 Lijuan Zhang1†
Lijuan Zhang1† Meng Tian
Meng Tian Jiamao Lin
Jiamao Lin Haiyong Wang
Haiyong Wang- 1Department of Pediatric Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China
- 2Department of Radiation Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
- 3Department of Medical Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
- 4Department of pathology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
- 5Department of Radiation Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
Estrogen receptor β (ERβ) can regulate cellular signaling through non-genomic mechanisms, potentially promoting resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). However, the mechanisms underlying the ERβ-mediated resistance to EGFR TKIs remain poorly understood. In this study, we investigated the role of the interaction between ERβ1 and ERβ5 in non-genomic signaling in lung adenocarcinoma. We established PC9 cell lines stably overexpressing ERβ1 or ERβ1/ERβ5. Immunofluorescence revealed that ERβ5 overexpression partly retained ERβ1 in the cytoplasm. Immunoblotting analyses revealed that EGFR pathway activation levels were higher in PC9/ERβ1/5 cells than those in PC9/ERβ1 or control PC9 cells. In the presence of estradiol, PI3K/AKT/mTOR pathway activation levels were higher in ERβ1/5-expressing cells than those in ERβ1-expressing cells. Additionally, PC9/ERβ1/5 cells were less prone to the cytotoxic and pro-apoptotic effects of gefitinib compared with PC9/ERβ1 or control PC9 cells. Cytoplasmic ERβ1 was associated with poor progression-free survival in lung cancer patients treated with EGFR TKIs. These results suggest that cytoplasmic ERβ1 was responsible for EGFR TKI resistance slightly through non-genomic mechanism in EGFR mutant lung adenocarcinoma.
Introduction
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have revolutionized non-small cell lung cancer (NSCLC) personalized treatment and have improved the survival and quality of life of EGFR-mutant NSCLC patients (1–3). However, the development of primary and acquired resistance to TKIs remains a significant clinical challenge. Several mechanisms underlying acquired resistance to EGFR TKIs have been identified, including the acquisition of EGFR T790M mutation, c-MET amplification, PIK3CA mutations, and phenotypic transformation into small cell lung cancer (4, 5). Nevertheless, the mechanisms involved in primary resistance are poorly understood (5–8).
Estrogen receptor β (ERβ) is the primary ER subtype expressed in lung cancer; upon binding to estrogen in the cytoplasm, ERβ activates non-genomic signaling pathways, including PI3K/AKT/mTOR and RAS/RAF/MEK/MAPK pathways, promoting cancer cell proliferation and apoptosis evasion (9, 10). Importantly, significant overlap exists between ERβ- and EGFR-regulated signaling pathways (11). Preclinical studies have shown that EGFR expression was downregulated in response to estradiol (E2); in contrast, ERβ antagonists upregulated EGFR expression, highlighting the crosstalk between ERβ and EGFR signaling (12). Hence, it is believed that non-genomic signaling events may modulate EGFR TKI resistance. ER belongs to the nuclear receptor superfamily of ligand-activated transcription factors. Since a nuclear localization of ERβ in cancer cells has been reported (13), the relevance of cytoplasmic ERβ in non-genomic signaling activation in cancer cells has attracted increasing attention in recent years.
Studies on endocrine-related cancers suggested that certain ERβ isoforms are associated with ERβ protein localization and patient prognosis (14–17). For example, ERβ1 (also known as wild-type ERβ) was primarily found in the nucleus of prostate cancer cells, whereas ERβ5 localized both in the cytoplasm and nucleus (16, 18, 19). Although ERβ5 lacks the ability to bind estrogen or form homodimers due to the absence of helix 12 in its C-terminal, it can heterodimerize with ERβ1 in the presence of estrogen (20).
The aim of this study was to assess the role of the interaction between ERβ1 and ERβ5 in non-genomic signaling in lung adenocarcinoma. To this end, we overexpressed ERβ1 and ERβ5 in EGFR exon 19 deletion-harboring lung adenocarcinoma cells and assessed their ability to form heterodimers, as well as the relevance of ERβ1/ERβ5 heterodimerization in non-genomic signaling and response to EGFR TKIs.
Materials and Methods
Cell Culture and Chemicals
The EGFR-mutant lung adenocarcinoma cells PC9, HCC827, H1975, and H1650 were kindly provided by Peking University Cancer Hospital. Cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and maintained at 37°C in a humidified 5% CO2 atmosphere.
Gefitinib was purchased from Selleck Chemicals (Selleck, USA) and diluted in dimethyl sulfoxide (DMSO) at a concentration of 10 mmol/L. Estradiol (E2) was purchased from Sigma-Aldrich (Sigma-Aldrich, Germany) and diluted in pure ethanol at a concentration of 10 mmol/L. Both drugs were aliquoted and stored at −80°C.
RNA Extraction and Quantitative Real-Time PCR
Total RNA was extracted using the RNAsimple Total RNA kit (Tiangen, China), and first-strand cDNA synthesis was performed using the PrimeScript™ RT Master Mix (Takara, Japan). The relative mRNA levels of ERβ1 and ERβ5 were measured using SYBR green PCR assays (Thermo Fisher Scientific, USA). The sequences of the primers used in qRT-PCR were as follows: ERβ1 forward primer: 5′-GTCAGGCATGCGAGTAACAA-3′, reverse primer: GGGAGCCCTCTTTGCTTTTA; ERβ5 forward primer: 5′-TGGTCACAGCGACCCAGGATG-3′, reverse primer: 5′-TTAGGGCGCGTACCTCGCATG-3′; GAPDH forward primer: 5′-GACCCCTTCATTGACCTCAAC-3′, reverse primer: 5′-CTTCTCCATGGTGGTGAAGA-3′. Cycle threshold (Ct) values were determined using the system and analysis software. The relative mRNA levels were determined by normalizing to the GAPDH mRNA levels.
Establishment of Cell Lines Stably Expressing Estrogen Receptor β1 and Estrogen Receptor β1/5
Lentiviral vectors expressing ERβ1 and ERβ5 were purchased from GenePharma (Shanghai, China). PC9 cells were infected with lentiviruses (MOI = 50) for three days. Subsequently, transduced cells were selected with 2 μg/ml of puromycin for one week. ERβ1-overexpressing single-cell clones were established (hereafter referred to as PC9/ERβ1 cells), and stable ERβ1 overexpression was confirmed by Western blot and qRT-PCR. PC9/ERβ1 cells were then infected with viruses carrying ERβ5 open reading frame (ORF), followed by selection with neomycin (600 mg/ml) for one week.
Immunofluorescence
ERβ expression was assessed by immunofluorescence (IF). Cells were fixed with 4% paraformaldehyde at room temperature for 20 min, followed by incubation with 0.5% Triton X-100 for 15 min. Non-specific binding was blocked with 5% bovine serum at 37°C for 30 min, before incubating with anti-ERβ (1:100; GeneTex, Cat No.: GTX70174, USA) and anti-ERβ1 (1:200; Abcam, Cat No.:ab187291, USA) primary antibodies. After incubation at 4°C overnight, cells were incubated with a secondary antibody conjugated with Alexa Fluor® 488 (1:500, Cell Signaling Technology, USA) for 1 h at room temperature. Subsequently, cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and samples were imaged using a confocal laser scanning microscope (Zeiss, Germany).
Immunoblotting Analysis
Cells were cultured in serum-free medium for 24 h and then treated with gefitinib (40 nM) or/and estradiol (20 nM) for another 8 h. Total protein was extracted from cells using cell lysis buffer (Beyotime, China) supplemented with a protease inhibitor cocktail (Roche, Germany); protein concentration was determined using the BCA protein assay (Beyotime, Beijing, China). The following primary antibodies (1:1,000) were used: anti-EGFR, anti-phospho-EGFR (Tyr1068), anti-AKT, anti-phospho-AKT (Ser473), anti-RPS6, anti-phosphor-RPS6 (Ser235/236), anti-P21, anti-CyclinD3, anti-cleaved-PARP (cPARP), and anti-beta-actin (Cell Signaling Technology, USA). Membranes were then incubated with peroxidase-linked anti-mouse or anti-rabbit secondary antibodies (1:5,000; Cell Signaling Technology, USA) for 2 h at room temperature. The expression levels of indicated proteins were quantified using quantity one.
Cell Viability and Colony Formation Assays
Cells were treated with estradiol (20 nM) during the experiment. Cell viability was assessed using a cell counting kit-8 (CCK8; Dojindo, Japan). Briefly, cells were seeded (3 × 103 cells/well) in sextuplicate in 96-well plates containing 100 μl medium and incubated for 24 h. Subsequently, cells were treated with increasing concentrations of the indicated drugs for an additional 72 h. After treatment, 10 μl of water-soluble tetrazolium salt (WST-8) was added to each well and incubated for 2 h. Optical absorbance at 450 nm was measured using a microplate reader. Relative viability was calculated using the following formula: Relative viability (%/control) = [A450 (treated) − A450 (blank)]/[A450 (control) − A450 (blank)].
For colony formation assays, cells were seeded into 6 cm cell culture dishes (500 cells/dish) and treated for two weeks with 40 nM gefitinib or DMSO (1/1,000 dilution). After washing twice with phosphate-buffered saline (PBS), cells were stained with crystal violet (Beyotime, China) for 20 min and washed with PBS.
Patients
The data from 103 Chinese patients with advanced lung adenocarcinoma were retrospectively reviewed. The inclusion criteria used for patient enrollment were as follows: (1) Pathological diagnosis of adenocarcinoma; (2) sufficient tissue for both EGFR and KRAS mutation detection and ERβ1 immunohistochemistry; (3) presence of EGFR mutations associated with sensitivity to EGFR TKIs, including 19 exon deletion and 21 exon point mutation, and absence of EGFR T790M or KRAS mutations; (4) patients treated with EGFR TKIs, including erlotinib, gefitinib, and icotinib; (5) available clinicopathological characteristics, including sex, age, disease stage, and smoking history. Treatment responses were classified according to the response evaluation criteria in solid tumors (RECIST), version 1.1. Progression-free survival (PFS) time was defined as the time between the first day of EGFR TKI treatment until radiologic progression or death. The study was approved by the Ethics Review Committee of the Shandong Cancer Hospital.
Epidermal Growth Factor Receptor and KRAS Mutation Detection and Immunohistochemistry for Estrogen Receptor β1
Amplification refractory mutation system (ARMS) was employed to detect different genetic variants, including EGFR (exon 19 deletions, L858R, and T790M) and KRAS mutations.
ERβ1 expression in lung adenocarcinoma tissue samples was assessed by immunohistochemistry (IHC). Informed consent to use biopsy tissues was obtained from all patients. Briefly, formalin-fixed, paraffin-embedded tissue sections (3 μm) were deparaffinized and stained according to standard procedures. Sections were probed with anti-ERβ1 mouse antibody (1:200; Abcam, USA); a biotinylated anti-mouse IgG secondary antibody was used. Brown staining in over 10% cancer cells with cytoplasm or/and nucleus was considered positive. No staining was observed in negative controls, including lung tissues probed with a non-immune primary antibody. Based on the localization of “positive” immunoreactivity in the cytoplasm, nucleus, or both, patients were grouped as cERβ1-, n/cERβ1-, or nERβ1-positive. IHC staining was evaluated independently by two investigators (LZ and MT) and a pathologist (JZ).
Statistical Analysis
Differences in the relative mRNA levels, cell viability, and apoptosis between different cell lines were analyzed using two-tailed Student’s t-tests. Difference of ERβ1 expression between male and female was analyzed using chi-square test. Patients’ survival was estimated using the Kaplan–Meier method, and comparisons between groups were conducted using log-rank tests. All statistical tests were two-tailed, and P-values <0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism 8.0 (Prism Software Inc., San Diego, USA).
Results
Estrogen Receptor β5 Affects Estrogen Receptor β1 Localization in Epidermal Growth Factor Receptor-Mutant Lung Adenocarcinoma Cancer Cells
ERβ5 has been identified as the predominant ERβ splice variant in non-malignant lung tissues. In this study, we found that ERβ5 mRNA levels were elevated in four lung adenocarcinoma cell lines harboring EGFR mutations (Figure 1A). We further assessed the role of ERβ5 in lung adenocarcinoma using PC9 cells, which harbor EGFR exon 19 deletions. Immunofluorescence analyses revealed that endogenous ERβ predominantly localized in the cell cytoplasm, and only low ERβ levels were detected in the nucleus (Figure 1B).
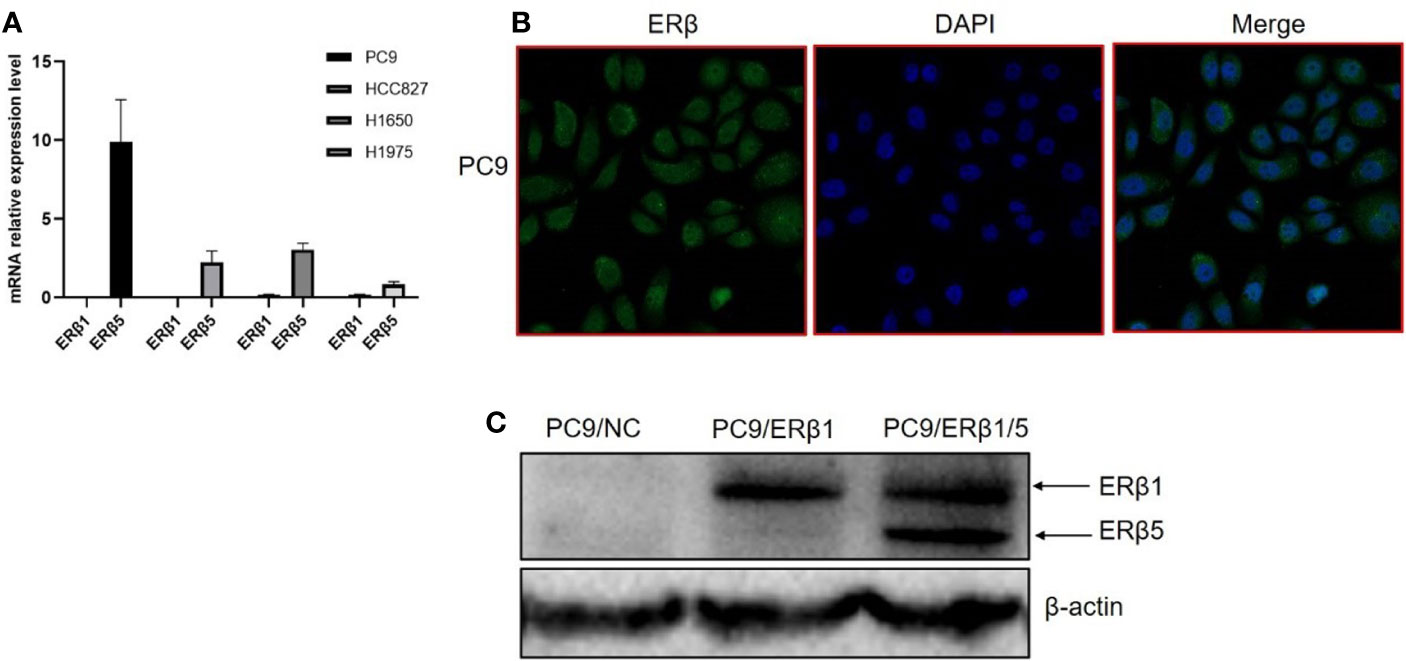
Figure 1 Construction of EGFR mutant lung cancer cell lines stably expressing ERβ1 and ERβ1/5. (A) mRNA level of ERβ1 and ERβ5 in four lung adenocarcinoma cell lines harboring EGFR mutation using specific primers determined by qRT PCR. Data shown as mean ± SD. (B) ERβ expression pattern was assessed by immunofluorescence in PC9. (C) Western blot for detecting the expression level of ERβ after stable transfection of ERβ1 and ERβ5. The upper stand refers to ERβ1 while the lower is ERβ5.
Next, we overexpressed ERβ1 in PC9 cells (PC9/ERβ1); we also overexpressed ERβ5 in PC9/ERβ1 cells (hereafter referred to as PC9/ERβ1/5). ERβ1 and ERβ5 overexpression was confirmed at the mRNA and protein levels by qRT-PCR and immunoblotting, respectively (Figure 1C; Table 1). Immunofluorescence using a non-variant specific antibody revealed that ERβ levels were elevated both in PC9/ERβ1 and PC9/ERβ1/5 cells; however, ERβ localization differed between the two cell lines. Although ERβ primarily localized in the cell nucleus in PC9/ERβ1 cells, in PC9/ERβ1/5 cells, it was found both in the cytoplasm and nucleus (Figures 2A, B).
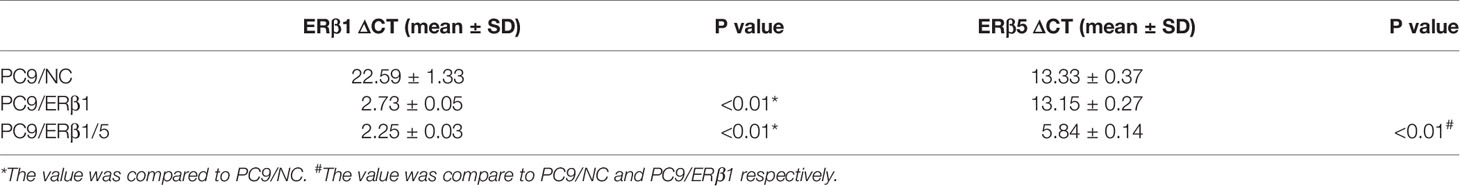
Table 1 ΔCT value of ERβ1 and ERβ5 in indicated cell lines.
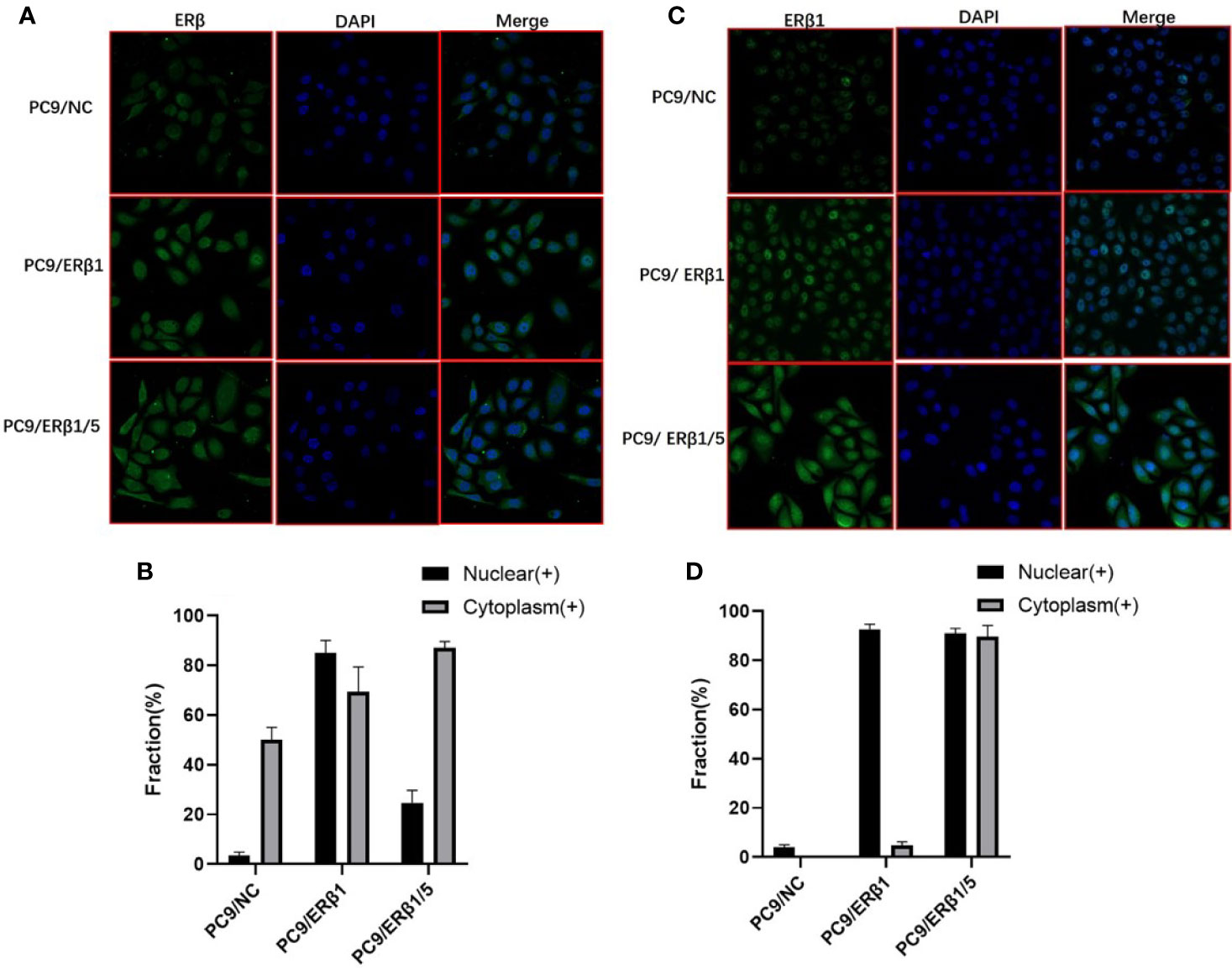
Figure 2 Different intracellular localization of ERβ in three cell lines (PC9/NC, PC9/ERβ1 and PC9/ERβ1/5. (A) ERβ expression was analyzed in established cell lines using immunofluorescence by using primary antibodies against ERβ. (B) The fraction of nuclear and cytoplasm ERβ positive cells in three cell lines. (C) ERβ1 expression was detected in the cell lines using immunofluorescence by using primary antibodies against ERβ1. (D) The fraction of nuclear and cytoplasm ERβ1 positive cells in three cell lines.
ERβ1 has the highest affinity for estradiol among all ERβ splice variants. Hence, we used an ERβ1-specific antibody to determine ERβ1 localization. ERβ1 predominantly localized in the cell nucleus in PC9/ERβ1 cells. However, in PC9/ERβ1/5 cells, we observed that ERβ1 was partly detained in the cytoplasm, suggesting that the expression of ERβ5 suppressed ERβ1 translocation from the cytoplasm to the nucleus (Figures 2C, D).
The Interaction Between Estrogen Receptor β1 and Estrogen Receptor β5 Regulates Downstream Signaling Events in the Presence of Estradiol
Next, we assessed the role of nuclear and cytoplasmic ERβ in transcriptional regulation and non-genomic signaling, respectively. The expression of the cell cycle regulator P21 is induced by the nuclear ERβ (18). In this study, we found that P21 expression levels were profoundly higher in PC9/ERβ1 cells compared to those in PC9/NC or PC9/ERβ1/5 cells, especially after stimulation with estradiol (Figure 3A).
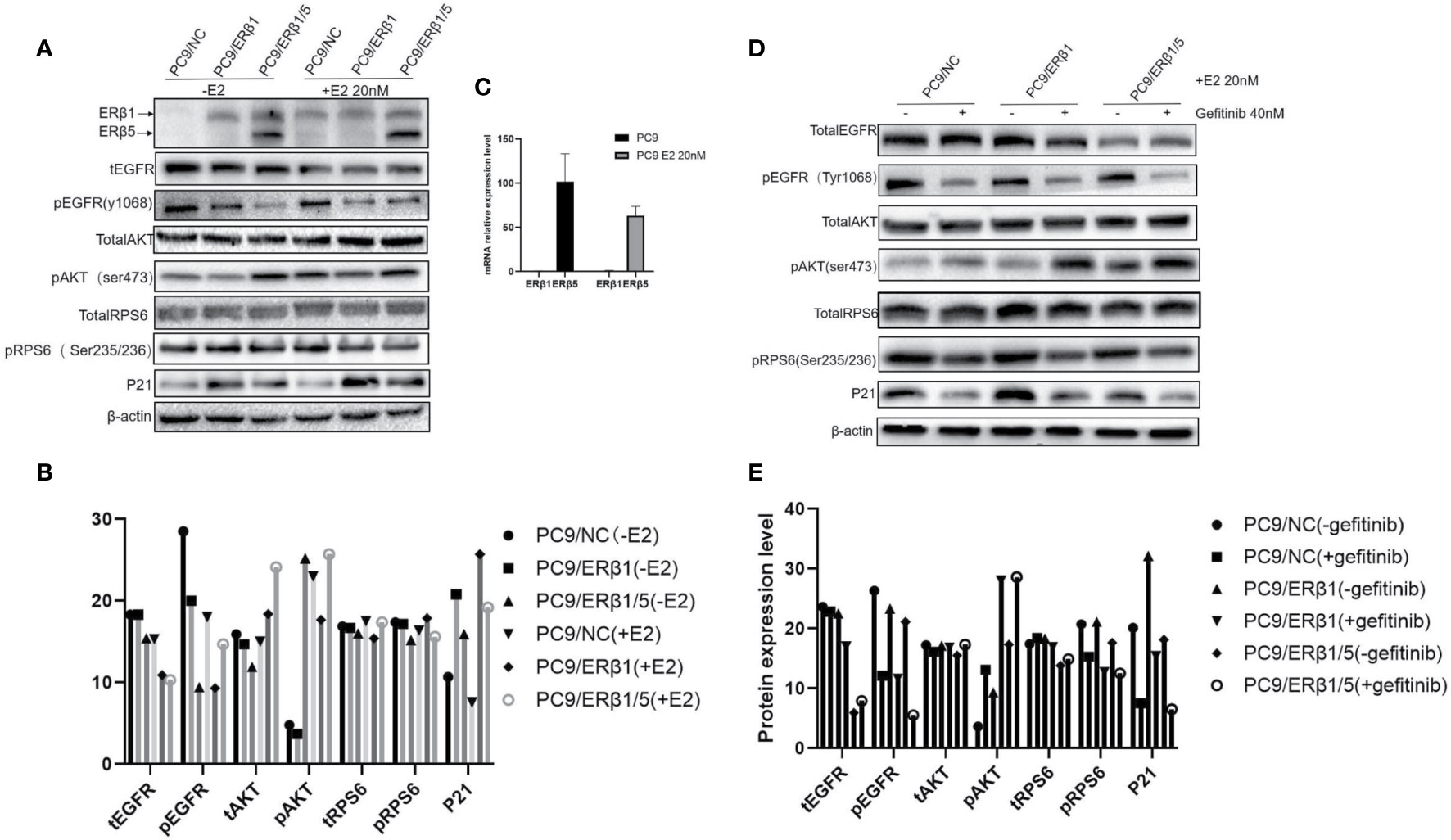
Figure 3 Interaction between ERβ1 and ERβ5 has a complex impact on downstream signaling under stimulation of estradiol. (A) Western blot was used to detect the indicated proteins involved in PI3K/AKT/mTOR signal pathway in the presence or absence of estradiol (20 nM). P21 was selected as marker for detecting ERβ nuclear transcriptional activity. (B) Indicated protein expression using quantity one for (A). (C) Relative mRNA expression level of ERβ1 and ERβ5 in the presence or absence of estradiol for PC9/NC. (D) The indicated cell lines were treated with gefitinib (40 nM) for 8 h in the presence of estradiol. Cell extracts were immunoblotted to detect the indicated proteins. (E) Indicated protein expression using quantity one for (D).
PI3K/AKT/mTOR signaling pathway is regulated by both EGFR and ERβ (11). To determine the PI3K/AKT/mTOR pathway activation status, we assessed both total and phosphorylated levels of EGFR, AKT, and RPS6. We found that phospho-EGFR levels were lower in PC9/ERβ1/5 cells than those in PC9/NC or PC9/ERβ1 cells. Although total and phospho-EGFR levels decreased in all groups after estradiol treatment, the decrease in phospho-EGFR levels was stronger in PC9/ERβ1 and PC9/ERβ1/5 cells than that in PC9/NC cells. The phospho-AKT levels were higher in PC9/ERβ1/5 cells than those in PC9/NC or PC9/ERβ1 cells, both at baseline and after estradiol treatment. The levels of phospho-RPS6, which functions downstream of mTOR, were similar among the groups (Figures 3A, B).
We also found that ERβ1 but not ERβ5 was upregulated in PC9/NC cells after estradiol treatment. Interestingly, qRT-PCR showed no changes in the ERβ1 mRNA levels after estradiol treatment, suggesting that the estradiol-mediated ERβ1 upregulation occurs at the post-transcriptional level (Figure 3C).
When cells were treated with gefitinib in addition to estradiol, phospho-EGFR levels were decreased in all groups, whereas phosphor-AKT levels were increased, especially in PC9/ERβ1 and PC9/ERβ1/5 cells. Similar to phospho-EGFR, phospho-RPS6 levels were decreased in all groups after gefitinib treatment. P21 was also downregulated in gefitinib-treated cells (Figures 3D, E).
PC9/Estrogen Receptor β1/5 Cells Are Less Prone to the Cytotoxic Effects of Gefitinib
To determine the effects of different ERβ splice variants in response to gefitinib, we performed cell viability and colony formation assays. We found that PC9/ERβ1/5 cells were less prone to the cytotoxic effects of gefitinib (40 nM) compared with PC9/NC or PC9/ERβ1 cells, although we found no significant differences in cell viability at low concentrations of gefitinib (Figure 4A). Additionally, the ability of gefitinib (40 nM) to inhibit colony formation was stronger in PC9/ERβ1 and PC9/NC than in PC9/ERβ1/5 cells (Figure 4B).
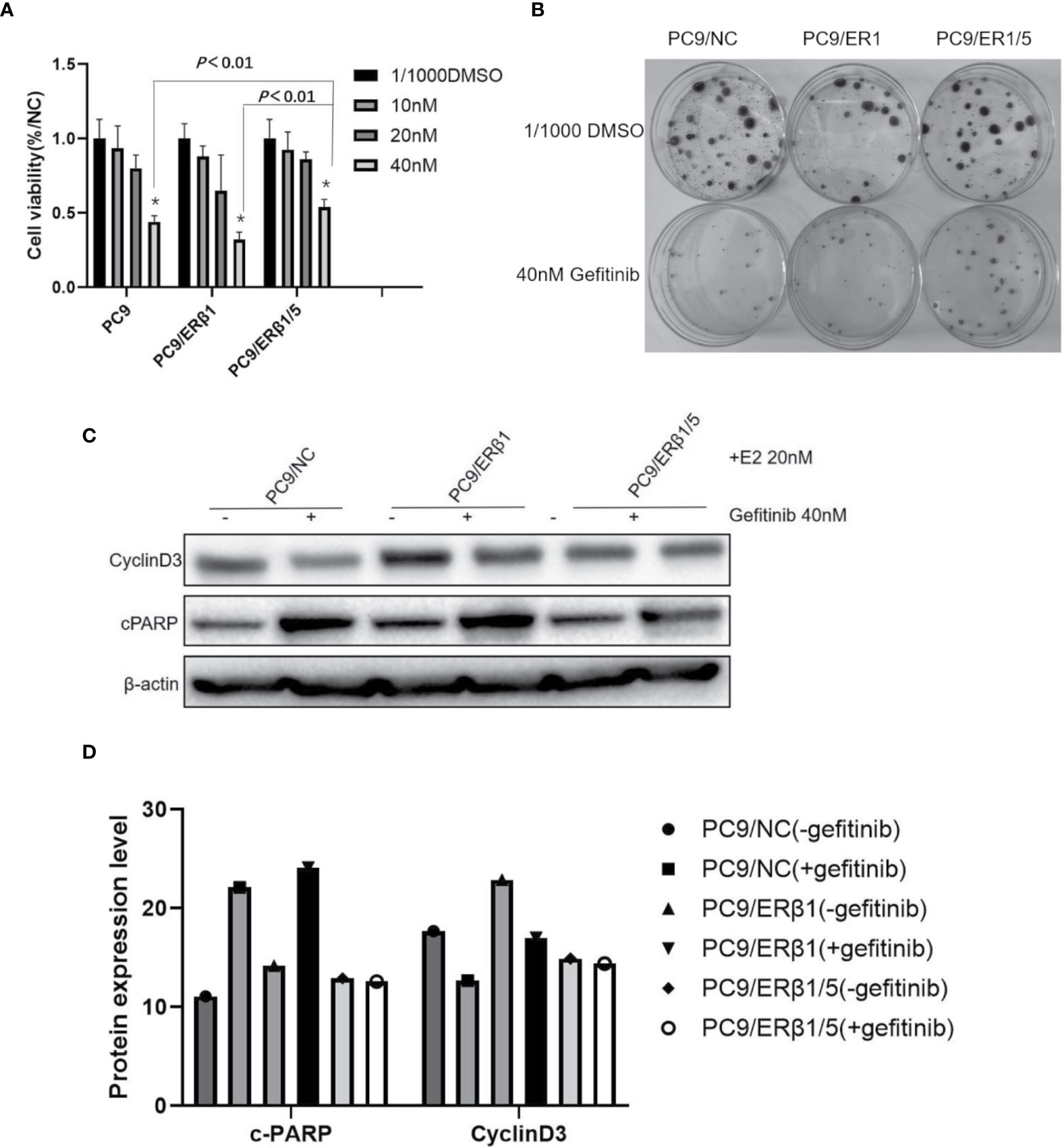
Figure 4 PC9/ERβ1/5 showed less effective to gefitinib compared to that of PC9/NC and PC/ERβ1 through anti-apoptosis effect. (A) Cell viability test for 72 h treatment at indicated concentration of Gefitinib (0, 10, 20, and 40 nM) in the presence of estradiol (20 nM). PC9/ERβ1/5 cells were less effective to gefitinib compared to PC9/NC and PC9/ER1 cells at concentration 40 nM (*P < 0.01). (B) Colony formation assay for 2 weeks in the presence of gefitinib 40 nM or 1/1,000 DMSO for three cell lines. (C) Cell apoptosis percentage after treatment of gefitinib (40 nM) for 24 h in the presence of estradiol (20 nM). Data shown as mean ± SD (* P < 0.01). (C) Western blot for detecting cleaved PARP and CyclinD3 for three cell lines with or without gefitinib (40 nM) treatment under the stimulation of estradiol (20 nM). (D) Indicated protein expression using quantity one for (C).
We found that cPARP levels were increased in all three groups after gefitinib treatment. However, the increase in cPARP levels was more substantial in PC9/NC and PC9/ERβ1 cells than in PC9/ERβ1/5 cells. Consistently, the decrease in cyclin D3 levels was more profound in PC9/NC and PC9/ERβ1 cells, while almost no change in cyclin D3 levels was observed in PC9/ERβ1/5 cells after gefitinib treatment (Figures 4C, D). These results suggest that PC9/ERβ1/5 cells are less sensitive to EGFR TKIs than PC9/NC and PC9/ERβ1 cells.
Estrogen Receptor β1 Expression and Intracellular Distribution Affect Progression-Free Survival in Patients With Advanced Epidermal Growth Factor Receptor-Mutant Lung Adenocarcinoma
In this study, we retrospectively analyzed the data from 103 stage IIIb–IV lung adenocarcinoma patients treated with EGFR TKIs at the Shandong Cancer Hospital between January 2014 and November 2017. All patients harbored EGFR mutations affecting response to EGFR TKIs, including exon 19 deletions (47; 45.6%) and exon 21 point mutations (55; 53.4%); one patient had G719X mutations, whereas no EGFR T790M or KRAS mutations were detected. The clinicopathological characteristics of the patients are summarized in Table 2. Most patients were never/light smokers (79; 76.7%) and women (65; 63.1%).
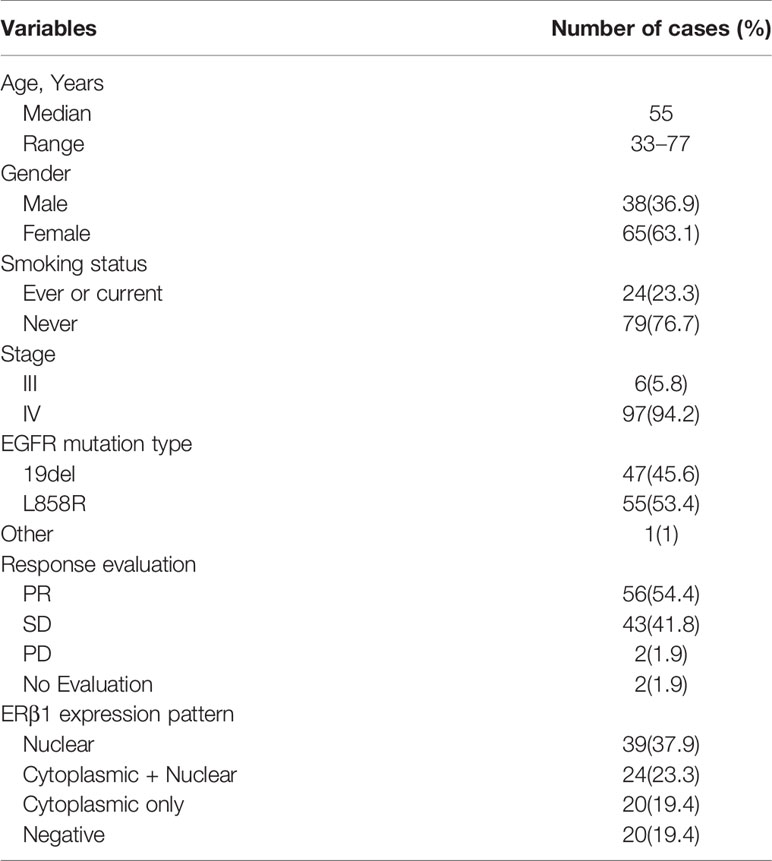
Table 2 Clinical and pathological characteristics of 103 patients with EGFR mutant lung adenocarcinomas.
ERβ1 expression was detected in 80.6% (83/103) of the patients, and the intracellular distribution pattern differed significantly (Figure 5A); strong nuclear accumulation (nERβ1) was observed in 37.9% (39/103) of the patients, nuclear/cytoplasmic accumulation (n/cERβ1) in 23.3% (24/103), and strong cytoplasmic accumulation (cERβ1) in 19.4% (20/103) of the patients (Table 2). There was no difference of ERβ1 pattern between male and female patients (Table 3).
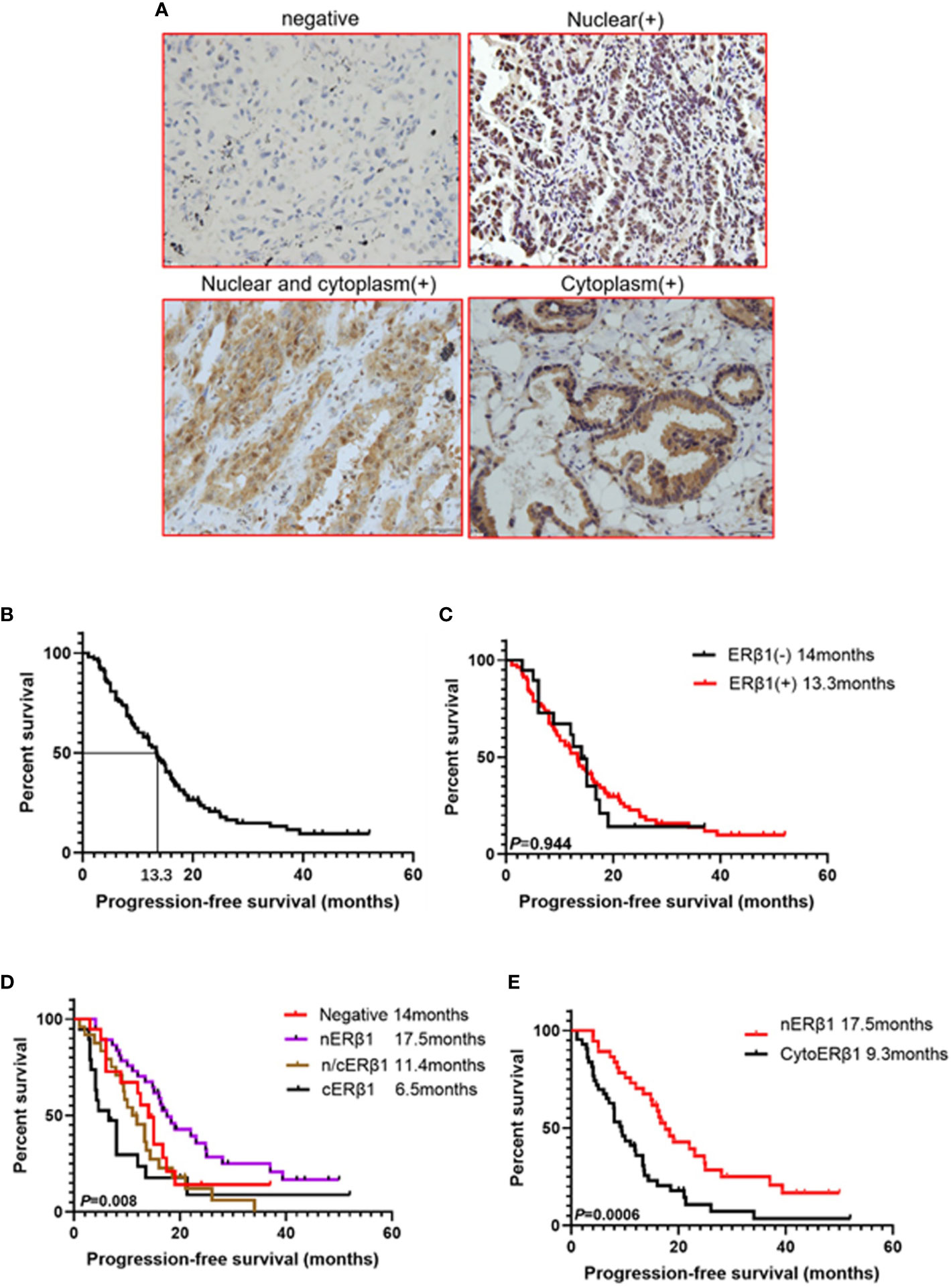
Figure 5 ERβ1 expression pattern had different effect on progression-free survival in advanced EGFR mutant lung adenocarcinomas. (A) Representative ERβ1 expression within lung adenocarcinoma tissues (Negative, nuclear, nuclear and cytoplasm, cytoplasm). (B). Kaplan–Meier curve showed progression free survival for all the patients (median survival: 13.3 months). (C) Kaplan–Meier curves illustrated progression-free survivals in the groups with ERβ1 positive (median survival:13.3 months) and ERβ1 negative (median survival: 14 months) (log-rank test, P = 0.944). (D) Comparison of progression free survival in four groups using log-rank test (P = 0.008). (E) Comparison of progression free survival between nuclear ERβ1 positive group (median survival: 17.5 months) and cytoERβ1 positive group (median survival: 9.3 months) (log-rank test, P = 0.0006).

Table 3 Expression pattern of ERβ1 stratified by sex.
At the time of data collection (Nov 20, 2017), four patients were lost to follow-up, and 79 patients (76.7%) presented with progressive disease. The median survival of the 99 patients was 13.3 months (Figure 5B). There was no significant difference in PFS between patients with ERβ1-positive and ERβ1-negative tumors (13.3 months vs. 14 months, P = 0.944) (Figure 5C). Interestingly, we also found significant differences in the median PFS of patients with different intracellular ERβ1distribution pattern (nERβ1, 17.5 months; n/cERβ1, 11.4 months; cERβ1, 6.5 months; negative, 14 months, P = 0.008) (Figure 5D). Because cytoplasmic ERβ1 is key for non-genomic signaling activation, we combined patients with n/cERβ1 expression and those with cERβ1 (cytoERβ1). Survival analysis showed that patients with cytoERβ1 expression (n = 43) had a shorter median PFS after EGFR TKI treatment (9.5 months) compared to those with nERβ1 expression (n =37; PFS, 17.5months; P = 0.0006) (Figure 5E).
Discussion
Approximately 20–30% of patients with EGFR activating mutations exhibit primary resistance to EGFR TKIs. The mechanism underlying resistance to EGFR TKIs, and primary resistance, in particular, are extremely complex and remain poorly understood. ERβ expression has been associated with response to EGFR TKIs. Notably, in a Japanese cohort study, strong ERβ expression predicted favorable clinical outcomes in patients with lung adenocarcinoma after treatment with EGFR TKIs. In contrast, we previously identified high cytoplasmic ERβ expression as a predictor of poor PFS (21, 22). Therefore, further elucidation of the expression pattern and intracellular distribution of ERβ is required to determine the effects of non-genomic signaling on EGFR signal transduction and clinical outcomes.
Several ERβ splicing variants have been identified, the most important of which are ERβ1 (wild-type ERβ), and ERβ2–5 (20, 23). ERβ1 is the only fully functional receptor in the ERβ family, and has the highest affinity for estradiol; other ERβ family members have weak to no ligand binding capacity, despite maintaining their ability to heterodimerize with ERβ1 (20). Therefore, assessing the function of ERβ splice variants other than ERβ1 is equally important. Notably, the crucial role of ERβ5 in lung cancer is becoming increasingly evident (17, 24).
In this study, we focused on the role of ERβ1 and ERβ5 in lung adenocarcinoma. Previous studies demonstrated that ERβ1 was predominantly localized in the cell nucleus and exerted anti-proliferative effects. In contrast, ERβ5 was found both in the cytoplasm and nucleus, and it has been implicated in cancer cell migration and invasion (17, 22, 25). Our results confirmed the elevated ERβ5 levels in EGFR-mutant lung cancer cells; in contrast, ERβ1 was lowly expressed. These results were consistent with those of a previous study showing that ERβ5 was the primary ERβ isoform expressed in non-malignant lung cells, and heterodimerized with ERβ1 (20). Similarly, we previously showed that ERβ5 formed complexes with ERβ1, confirming their ability to interact (22).
In this study, we also found that ERβ1 was predominantly localized in the cell nucleus. However, the forced overexpression of ERβ5 partly retained ERβ1 in the cytoplasm. Hence, the presence of ERβ5 can explain previous findings of ERβ1 localization in the nucleus and cytoplasm in cancer cells.
Total and phospho-EGFR levels were decreased after estradiol treatment, highlighting the crosstalk between EGFR and ERβ signaling pathways (12). P21 is an essential cell cycle regulator, playing important tumor-suppressing roles (26). Importantly, P21 expression was induced by ERβ (18, 25). In this study, we confirmed that ERβ1 increased P21 levels, suggesting a role of ERβ1 in transcriptional regulation in lung cancer cells. Consistently, ERβ1 exerted anti-proliferative effects in other cancer cells (15, 18). However, when ERβ1 and ERβ5 were co-expressed, P21 levels were lower compared with those in PC9/ERβ1 cells, suggesting that ERβ5 impairs the transcriptional abilities of ERβ1. However, in the presence of estradiol, PI3K/AKT/mTOR signaling pathway activation levels were higher in ERβ1/5-expressing lung cancer cells than those in ERβ1-expressing cells, suggesting that the interaction between ERβ1 and ERβ5 potentiated the effects of ERβ1 in non-genomic signaling. Hence, we believe that ERβ1 translocation from the nucleus to cytoplasm in the presence of ERβ5 was essential in determining its biological function, reflecting the bi-faceted role of ERβ in cancer (27).
mTOR signaling was inhibited by gefitinib treatment in all groups, although phospho-AKT levels were increased. Consistently, gefitinib treatment exerted cytotoxic effects in all cell groups. However, ERβ1/ERβ5 co-expression rendered cells less prone to the cytotoxic and pro-apoptotic effects of gefitinib. These results confirmed the critical role of ERβ1/ERβ5 complexes in estrogen receptor-mediated non-genomic signaling. We also found that estradiol upregulated ERβ1 but not ERβ5 at the post-transcriptional level, confirming the high affinity of ERβ1 for estradiol.
In this study, we also investigated the effect of the ERβ1 expression pattern on PFS in EGFR-mutant lung adenocarcinoma patients. We found that patients with nuclear ERβ1 expression exhibited a relatively longer PFS after EGFR TKI treatment, whereas cytoplasmic ERβ1 was associated with shorter PFS after EGFR TKI treatment. These results highlight the clinical relevance of our findings from in vitro experiments in EGFR-mutant lung cancer cell lines. Importantly, nuclear ERβ1 expression in lung cancer tissues was associated with tumor-suppressing effects, whereas cytoplasmic ERβ1 promoted EGFR TKI resistance to some extent. Although cytoERβ1 was associated with a lower response to EGFR TKIs, the median PFS of patients with cytoERβ1 was 9.5 months, suggesting that EGFR mutations remain the most powerful predictor for EGFR TKI treatment response. The findings reported here need to be confirmed in large cohort prospective studies. Additionally, the relationship between ERβ1/ERβ5 ratio and response to EGFR TKIs merits further investigation. Our study has potential clinical relevance for guiding anti-estrogen agents’ treatment in future. Recently two clinical trials evaluating efficacy and safety of EGFR TKI plus anti-estrogen treatment showed no survival improvement in combination group compared to that of EGFR TKI alone (28, 29). However, the status of ERβ expression and its predictive value for anti-estrogen therapy were not evaluated in both trials.
In conclusion, we showed that ERβ1 localized in the cell cytoplasm by interacting with ERβ5, inducing non-genomic signaling activation, and promoting EGFR TKI treatment resistance in EGFR-mutant lung adenocarcinoma. Hence, these results suggest that cytoplasmic ERβ1 was responsible for EGFR TKI resistance slightly through non-genomic mechanism in EGFR mutant lung adenocarcinoma.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials; further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Shandong Cancer Hospital and Institute. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
LZ and MT did the experimental work and participated in the analysis of the results. ZL supervised the research and secured funding for the project. JZ interpreted the histological findings. HW and JL wrote the paper draft, and all authors contributed to the analysis of the results. All authors contributed to the article and approved the submitted version.
Funding
This study was supported jointly by the National Natural Science Foundation of China (Grant No. 81602031), The Natural Science Foundation of Shandong Province (Grant No. ZR2016HB12), The National Natural Science Foundation of China (Grant No. 81904186), Special Funds for Taishan Scholars Project (Grant No. tsqn201812149), Academic Promotion Programme of Shandong First Medical University (Grant No. 2019RC004) and Key research and development plan project of Shandong Province (Grant No. 2017GSF18158).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361:947–57. doi: 10.1056/NEJMoa0810699
2. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol (2011) 12:735–42. doi: 10.1016/S1470-2045(11)70184-X
3. Lin JJ, Cardarella S, Lydon CA, Dahlberg SE, Jackman DM, Jänne PA, et al. Five-Year Survival in EGFR-Mutant Metastatic Lung Adenocarcinoma Treated with EGFR-TKIs. J Thorac Oncol (2016) 11:556–65. doi: 10.1016/j.jtho.2015.12.103
4. Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Jänne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol (2010) 28:357–60. doi: 10.1200/JCO.2009.24.7049
5. Suda K, Bunn PA Jr, Rivard CJ, Mitsudomi T, Hirsch FR. Primary Double-Strike Therapy for Cancers to Overcome EGFR Kinase Inhibitor Resistance: Proposal from the Bench. J Thorac Oncol (2017) 12:27–35. doi: 10.1016/j.jtho.2016.09.003
6. Lee JK, Shin JY, Kim S, Lee S, Park C, Kim JY, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol (2013) 24:2080–7. doi: 10.1093/annonc/mdt127
7. Xia H, Xue X, Ding H, Ou Q, Wu X, Nagasaka M, et al. Evidence of NTRK1 Fusion as Resistance Mechanism to EGFR TKI in EGFR+ NSCLC: Results From a Large-Scale Survey of NTRK1 Fusions in Chinese Patients With Lung Cancer. Clin Lung Cancer (2020) 21:247–54. doi: 10.1016/j.cllc.2019.09.004
8. Lei L, Wang WX, Zhu YC, Li JL, Fang Y, Wang H, et al. Potential mechanism of primary resistance to icotinib in patients with advanced non-small cell lung cancer harboring uncommon mutant epidermal growth factor receptor: A multi-center study. Cancer Sci (2020) 111:679–86. doi: 10.1111/cas.14277
9. Omoto Y, Kobayashi Y, Nishida K, Tsuchiya E, Eguchi H, Nakagawa K, et al. Expression, function, and clinical implications of the estrogen receptor beta in human lung cancers. Biochem Biophys Res Commun (2001) 285:340–7. doi: 10.1006/bbrc.2001.5158
10. Zhang G, Liu X, Farkas AM, Parwani AV, Lathrop KL, Lenzner D, et al. Estrogen receptor beta functions through nongenomic mechanisms in lung cancer cells. Mol Endocrinol (2009) 23:146–56. doi: 10.1210/me.2008-0431
11. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (2004) 304:1497–500. doi: 10.1126/science.1099314
12. Stabile LP, Lyker JS, Gubish CT, Zhang W, Grandis JR, Siegfried JM. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res (2005) 65:1459–70. doi: 10.1158/0008-5472.CAN-04-1872
13. Williams C, DiLeo A, Niv Y, Gustafsson JÅ. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett (2016) 372:48–56. doi: 10.1016/j.canlet.2015.12.009
14. Gustafsson JA, Strom A, Warner M. Update on ERbeta. J Steroid Biochem Mol Biol (2019) 191:105312. doi: 10.1016/j.jsbmb.2019.02.007
15. Liu J, Sareddy GR, Zhou M, Viswanadhapalli S, Li X, Lai Z, et al. Differential Effects of Estrogen Receptor β Isoforms on Glioblastoma Progression. Cancer Res (2018) 78:3176–89. doi: 10.1158/0008-5472.CAN-17-3470
16. Ciucci A, Zannoni GF, Travaglia D, Petrillo M, Scambia G, Gallo D. Prognostic significance of the estrogen receptor beta (ERβ) isoforms ERβ1, ERβ2, and ERβ5 in advanced serous ovarian cancer. Gynecol Oncol (2014) 132:351–9. doi: 10.1016/j.ygyno.2013.12.027
17. Bai Y, Shen W, Zhang L, Yang Z, Xiong L, Tang H, et al. Oestrogen receptor β5 and epidermal growth factor receptor synergistically promote lung cancer progression. Autoimmunity (2018) 51:157–65. doi: 10.1080/08916934.2018.1486825
18. Dey P, Jonsson P, Hartman J, Williams C, Ström A, Gustafsson JÅ. Estrogen receptors β1 and β2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol Endocrinol (2012) 26:1991–2003. doi: 10.1210/me.2012.1227
19. Leung YK, Lam HM, Wu S, Song D, Levin L, Cheng L, et al. Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocr Relat Cancer (2010) 17:675–89. doi: 10.1677/ERC-09-0294
20. Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proc Natl Acad Sci U S A (2006) 103:13162–7. doi: 10.1073/pnas.0605676103
21. Nose N, Uramoto H, Iwata T, Hanagiri T, Yasumoto K. Expression of estrogen receptor beta predicts a clinical response and longer progression-free survival after treatment with EGFR–TKI for adenocarcinoma of the lung. Lung Cancer (2010) 71:350–5. doi: 10.1016/j.lungcan.2010.06.009
22. Wang Z, Li Z, Ding X, Shen Z, Liu Z, An T, et al. ERβ localization influenced outcomes of EGFR-TKI treatment in NSCLC patients with EGFR mutations. Sci Rep (2015) 5:11392. doi: 10.1038/srep11392
23. Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochem Biophys Res Commun (1998) 247:75–8. doi: 10.1006/bbrc.1998.8738
24. Tang H, Bai Y, Xiong L, Zhang L, Wei Y, Zhu M, et al. Interaction of estrogen receptor β5 and interleukin 6 receptor in the progression of non-small cell lung cancer. J Cell Biochem (2018):1–11. doi: 10.1002/jcb.27510
25. Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Ström A, et al. Tumor repressive functions of estrogen receptor beta in SW480 colon cancer cells. Cancer Res (2009) 69:6100–6. doi: 10.1158/0008-5472.CAN-09-0506
26. Al Bitar S, Gali-Muhtasib H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers (Basel) (2019) 11:1475. doi: 10.3390/cancers11101475
27. Leygue E, Murphy LC. A bi-faceted role of estrogen receptor β in breast cancer. Endocr Relat Cancer (2013) 20:R127–139. doi: 10.1530/ERC-12-0389
28. Garon EB, Siegfried JM, Stabile LP, Young PA, Marquez-Garban DC, Park DJ, et al. Randomized phase II study of fulvestrant and erlotinib compared with erlotinib alone in patients with advanced or metastatic non-small cell lung cancer. Lung Cancer (2018) 123:91–8. doi: 10.1016/j.lungcan.2018.06.013
29. Mazieres J, Barlesi F, Rouquette I, Molinier O, Besse B, Monnet I, et al. Randomized Phase II Trial Evaluating Treatment with EGFR-TKI Associated with Antiestrogen in Women with Nonsquamous Advanced-Stage NSCLC: IFCT-1003 LADIE Trial. Clin Cancer Res (2020) 26(13):3172–81. doi: 10.1158/1078-0432.CCR-19-3056
Keywords: estrogen receptor β, lung adenocarcinoma, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), non-genomic mechanism, resistance
Citation: Zhang L, Tian M, Lin J, Zhang J, Wang H and Li Z (2021) Estrogen Receptor β1 Expression Patterns Have Different Effects on Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors’ Treatment Response in Epidermal Growth Factor Receptor Mutant Lung Adenocarcinoma. Front. Oncol. 10:603883. doi: 10.3389/fonc.2020.603883
Received: 08 September 2020; Accepted: 04 December 2020;
Published: 29 January 2021.
Edited by:
Timothy F. Burns, University of Pittsburgh, United StatesReviewed by:
Edward Garon, UCLA David Geffen School of Medicine, United StatesLaura P. Stabile, University of Pittsburgh, United States
Copyright © 2021 Zhang, Tian, Lin, Zhang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiyong Wang, d2FuZ2hhaXlvbmc2Njg4QDEyNi5jb20=; Zhenxiang Li, bGl6eDAxMDhAMTYzLmNvbQ==
†These authors have contributed equally to this work