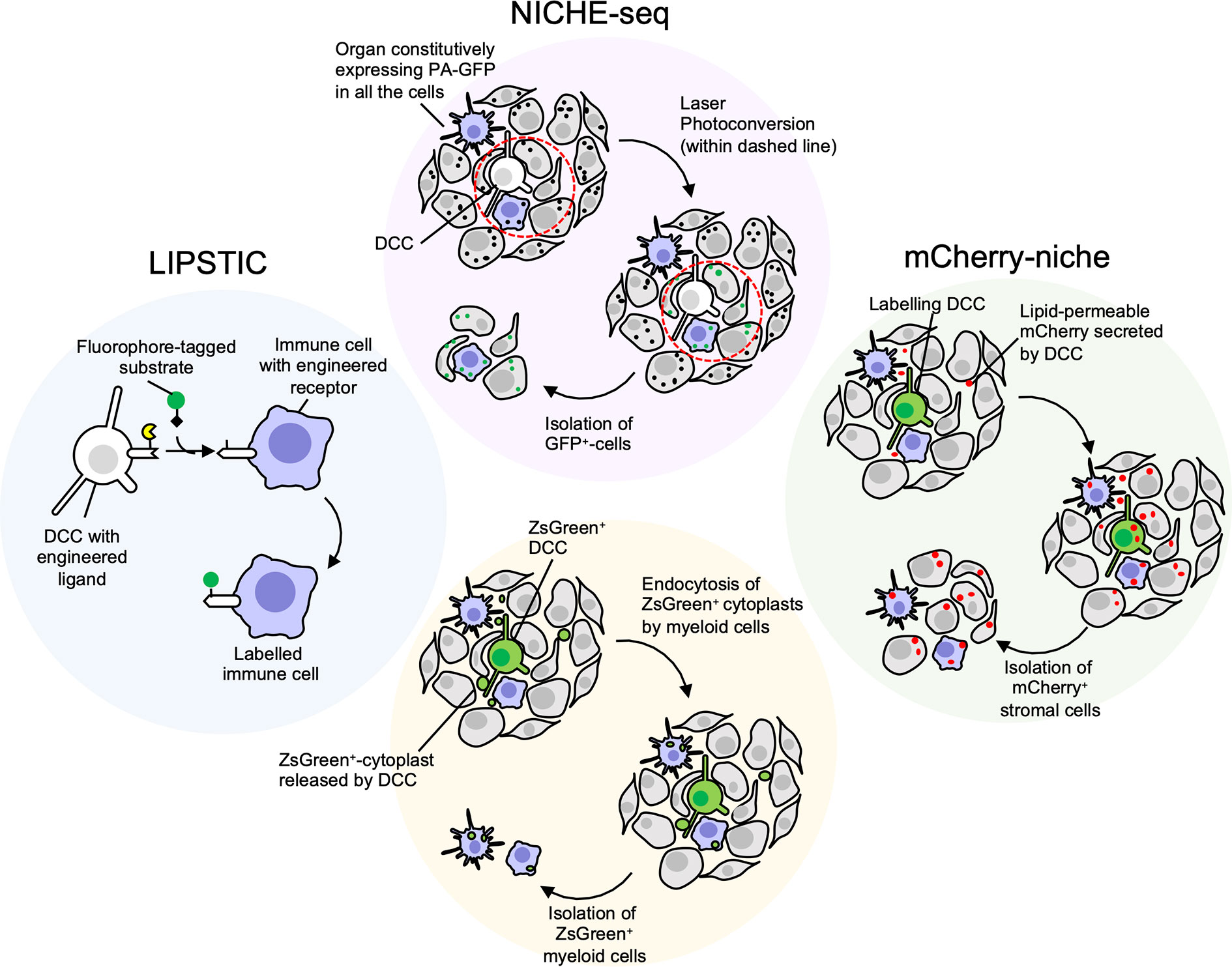
Figure 2 Niche-labeling techniques for characterization of immune metastatic tumor microenvironment (TME). White/Green: cancer cells; Grey: stromal cells; Purple: immune stromal cells. NICHE-seq (172) employs transgenic mice constitutively expressing photoactivatable GFP (PA-GFP), a fluorescent protein that increases its emission after excitation with 413 nm light (black dots: dark state; green dots: fluorescent state after photoconversion). Once a disseminated cancer cell (DCC) is located in the metastatic organ the surrounding niche can be irradiated and GFP+ cells isolated. Main limitations: i) the difficulties to spatially locate few scattered dormant DCCs (DDCCs) throughout entire organs; ii) the accessibility of those organs for photoconversion in vivo. The latter issue can be overcome with ex vivo photoconversion of freshly explanted organs. LIPSTIC (labeling Immune Partnerships by SorTagging Intercellular Contacts) (173) is an intercellular enzymatic labeling technique that exploits Staphylococcus aureus transpeptidase sortase A (SrtA, in yellow). Here, SrtA transfers a substrate containing “LPXTG” motif (black diamond), fused with biotin or fluorophore, to five N-terminal glycine residues tag (G5). This transfer requires proximity of SrtA and G5, thus a receptor and its membrane-bound ligand are fused with either SrtA and G5 in different cells. If these cells, that could be DDCCs and stromal cells, lie in close proximity at the metastatic site, stromal cells surrounding DDCCs are labeled and can be isolated for further analysis. Main limitations: i) the stromal lineage of interest must be genetically engineered a priori with tagged receptor or ligand, making this technique not suitable for unbiased identification of niche stromal cells; ii) the cells must be in close proximity for the reaction to happen. Cherry-niche (172) was developed to overcome these limitations. Here, the mCherry protein is engineered with a lipid-permeable domain (sLP-mCherry). DCCs expressing the sLP-mCherry release the protein in the extracellular space and the protein is uptaken by neighboring cells that can be isolated and analysed. Similarly, Krummel lab observed that blebs (cytoplasts, green dots) released by disseminated ZsGreen+-melanoma cells are endocytosed by resident myeloid cells (159). This approach is limited to DCCs releasing a significant amount of blebs and to stromal cells with efficient endocytic capacity.
To overcome this limitation, Headley and colleagues engineered melanoma cells to express cytoplasmic Zs-green, a highly brilliant fluorescent protein, that is also incorporated in tumor cell fragments (cytoplasts). By endocyting these fragments, neighboring cells become fluorescent themselves and can be visualised or isolated (159). The efficacy of this method depends on the amount of microvesicles the tumor cells release and on the ability of the neighbouring cells to internalise/phagocyte them, therefore limiting cell detection mostly to myeloid immune cells.
In an alternative method called LIPSTIC, a receptor-ligand interaction can be marked by the transferring of a biotin-tag on the recipient cells (173). Here, “donor” T cells expressing a CD40L fused to Sortase A interacted with B-cells engineered to express an “acceptor” domain fused to the CD40 receptor. When the receptor-ligand interaction occurs in presence of a fluorescent or a biotinylated substrate, the acceptor cells are labeled. This strategy implies a physical interaction between cells, and a ligand-receptor pair previously engineered and expressed by the right cell lineage(s). Moreover, the ectopic expression of endogenous ligand-receptors may cause unwanted biological effects, thus suggesting the need to engineer more neutral synthetic systems.
Another approach that we have recently developed, named Cherry-niche, allows engineered cancer cells to label their surroundings by transferring a modified red-fluorescent protein (174). Neighbouring cells of the cancer cells endocyte this protein and become fluorescent. Thanks to its liposoluble features, Cherry-niche does not require direct cell-cell contact nor a-priori knowledge of the recipient cells, as all the surrounding cells have the potential to internalise the fluorescent tag. Importantly, in in vivo organs, such as in the lung, the bulk of the labeling is limited to the close proximity of the metastatic cells, highlighting the potential of Cherry-niche to specifically reveal the cancer neighbouring cells.
Conclusions
In the last decade tremendous advancements have been achieved in oncology following the development of cutting-edge techniques. Among the different aspects of cancer biology, survival, quiescence and outgrowth of DCCs remained underexplored due to experimental hurdles such as faithfully modeling of metastatic organs in vitro and labeling of metastatic TME in vivo. In this review we presented recent techniques that in our opinion will give great impulse towards these directions. Despite this, our knowledge of DDCCs in human patients is extremely limited. This is mostly due to the current lack of techniques to track single or small clusters of DDCCs, together with ethical and technical issues with collecting and analysing metastatic organs in cured healthy patients. A notable exception is the bone marrow, a frequent site of relapse for several cancers. From this tissue, single DDCCs have been isolated, profiled (175) and provided clinical evidence of the existence of DDCCs in patients with no evidence of disease (176, 177). Isolation of circulating tumor cells or circulating tumor-derived factors from blood biopsies holds great potential to bypass the aforementioned limitations (178), although work is still needed to identify DCCs with metastasis-forming ability from a heterogeneous population of DCCs. Moreover, DDCCs do not effectively respond to chemotherapies or radiation therapies as a consequence of quiescence and because of the protective role of microenvironment (45, 179), thus, immunotherapy holds great hopes for clearing organs from DDCCs before relapse. In the light of this, it will be of utmost importance to exploit the most recent techniques to deepen our knowledge of DDCCs-immune cells crosstalk at the metastatic site.
Author Contributions
LO and MM equally contributed to conceiving and writing the review. All authors contributed to the article and approved the submitted version.
Funding
LO was funded by a Barts Charity Lectureship (grant MGU045). MM also received funding from Marie Curie Actions-Intra-European Fellowships no. 625496 and BIRD Seed grant from Department of Molecular Medicine (University of Padua).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Dr. Victoria L Bridgeman for reviewing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.594514/full#supplementary-material
Supplementary Table 1 | Lineage markers to identify cell populations in the murine TME. TME components have been roughly categorised here in broad stromal cell types (immune, endothelial, mesenchymal and parenchymal). CD45 can be used to identify all the immune cells. To further discriminate subpopulation within the immune compartment it needs to be combined with other markers. For example, a combination of CD45, CD11b and Ly6G will distinctively identify the neutrophils. This list represents a very simplified guide to roughly discriminate the most abundant cellular components in the TME, therefore not including all type of cells that might be found. We recommend to refine the marker combination to use according to specific experimental needs. Moreover, new findings may need to be considered to implement or update this list.
References
1. Summers MA, McDonald MM, Croucher PI. Cancer Cell Dormancy in Metastasis. Cold Spring Harb Perspect Med (2020) 10:a037556. doi: 10.1101/cshperspect.a037556
2. Dillekås H, Demicheli R, Ardoino I, Jensen SA, Biganzoli E, Straume O. The recurrence pattern following delayed breast reconstruction after mastectomy for breast cancer suggests a systemic effect of surgery on occult dormant micrometastases. Breast Cancer Res Treat (2016) 158:169–78. doi: 10.1007/s10549-016-3857-1
3. Uhr J, Pantel K. Controversies in clinical cancer dormancy. Proc Natl Acad Sci (2011) 108:12396–400. doi: 10.1073/pnas.1106613108
4. Gomis RR, Gawrzak S. Tumor cell dormancy. Mol Oncol (2017) 11:62–78. doi: 10.1016/j.molonc.2016.09.009
5. Kim K, Marquez-Palencia M, Malladi S. Metastatic Latency, a Veiled Threat. Front Immunol (2019) 10:1836. doi: 10.3389/fimmu.2019.01836
6. Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med (2017) 377:1836–46. doi: 10.1056/NEJMoa1701830
7. Zhang XH-F, Giuliano M, Trivedi MV, Schiff R, Osborne CK. Metastasis Dormancy in Estrogen Receptor–Positive Breast Cancer. Clin Cancer Res (2013) 19:6389–97. doi: 10.1158/1078-0432.CCR-13-0838
8. Demicheli R, Retsky MW, Hrushesky WJ, Baum M. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat Clin Pract Oncol (2007) 4:699–710. doi: 10.1038/ncponc0999
9. Demicheli R, Desmedt C, Piccart M, Biganzoli E. Tumor dormancy at bedside: A late awakening. Breast (2019) 45:61–3. doi: 10.1016/j.breast.2019.03.001
10. Weigl J, Hauner H, Hauner D. Can Nutrition Lower the Risk of Recurrence in Breast Cancer. Breast Care (2018) 13:86–91. doi: 10.1159/000488718
11. Chambers AF. Influence of diet on metastasis and tumor dormancy. Clin Exp Metastasis (2009) 26:61–6. doi: 10.1007/s10585-008-9164-4
12. Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, Dougan M, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med (2018) 10:eaan3464. doi: 10.1126/scitranslmed.aan3464
13. Friberg S, Nyström A. Cancer Metastases: Early Dissemination and Late Recurrences. Cancer Growth Metastasis (2015) 8:43–9. doi: 10.4137/CGM.S31244
14. Stephens J, Everson G, Elliott C, Kam I, Wachs M, Haney J, et al. Fatal transfer of malignant melanoma from multiorgan donor to four allograft recipients. Transplantation (2000) 70:232–6.
15. Strauss DC, Thomas JM. Transmission of donor melanoma by organ transplantation. Lancet Oncol (2010) 11:790–6. doi: 10.1016/S1470-2045(10)70024-3
17. Panigrahy D, Gartung A, Yang J, Yang H, Gilligan MM, Sulciner ML, et al. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. J Clin Invest (2019) 129:2964–79. doi: 10.1172/JCI127282
18. Giancotti FG. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell (2013) 155:750–64. doi: 10.1016/j.cell.2013.10.029
19. Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase Receptor and Fibronectin Regulate the ERKMAPK to p38MAPK Activity Ratios That Determine Carcinoma Cell Proliferation or Dormancy In Vivo. Mol Biol Cell (2001) 12:863–79. doi: 10.1091/mbc.12.4.863
20. Montagner M, Bhome R, Hooper S, Chakravarty P, Qin X, Sufi J, et al. Crosstalk with lung epithelial cells regulates Sfrp2 -mediated latency in breast cancer dissemination. Nat Cell Biol (2020) 22(3):289–96. doi: 10.1038/s41556-020-0474-3
21. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature (2016) 540:588–92. doi: 10.1038/nature20609
22. Fluegen G, Alvaro A-V, Wang Y, Padgen MR, Williams JK, Nobre ARR, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol (2017) 19:120–32. doi: 10.1038/ncb3465
23. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol (2013) 15:807–17. doi: 10.1038/ncb2767
24. Montagner M, Sahai E. In vitro Models of Breast Cancer Metastatic Dormancy. Front Cell Dev Biol (2020) 8:37. doi: 10.3389/fcell.2020.00037
25. Aguirre-Ghiso JA, Sosa MS. Emerging Topics on Disseminated Cancer Cell Dormancy and the Paradigm of Metastasis. Annu Rev Cancer Biol (2018) 2:377–93. doi: 10.1146/annurev-cancerbio-030617-050446
26. Linde N, Fluegen G, Julio AA-G. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv Cancer Res (2016) 132:45–71. doi: 10.1016/bs.acr.2016.07.002
27. Gawrzak S, Rinaldi L, Gregorio S, Arenas EJ, Salvador F, Urosevic J, et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER + breast cancer. Nat Cell Biol (2018) 20:211–21. doi: 10.1038/s41556-017-0021-z
28. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat Cell Biol (2013) 15:1351–61. doi: 10.1038/ncb2861
29. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med (2011) 208:2641–55. doi: 10.1084/jem.20110840
30. Hartsough MT, Morrison DK, Salerno M, Palmieri D, Ouatas T, Mair M, et al. Nm23-H1 Metastasis Suppressor Phosphorylation of Kinase Suppressor of Ras via a Histidine Protein Kinase Pathway. J Biol Chem (2002) 277:32389–99. doi: 10.1074/jbc.M203115200
31. Palmieri D, Halverson DO, Ouatas T, Horak CE, Salerno M, Johnson J, et al. Medroxyprogesterone Acetate Elevation of Nm23-H1 Metastasis Suppressor Expression in Hormone Receptor–Negative Breast Cancer. JNCI J Natl Cancer Inst (2005) 97:632–42. doi: 10.1093/jnci/dji111
32. Hickson JA, Huo D, Griend DJV, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 Kinases MKK4 and MKK6 Suppress Metastatic Colonization in Human Ovarian Carcinoma. Cancer Res (2006) 66:2264–70. doi: 10.1158/0008-5472.CAN-05-3676
33. Balz LM, Bartkowiak K, Andreas A, Pantel K, Niggemann B, Zänker KS, et al. The interplay of HER2/HER3/PI3K and EGFR/HER2/PLC-γ1 signalling in breast cancer cell migration and dissemination. J Pathol (2012) 227:234–44. doi: 10.1002/path.3991
34. Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell (2009) 16:67–78. doi: 10.1016/j.ccr.2009.05.017
35. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The Osteogenic Niche Promotes Early-Stage Bone Colonization of Disseminated Breast Cancer Cells. Cancer Cell (2015) 27:193–210. doi: 10.1016/j.ccell.2014.11.017
36. Wang H, Tian L, Liu J, Goldstein A, Bado I, Zhang W, et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell (2018) 34:823–39.e7. doi: 10.1016/j.ccell.2018.10.002
37. Paszek MJ, Christopher CD, Rossier O, Bainer R, Mouw JK, Godula K, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature (2014) 511:319–25. doi: 10.1038/nature13535
38. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res (2008) 68:6241–50. doi: 10.1158/0008-5472.CAN-07-6849
39. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, et al. Metastatic growth from dormant cells induced by a Col-I-enriched fibrotic environment. Cancer Res (2010) 70:5706–16. doi: 10.1158/0008-5472.CAN-09-2356
40. Montagner M, Dupont S. Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate. Cells (2020) 9:250. doi: 10.3390/cells9010250
41. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361:eaao4227. doi: 10.1126/science.aao4227
42. Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol (2018) 20:966–78. doi: 10.1038/s41556-018-0138-8
43. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell (2011) 20:701–14. doi: 10.1016/j.ccr.2011.11.002
44. Chen Q, Zhang XH, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell (2011) 20:538–49. doi: 10.1016/j.ccr.2011.08.025
45. Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol (2019) 21:238–50. doi: 10.1038/s41556-018-0267-0
46. Gao H, Chakraborty G, Ai L-L, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell (2012) 150:764–79. doi: 10.1016/j.cell.2012.06.035
47. Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, et al. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep (2016) 6:36520. doi: 10.1038/srep36520
48. Montagner M, Martello G, Piccolo S. Monitoring smad activity in vivo using the xenopus model system. Methods Mol Biol (2016) 1344:245–59. doi: 10.1007/978-1-4939-2966-5_15
49. Oskarsson T, Acharyya S, Zhang XHFF, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med (2011) 17:867–74. doi: 10.1038/nm.2379
50. Malanchi I, Albert S-M, Susanto E, Peng H, Lehr H-A, Delaloye J-F, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2011) 481:85–9. doi: 10.1038/nature10694
51. Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med (2018) 216(2):428–49. doi: 10.1084/jem.20180661
52. Capulli M, Hristova D, Valbret Z, Carys K, Arjan R, Maurizi A, et al. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br J Cancer (2019) 121:157–71. doi: 10.1038/s41416-019-0501-y
53. Gao H, Chakraborty G, Zhang Z, Akalay I, Gadiya M, Gao Y, et al. Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell (2016) 166:47–62. doi: 10.1016/j.cell.2016.06.009
54. Chen D, Sun Y, Wei Y, Zhang P, Rezaeian A, Julie T-F, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med (2012) 18:1511–7. doi: 10.1038/nm.2940
55. La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun (2019) 10:3668. doi: 10.1038/s41467-019-11640-9
56. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE, Vera-Ramirez L, et al. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun (2018) 9:1944. doi: 10.1038/s41467-018-04070-6
57. Pommier A, Anaparthy N, Memos N, Kelley Z, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science (2018) 360:eaao4908. doi: 10.1126/science.aao4908
58. Vera-Ramirez L. Cell-intrinsic survival signals. The role of autophagy in metastatic dissemination and tumor cell dormancy. Semin Cancer Biol (2020) 60:28–40. doi: 10.1016/j.semcancer.2019.07.027
59. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-Induced Lysyl Oxidase Is a Critical Mediator of Bone Marrow Cell Recruitment to Form the Premetastatic Niche. Cancer Cell (2009) 15:35–44. doi: 10.1016/j.ccr.2008.11.012
60. Barney LE, Hall CL, Schwartz AD, Parks AN, Sparages C, Galarza S, et al. Tumor cell–organized fibronectin maintenance of a dormant breast cancer population. Sci Adv (2020) 6:eaaz4157. doi: 10.1126/sciadv.aaz4157
61. Sowder ME, Johnson RW. Bone as a Preferential Site for Metastasis. JBMR Plus (2019) 3:e10126. doi: 10.1002/jbm4.10126
62. Cackowski F, Eber MR, Rhee J, Decker A, Yumoto K, Berry JE, et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J Cell Biochem (2017) 118:891–902. doi: 10.1002/jcb.25768
63. Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood (2019) 134:30–43. doi: 10.1182/blood.2018880930
64. Shiozawa Y, Pedersen EA, Taichman RS. GAS6/Mer axis regulates the homing and survival of the E2A/PBX1-positive B-cell precursor acute lymphoblastic leukemia in the bone marrow niche. Exp Hematol (2010) 38:132–40. doi: 10.1016/j.exphem.2009.11.002
65. Taichman RS, Patel LR, Bedenis R, Wang J, Weidner S, Schumann T, et al. GAS6 Receptor Status Is Associated with Dormancy and Bone Metastatic Tumor Formation. PloS One (2013) 8:e61873. doi: 10.1371/journal.pone.0061873
66. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest (2011) 121:1298–312. doi: 10.1172/JCI43414
67. Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med (2016) 8:340ra73–340ra73. doi: 10.1126/scitranslmed.aad4059
68. Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz Á, et al. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell (2019) 24:769–784.e6. doi: 10.1016/j.stem.2019.02.018
69. Zhang XH-F, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of Bone Metastasis Seeds by Mesenchymal Signals in the Primary Tumor Stroma. Cell (2013) 154:1060–73. doi: 10.1016/j.cell.2013.07.036
70. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun (2015) 6:8983. doi: 10.1038/ncomms9983
71. Ottewell PD, Wang N, Brown HK, Fowles CA, Croucher PI, Eaton CL, et al. OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int J Cancer (2015) 137:968–77. doi: 10.1002/ijc.29439
72. Ooi LL, Zhou H, Kalak R, Zheng Y, Conigrave AD, Seibel MJ, et al. Vitamin D Deficiency Promotes Human Breast Cancer Growth in a Murine Model of Bone Metastasis. Cancer Res (2010) 70:1835–44. doi: 10.1158/0008-5472.CAN-09-3194
73. Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci (2009) 106:10290–5. doi: 10.1073/pnas.0904227106
74. Shibue T, Brooks MW, Weinberg RA. An Integrin-Linked Machinery of Cytoskeletal Regulation that Enables Experimental Tumor Initiation and Metastatic Colonization. Cancer Cell (2013) 24:481–98. doi: 10.1016/j.ccr.2013.08.012
75. Cox TR, Bird D, Baker A-M, Barker HE, Ho MW-Y, Lang G, et al. LOX-Mediated Collagen Crosslinking Is Responsible for Fibrosis-Enhanced Metastasis. Cancer Res (2013) 73:1721–32. doi: 10.1158/0008-5472.CAN-12-2233
76. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WEF, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med (2010) 16:116–22. doi: 10.1038/nm.2072
77. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
79. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
80. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA (2010) 107:21248–55. doi: 10.1073/pnas.1015855107
81. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature (2015) 528:413–7. doi: 10.1038/nature16140
82. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol (2015) 17:816–26. doi: 10.1038/ncb3169
83. Seubert B, Grünwald B, Kobuch J, Cui H, Schelter F, Schaten S, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology (2015) 61:238–48. doi: 10.1002/hep.27378
84. Chafe SC, Lou Y, Sceneay J, Vallejo M, Hamilton MJ, McDonald PC, et al. Carbonic Anhydrase IX Promotes Myeloid-Derived Suppressor Cell Mobilization and Establishment of a Metastatic Niche by Stimulating G-CSF Production. Cancer Res (2015) 75:996–1008. doi: 10.1158/0008-5472.CAN-14-3000
85. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell (2016) 30:243–56. doi: 10.1016/j.ccell.2016.06.021
86. Liu Y, Cao X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
87. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer (2017) 17:302–17. doi: 10.1038/nrc.2017.6
88. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-Mediated Metastasis: Communication from a Distance. Dev Cell (2019) 49:347–60. doi: 10.1016/j.devcel.2019.04.011
89. Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature (2015) 527:329–35. doi: 10.1038/nature15756
90. Keklikoglou I, Cianciaruso C, Güç E, Squadrito ML, Spring LM, Tazzyman S, et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat Cell Biol (2019) 21:190–202. doi: 10.1038/s41556-018-0256-3
91. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discovery (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
92. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N. Miller WH. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol (2020) 27:S87–97. doi: 10.3747/co.27.5223
93. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
94. Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, et al. Advances in cancer immunotherapy 2019 – latest trends. J Exp Clin Cancer Res (2019) 38:268. doi: 10.1186/s13046-019-1266-0
95. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183:1161–72. doi: 10.1084/jem.183.3.1161
96. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell derived exosomes. Nat Med (1998) 4:594–600. doi: 10.1038/nm0598-594
97. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
98. Poggio M, Hu T, Pai C-C, Chu B, Belair CD, Chang A, et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell (2019) 177:414–27.e13. doi: 10.1016/j.cell.2019.02.016
99. Kim DH, Kim H, Choi YJ, Kim SY, Lee J-E, Sung KJ, et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp Mol Med (2019) 51:1–13. doi: 10.1038/s12276-019-0295-2
100. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
101. Baxevanis CN, Perez SA. Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State. Vaccines (2015) 3:597–619. doi: 10.3390/vaccines3030597
102. Goddard ET, Bozic I, Riddell SR, Ghajar CM. Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol (2018) 20:1240–9. doi: 10.1038/s41556-018-0214-0
103. Manjili MH. The premise of personalized immunotherapy for cancer dormancy. Oncogene (2020) 39:4323–30. doi: 10.1038/s41388-020-1295-3
104. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discovery (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
105. Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol (2015) 15:73–86. doi: 10.1038/nri3789
106. Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun (2018) 9:21. doi: 10.1038/s41467-017-02481-5
107. De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res (2016) 76:6778–84. doi: 10.1158/0008-5472.CAN-16-0608
108. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
109. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med (2016) 8:361ra138–361ra138. doi: 10.1126/scitranslmed.aag1711
110. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer (2016) 16:431–46. doi: 10.1038/nrc.2016.52
111. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
112. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
113. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol (2015) 35:S185–98. doi: 10.1016/j.semcancer.2015.03.004
114. López-Soto A, Huergo-Zapico L, Galván JA, Rodrigo L, de Herreros AG, Astudillo A, et al. Epithelial–Mesenchymal Transition Induces an Antitumor Immune Response Mediated by NKG2D Receptor. J Immunol (2013) 190:4408–19. doi: 10.4049/jimmunol.1202950
115. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils Suppress Intraluminal NK Cell–Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discovery (2016) 6:630–49. doi: 10.1158/2159-8290.CD-15-1157
116. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell (2016) 165:45–60. doi: 10.1016/j.cell.2016.02.025
117. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi: 10.1038/ni1102-991
118. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
119. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature (2007) 450:903–7. doi: 10.1038/nature06309
120. Teng MWL, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, et al. Opposing Roles for IL-23 and IL-12 in Maintaining Occult Cancer in an Equilibrium State. Cancer Res (2012) 72:3987–96. doi: 10.1158/0008-5472.CAN-12-1337
121. Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature (2019) 565:366–71. doi: 10.1038/s41586-018-0812-9
122. Eyles J, Puaux A-L, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest (2010) 120:2030–9. doi: 10.1172/JCI42002
123. Farrar JD, Katz KH, Windsor J, Thrush G, Scheuermann RH, Uhr JW, et al. Cancer Dormancy. VII. A Regulatory Role for CD8+ T Cells and IFN-γ in Establishing and Maintaining the Tumor-Dormant State. J Immunol (1999) 162:2842–9.
124. Lan Q, Peyvandi S, Duffey N, Huang Y-T, Barras D, Held W, et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene (2019) 38:2814–29. doi: 10.1038/s41388-018-0624-2
125. Gómez-Cuadrado L, Tracey N, Ma R, Qian B, Brunton VG. Mouse models of metastasis: progress and prospects. Dis Model Mech (2017) 10:1061–74. doi: 10.1242/dmm.030403
126. Hosseini H, Obradović MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature (2016) 540:552–8. doi: 10.1038/nature20785
127. Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell (2008) 13:58–68. doi: 10.1016/j.ccr.2007.12.003
128. Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene (2000) 19:1002–9. doi: 10.1038/sj.onc.1203273
129. Allen TM, Brehm MA, Bridges S, Ferguson S, Kumar P, Mirochnitchenko O, et al. Humanized immune system mouse models: progress, challenges and opportunities. Nat Immunol (2019) 20:770–4. doi: 10.1038/s41590-019-0416-z
130. Wege AK. Humanized Mouse Models for the Preclinical Assessment of Cancer Immunotherapy. BioDrugs (2018) 32:245–66. doi: 10.1007/s40259-018-0275-4
131. Rao SS, Kondapaneni RV, Narkhede AA. Bioengineered models to study tumor dormancy. J Biol Eng (2019) 13:3. doi: 10.1186/s13036-018-0137-0
132. Pradhan S, Slater JH. Tunable hydrogels for controlling phenotypic cancer cell states to model breast cancer dormancy and reactivation. Biomaterials (2019) 215:119177. doi: 10.1016/j.biomaterials.2019.04.022
133. Aguado BA, Bushnell GG, Rao SS, Jeruss JS, Shea LD. Engineering the pre-metastatic niche. Nat BioMed Eng (2017) 1:77. doi: 10.1038/s41551-017-0077
134. Rijal G, Li W. 3D scaffolds in breast cancer research. Biomaterials (2016) 81:135–56. doi: 10.1016/j.biomaterials.2015.12.016
135. Datta P, Dey M, Ataie Z, Unutmaz D, Ozbolat IT. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis Oncol (2020) 4:1–13. doi: 10.1038/s41698-020-0121-2
136. Urciuolo A, Poli I, Brandolino L, Raffa P, Scattolini V, Laterza C, et al. Intravital three-dimensional bioprinting. Nat BioMed Eng (2020) 4:901–15. doi: 10.1038/s41551-020-0568-z
137. Sleeboom JJF, Amirabadi HE, Nair P, Sahlgren CM, den Toonder JMJ. Metastasis in context: modeling the tumor microenvironment with cancer-on-a-chip approaches. Dis Model Mech (2018) 11(3). doi: 10.1242/dmm.033100
138. Wan L, Neumann CA, LeDuc PR. Tumor-on-a-chip for integrating a 3D tumor microenvironment: chemical and mechanical factors. Lab Chip (2020) 20:873–88. doi: 10.1039/C9LC00550A
139. Lee J, Li M, Milwid J, Dunham J, Vinegoni C, Gorbatov R, et al. Implantable microenvironments to attract hematopoietic stem/cancer cells. Proc Natl Acad Sci (2012) 109:19638–43. doi: 10.1073/pnas.1208384109
140. Azarin SM, Yi J, Gower RM, Aguado BA, Sullivan ME, Goodman AG, et al. In vivo capture and label-free detection of early metastatic cells. Nat Commun (2015) 6:8094. doi: 10.1038/ncomms9094
141. Rao SS, Bushnell GG, Azarin SM, Spicer G, Aguado BA, Stoehr JR, et al. Enhanced Survival with Implantable Scaffolds That Capture Metastatic Breast Cancer Cells In Vivo. Cancer Res (2016) 76:5209–18. doi: 10.1158/0008-5472.CAN-15-2106
142. Abarrategi A, Foster K, Hamilton A, Mian SA, Passaro D, Gribben J, et al. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J Clin Invest (2017) 127:543–8. doi: 10.1172/JCI89364
143. Passaro D, Abarrategi A, Foster K, Ariza-McNaughton L, Bonnet D. Bioengineering of Humanized Bone Marrow Microenvironments in Mouse and Their Visualization by Live Imaging. JoVE J Vis Exp (2017) (126):e55914. doi: 10.3791/55914
144. Carpenter RA, Kwak J-G, Peyton SR, Lee J. Implantable pre-metastatic niches for the study of the microenvironmental regulation of disseminated human tumour cells. Nat BioMed Eng (2018) 2:915–29. doi: 10.1038/s41551-018-0307-x
145. Hoshiba T. Decellularized Extracellular Matrix for Cancer Research. Materials (2019) 12(8):1311. doi: 10.3390/ma12081311
146. Ferreira LP, Gaspar VM, Mano JF. Decellularized Extracellular Matrix for Bioengineering Physiomimetic 3D in Vitro Tumor Models. Trends Biotechnol (2020) 0. doi: 10.1016/j.tibtech.2020.04.006
147. Xiong G, Flynn TJ, Chen J, Trinkle C, Xu R. Development of an ex vivo breast cancer lung colonization model utilizing a decellularized lung matrix. Integr Biol (2015) 7:1518–25. doi: 10.1039/c5ib00157a
148. Dunne LW, Huang Z, Meng W, Fan X, Zhang N, Zhang Q, et al. Human decellularized adipose tissue scaffold as a model for breast cancer cell growth and drug treatments. Biomaterials (2014) 35:4940–9. doi: 10.1016/j.biomaterials.2014.03.003
149. Pinto ML, Rios E, Silva AC, Neves SC, Caires HR, Pinto AT, et al. Decellularized human colorectal cancer matrices polarize macrophages towards an anti-inflammatory phenotype promoting cancer cell invasion via CCL18. Biomaterials (2017) 124:211–24. doi: 10.1016/j.biomaterials.2017.02.004
150. Palma E, Doornebal EJ, Chokshi S. Precision-cut liver slices: a versatile tool to advance liver research. Hepatol Int (2019) 13:51–7. doi: 10.1007/s12072-018-9913-7
151. Liu G, Betts C, Cunoosamy DM, Åberg PM, Hornberg JJ, Sivars KB, et al. Use of precision cut lung slices as a translational model for the study of lung biology. Respir Res (2019) 20:162. doi: 10.1186/s12931-019-1131-x
152. Schwarz N, Uysal B, Welzer M, Bahr JC, Layer N, Löffler H, et al. Long-term adult human brain slice cultures as a model system to study human CNS circuitry and disease. eLife (2019) 8:e48417. doi: 10.7554/eLife.48417
153. Yu Y, Alkhawaji A, Ding Y, Mei J. Decellularized scaffolds in regenerative medicine. Oncotarget (2016) 7:58671–83. doi: 10.18632/oncotarget.10945
154. Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE, et al. Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods (2008) 5:1019–21. doi: 10.1038/nmeth.1269
155. Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P. Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochem Cell Biol (2008) 130:1147–54. doi: 10.1007/s00418-008-0529-1
156. Kitamura T, Pollard JW, Vendrell M. Optical Windows for Imaging the Metastatic Tumour Microenvironment in vivo. Trends Biotechnol (2017) 35:5–8. doi: 10.1016/j.tibtech.2016.05.001
157. Ellenbroek SIJ, van Rheenen J. Imaging hallmarks of cancer in living mice. Nat Rev Cancer (2014) 14:406–18. doi: 10.1038/nrc3742
158. Entenberg D, Rodriguez-Tirado C, Kato Y, Kitamura T, Pollard JW, Condeelis J. In vivo subcellular resolution optical imaging in the lung reveals early metastatic proliferation and motility. IntraVital (2015) 4:1–11. doi: 10.1080/21659087.2015.1086613
159. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature (2016) 531:513–7. doi: 10.1038/nature16985
160. Shang M, Soon RH, Lim CT, Khoo BL, Han J. Microfluidic modelling of the tumor microenvironment for anti-cancer drug development. Lab Chip (2019) 19:369–86. doi: 10.1039/C8LC00970H
161. Zhang B, Korolj A, Lai BFL, Radisic M. Advances in organ-on-a-chip engineering. Nat Rev Mater (2018) 3:257–78. doi: 10.1038/s41578-018-0034-7
162. Miller CP, Shin W, Ahn EH, Kim HJ, Kim D-H. Engineering Microphysiological Immune System Responses on Chips. Trends Biotechnol (2020) 38:857–72. doi: 10.1016/j.tibtech.2020.01.003
163. Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, et al. Laser-capture microdissection. Nat Protoc (2006) 1:586–603. doi: 10.1038/nprot.2006.85
164. Legres LG, Janin A, Masselon C, Bertheau P. Beyond laser microdissection technology: follow the yellow brick road for cancer research. Am J Cancer Res (2014) 4:1–28.
165. Kielbassa K, Vegna S, Ramirez C, Akkari L. Understanding the Origin and Diversity of Macrophages to Tailor Their Targeting in Solid Cancers. Front Immunol (2019) 10:2215. doi: 10.3389/fimmu.2019.02215
166. Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol Mech Dis (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
167. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
168. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell (2018) 33:463–79.e10. doi: 10.1016/j.ccell.2018.01.011
169. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discovery (2020) 10:1330–51. doi: 10.1158/2159-8290.CD-19-1384
170. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
171. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal Center Dynamics Revealed by Multiphoton Microscopy with a Photoactivatable Fluorescent Reporter. Cell (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
172. Medaglia C, Giladi A, Stoler-Barak L, Giovanni MD, Salame TM, Biram A, et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science (2017) 358:1622–6. doi: 10.1126/science.aao4277
173. Pasqual G, Chudnovskiy A, Tas JMJ, Agudelo M, Schweitzer LD, Cui A, et al. Monitoring T cell–dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature (2018) 553:496–500. doi: 10.1038/nature25442
174. Ombrato L, Nolan E, Kurelac I, Mavousian A, Bridgeman VL, Heinze I, et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature (2019) 572:603–8. doi: 10.1038/s41586-019-1487-6
175. Pantel K, Catherine A-P. Bone marrow as a reservoir for disseminated tumor cells: a special source for liquid biopsy in cancer patients. Bonekey Rep (2014) 3:584. doi: 10.1038/bonekey.2014.79
176. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun (2015) 6:6170. doi: 10.1038/ncomms7170
177. Chéry L, Lam HM, Coleman I, Lakely B, Coleman R, Larson S, et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget (2014) 5:9939–51. doi: 10.18632/oncotarget.2480
178. Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease — latest advances and implications for cure. Nat Rev Clin Oncol (2019) 16:409–24. doi: 10.1038/s41571-019-0187-3
Keywords: dormancy, metastasis, tumor microenviroment (TME), immune cells, labeling techniques
Citation: Ombrato L and Montagner M (2020) Technical Advancements for Studying Immune Regulation of Disseminated Dormant Cancer Cells. Front. Oncol. 10:594514. doi: 10.3389/fonc.2020.594514
Received: 13 August 2020; Accepted: 14 October 2020;
Published: 04 November 2020.
Edited by:
Angélica Santiago Gómez, The University of Manchester, United KingdomReviewed by:
Ana Luisa Correia, University of Basel, SwitzerlandHung-Ming Lam, University of Washington, United States
Copyright © 2020 Ombrato and Montagner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Montagner, bWFyY28ubW9udGFnbmVyQHVuaXBkLml0; Luigi Ombrato, bC5vbWJyYXRvQHFtdWwuYWMudWs=