 Lorenz Thurner1*
Lorenz Thurner1* Sylvia Hartmann2
Sylvia Hartmann2 Frank Neumann1
Frank Neumann1 Markus Hoth3Stephan Stilgenbauer1
Markus Hoth3Stephan Stilgenbauer1 Ralf Küppers4,5
Ralf Küppers4,5 Klaus-Dieter Preuss1
Klaus-Dieter Preuss1 Moritz Bewarder1
Moritz Bewarder1- 1Department of Internal Medicine I, José Carreras Center for Immuno- and Gene Therapy, Saarland University Medical School, Homburg, Germany
- 2Dr. Senckenberg Institute of Pathology, Goethe University, Frankfurt a. Main, Germany
- 3Department of Biophysics, Center for Integrative Physiology and Molecular Medicine, School of Medicine, Saarland University, Homburg, Germany
- 4Medical School, Institute of Cell Biology (Cancer Research), University of Duisburg-Essen, Essen, Germany
- 5Deutsches Konsortium für translationale Krebsforschung (DKTK), Partner Site Essen, Essen, Germany
The B-cell receptor (BCR) signaling pathway is a crucial pathway of B cells, both for their survival and for antigen-mediated activation, proliferation and differentiation. Its activation is also critical for the genesis of many lymphoma types. BCR-mediated lymphoma proliferation may be caused by activating BCR-pathway mutations and/or by active or tonic stimulation of the BCR. BCRs of lymphomas have frequently been described as polyreactive. In this review, the role of specific target antigens of the BCRs of lymphomas is highlighted. These antigens have been found to be restricted to specific lymphoma entities. The antigens can be of infectious origin, such as H. pylori in gastric MALT lymphoma or RpoC of M. catarrhalis in nodular lymphocyte predominant Hodgkin lymphoma, or they are autoantigens. Examples of such autoantigens are the BCR itself in chronic lymphocytic leukemia, LRPAP1 in mantle cell lymphoma, hyper-N-glycosylated SAMD14/neurabin-I in primary central nervous system lymphoma, hypo-phosphorylated ARS2 in diffuse large B-cell lymphoma, and hyper-phosphorylated SLP2, sumoylated HSP90 or saposin C in plasma cell dyscrasia. Notably, atypical posttranslational modifications are often responsible for the immunogenicity of many autoantigens. Possible therapeutic approaches evolving from these specific antigens are discussed.
B-Cell Development and Differentiation in the Context of Lymphoma Genesis and Autoreactivity
B Cell Development and Generation of B-Cell-Receptor Diversity
B-lymphocytes are part of the adaptive immune system. Their main function is the production of antigen-specific antibodies during humoral immune responses. They also function as antigen presenting cells (APC) for T helper cells and can have regulatory tasks. In the course of immune responses, activated B cells can differentiate into memory B cells or antibody-secreting plasma cells. B cell development is initiated when hematopoietic stem cell-derived common lymphoid progenitors in the bone marrow differentiate into pro-B cells. Here, mediated by the lymphocyte-specific recombinases RAG1 and RAG2, and other DNA-modifying enzymes such as KU70/KU80 (1) and artemis (2), a V(D)J gene recombination of individual immunoglobulin (Ig) variable (V), diversity (D), and joining (J) genes is initiated (3). First, the Ig heavy chain is assembled through a DH to JH recombination, followed by a VH to DHJH joining. Multiple genes of each of the three types of genes are available for recombination, causing combinatorial diversity. As further diversification mechanisms, individual nucleotides can be deleted from the joining ends of the rearranging genes, or non-germline-encoded nucleotides (N nucleotides) are randomly inserted between the VH, DH, and JH genes by terminal deoxynucleotidyltransferase (TdT) (4, 5). These processes of combinatorial and junctional diversity represent key mechanisms enabling a large variety of possible B cell receptor (BCR) reactivities given a relatively limited number of genes for immunoglobulins. After VH to DHJH joining, the heavy chain rearrangement is expressed as a pre-BCR with a non-rearranged surrogate light chain, and tested for functionality (this is needed, as about two thirds of rearrangements are out-of-frame and hence unproductive). If the first rearrangement is non-productive, a further attempt is made on the second heavy chain allele. Pre-B cells with a completed heavy chain rearrangement then perform Ig light chain gene rearrangements, beginning at the Igκ locus. The same diversification mechanisms as described for the heavy chain take place, with the exception that the light chains lack a D gene, so that VL genes are directly joined to JL genes. If the first rearranged VκJκ light-chain gene is not functional, further rearrangements can occur on the same allele, or on the second Igκ locus. If all these attempts fail, rearrangements of the Igλ locus occur. The combination of a rearranged heavy chain with a rearranged light chain (Igκ or Igλ) represents a further mechanism of combinatorial BCR diversity. After successful light chain rearrangement the differentiation stage of immature B cell is reached and the BCR is expressed as an IgM surface receptor. These cells are then selected against autoreactivity of the BCR (further discussed below), and the cells surviving this selection process exit the bone marrow and become mature, naive B cells, co-expressing the BCR as IgM and IgD molecules, mediated by differential splicing of the IgH transcripts.
Oncogenic Translocations During the V(D)J Rearrangement
The rearrangement processes of Ig heavy and light chain genes, which are accompanied by DNA double strand breaks, bear the inherent risk of causing oncogenic chromosomal translocations of protooncogenes, which bring the translocated oncogenes under control of the Ig enhancers. As these are highly active in B cells, this causes a constitutive, deregulated expression of the oncogenes. These translocations are often characteristic for certain B-cell Non-Hodgkin’s lymphoma entities: In mantle cell lymphoma (MCL) the gene encoding cyclin D1 (CCND1) is characteristically translocated into the IgH locus (t(11;14) (q13;q32)) and in follicular lymphoma (FL) BCL2-IgH translocations (t(14;18)(q31;q21)) are seen in more than 90% of cases (6). Despite these translocations, a functional BCR is regularly still expressed by the respective lymphomas, strongly indicating that the cells, despite carrying these oncogenic translocations, still depend on expression of a BCR (7).
Activation of Mature B Cells and Germinal Center Reaction
If mature B cells are activated through binding of an antigen to the BCR, and if T cell help is available, a T cell-dependent humoral immune response is initiated. After an initial encounter of antigen-specific B cells and cognate T cells in the T cell area of secondary lymphoid organs (e.g. lymph nodes), antigen-activated B and T lymphocytes migrate into B cell follicles and establish germinal centers (GCs). In the dark zone of these structures, the activated B cells proliferate (8). These dark zone GC B cells also activate the process of somatic hypermutation (SHM), which introduces mutations at a very high rate (10-3 to 10-4 mutations/bp per cell division (9)) into the Ig heavy and light chain V region genes. The key enzyme for this process is activation-induced cytidine deaminase (AID) (10, 11). As the mutations are largely random, most will be disadvantageous and will result in death of the respective B cells. Only B cells expressing a BCR with improved affinity will be positively selected through interactions with follicular dendritic cells and follicular T helper cells. This interaction takes place in the light zone of the GC, where the GC B cells are mostly non-proliferating. GC B cells typically undergo multiple rounds of proliferation/mutation and selection, resulting in a stepwise improvement of BCR affinity. In the course of the GC reaction, many B cells undergo class switch recombination (CSR) to change the isotype of the Ig heavy chain from IgM and IgD to IgG, IgA, or IgE (10). Also for this process, AID is an essential enzyme. Migration of the B cells within the GC is controlled by dynamic expression of the chemokine receptors CXCR4 (highly expressed on B cells in the dark zone) and CXCR5 (highly expressed on B cells in the light zone), and gradients of their ligands CXCL12 and CXCL13, respectively (12).
The transcription factor BCL6 is the master regulator of the GC B cell gene expression program (13). BCL6 activates PAX5, BACH2, and MITF, and it inhibits the plasma cell master regulators IRF4, BLIMP1 and (indirectly) XBP1 (14). Strong BCR activation leads to a shift from BCL6 dominance to upregulation of BLIMP1 (PRDM1) (14). BLIMP1 represses transcription of BCL6 and PAX5, and induces expression of IRF4 (MUM1) and XBP1, leading to differentiation of GC B cells into plasma cells. Other positively selected GC B cells differentiate into long-lived memory B cells, but the responsible transcription factor networks are less well understood (15).
Mechanisms of Loss of Immunological Self-Tolerance
The mechanisms of BCR diversity inevitably have the side effect of generating also autoreactive BCRs (16, 17). Immature B cells with strongly autoreactive BCR are usually deleted (18), which is referred to as central tolerance. Furthermore, B cells with autoreactive BCRs can change into an anergic state (19, 20) and immature B cells with autoreactive BCRs can modify their light chain genes by new rearrangements, which is called receptor editing, and thus escape clonal deletion (21–23). Failure of the tolerance process leads to the generation of naive mature autoreactive B cells (24–26). Furthermore, peripheral self-reactive B cells receiving proliferative signals via MHCII/T cell receptor (TCR) and CD40/CD40L interactions can be depleted in a Fas/FasL-dependent manner (27–29). Altered pro-inflammatory, B-cell-stimulating signals such as BAFF, IL-6 or CpG or anti-inflammatory, immunosuppressive signals such as IL-10 can influence these peripheral self-tolerance checkpoints (29–31).
The presence of certain types of HLA (32) is a crucial prerequisite for most autoimmune phenomena. In addition, there are a large number of single nucleotide polymorphisms (SNPs) or mutations in susceptibility genes associated with autoreactivity, including PTPN22, CTLA4, A20, TLR7, TLR9, MYD88, CD40/CD40L, ICOS/ICOSL or genes in the BCR signaling pathway (33–39). In addition, external factors can create an inflammatory environment, reverse the segregation of certain antigens, or activate autoreactive bystander cells. In the presence of certain HLA types, immune responses against certain infectious pathogens can lead to autoreactivity via molecular mimicry (40, 41).
Another mechanism of loss of self-tolerance is the occurrence of alterations in self-proteins, either by somatic mutations or by atypical secondary modifications (42). The secondary occurrence of RPC1 autoantibodies and scleroderma in patients with a precancerous disease or cancer with somatically mutated POLR3A gene are examples (43). Besides somatically mutated neoantigens, posttranslational modifications (PTM) can characteristically lead to antigen-specific breaks of tolerance, (44) such as modified wheat gliadin in celiac disease (45), N-terminal acetylated myelin basic protein in multiple sclerosis (46), citrullinated fibrin/vimentin in rheumatoid arthritis (47, 48), phosphorylated SR proteins in systemic lupus erythematosus (49, 50), or phosphorylated enolase in pancreatic carcinoma (51–53) (Table 1). Not all of these autoantibodies differentiate between modified antigens and wildtype isoforms. It is assumed that PTM-specific T cells, in contrast to non-PTM-specific T cells, escape central negative selection in the thymus (59).

Table 1 Examples of post-translationally modified B-cell receptor (BCR) antigens in diseases other than lymphoma.
Germinal Center Reaction and Lymphoma Genesis
The two processes modifying IgG genes in GC B cells – SHM and CSR – have not only very important roles for an efficient humoral immune response, but they also bear an inherent risk for mutations. SHM is not completely restricted to the IgV genes and can also target some non-Ig genes, including the proto-oncogene BCL6. This off-target SHM is particularly extensive in some types of lymphomas, including diffuse large B cell lymphoma (DLBCL), and is therefore termed aberrant SHM (60–62). Both SHM and CSR are mechanistically linked to DNA strand breaks, which is why both of them can also cause chromosomal translocations (63). Translocation of BCL6 or MYC into the Ig loci are prototypical examples of such translocations mediated by misguided SHM or CSR (64). Notably, also the translocation events in GC B cells are mostly targeted to the non-expressed Ig alleles (as described earlier for V(D)J recombination-associated translocations), indicating that also at this stage of B cell differentiation, the occurrence of an oncogenic translocation does not inevitably prevent the selection for expression of a functional BCR by the lymphoma cells. Two further vulnerabilities of GC B cells for lymphoma genesis are the intrinsically high and fast proliferation activity of GC B cells, and the transient down-regulation of DNA damage responses. This allows SHM to occur without automatic induction of apoptosis (65). All these features together likely explain why about 90% of lymphomas are of B cell origin, mostly induced during a GC reaction.
Key signaling pathways frequently affected in lymphoma genesis are the following ones: the BCR- pathway with CD79B and/or MYD88 mutations in the activated B cell (ABC) type of DLBCL (66), the latter also being typically involved in lymphoplasmocytic lymphoma (67), the canonical and the alternative NF-κB signaling pathway in a variety of different lymphomas including classical Hodgkin lymphoma (68–70), the NOTCH1 signaling pathway in chronic lymphocytic leukemia (CLL) (71) and a DLBCL subgroup with poor prognosis (72), the NOTCH2 signaling pathway in splenic MZL (73), as well as the JAK-STAT pathway, especially in classical Hodgkin lymphoma (74) and in primary mediastinal B-cell lymphoma (75). Furthermore, frequent mutations are described in genes encoding factors of the apoptosis signaling pathway (76) and in genes encoding for important molecules of immune surveillance (77–79).
Typically, the malignant lymphoma cells retain many characteristics of their origin counterparts, including their morphology, surface markers and gene expression profiles (7). For example, the differentiation between GC B cell-like (GCB) and ABC type of DLBCL is based on gene expression profiles (80) and immunophenotypical profiles (81). These original cell characteristics, which transformed cells can retain as established cell lines even after decades of cell culture, mostly also include the expression of the BCR. Subgroups of some lymphomas (e.g. Burkitt lymphoma (BL), primary central nervous system lymphoma (PCNSL), DLBCL, marginal zone lymphoma (MZL), MCL, and CLL) express a functional BCR, partly despite persistent AID expression with variable persistent SHM. This indicates a certain dependence or a selection advantage by BCR expression, possibly even a permanent BCR stimulation by an antigen in subgroups of the above mentioned lymphoma. In addition to the typical translocations as well as activating mutations of proto-oncogenes and inactivating mutations of tumor suppressor genes, the involvement of the BCR in lymphoma genesis was suspected early on (7, 82). The strong upregulation of the NF-κB signaling pathway in many B-cell lymphomas could also be partly explained by BCR activation.
Two principal types of BCR signaling are being distinguished. Tonic signaling is a constitutive and presumably antigen-independent signaling that is crucial for B-cell survival. In contrast, crosslinking of the BCR by direct binding to the cognate antigen induces activation of the B cell and plays an important role in humoral responses inducing B cell proliferation, AID expression, affinity maturation and differentiation. Whereas tonic signaling mainly relies on the PI3K/AKT pathway, the NF-κB pathway plays a major role in antigen-mediated active BCR signaling.
If the concept of two types of BCR signaling is applied to lymphoma, a tonic BCR stimulation pattern plays a major role in GCB-DLBCL, mainly mediated by a Y188 mutation within CD79A, and in a relevant subgroup of BL accompanied by mutations in TCF3 and ID3, and activation of the PI3K pathway (83, 84). Active BCR signaling in lymphomas shares similarities with BCR stimulation by exogenous cognate antigens and plays an important role in ABC-DLBCL, where it is called chronic active BCR signaling (80). In ABC-DLBCL, mutations of components of the BCR pathway, including members of the CARD11/BCL10/MALT1 (CBM) complex (85, 86), and of MYD88 (87) were frequently found. In ABC-DLBCL (88) and in CLL (89) constitutive BCR clustering is observed as it is seen in normal B cells after BCR binding of an antigen. For a particular genotypic subgroup of ABC-DLBCL with MYD88 L265P and CD79B mutations, an interaction of the BCR with MYD88 was reported, mediated by TLR9 (90), which is located in the endosome and normally senses CpG DNA. This was named My-T-BCR supercomplex (91).
Specific BCR Antigens in Lymphoproliferative Diseases
When considering BCR stimulation by antigens in lymphomas, several questions arise: is the BCR polyreactive or specific for one antigen? Are there random antigens for each individual patient with lymphoma or is there an over-representation of certain antigens? If so, are these over-representations entity-specific? And what are the possible underlying causes of the misdirected immune responses? We will highlight antigens of infectious origin as possible triggers of (mainly indolent) lymphomas. Subsequently, the involvement of autoantigens and underlying mechanisms of autoreactivity will be discussed.
The Role of Infectious Agents in B-Cell Neoplasia
The suspected relevant BCR target antigens could be antigens of persistent or recurrent infections. The most prominent example for this is the involvement of Helicobacter pylori in the pathogenesis of MALT lymphoma (a subtype of MZL) of the stomach (92–95). However the BCR were found to resemble rheumatoid factor (96, 97) or in other reports were polyreactive to autoantigens including IgG and Helicobacter sonicate (95). This indicates a mainly indirect role of bacterial infection for triggering lymphoma growth, presumably mediated to H. pylori-stimulated T helper cells. Regarding the gastrointestinal tract, Campylobacter jejuni was also associated with the genesis of lymphoproliferative diseases in the small intestine (98). It was furthermore speculated that Campylobacter jejuni may also contribute to duodenal FL, as it is typically restricted to this anatomic site. Considering the usually favorable outcome, this is often managed with a watch & wait strategy. Beside gastric MALT lymphoma other entities of MZL are triggered by chronic infections. For MALT lymphoma of the ocular adnexae, a strong association with Chlamydia psittaci was reported in specific regions (99), and for primary cutaneous MZL, Borrelia spec. infections were reported to be potentially causative. Splenic MZL shows an over-representation of the IGHV1-2*04 gene, has recurrent mutations in NOTCH2, and in a fraction of cases its development may be triggered by hepatitis C virus (HCV) (73, 100, 101).
Successful therapeutic concepts for infection caused MZL have been demonstrated by eradication of H. pylori with proton pump inhibitors with or without bismuth, in combination with clarithromycin and metronidazole or amoxicillin for gastric MALT lymphomas. These drugs have been incorporated into the current therapeutic standard of ESMO/EHA for gastric MALT lymphomas regardless of stage (94, 102, 103). Furthermore, the efficacy of antibiotic eradication of Chlamydia psittaci for ocular adnexal MALT lymphomas by doxycycline or clarithromycine has been demonstrated (104, 105). Moreover, preliminary reports about successful treatment of Borrelia-spec. associated primary cutaneous MZL were published (106). Similarly, the eradication of HCV and thus elimination of viral antigens as triggers of lymphoma BCRs can lead to regression of HCV-associated splenic MZL, and this is currently recommended as first line therapy in the current ESMO/EHA guidelines (101, 103). In summary, the examples presented here highlight the potential of lymphoma regression upon anti-viral or anti-bacterial treatment. This is a strong argument for a causative role of the respective infections for sustained triggering of lymphoma growth.
Using BCR expression cloning and subsequent antigen screenings, we could extend this list of infection-triggered lymphomas. We identified a specific antigen of a common bacteria as BCR antigen of nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), a rare type of B-cell lymphoma, which frequently manifests at cervical lymph nodes and with regular expression of functional BCRs. This target antigen was DNA-directed RNA polymerase beta’ (RpoC) of the Gram-negative cocci Moraxella catarrhalis (107). Moraxella catarrhalis is known to co-express MID/hag, a superantigen activating IgD+ B cells by binding to the Fc domain of IgD. RpoC and MID/hag additively activate the BCR and the NF-κB pathways and induce proliferation of lymphocyte predominant (LP) tumor cells of NLPHL with RpoC-specific BCRs. In particular, RpoC was a frequent antigen of BCRs of IgD+ LP cells, whose IgHV genes had extraordinarily long complementary determining region 3 (CDR3s). Moreover, patients showed a predominance of HLA-DRB1*04/07, suggesting existence of a permissive MHC-II haplotype (107, 108). Interestingly, this haplotype is also known from autoimmunity for its association with rheumatoid arthritis as shared epitope (109). Patients had high-titer, and light-chain-restricted anti-RpoC serum-antibodies, further supporting infection of the patients by M. catarrhalis and mislead immune responses against this bacteria (Figure 1). These results suggest to conduct clinical trials examining a potential effect of antibiotic therapy for relapsed or refractory IgD+ NLPHL. Moreover, if no B-cell depletion was therapeutically induced in the patients, active vaccination might make sense after local therapy. For this potential strategy the target antigens, i.e. RpoC and MID/Hag of the lymphoma BCRs must not be present in the vaccine, to avoid stimulation of remaining LP cells.
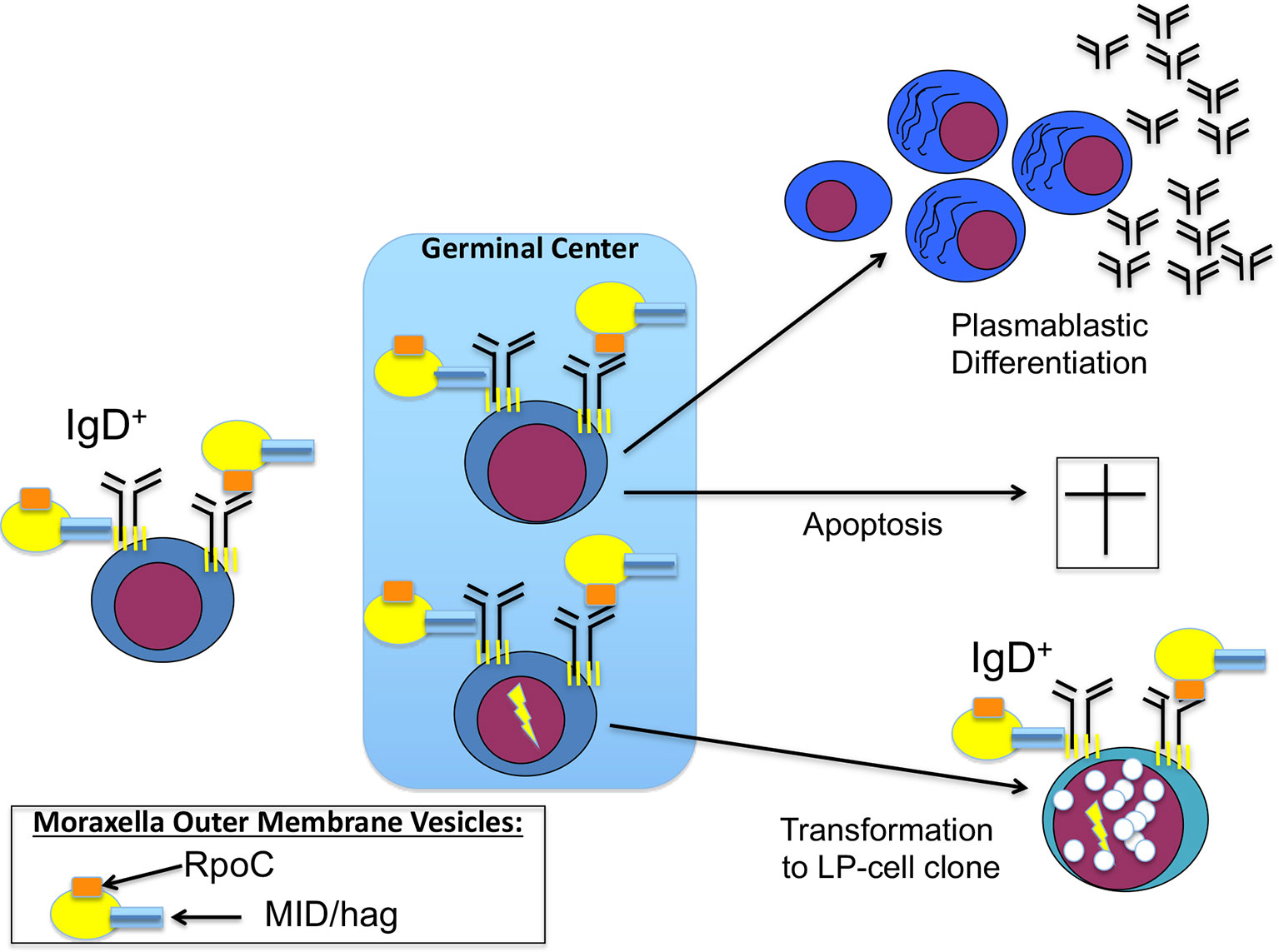
Figure 1 Contribution of Moraxella catarrhalis to IgD+ nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) pathogenesis: costimulation of IgD-positive B cells by Moraxella catarrhalis RNA polymerase beta’ (RpoC) via the Fab fragment and MID/hag via the Fc fragment of the B-cell receptor (BCR). Naive IgD+ B cells with a BCR specific for RpoC encounter M. catarrhalis outer membrane vesicles. Binding of RpoC to the Fab and of MID/hag to the Fc of membrane IgD induces activation of RpoC-specific IgD+ B cells, which is supported by CD4+ T cells particularly in patients with an HLA-DRB1*04 haplotype. The persistent/recurrent presence of M. catarrhalis presumably induces a germinal center (GC) reaction resulting in differentiation of memory B cells and plasmablasts and production of class-switched anti-RpoC serum antibodies and apoptosis of some GC B cells due to disadvantageous mutations. Subsequently, transformation into lymphocyte predominant (LP) cells may occur, accompanied by mutations in proto-oncogenes and tumor suppressor genes, and by chromosomal translocations (e.g. BCL6 translocations).
Regarding aggressive lymphomas like BL, potential infectious triggers of the BCR are discussed for many years. Expression of sIg is a hallmark of all types of BL, despite MYC-involving translocations with one Ig gene allele as t(8;14), t(2;8) or t(8;22). However, for BL no direct BCR stimulation by an antigen has been suspected, but actually tonic BCR pathway activation amplified by mutations in ID3 and TCF3 genes has been reported in BL (84). CRISPR-screening identified CD79B-dependency in the BL Ramos cell line (110). For endemic BL a frequently preceding coincidence of malaria and latent EBV infection was observed, which both likely contribute to BL pathogenesis (111, 112). However, in this case EBV is not a BCR stimulating antigen, but it infects B cells and can contribute to their transformation through expression of EBV-encoded genes in latently infected B cells. For BL, in general EBV latency phase I is observed with expression of just EBNA1, so that the pathogenetic role of EBV in BL is still not fully understood (113). In sporadic BL the frequent extranodal manifestation in the appendix vermiformic and ileocoeliac junction area raised speculations about a possible infectious trigger, but a causative infectious agent has not yet been identified.
The role of infections can go far beyond direct BCR stimulation and influence lymphoma genesis in other ways. The BCR often does not seem to play a significant role in EBV-associated B-cell lymphomas, e.g. in classical Hodgkin lymphoma, functional BCR are often lost. In classical Hodgkin lymphoma, typically EBV latency II is present with expression of EBNA1, and LMP1 and LMP2a. In post-transplant lymphoproliferative disease (PTLD), EBV latency III is observed with expression of EBNA1, -2A, -3A, -3B, -3C, and LMP1 and LMP2a (113). LMP2a contains an ITAM mimicking motive (potentially) relevant for proximal BCR pathway activation, and LMP1 is a viral oncogenic mimic of CD40, recruiting among others the signaling factors TRAF2 and TRAF3, but in contrast to CD40 not TRAF3 (114–120). Besides EBV, other viruses can play important roles in lymphoma genesis by transformation of lymphocytes by latent viral infections, such as HTLV1 in adult T-cell leukemia (121) and HHV8 (122) in primary effusion lymphoma. Another important mechanism is immunosuppression by HIV attenuating control of EBV- or HHV8-infected B cells (123).
The Role of Autoantigens in Lymphoproliferative Diseases
Autoantigens in Indolent Lymphoma
Endogenous immunogenic proteins could contribute to permanent growth advantages of lymphoma cells with the appropriate BCR autoreactivity by their inexhaustible supply. MZL is a CD5- and CD10- indolent lymphoma often accompanied by a paraprotein. Extranodal MZL is frequently associated with infectious triggers as described above. Beside recurrent mutations in MLL2, NOTCH2, PTPRD, and KLF2, nodal MZL correlates (shows) over-represented usage of IGHV4-34 in around 30% of cases (124), which is known to be linked with autoreactivity. This autoreactivity is also demonstrated by MZL emerging from Sjögren’s disease (125).
FL is a CD5- and CD10+ indolent lymphoma characterized by the presence of t(14;18)(q32;q21) leading to overexpression of BCL2. Regarding the BCR pathway Freda Stevenson et al. described a gain of N-glycosylation sites within the IgV genes by SHM leading to chronic activation of the BCR pathway by binding of N-hyperglycosylated BCRs to lectins in the lymphoma microenvironment (126). Subtypes of FL with a distinct manifestation and clinical course may have a different underlying biology. Here, pediatric FL, with regular cervical nodal manifestation and without BCL2 translocation, and duodenal FL have to be mentioned. Both characteristically do not spread beyond initial local manifestations.
CLL is the most common hematological cancer in adults in the Western world and clinically shows considerable heterogeneity (127). It is characterized by a population of ≥ 5,000 clonal B cells/µl in the peripheral blood. Nodal, extranodal or splenic manifestation with < 5,000 clonal B cells/µl is called small lymphocytic lymphoma (SLL). The monoclonal tumor cells express CD5, CD23, CD200, and low levels of sIg, and lack CD10 expression. By analysis of IGHV genes of a very large number of CLL cases, it became clear that unrelated CLL patients can have highly similar if not identical BCRs (128). This phenomenon of groups of CLL with highly similar IGHV and IGHL gene rearrangements is termed BCR stereotypy. It is considered the strongest evidence that antigen selection plays an important role in the pathobiology of CLL. CLL patients whose disease belong to a specific stereotypic subset often show similar clinical and biological characteristics, including disease progression. Interestingly, for several of the stereotypic groups, autoantigen specificity of the BCR has been demonstrated (129–131). There is evidence to suggest that these BCR enable specific recognition of an (auto)antigen, which leads to increased proliferation of the malignant B-cell clone and thus plays a crucial role in the pathophysiology of CLL (132). Indeed, inhibitors of BCR signaling pathway are of great importance in clinical practice for CLL patients (133). OxLDL, Fam32a, SMCHD1, MAZ, vimentin, myosin chains, and pUL32 have been identified as (auto)antigens that can specifically bind to CLL BCRs (134–137). The mutation status of the BCR of CLL clones represents a strong prognostic marker. CLL with no or few somatic BCR mutations within their rearranged IGHV genes (“unmutated” CLL, U-CLL) experience a significantly more aggressive disease than patients with >2% mutation load (“mutated” CLL, M-CLL) (138). However, this subdivision might be more complex regarding the clinical heterogeneity of CLL. If the findings of the subsets are combined with the findings of U-CLL vs. M-CLL, there are three categories: CLLs with stereotypic BCRs (about 1/3 of the cases and mostly U-CLL), CLLs with specific IGHV genes (U- and M-CLL) and those with heterogeneous and no particular IG features (mainly M-CLL). U-CLL have polyreactive BCRs specific for autologous neoantigens (e.g. myosin chains, vimentin, oxLDL, PC9, Fam32A, SMCHD1, and MAZ) (134), while the BCRs of some M-CLL react with foreign antigens, such as yeast derived glucans (139), or autoantigens as Fc parts of rheumatoid factors (140).
There are also indications that CLL cells show antigen-independent, cell-autonomous signaling (141, 142); a behavior that has not been shown in normal B cells and other B-cell malignancies. This cell-autonomous signaling is based on the recognition and self-association of the BCR of CLL cells to itself, the ultimate autoantigen. Various CLL-derived BCR bind to specific, different epitopes of themselves and thus initiate intracellular signal transduction. The avidity of BCR self-recognition seems to have an influence on the course and severity of the disease. In summary, for CLL different models and ideas exist for the significance of the BCR in CLL. It is likely that all models of BCR reactivity have their justification, whereby, depending on the situation, one or the other mechanism may be more important. All models emphasize the importance of BCR antigen recognition in conjunction with BCR auto-stimulation in addition to genetic lesions in the pathogenesis of CLL. However, it should not be forgotten that additional effects with an influence on the pathogenesis have to be considered, such as the existence of specific effector functions for IgM and IgD. Nevertheless, the use of inhibitors of the BCR signaling pathway (e.g. BTK inhibitors) and thus the proliferation of CLL cells has significantly improved the therapeutic options and led to permanent remissions, even in high-risk and refractory CLL patients.
Hairy cell leukemia (HCL) is a rare indolent lymphoma typically affecting middle-aged to old males. Beside its name-giving feature of protruding villi on the surface of the leukemic cells, it has an immunophenotype characterized by expression of CD103, CD11c, CD22, and CD123, and lack of CD5 and CD10 expression. Expression of CD25 distinguishes a classical and a variant form of HCL (143). Classic HCL always carry BRAF mutations (144). HCL cells express a BCR and the IGHV genes IGHV3-21, IGHV3-30, and IGHV3-33 are overrepresented. HCL is frequently associated with Igλ light chains (145, 146). Variant HCL lack BRAF mutations, and often use the IGHV4-34 gene.
MCL is a rare B-cell neoplasia, which accounts for about 6-8% of all Non-Hodgkin lymphoma (127). Male, elderly patients are over-represented, and extra-lymphatic manifestations are common (147, 148). MCL cells typically show a CD5+, CD23-, CD200- immunophenotype with strong expression of CyclinD1, due to translocation of the CCND1 gene into the IgH locus (t(11;14)(q13;q32)) (149). Regarding its IGHV mutational and DNA methylation status, MCL can be distinguished into pre- and post-GC-derived cases (150). Over-representation of specific IGHV gene groups and stereotypic rearrangements has been described similar to CLL, but with a lower frequency (151–153). In accordance with this, a strong BCR and NF-κB pathway activation in MCL was reported (154), and antigen-induced activation was stronger compared to other B-cell neoplasia (155). Pharmacological targeting of the BCR pathway by inhibition of PI3K or BTK is established for relapsed/refractory MCL (156–159). Recently, we identified human LDL receptor-related protein associated-protein 1 (LRPAP1) as frequent autoantigen of recombinant BCRs in MCL cases (8/21) and two of seven MCL cell lines (MAVER1 and Z138) (160). LRPAP1 consists of 357 amino acids and has a molecular weight of 39 kDa. LRPAP1 functions as antagonist and chaperon of the family of LDL-receptors and it takes part in Megalin/Cubilin endocytosis (161, 162). Immunization of rats with LRPAP1 results in Heymann-Nephritis (163, 164).
Autoantigens in Aggressive Lymphomas
Regarding autoantigenic targets of BCRs of aggressive lymphomas, several examples exist. DLBCL is the most common aggressive B-cell Non-Hodgkin lymphoma. According to the WHO classification, DLBCL can be classified based on gene expression profiling into ABC–like type, GCB–like type and primary mediastinal B-cell lymphoma (80, 165). In contrast to relatively well studied genetic or epigenetic pathway alterations, little is known about specific external stimuli of distinct subgroups of DLBCL (166, 167). In particular, DLBCL of the ABC-type or the recently specified MCD-type or cluster 5 harbor recurrent mutations in MYD88 and CD79B with dependency on constitutive BCR signaling (72, 88, 168, 169). For systemic DLBCL a cis and trans stimulation of the BCR by a so far non-characterized autoantigen was reported for the HBL1 cell line. Moreover, an anti-idiotype reactivity of the BCR of the TMD8 cell line against an epitope within its own FR2 (V37R38) was described, and for the U2932 and OCI-LY10 cell lines BCR reactivity against apoptotic cell debris was reported (169–172). Recently, Arsenite resistance protein 2 (ARS2) was identified as the BCR target of ABC-DLBCL. Compared to controls, ARS2 was hypo-phosphorylated exclusively in cases and cell lines with ARS2-specific BCRs (Figure 2 and Table 2). In a validation cohort, hypo-phosphorylated ARS2 was found in 8/31 ABC-DLBCL, but only 1/20 GBC-DLBCL. Incubation with ARS2 induced BCR-pathway activation and increased proliferation, while an ARS2/ETA’ toxin conjugate induced killing of cell lines with ARS2-reactive BCRs.
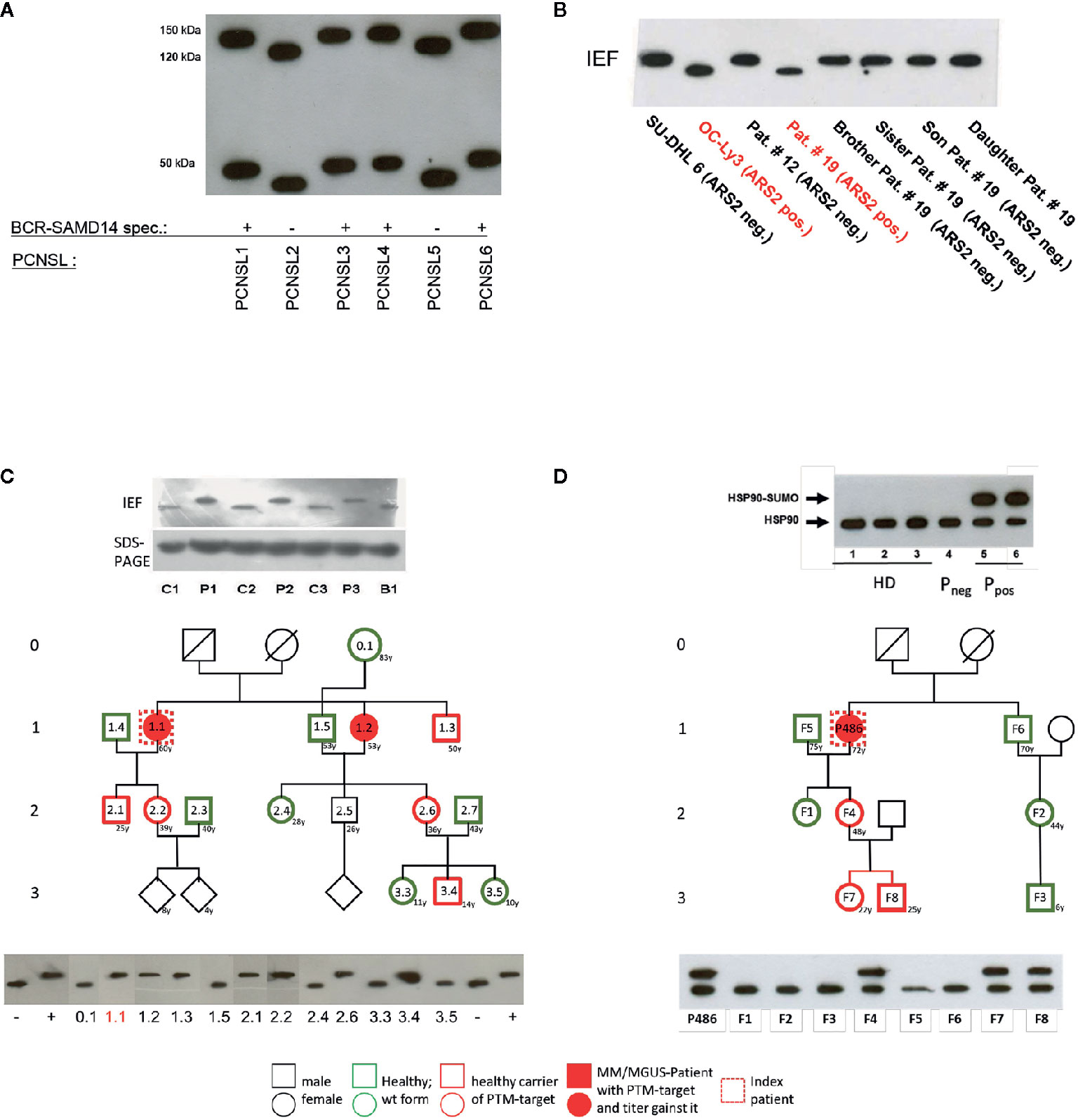
Figure 2 Examples of posttranslational modification of lymphoma B-cell receptor (BCR) target antigens: (A) Representative Western blot of hyper-N-glycosylated Neurabin-I and SAMD14 in patients with PCNSL first reported by Thurner et al (173). Patients with primary central nervous system lymphoma (PCNSL) and SAMD14/Neurabin-I reactive lymphoma BCRs had exclusively hyper-N-glycosylated isoforms of both antigens. (B) Representative isoelectric focusing (IEF) of hypophosphorylated Arsenite resistance protein 2 (ARS2) in diffuse large B cell lymphoma (DLBCL) first reported by Thurner et al (174). DLBCL cell lines and peripheral blood lysates of DLBCL of patients and family members. ARS2 was found to be hypo-phosphorylated in a cell line and a patient, but this phenotype of an atypical posttranslational modifications (PTM) was not inherited in a Mendelian manner. (C) Representative analysis of hyperphosphorylated SLP2 (paratarg-7), which was first reported by Preuss et al (171). Hyperphosphorylated SLP2 was detected by IEF and not by SDS-PAGE. The hyperphosphorylation introduces an additional phosphate group in the molecule leading to a different isoelectric point of the protein; the increase in molecular weight is too small to be detected by SDS PAGE. Shown are immunoblots incubated with anti-human-STOML2 (paratarg-7). P1-3: MM/monoclonal gammopathy of undetermined significance (MGUS) patients with immunoreactivity against paratarg-7; C1-3: MM/MGUS patients without SLP2 (paratarg-7) immunoreactivity; B1: healthy blood donor. Inheritance: SLP2- phosphorylation state in patient families (example). The family of a relevant patient was analyzed for its SLP2 phosphorylation state by IEF. Carriers of hyperphosphorylated SLP2 (patient and persons of risk) were indicated in red. (D) Representative SDS PAGE of sumoylated HSP90 first reported by Preuss et al (175). Sumoylation of HSP90 does not change the isoelectric point of the molecule but leads to an increase in molecular weight which is detected by SDS PAGE. HD: healthy donor; Pneg: MM/MGUS patient without immunoreactivity against HSP90-SUMO; Ppos: MM/MGUS patients with immunoreactivity against HSP90-SUMO.Inheritance: HSP90 sumoylation state in patient family (example). The family of a relevant patient was analyzed for HSP90-SUMO by SDS PAGE. Carriers of HSP90-SUMO (patient and persons of risk) are indicated in red.
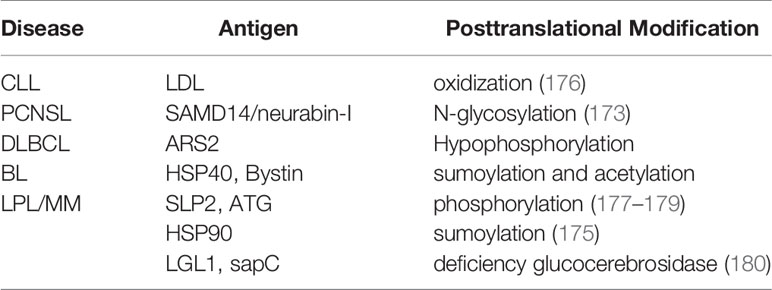
Table 2 Post-translationally modified B-Cell Receptor (BCR) antigens in lymphoma.
PCNSL represents a specific extranodal subtype of DLBCL with molecular similarities to systemic DLBCL of MCD or C5 type with frequent mutations in MYD88 and CD79 (195, 202). PCNSL show strong over-representation of IGVH4-34, and poly-reactivity against a plethora of antigens was reported (196, 203). In addition, sterile a-motif domain containing protein 14 (SAMD14) and neural tissue-specific F-actin binding protein I (neurabin-I) with a homologous SAM domain were identified as specific auto-antigenic targets of recombinant BCRs of PCNSL and SAMD14/neurabin-I specific autoantibodies were detected in sera and cerebrospinal fluid of patients. In the respective cases, SAMD14 and neurabin-I were atypically hyper-N-glycosylated (SAMD14 at ASN339 and neurabin-I at ASN1277), explaining their auto-immunogenicity (Figure 2) (173). Primary intraocular lymphoma (PIOL) is biologically closely related to PCNSL and can progress after a various period of time into PCNSL (198), but it is not clear whether SAMD14/neurabin-I are altered as well in this subgroup, or if a different antigenic trigger exists.
Although tonic BCR activation is characteristic for BL (84), preliminary results suggest the involvement of post-translationally modified specific autoantigens that contribute to pathogenesis in at least a subgroup of sporadic EBV-negative BL (191).
Autoantigens in Plasma Cell Dyscrasia
Multiple myeloma (MM) accounts for 1% of all malignancies, and for over 10% of hematological malignancies. The disease is characterized by neoplastic proliferation of a single plasma cell clone producing a large amount of a monoclonal antibody termed paraprotein, M-protein or M-component (204). Malignant gammopathies are often preceded by monoclonal gammopathy of undetermined significance (MGUS), a benign disorder with a strikingly elevated monoclonal Ig level in individuals lacking evidence of MM or other lymphoproliferative malignancies. Long-term follow-up of patients with MGUS reveals a 1% to 3% annual risk of developing MM or, to a lesser extent, other lymphoproliferative malignancies (204). In MGUS and plasma cell dyscrasia hyperphosphorylated SLP2 and sumoylated HSP90 were found to be the targets of paraproteins (171, 175) both in MM and lymphoplasmocytic lymphoma (LPL) (Tables 2 and 3). These “paratargets” with their atypical PTMs were found with different frequencies in different ethnics (205). Of interest, PTMs for both antigens had an autosomal dominant pattern of inheritance (Figure 2), and pedigrees with family members as carriers and affected with MGUS or plasma cell dyscrasia were described for both hyperphosphorylated SLP2 and sumoylated HSP90 (175, 179). Interestingly, SLP2-reactive paraproteins do not differentiate between the normally phosphorylated SLP2 and the hyperphosphorylated SLP2 isoform in contrast to HSP90-reactive paraprotein, which is specific for the sumoylated isoform.
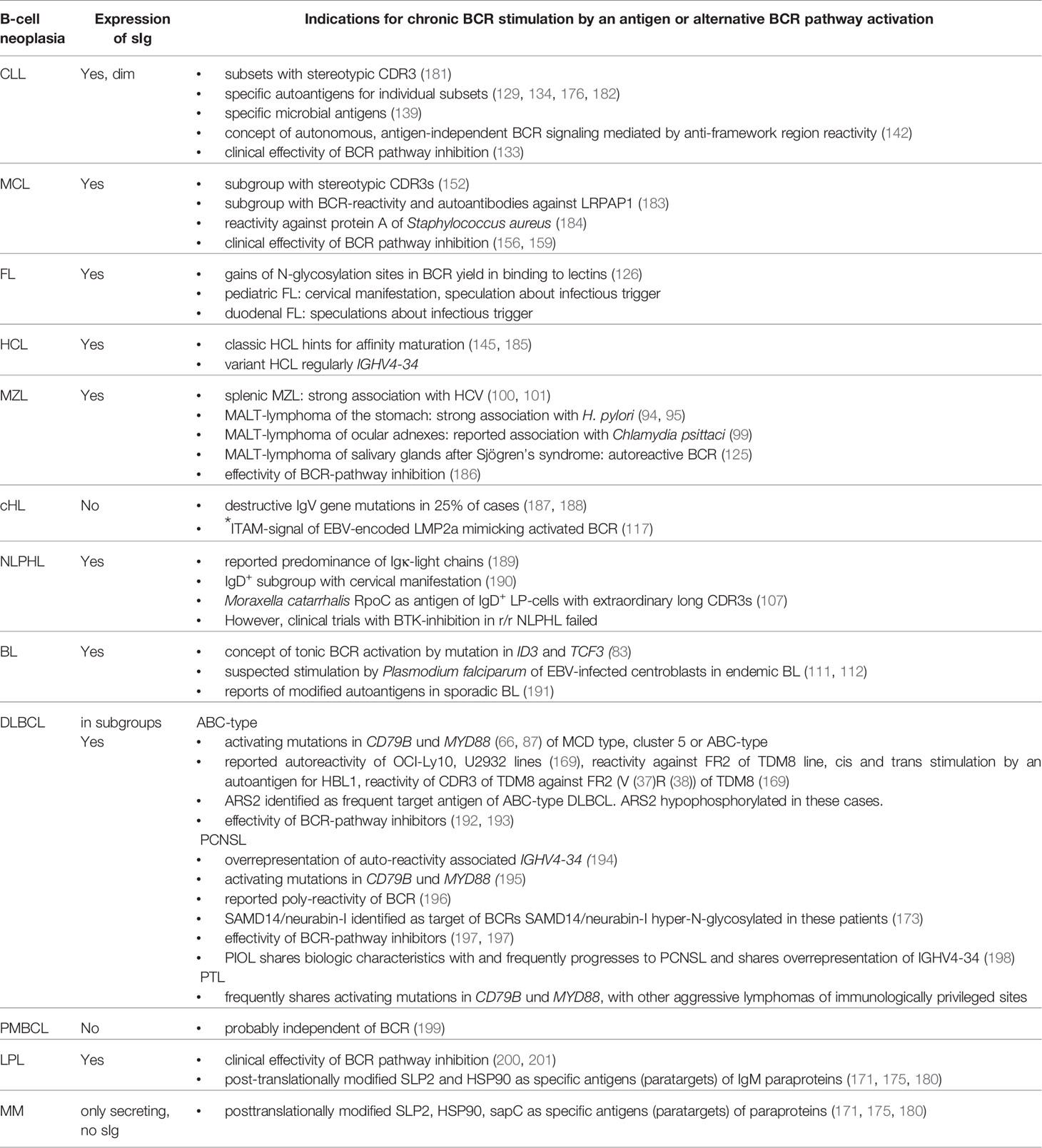
Table 3 B cell lymphoma and B-cell receptor (BCR) antigens.
The question as to why the respective paraprotein antigen is present in post-translationally modified form in this group of people and to what extent this influences the development and progress of the disease remains unsolved. It is remarkable that these post-translationally modified antigens were detected almost exclusively in MM/MGUS patients and their blood relatives as well as in approx. 1-2% of the healthy population; in all other examined subjects (patients and healthy persons) the antigen is present in wild-type form, i.e. unchanged and does not induce an immune response. Nair et al. had different findings and described glycosphingosine as a frequent antigenic target structure of paraproteins in sporadic MGUS and MM as well as in monoclonal gammopathies associated with Gaucher disease (180, 206). Data from our laboratory rather suggest post-translationally modified saposin C as a paraprotein target structure in Gaucher-associated MM/MGUS (207).
Role of T-Helper Cell Co-Stimulation in Lymphoproliferative B-Cell-Diseases
Sequence and structure of the BCR antigens of malignant plasma cells found to date, whether post-translationally modified or not, indicated the need for the involvement of T-helper cells for stimulation at the beginning and during the course of pathogenesis. Furthermore, several studies provided evidence for a causal relationship between MGUS/MM and chronic antigenic stimulation (208). In addition, when SLP2 is used as a model antigen in MM, the patient’s paraprotein binds to both the wild type SLP2 and the actually immunogenic post-translationally hyper-phosphorylated variant of SLP2. Thus, SLP2-specific B cells cannot be the initiators of the postulated chronic stimulation or pathogenesis.
In vitro stimulation of CD4+ T-helper cells of MGUS/MM patients with a paratarg-7-specific paraprotein induced distinct paratarg-7-specific responses: 65% of these patients had a paratarg-7-specific TH1 response. 89% of these TH1 cells specifically recognized the modified hyperphosphorylated SLP2. 42% of the stimulated patients also had modification-specific TH2 cells (Figures 3A, B). Hence, with the T-helper cells the contribution of the adaptive immune system was found, which specifically recognize the post-translational modification and thus are at the beginning of chronic stimulation. Further characterization of the hyperphosphorylated SLP2-specific T-helper cells showed that there are (at least) six HLA-DR subtypes, named “permissive”, that can present phosphorylated peptide epitopes to the T helper cells for stimulation. Compared to a healthy reference population, patients with SLP2-specific MM express these six permissive DR subtypes significantly more frequently. Thus, expression of a hyperphosphorylated-SLP2-permissive DR subtype is, besides posttranslational modifications, the second important prerequisite for the development of this disease (209).
The interaction of these modification-specific T-helper cells from patients with non-modification-specific paraprotein and their corresponding B cells is a new type of epitope spreading. In contrast to classical epitope spreading, which extends horizontally across the amino acid sequence of an antigen, this is a vertically spread modification of the same amino acid.
Physiologically, however, both phenomena are based on the same fact that the epitope of an antigen that is specifically bound by the BCR of a B cell does not have to be the same epitope that the B cell presents to the T-helper cells in its MHC-II molecules after antigen processing. When a T-helper cell finds its antigenic epitope in the appropriate MHC-II context on the B cell, it provides the help necessary for the maturation of this B cell. Applied to the situation with SLP2, this means that even those SLP2-specific B cells whose BCR does not differentiate between the modified version of this antigen phosphorylated on serine17 and the non-phosphorylated wild-type, can be stimulated by modification-specific T-helper cells (Figures 3C, D).
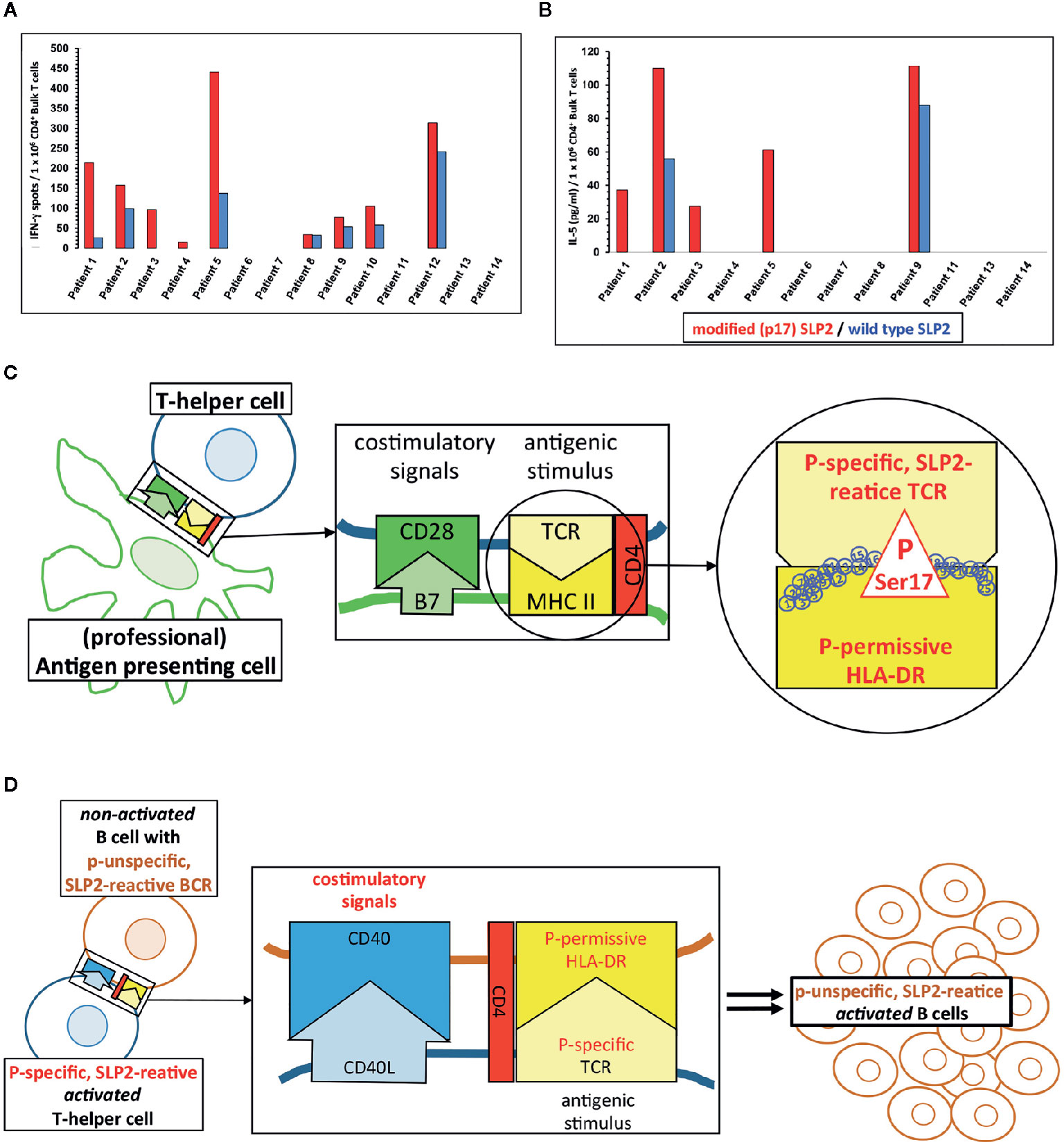
Figure 3 Paratarg-7-specific T-helper cells in myeloma/monoclonal gammopathy of undetermined significance (MGUS) patients with a SLP2-specific paraprotein as a new type of epitope spreading. (A) Representative IFN-g ELISPOTs first reported by Neumann et al. showed by in vitro stimulation for SLP2(paratarg-7)-specific TH1 cells in 9/14 patients specific responses to the antigen. 8/9 patients had a significant (p << 0.05.) stronger response against the phosphorylated variant of the SLP2-peptides used for stimulation (red columns) compared to the peptides derived from the non-phosphorylated wild type (blue columns) (209). (B) T-helper cells from 5/12 patients showed a significant stronger TH2 response against the modified peptides compared to the wild type peptides. Again, these are the results of in vitro stimulation of myeloma/MGUS patients’ T-helper cells with a paratarg-7/SLP2-specific paraprotein using overlapping 15 amino acids long peptides covering the first 30 amino acids of the SLP2 sequence. Wild-type peptides and peptides with a phosphorylated Ser17 of the posttranslational modifications (PTM) variant were used. Subsequently, the culture supernatant was analyzed by ELISA for the TH2 cytokine IL-5 (209). (C) T-helper cells with a T-cell receptor (TCR) specific for Ser17-phosphorylated version of SLP2 are primed by antigen-presenting cells, equipped with the corresponding permissive MHC-II molecules offering all necessary costimulatory signals for full maturation. (D) Subsequently, T-helper cells with these properties (phosphospecific SLP2-reactive) can stimulate all B cells presenting a phosphorylated Ser17 epitope. The specificity of the receptor of these B cells for the hyperphosphorylated isoform of the antigen is not important. Thus, B cells are also stimulated whose BCRs bind the unmodified wild type of SLP2. This form of epitope spreading comprises the same amino acid of the antigen, but with the difference of posttranslational modification. Therefore, this type of spreading is vertically oriented. For some other posttranslationally modified antigens, the lymphoma BCRs are specific for modified isoform/variant depending on the PTM, i.e. HSP90-SUMO.
Since all posttranslational paratarg modifications found to date (including SLP2) are always consistently expressed in all cells of the organism, the B cells of the respective patients with a paratarg-specific BCR as well as all other antigen-presenting cells (e.g. macrophages or DC) can present only the modified variant to their T-helper cells. This can be seen by the fact that, except in the TH1 response of patient 8, the induced SLP2-specific T-cell responses were significantly modification-specific after in vitro stimulation of the T-helper cells (Figures 3A, B).
B-Cell Lymphoma Without a Role of BCR Antigens
For some other lymphoma entities, stimulation of the BCR by antigens is likely not important. These are for example classical Hodgkin lymphoma, whose malignant Reed-Sternberg und Hodgkin cells have lost their B-cell-phenotype including functional BCR genes (187), PTLD with crippled BCR genes (210), or primary mediastinal B cell lymphoma (211, 212), which usually does not express sIg either (Figure 4).
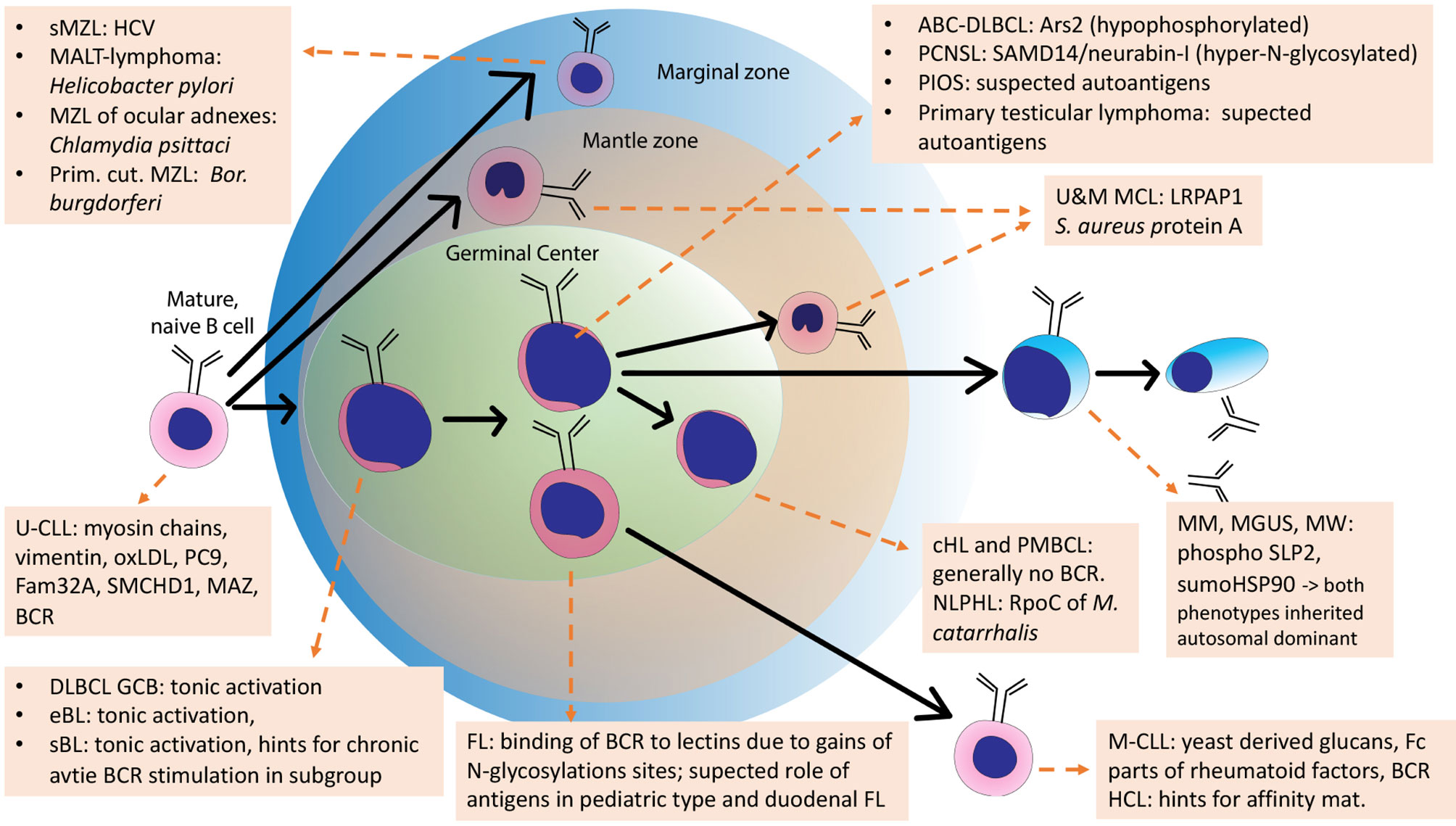
Figure 4 Schematic overview of the development of normal B cells and their malignant counterparts. Arrows: Normal B-cell differentiation; dotted arrows: normal B-cells as cell of origins of specific lymphomas. This scheme is an adaption of the scheme from Küppers et al., 2005, with added B-cell receptor (BCR) antigens identified in the meantime.
Suspected Role of Specific Antigens in T Cell Neoplasia
The search for TCR antigens is much more complicated as it requires screening of peptides presented on correct HLAs. This is probably the main reason why no TCR target antigens of PTCL have been found. For certain peripheral T cell lymphomas (PTCL) there is evidence for a potential role of antigen stimulation in their pathogenesis. One example is angioimmunoblastic T cell lymphoma, in which clonal B cell populations and paraproteins are often also found. It is unclear if the paraproteins in AITL might be a simple epiphenomenon of accompanying EBV+ B-cell-clones, or if they target the same antigen as the TCRs of the T follicular helper cells of origin of AITL. Another example is enteropathy-associated T cell lymphoma, which is rare and typically occurs after a long period of celiac disease. Breast implant-associated anaplastic T cell lymphoma usually shows a significantly more favorable course than ALK-negative ALCL and the role of antigen stimulation is partly shown by remission after explanation of the breast implants alone. However, a TCR-reactivity against components of breast implants has not been shown.
Lymphoma BCR and Therapeutic Implications
Inhibition of the BCR Pathway
In the treatment of B-cell lymphomas and leukemias and increasingly in the treatment of autoimmune diseases, the inhibition of the main signaling pathway of B cells, the BCR pathway, plays a crucial role. SYK inhibitors make pathogenetically sense because the SYK kinase lies relatively far upstream in this pathway. SYK inhibitors showed in vitro and in vivo activity against B-cell lymphomas and various other hematological neoplasias. However, SYK inhibitors have not yet been able to gain clinical importance in the treatment of B-cell lymphomas (213). Interestingly, SYK inhibitors were investigated for autoimmune diseases with major B-cell involvement, including rheumatoid arthritis (214), and the first FDA approval of a SYK inhibitor, fostamatinib was granted for treatment of immune thrombocytopenia (ITP) (215). The BTK inhibitor ibrutinib initially represented a new standard in the therapy of r/r CLL showing even enduring responses in CLL with TP53 mutations or del17p (133), and later it was shown that even in the therapy of CLL with mutated IGHV status the results were superior to the previous gold-standard immunochemotherapy concepts (216). In these cases, the accumulation of cancer cells is usually slowed down, but no complete remission in terms of negative minimally residual disease (MRD) is achieved. The C481S mutation in BTK and various PLCg2 mutations as well as mutations in the PI3K signaling pathway have been identified as resistance mechanisms (217, 218). Further indications for which ibrutinib is approved are LPL in combination with rituximab (201) and r/r MCL (156), for which the combination with the BCL2 inhibitor venetoclax was particularly impressive (159). In aggressive lymphomas, BTK inhibitors appear to be of particular benefit in lymphomas with activating mutations in MYD88 and CD79B (91). In PCNSL, BTK inhibitors have been used as monotherapy (197). Unfortunately, in combination with immunochemotherapy, increased mold infections were observed (219). In a DLBCL first line trial, the combination of ibrutinib with immunochemotherapy led to increased toxicity, so that immunochemotherapy could often not be completed in a relevant proportion of patients, but a subgroup analysis showed a significant improvement of overall survival for younger patients (193). Acalabrutinib is a second generation BTK inhibitor, which is also approved in CLL and shows a different spectrum of side effects as compared to ibrutinib (220–222). In contrast to ibrutinib and acalabrutinib, the non-covalent BTK inhibitors Loxo-305, Vecabrutinib and ARQ 531 do not require the presence of the C481 wild type configuration, but may also be active in C481S BTK mutated disease (223–225). Similar to SYK inhibitors, BTK inhibitors are also being investigated in autoimmune diseases and some are approved for this purpose as for ITP, multiple sclerosis or graft-versus-host disease (GVHD) (226–228). A further prominent target for inhibition is PI3K with idelalisib, copanlisib and duvelisib approved for r/r CLL/SLL and FL, the first inhibits selectively PI3kdelta, the latter inhibits PI3Kalpha and PI3Kdelta and the last PI3Kdelta and PI3Kgamma (229–231). However, the use of idelalisib was decreased due to toxicity problems, mainly related to autoimmune phenomena such as pneumonitis and colitis (232). Further potential targets for inhibition are Lyn and the components of the CBM complex, i.e. CARD11, BCL10, and MALT1 (233).
It is presently unclear why some types of lymphomas respond well to BCR inhibiting treatment and others not. Perhaps, in non-responding lymphomas, BCR pathway activity is less essential for lymphoma cell survival and proliferation, or this pathway is activated by genetic and/or epigenetic mechanisms further downstream, so that upstream inhibition of BTK does not cause a downregulation of this pathway, an example is r/r FL lymphoma with mutation in CARD11 showing poor response to ibrutinib (234).
Attenuation of BCR Activation by Eradication of Antigenic Trigger
A possible therapeutic strategy for antigen-driven lymphomas is to remove the antigenic trigger. This can be possible for infectious antigenic triggers as typically in MZL by antibiotics or antiviral therapy (101, 103). A similar approach of antibiotic eradication could be investigated for Moraxella spec.-reactive NLPHL in clinical trials for patients with relapsed/refractory IgD+ NLPHL, or a consolidating vaccination against Moraxella catarrhalis in early stage NLPHL patients who have only been treated locally and have not received therapeutic B-cell depletion. Of course, this vaccination must not contain the antigenic triggers themselves.
Attenuation of Stimulation by Modulation of Immunogenic PTMs
For lymphoplasmocytic lymphoma, for ABC-DLBCL, and for PCNSL, the search for specific substances to modulate the immunogenic PTMs of hyper-phosphorylated SLP2 and sumoylated HSP90 in LPL, of N-hyperglycosylated SAMD14/neurabin-I in PCNSL and of hypophosphorylated ARS2 in DLBCL would be useful. The aim would be to reverse the PTMs as permanently as possible and thus weaken the immunogenic stimulus. These substances could, for example, be investigated in secondary prophylaxis.
Targeting Lymphoma-Cells by Their BCR-Reactivity
The specific BCR antigens identified so far could possibly be used as basis for therapeutic approaches using retrograde BCR targeting - which has been proposed as the BAR (BCR antigen for reverse targeting) concept. This approach has parallels of targeting lymphoma BCRs by anti-idiotypes (235) and exploits the entity-specific BCR reactivity of lymphoma clones. Possibilities would be, for example, immunotoxins consisting of fusion proteins with the epitope region of the target antigen coupled to a toxin or an enhancer of the immune response (236–238), or T or NK cells with chimeric antigen receptors with the epitope region of the target antigens as extracellular capture domain (Figure 5) (239). This approach was demonstrated in vitro using immunotoxins consisting of the epitope of the respective BCR target antigen and a shortened form of Pseudomonas aeruginosa exotoxin A (ETA’). The role of pre-existing serum autoantibodies against the corresponding target antigens is critical here. After infusion of immunotoxins, consisting of epitope region and toxin, deposits of toxic immune complexes could develop. When using chimeric antigen receptor (CAR) T cells with an epitope region of the lymphoma BCR target antigen as part of the CAR ectodomain, the antibodies could possibly cause strong stimulation with cytokine release syndrome. In vivo, however, such CAR T-cell constructs appear to function despite the presence of autoantibodies (239). A standard anti-CD19-scFv/CD28/CD3ζ second-generation CAR construct might be used as a basis. The target antigen epitopes are combined or replace the anti CD19 scFv.
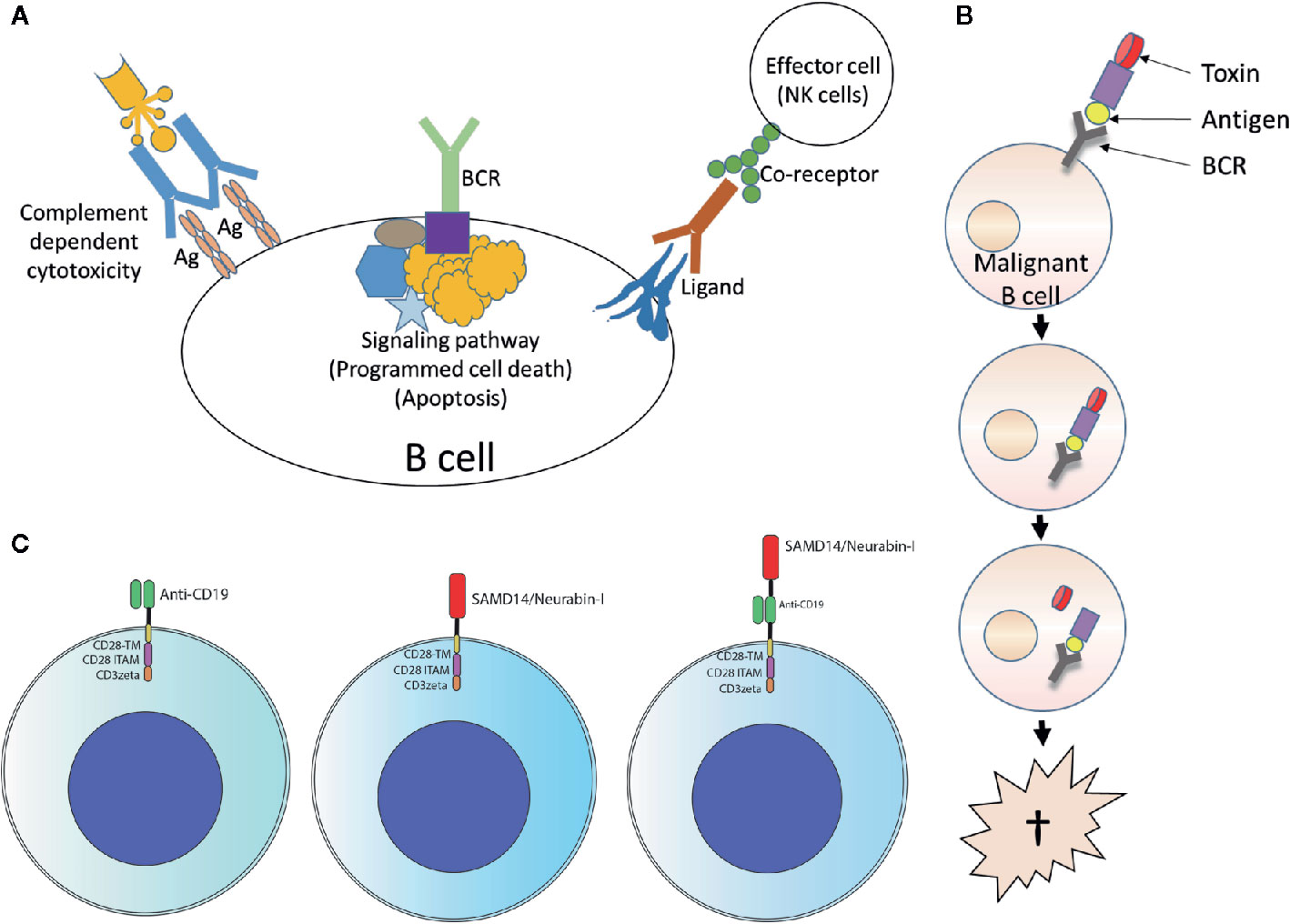
Figure 5 Usage of lymphoma B-cell receptor (BCR) antigens for targeting lymphoma. (A) Forward targeting: Classical antibody therapy cannot differentiate between malignant and benign B cells. mAbs against B cell surface antigens such as CD20 bind to their target present on all B cells and activate complement, antibody-dependent cell-mediated cytotoxicity or direct cell death. The same is also true for antibodies against receptors with immunomodulatory functions. Independently of this, tumor cell death could also be induced antibody-independently by drugs that interfere with critical signaling pathways (such as ibrutinib, which interferes with BTK, a step in the BCR signaling pathway). Ag, antigen. (B) Reverse targeting: The BAR (BCR antigen for reverse targeting) concept is based on the highly specific interaction of a BCR found exclusively on malignant B cells with its highly specific target antigen; benign B cells do not possess this BCR. Synthetic conjugates of BCR antigen with a toxin (BAR toxin) bind exclusively to the malignant cells, are internalized and release the toxin that kills the cell. (C) CARs: Conventional CAR with CD19 scFv/CD28 4-1BB CD3ζ CAR backbone (left); the anti CD19 scFv was exchanged by the frequent BCR antigen of MCL resulting in the construct SAMD14/neurabin-I/CD28 4-1BB CD3ζ CAR backbone (middle) or combined with anti CD19 scFv (right).
Author Contributions
LT wrote the manuscript. K-DP wrote the section on plasma cell diseases, FN on the role of Th cells, SH on NLPHL, and MB and K-DP on reverse targeting. RK and SS revised the manuscript and contributed significant and very important additional information. MH revised the manuscript and contributed information concerning effector cells and therapeutic mechanisms. All authors contributed to the article and approved the submitted version.
Funding
We thank the following funding organizations for supporting lymphoma research of the authors relevant for this review: HOMFOR grants to LT and MB, Deutsche Forschungsgemeinschaft (grants KU1315/9-2 and KU1315/14-1 to RK), Deutsche Krebshilfe (grant 70112112 to RK), Wilhelm Sander Foundation (grant 2019.056.1 to LT, K-DP, FN, and SH and grant 2018.101.1 to RK), Deutsche José Carreras Leukämie Stiftung (grant 02-R/2020 to RK), the Hairy Cell Leukemia Foundation (to RK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Sci (80) (1990) 248:1517–23. doi: 10.1126/science.2360047
2. Moshous D, Callebaut I, De Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell (2001) 105:177–86. doi: 10.1016/S0092-8674(01)00309-9
3. Hozumi N, Tonegawa S. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc Natl Acad Sci U S A (1976) 73:3628–32. doi: 10.1073/pnas.73.10.3628
4. Komori T, Okada A, Stewart V, Alt FW. Lack of N regions in antigen receptor variable region genes of TdT-deficient lymphocytes. Science (1993) 261:1171–5. doi: 10.1126/science.8356451
5. Gilfillan S, Dierich A, Lemeur M, Benoist C, Mathis D. Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science (1993) 261:1175–8. doi: 10.1126/science.8356452
6. Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science (1985) 229:1390–3. doi: 10.1126/science.3929382
7. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. NatRevCancer (2005) 5:251–62. doi: 10.1038/nrc1589
8. MacLennan ICM. Germinal Centers. Annu Rev Immunol (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
9. Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol (2006) 6(8):573–83. doi: 10.1038/nri1896
10. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell (2000) 102:553–63. doi: 10.1016/S0092-8674(00)00078-7
11. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell (2000) 102:565–75. doi: 10.1016/S0092-8674(00)00079-9
12. Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol (2004) 5(9):943–52. doi: 10.1038/ni1100
13. Basso K, Dalla-Favera R. BCL6. master regulator of the germinal center reaction and key oncogene in B Cell lymphomagenesis. Adv Immunol (2010) 105:193–210. doi: 10.1016/S0065-2776(10)05007-8
14. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity (2003) 19(4):607–20. doi: 10.1016/S1074-7613(03)00267-X
15. Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol (2020) 21:1082–93. doi: 10.1038/s41590-020-0713-6
16. Rajewsky K. Clonal selection and learning in the antibody system. Nature (1996) 381:751–8. doi: 10.1038/381751a0
17. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Sci (80) (2003) 301:1374–7. doi: 10.1126/science.1086907
18. Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature (1989) 337:562–6. doi: 10.1038/337562a0
19. Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature (1988) 334:676–82. doi: 10.1038/334676a0
20. Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, Weigert M. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature (1991) 349:331–4. doi: 10.1038/349331a0
21. Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat Immunol (2004) 5:645–50. doi: 10.1038/ni1076
22. Köhler F, Hug E, Eschbach C, Meixlsperger S, Hobeika E, Kofer J, et al. Autoreactive B Cell Receptors Mimic Autonomous Pre-B Cell Receptor Signaling and Induce Proliferation of Early B Cells. Immunity (2008) 29:912–21. doi: 10.1016/j.immuni.2008.10.013
23. Eschbach C, Bach MP, Fidler I, Pelanda R, K??hler F, Rajewsky K, et al. Efficient generation of B lymphocytes by recognition of self-antigens. Eur J Immunol (2011) 41:2397–403. doi: 10.1002/eji.201041344
24. Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol (2008) 20:632–8. doi: 10.1016/j.coi.2008.09.001
25. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med (2005) 201:703–11. doi: 10.1084/jem.20042251
26. Samuels J, Ng Y, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med (2005) 201:1659–67. doi: 10.1084/jem.20042321
27. Rathmell JC, Cooke MP, Ho WY, Grein J, Townsend SE, Davis MM, et al. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature (1995) 376:181–4. doi: 10.1038/376181a0
28. Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: Dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell (1996) 87:319–29. doi: 10.1016/S0092-8674(00)81349-5
29. Hervé M, Isnardi I, Ng Y, Bussel JB, Ochs HD, Cunningham-Rundles C, et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med (2007) 204:1583–93. doi: 10.1084/jem.20062287
30. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature (1995) 374:546–9. doi: 10.1038/374546a0
31. Glocker EO, Kotlarz D, Klein C, Shah N, Grimbacher B. IL-10 and IL-10 receptor defects in humans. Ann N Y Acad Sci (2011) 1246:102–7. doi: 10.1111/j.1749-6632.2011.06339.x
32. Fernando MMA, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, et al. Defining the role of the MHC in autoimmunity: A review and pooled analysis. PloS Genet (2008) 4. doi: 10.1371/journal.pgen.1000024
33. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36:337–8. doi: 10.1038/ng1323
34. Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol (Baltimore Md 1950) (2007) 179:4704–10. doi: 10.4049/jimmunol.179.7.4704
35. Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol (2014) 10:602–11. doi: 10.1038/nrrheum.2014.109
36. Ueda H, Howson JMM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423:506–11. doi: 10.1038/nature01621
37. Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The Ubiquitin Modifying Enzyme A20 Restricts B Cell Survival and Prevents Autoimmunity. Immunity (2010) 33:181–91. doi: 10.1016/j.immuni.2010.07.017
38. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature (2002) 416:603–7. doi: 10.1038/416603a
39. Jackson SW, Kolhatkar NS, Rawlings DJ. B cells take the front seat: Dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr Opin Immunol (2015) 33:70–7. doi: 10.1016/j.coi.2015.01.018
40. Rees JH. Campylobacter jejuni Infection and Guillain–Barré Syndrome. NEJM (1995) 333:1374–9. doi: 10.1056/NEJM199511233332102
41. Guilherme L, Kalil J, Cunningham MM. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity (2006) 39:31–9. doi: 10.1080/08916930500484674
42. Sahin U, Tureci O, Schmitt H, Cochlovius B, Johannes T, Schmits R, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. ProcNatlAcadSciUSA (1995) 92:11810–3. doi: 10.1073/pnas.92.25.11810
43. Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Sci (80) (2014) 343:152–7. doi: 10.1126/science.1246886
44. Doyle HA, Mamula MJ. Autoantigenesis: The evolution of protein modifications in autoimmune disease. Curr Opin Immunol (2012) 24:112–8. doi: 10.1016/j.coi.2011.12.003
45. Arentz-Hansen H, Korner R, Molberg O, Quarsten H, Vader W, Kooy YM, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. JExpMed (2000) 191:603–12. doi: 10.1084/jem.191.4.603
46. Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L. Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature (1986) 324:258–60. doi: 10.1038/324258a0
47. Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. JClinInvest (1998) 101:273–81. doi: 10.1172/JCI1316
48. van Venrooij WJ, Pruijn GJ. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res (2000) 2:249–51. doi: 10.1186/ar95
49. Neugebauer KM, Merrill JT, Wener MH, Lahita RG, Roth MB. SR proteins are autoantigens in patients with systemic lupus erythematosus. Importance of phosphoepitopes. Arthritis Rheum (2000) 43:1768–78. doi: 10.1002/1529-0131(200008)43:8<1768::AID-ANR13>3.0.CO;2-9
50. Utz PJ, Hottelet M, Schur PH, Anderson P. Proteins phosphorylated during stress-induced apoptosis are common targets for autoantibody production in patients with systemic lupus erythematosus. JExpMed (1997) 185:843–54. doi: 10.1084/jem.185.5.843
51. Cappello P, Tomaino B, Chiarle R, Ceruti P, Novarino A, Castagnoli C, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by α-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer (2009) 125:639–48. doi: 10.1002/ijc.24355
52. Tomaino B, Cappello P, Capello M, Fredolini C, Sperduti I, Migliorini P, et al. Circulating autoantibodies to phosphorylated α-enolase are a hallmark of pancreatic cancer. J Proteome Res (2011) 10:105–12. doi: 10.1021/pr100213b
53. Zhou W, Capello M, Fredolini C, Piemonti L, Liotta LA, Novelli F, et al. Mass spectrometry analysis of the post-translational modifications of alpha-enolase from pancreatic ductal adenocarcinoma cells. J Proteome Res (2010) 9:2929–36. doi: 10.1021/pr901109w
54. Mor-Vaknin N, Kappes F, Dick AE, Legendre M, Damoc C, Teitz-Tennenbaum S, et al. DEK in the synovium of patients with juvenile idiopathic arthritis: Characterization of DEK antibodies and posttranslational modification of the DEK autoantigen. Arthritis Rheum (2011) 63(2):556–67. doi: 10.1002/art.30138
55. Wållberg M, Bergquist J, Achour A, Breij E, Harris RA. Malondialdehyde modification of myelin oligodendrocyte glycoprotein leads to increased immunogenicity and encephalitogenicity. Eur J Immunol (2007) 37:1986–95. doi: 10.1002/eji.200636912
56. Vanacore RM, Ham AJL, Cartailler JP, Sundaramoorthy M, Todd P, Pedchenko V, et al. A role for collagen IV cross-links in conferring immune privilege to the goodpasture autoantigen: Structural basis for the crypticity of B cell epitopes. J Biol Chem (2008) 283(33):22737–48. doi: 10.1074/jbc.M803451200
57. Vanacore R, Ham AJL, Voehler M, Sanders CR, Conrads TP, Veenstra TD, et al. A Sulfilimine Bond Identified in Collagen IV. Sci (80) (2009) 325(5945):1230–4. doi: 10.1126/science.1176811
58. Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest (2009) 119(6):1668–77. doi: 10.1172/JCI38468
59. Raposo B, Merky P, Lundqvist C, Yamada H, Urbonaviciute V, Niaudet C, et al. T cells specific for post-translational modifications escape intrathymic tolerance induction. Nat Commun (2018) 9:353. doi: 10.1038/s41467-017-02763-y
60. Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature (2001) 412:341–6. doi: 10.1038/35085588
61. Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, et al. BCL-6 mutations in normal germinal center B cells: Evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci (1998) 95(20):11816–21. doi: 10.1073/pnas.95.20.11816
62. Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet (2008) 40:108–12. doi: 10.1038/ng.2007.35
63. Dorsett Y, Robbiani DF, Jankovic M, Reina-San-Martin B, Eisenreich TR, Nussenzweig MC. A role for AID in chromosome translocations between c-myc and the IgH variable region. J Exp Med (2007) 204(9):2225–32. doi: 10.1084/jem.20070884
64. Küppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene (2001) 20:5580–94. doi: 10.1038/sj.onc.1204640
65. Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature (2004) 432:635–9. doi: 10.1038/nature03147
66. Davis RE, Lenz G, Wright G, Xiao W, Zhao H, Staudt LM, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature (2010) 463:88–92. doi: 10.1038/nature08638
67. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N Engl J Med (2012) 367:826–33. doi: 10.1056/NEJMoa1200710
68. Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci U.S.A. (1991) 88:5413–7. doi: 10.1073/pnas.88.12.5413
69. Schmitz R, Hansmann M-L, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med (2009) 206:981–9. doi: 10.1084/jem.20090528
70. Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature (2009) 459:712–6. doi: 10.1038/nature07969
71. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475:101–5. doi: 10.1038/nature10113
72. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
73. Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med (2012) 209(9):1537–51. doi: 10.1084/jem.20120904
74. Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood (2018) 131(22):2454–65. doi: 10.1182/blood-2017-11-814913
75. Guiter C, Dusanter-Fourt I, Copie-Bergman C, Boulland M, Le Gouvello S, Gaulard P, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood (2004) 104(2):543–9. doi: 10.1182/blood-2003-10-3545
76. MacPherson D, Kim J, Kim T, Rhee BK, Van Oostrom CTM, DiTullio RA, et al. Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J (2004) 23:3689–99. doi: 10.1038/sj.emboj.7600363
77. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood (2010) 116(17):3268–77. doi: 10.1182/blood-2010-05-282780
78. Berghoff AS, Ricken G, Widhalm G, Rajky O, Hainfellner JA, Birner P, et al. PD1 (CD279) and PD-L1 (CD274, B7H1) expression in primary central nervous system lymphomas (PCNSL). Clin Neuropathol (2014) 33:42–9. doi: 10.5414/NP300698
79. Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, et al. Combined Genetic Inactivation of β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell (2011) 20(6):728–40. doi: 10.1016/j.ccr.2011.11.006
80. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
81. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood (2004) 103:275–82. doi: 10.1182/blood-2003-05-1545
82. Dameshek W, Schwartz RS. Leukemia and auto-immunization- some possible relationships. Blood (1959) 14:1151–8. doi: 10.1182/blood.V14.10.1151.1151
83. Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet (2012) 44:1316–20. doi: 10.1038/ng.2469
84. Corso J, Pan KT, Walter R, Doebele C, Mohr S, Bohnenberger H, et al. Elucidation of tonic and activated B-cell receptor signaling in Burkitt’s lymphoma provides insights into regulation of cell survival. Proc Natl Acad Sci U.S.A. (2016). doi: 10.1073/pnas.1601053113
85. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature (2006). doi: 10.1038/nature04687
86. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Sci (80) (2008) 319(5870):1676–9. doi: 10.1126/science.1153629
87. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470:115–21. doi: 10.1038/nature09671
88. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature (2010) 463:88–92. doi: 10.1038/nature08638
89. Gomes de Castro MA, Wildhagen H, Sograte-Idrissi S, Hitzing C, Binder M, Trepel M, et al. Differential organization of tonic and chronic B cell antigen receptors in the plasma membrane. Nat Commun (2019) 10:820. doi: 10.1038/s41467-019-08677-1
90. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi: 10.1038/35047123
91. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature (2018) 560:387–91. doi: 10.1038/s41586-018-0290-0
92. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet (1991) 338(8776):1175–6. doi: 10.1016/0140-6736(91)92035-Z
93. Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med (1994) 330:1267–71. doi: 10.1056/NEJM199405053301803
94. Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de BM, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet (1993) 342:575–7. doi: 10.1016/0140-6736(93)91409-F
95. Craig VJ, Arnold I, Gerke C, Huynh MQ, Wundisch T, Neubauer A, et al. Gastric MALT lymphoma B cells express polyreactive, somatically mutated immunoglobulins. Blood (2010) 115:581–91. doi: 10.1182/blood-2009-06-228015
96. Bende RJ, Aarts WM, Riedl RG, De Jong D, Pals ST, Van Noesel CJM. Among B cell non-Hodgkin’s lymphomas, MALT lymphomas express a unique antibody repertoire with frequent rheumatoid factor reactivity. J Exp Med (2005) 201(8):1229–41. doi: 10.1084/jem.20050068
97. Hoogeboom R, Bende RJ, Van Noesel CJM. MALT lymphoma-derived rheumatoid factors are nonpolyreactive high-affinity antibodies. Blood (2010) 116(10):1818–9. doi: 10.1182/blood-2010-03-274613
98. Lecuit M, Abachin E, Martin A, Poyart C, Pochart P, Suarez F, et al. Immunoproliferative Small Intestinal Disease Associated with Campylobacter jejuni. N Engl J Med (2004) 350:239–48. doi: 10.1056/NEJMoa031887
99. Chanudet E, Zhou Y, Bacon CM, Wotherspoon AC, Muller-Hermelink HK, Adam P, et al. Chlamydia psittaci is variably associated with ocular adnexal MALT lymphoma in different geographical regions. JPathol (2006) 209:344–51. doi: 10.1002/path.1984
100. Quinn ER, Chan CH, Hadlock KG, Foung SK, Flint M, Levy S. The B-cell receptor of a hepatitis C virus (HCV)-associated non-Hodgkin lymphoma binds the viral E2 envelope protein, implicating HCV in lymphomagenesis. Blood (2001) 98:3745–9. doi: 10.1182/blood.V98.13.3745
101. Hermine O, Lefrere F, Bronowicki JP, Mariette X, Jondeau K, Eclache-Saudreau V, et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. NEnglJMed (2002) 347:89–94. doi: 10.1056/NEJMoa013376
102. Roggero E, Zucca E, Pinotti G, Pascarella A, Capella C, Savio A, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med (1995) 122(10):767–9. doi: 10.7326/0003-4819-122-10-199505150-00006
103. Zucca E, Arcaini L, Buske C, Johnson PW, Ponzoni M, Raderer M, et al. Marginal zone lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol (2020) 31(1):17–29. doi: 10.1016/j.annonc.2019.10.010
104. Ferreri AJM, Govi S, Pasini E, Mappa S, Bertoni F, Zaja F, et al. Chlamydophila Psittaci eradication with doxycycline as first-line targeted therapy for ocular adnexae lymphoma: Final results of an international phase II trial. J Clin Oncol (2012) 30(24):2988–94. doi: 10.1200/JCO.2011.41.4466
105. Ferreri AJM, Sassone M, Kiesewetter B, Govi S, Scarfò L, Donadoni G, et al. High-dose clarithromycin is an active monotherapy for patients with relapsed/refractory extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT): The HD-K phase II trial. Ann Oncol (2015) 26(8):1760–5. doi: 10.1093/annonc/mdv214
106. Roggero E, Zucca E, Mainetti C, Bertoni F, Valsangiacomo C, Pedrinis E, et al. Eradication of Borrelia burgdorferi infection in primary marginal zone B-cell lymphoma of the skin. Hum Pathol (2000) 31(2):263–8. doi: 10.1016/S0046-8177(00)80233-6
107. Thurner L, Hartmann S, Fadle N, Regitz E, Kemele M, Kim YJ, et al. Lymphocyte predominant cells detect Moraxella catarrhalis-derived antigens in nodular lymphocyte-predominant Hodgkin lymphoma. Nat Commun (2020) 11:2465. doi: 10.1038/s41467-020-16375-6
108. Thurner L, Hartmann S, Kemele M, Regitz E, Fadle N, Preuss K-D, et al. Moraxella catarrhalis-a bacterial involvement in the origin of nodular lymphocyte predominant hodgkin lymphoma of IGD type? Haematologica (2015) 10566:322.
109. Angelini G, Morozzi G, Delfino L, Pera C, Falco M, Marcolongo R, et al. Analysis of HLA DP, DQ, and DR allesles in adult Italian rheumatoid arthritis patients. Hum Immunol (1992) 34(2):135–41. doi: 10.1016/0198-8859(92)90039-P
110. He X, Kläsener K, Iype JM, Becker M, Maity PC, Cavallari M, et al. Continuous signaling of CD 79b and CD 19 is required for the fitness of Burkitt lymphoma B cells. EMBO J (2018) 37:e97980. doi: 10.15252/embj.201797980
111. Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt’s lymphoma: a polymicrobial disease? Nat Rev Microbiol (2005) 3:182–7. doi: 10.1038/nrmicro1089
112. Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell (2015) 162:727–37. doi: 10.1016/j.cell.2015.07.019
113. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-barr virus-associated lymphomas. Philos Trans R Soc B Biol Sci (2017) 372(1732):20160271. doi: 10.1098/rstb.2016.0271
114. Kilger E, Kieser A, Baumann M, Hammerschmidt W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J (1998) 17:1700–9. doi: 10.1093/emboj/17.6.1700
115. Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. NatRevImmunol (2001) 1:75–82. doi: 10.1038/35095584
116. Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell (2012) 148:739–51. doi: 10.1016/j.cell.2011.12.031
117. Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity (1998) 9:405–11. doi: 10.1016/S1074-7613(00)80623-8
118. Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, et al. B cell receptor signal strength determines B cell fate. NatImmunol (2004) 5:317–27. doi: 10.1038/ni1036
119. Mancao C, Hammerschmidt W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood (2007) 110:3715–21. doi: 10.1182/blood-2007-05-090142
120. Graham JP, Moore CR, Bishop GA. Roles of the TRAF2/3 Binding Site in Differential B Cell Signaling by CD40 and Its Viral Oncogenic Mimic, LMP1. J Immunol (2009) 183(5):2966–73. doi: 10.4049/jimmunol.0900442
121. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci (1980). doi: 10.1073/pnas.77.12.7415
122. Horenstein MG, Nador RG, Chadburn A, Hyjek EM, Inghirami G, Knowles DM, et al. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8. Blood (1997). doi: 10.1182/blood.V90.3.1186.1186_1186_1191
123. Dunleavy K, Wilson WH. How I treat HIV-associated lymphoma. Blood (2012). doi: 10.1182/blood-2011-08-373738
124. Spina V, Khiabanian H, Messina M, Monti S, Cascione L, Bruscaggin A, et al. The genetics of nodal marginal zone lymphoma. Blood (2016) 128(10):1362–73. doi: 10.1182/blood-2016-02-696757
125. Martin T, Weber JC, Levallois H, Labouret N, Soley A, Koenig S, et al. Salivary gland lymphomas in patients with Sjogren’s syndrome may frequently develop from rheumatoid factor B cells. Arthritis Rheum (2000) 43:908–16. doi: 10.1002/1529-0131(200004)43:4<908::AID-ANR24>3.0.CO;2-K
126. Coelho V, Krysov S, Ghaemmaghami AM, Emara M, Potter KN, Johnson P, et al. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. ProcNatlAcadSciUSA (2010) 107:18587–92. doi: 10.1073/pnas.1009388107
127. Swerdlow SH, Campo E, Pileri SA, Lee Harris N, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
128. Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med (2004) 200(4):519–25. doi: 10.1084/jem.20040544
129. Binder M, Léchenne B, Ummanni R, Scharf C, Balabanov S, Trusch M, et al. Stereotypical chronic lymphocytic leukemia B-cell receptors recognize survival promoting antigens on stromal cells. PloS One (2010) 5(12):e15992. doi: 10.1371/journal.pone.0015992
130. Chu CC, Catera R, Zhang L, Didier S, Agagnina BM, Damle RN, et al. Many chronic lymphocytic leukemia antibodies recognize apoptotic cells with exposed nonmuscle myosin heavy chain IIA: Implications for patient outcome and cell of origin. Blood (2010) 115(19):3907–15. doi: 10.1182/blood-2009-09-244251
131. Catera R, Silverman GJ, Hatzi K, Seiler T, Didier S, Zhang L, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol Med (2008) 14:665–74. doi: 10.2119/2008-00102.Catera
132. Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: Lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol (2003) 21:841–94. doi: 10.1146/annurev.immunol.21.120601.141018
133. Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med (2014) 371:213–23. doi: 10.1056/NEJMoa1400376
134. Zwick C, Fadle N, Regitz E, Kemele M, Stilgenbauer S, Buhler A, et al. Autoantigenic targets of B-cell receptors derived from chronic lymphocytic leukemias bind to and induce proliferation of leukemic cells. Blood (2013) 99(7):1138–48. doi: 10.1182/blood-2012-08-447904
135. Borche L, Lim A, Binet J, Dighiero G. Evidence that chronic lymphocytic leukemia B lymphocytes are frequently committed to production of natural autoantibodies. Blood (1990) 76(3):562–69. doi: 10.1182/blood.v76.3.562.562
136. Hervé M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, et al. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest (2005) 115(6):1636–43. doi: 10.1172/JCI24387
137. Steininger C, Widhopf GF, Ghia EM, Morello CS, Vanura K, Sanders R, et al. Recombinant antibodies encoded by IGHV1-69 react with pUL32, a phosphoprotein of cytomegalovirus and B-cell superantigen. Blood (2012) 119(10):2293–301. doi: 10.1182/blood-2011-08-374058
138. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V Gene Mutation Status and CD38 Expression As Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood (1999) 94(6):1840–7. doi: 10.1182/blood.v94.6.1840
139. Hoogeboom R, van Kesse KPM, Hochstenbach F, Wormhoudt TA, Reinten RJA, Wagner K, et al. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J Exp Med (2013) 210(1):59–70. doi: 10.1084/jem.20121801
140. Kostareli E, Gounari M, Janus A, Murray F, Brochet X, Giudicelli V, et al. Antigen receptor stereotypy across B-cell lymphoproliferations: The case of IGHV4-59/IGKV3-20 receptors with rheumatoid factor activity. Leukemia (2012) 1127–1131(2012). doi: 10.1038/leu.2011.311
141. Binder M, Müller F, Frick M, Wehr C, Simon F, Leistler B, et al. CLL B-cell receptors can recognize themselves: Alternative epitopes and structural clues for autostimulatory mechanisms in CLL. Blood (2013) 121(1):239–41. doi: 10.1182/blood-2012-09-454439
142. Minden MDV, Übelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature (2012) 13;489(7415):309–12. doi: 10.1038/nature11309
143. Shao H, Calvo KR, Grönborg M, Tembhare PR, Kreitman RJ, Stetler-Stevenson M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: Development and validation of diagnostic criteria. Leuk Res (2013) 37(4):401–9. doi: 10.1016/j.leukres.2012.11.021
144. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med (2011) 364:2305–15. doi: 10.1056/NEJMoa1014209
145. Forconi F, Sozzi E, Rossi D, Sahota SS, Amato T, Raspadori D, et al. Selective influences in the expressed immunoglobulin heavy and light chain gene repertoire in hairy cell leukemia. Haematologica (2008) 93(5). doi: 10.3324/haematol.12282
146. Sozzi E, Amato T, Sahota SS, Nuti S, Raspadori D, Sicuranza A, et al. Lack of allelic exclusion by secondary rearrangements of tumour B-cell receptor light chains in hairy cell leukaemia. Hematol Oncol (2011) 29(1):31–7. doi: 10.1002/hon.952
147. Swerdlow SH, Habeshaw JA, Murray LJ, Dhaliwal HS, Lister TA, Stansfeld AG. Centrocytic lymphoma: a distinct clinicopathologic and immunologic entity. A multiparameter study of 18 cases at diagnosis and relapse. AmJPathol (1983) 113(2):181–97.
148. Tiemann M, Schrader C, Klapper W, Dreyling MH, Campo E, Norton A, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): A clinicopathological study from the European MCL Network. Br J Haematol (2005) 131(1):29–38. doi: 10.1111/j.1365-2141.2005.05716.x
149. Williams ME, Westermann CD, Swerdlow SH. Genotypic characterization of centrocytic lymphoma: frequent rearrangement of the chromosome 11 bcl-1 locus. Blood (1990) 1;76(7):1387–91. doi: 10.1182/blood.V76.7.1387.1387
150. Queirós AC, Beekman R, Vilarrasa-Blasi R, Duran-Ferrer M, Clot G, Merkel A, et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell (2016) 30:806–21. doi: 10.1016/j.ccell.2016.09.014
151. Kienle D, Kr??ber A, Katzenberger T, Ott G, Leupolt E, Barth TFE, et al. VH mutation status and VDJ rearrangement structure in mantle cell lymphoma: Correlation with genomic aberrations, clinical characteristics, and outcome. Blood (2003) 102:3003–9. doi: 10.1182/blood-2003-05-1383
152. Hadzidimitriou A, Agathangelidis A, Darzentas N, Murray F, Delfau-Larue MH, Pedersen LB, et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood (2011) 118:3088–95. doi: 10.1182/blood-2011-03-343434
153. Walsh SH, Thorsélius M, Johnson A, Söderberg O, Jerkeman M, Björck E, et al. Mutated VH genes and preferential VH3-21 use define new subsets of mantle cell lymphoma. Blood (2003) 101:4047–54. doi: 10.1182/blood-2002-11-3479
154. Saba NS, Liu D, Herman SEM, Underbayev C, Tian X, Behrend D, et al. Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood (2016) 128:82–93. doi: 10.1182/blood-2015-11-681460
155. Myklebust JH, Brody J, Kohrt HE, Kolstad A, Czerwinski DK, Wälchli S, et al. Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood (2017) 129:759–70. doi: 10.1182/blood-2016-05-718494
156. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2013) 369:507–16. doi: 10.1056/NEJMoa1306220
157. Kahl BS, Spurgeon SE, Furman RR, Flinn IW, Coutre SE, Brown JR, et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood (2014) 123:3398–405. doi: 10.1182/blood-2013-11-537555
158. Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: An international, randomised, open-label, phase 3 study. Lancet (2016) 387(10020):770–8. doi: 10.1016/S0140-6736(15)00667-4
159. Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N Engl J Med (2018) 378:1211–23. doi: 10.1056/NEJMoa1715519
160. Thurner L, Hartmann S, Fadle N, Kemele M, Bock T, Bewarder M, et al. LRPAP1 is a frequent proliferation-inducing antigen of BCRs of mantle cell lymphomas and can be used for specific therapeutic targeting. Leukemia (2019) 33:148–58. doi: 10.1038/s41375-018-0182-1
161. Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS. 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. J Biol Chem (1991) 266:21232–8.
162. Williams SE, Ashcom JD, Argraves WS, Strickland DK. A novel mechanism for controlling the activity of alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J Biol Chem (1992) 267:9035–40.
163. Striekland DK, Ashcom JD, Williams S, Battey F, Behre E, McTigue K, et al. Primary structure of alpha 2-macroglobulin receptor-associated protein. Human homologue of a Heymann nephritis antigen. J Biol Chem (1991) 266:13364–9. doi: 10.1083/jcb.200302069
164. Orlando RA, Farquhar MG. Functional domains of the receptor-associated protein (RAP). Proc Natl Acad Sci U S A (1994) 91:3161–5. doi: 10.1073/pnas.91.8.3161
165. Rosenwald A, Staudt LM. Gene expression profiling of diffuse large B-cell lymphoma. Leuk Lymphoma (2003) 44 Suppl 3:S41–7. doi: 10.1080/10428190310001623775
166. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171(2):481–94. doi: 10.1016/j.cell.2017.09.027
167. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N Engl J Med (2008) 359:2313–23. doi: 10.1056/NEJMoa0802885
168. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24:679–90. doi: 10.1038/s41591-018-0016-8
169. Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U.S.A. (2015) 112:13447–54. doi: 10.1073/pnas.1514944112
170. Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Büssow K, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest (2005) 115:1352–60. doi: 10.1172/JCI200523661
171. Preuss KD, Pfreundschuh M, Ahlgrimm M, Fadle N, Regitz E, Murawski N, et al. A frequent target of paraproteins in the sera of patients with multiple myeloma and MGUS. IntJCancer (2009) 125:656–61. doi: 10.1002/ijc.24427
172. Thurner L, Müller A, Cérutti M, Martin T, Pasquali JL, Gross WL, et al. Wegener’s granuloma harbors B lymphocytes with specificities against a proinflammatory transmembrane protein and a tetraspanin. J Autoimmun (2011) 36:87–90. doi: 10.1016/j.jaut.2010.09.002
173. Thurner L, Preuss K-D, Bewarder M, Kemele M, Fadle N, Regitz E, et al. Hyper N-glycosylated SAMD14 and neurabin-I as driver CNS autoantigens of PCNSL. Blood (2018). doi: 10.1182/blood-2018-03-836932
174. Thurner L, Hartmann S, Bewarder M, Fadle N, Regitz E, Schormann C, et al. Identification of the atypically modified autoantigen Ars2 as the target of B-cell receptors from activated B cell-type diffuse large B-cell lymphoma. Haematologica (2020) haematol.2019.241653. doi: 10.3324/haematol.2019.241653
175. Preuss KD, Pfreundschuh M, Fadle N, Regitz E, Kubuschok B. Sumoylated HSP90 is a dominantly inherited plasma cell dyscrasias risk factor. JClinInvest (2015) 125:316–23. doi: 10.1172/JCI76802
176. Myhrinder AL, Hellqvist E, Sidorova E, S??derberg A, Baxendale H, Dahle C, et al. A new perspective: Molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood (2008) 111(7):3838–48. doi: 10.1182/blood-2007-11-125450
177. Grass S, Preuss KD, Wikowicz A, Terpos E, Ziepert M, Nikolaus D, et al. Hyperphosphorylated paratarg-7: a new molecularly defined risk factor for monoclonal gammopathy of undetermined significance of the IgM type and Waldenstrom macroglobulinemia. Blood (2011) 117:2918–23. doi: 10.1182/blood-2010-09-306076
178. Preuss KD, Pfreundschuh M, Fadle N, Regitz E, Raudies S, Murwaski N, et al. Hyperphosphorylation of autoantigenic targets of paraproteins is due to inactivation of PP2A. Blood (2011) 118:3340–6. doi: 10.1182/blood-2011-04-351668
179. Grass S, Preuss KD, Pfreundschuh M. Autosomal-dominant inheritance of hyperphosphorylated paratarg-7. Lancet Oncol (2010) 11:12. doi: 10.1016/S1470-2045(09)70331-6
180. Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N Engl J Med (2016) 374:555–61. doi: 10.1056/NEJMoa1508808
181. Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood (2007) 109:259–70. doi: 10.1182/blood-2006-03-012948
182. Chu CC, Catera R, Hatzi K, Yan XJ, Zhang L, Wang XB, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood (2008) 112(13):5122–9. doi: 10.1182/blood-2008-06-162024
183. Thurner L, Nimmesgern A, Neumann F, Preuss K-D, Schuster SJ, Regitz E, et al. LRPAP1 is a frequent proliferation-inducing antigen of BCRs of mantle cell lymphomas and can be used for specific therapeutic targeting. Leukemia (2018) 33:148–158(2019). doi: 10.1038/s41375-018-0182-1
184. Fichtner M, Spies E, Seismann H, Riecken K, Engels N, Gösch B, et al. Complementarity Determining Region-Independent Recognition Of A Superantigen By B-Cell Antigen Receptors Of Mantle Cell Lymphoma. Haematologica (2016) 101:e378–81. doi: 10.3324/haematol.2016.141929
185. Forconi F, Sahota SS, Raspadori D, Ippoliti M, Babbage G, Lauria F, et al. Hairy cell leukemia: At the crossroad of somatic mutation and isotype switch. Blood (2004) 104(10):3312–7. doi: 10.1182/blood-2004-03-0950
186. Noy A, de Vos S, Thieblemont C, Martin P, Flowers C, Morschhauser F, et al. Single-Agent Ibrutinib Demonstrates Efficacy and Safety in Patients with Relapsed/Refractory Marginal Zone Lymphoma: A Multicenter, Open-Label, Phase 2 Study. Blood (2016) 128(22):1213. doi: 10.1182/blood.V128.22.1213.1213
187. Kuppers R, Rajewsky K, Zhao M, Simons G, Laumann R, Fischer R, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci (1994) 91(23):10962–6. doi: 10.1073/pnas.91.23.10962
188. Kanzler H. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med (1996) 184(4):1495–505. doi: 10.1084/jem.184.4.1495
189. Schmid C, Sargent C, Isaacson PG. L and H cells of nodular lymphocyte predominant Hodgkin’s disease show immunoglobulin light-chain restriction. AmJPathol (1991) 139:1281–9.
190. Prakash S, Fountaine T, Raffeld M, Jaffe ES, Pittaluga S. IgD positive L&H cells identify a unique subset of nodular lymphocyte predominant Hodgkin lymphoma. AmJSurgPathol (2006) 30:585–92. doi: 10.1097/01.pas.0000194741.87798.45
191. Thurner L, Hartmann S, Bock T, Fadle N, Kemele M, Regitz E, et al. Identification of Posttranvslationally Modified Neoantigens As Targets of B Cell Receptors of Burkitt Lymphoma. Blood (2018) 132(Suppl_1):1588–8. doi: 10.1182/blood-2018-99-114209
192. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med (2015) 21:1–6. doi: 10.1038/nm.3884
193. Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non–germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol (2019) 37(15):1285–95. doi: 10.1200/JCO.18.02403
194. Thompsett AR, Ellison DW, Stevenson FK, Zhu D. V(H) gene sequences from primary central nervous system lymphomas indicate derivation from highly mutated germinal center B cells with ongoing mutational activity. Blood (1999) 94:1738–46. doi: 10.1182/blood.V94.5.1738.417k10_1738_1746
195. Montesinos-Rongen M, Schafer E, Siebert R, Deckert M. Genes regulating the B cell receptor pathway are recurrently mutated in primary central nervous system lymphoma. Acta Neuropathol (2012) 124:905–6. doi: 10.1007/s00401-012-1064-7
196. Montesinos-Rongen M, Purschke FG, Brunn A, May C, Nordhoff E, Marcus K, et al. Primary Central Nervous System (CNS) Lymphoma B Cell Receptors Recognize CNS Proteins. JImmunol (2015) 195:1312–9. doi: 10.4049/jimmunol.1402341
197. Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib unmasks critical role of bruton tyrosine kinase in primary CNS lymphoma. Cancer Discovery (2017) 7(9):1018–29. doi: 10.1158/2159-8290.CD-17-0613
198. Coupland SE, Hummel M, Müller HH, Stein H. Molecular analysis of immunoglobulin genes in primary intraocular lymphoma. Invest Ophthalmol Vis Sci (2005) 46:3507–14. doi: 10.1167/iovs.05-0401
199. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization (WHO) classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
200. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N Engl J Med (2015) 372(15):1430–40. doi: 10.1056/NEJMoa1501548
201. Dimopoulos MA, Tedeschi A, Trotman J, Garcia-Sanz R, Macdonald D, Leblond V, et al. Phase 3 trial of Ibrutinib plus rituximab in Waldenstrom’s macroglobulinemia. N Engl J Med (2018) 378(25):2399–410. doi: 10.1056/NEJMoa1802917
202. Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol (2011) 122:791–2. doi: 10.1007/s00401-011-0891-2
203. Montesinos-Rongen M, Kuppers R, Schluter D, Spieker T, Van RD, Schaller C, et al. Primary central nervous system lymphomas are derived from germinal-center B cells and show a preferential usage of the V4-34 gene segment. AmJPathol (1999) 155:2077–86. doi: 10.1016/S0002-9440(10)65526-5
204. Kyle RA, Therneau TM, Vincent Rajkumar S, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med (2002) 346:564–9. doi: 10.1056/NEJMoa01133202
205. Grass S, Iida S, Wikowicz A, Preuss KD, Inagaki A, Shimizu K, et al. Risk of Japanese carriers of hyperphosphorylated paratarg-7, the first autosomal-dominantly inherited risk factor for hematological neoplasms, to develop monoclonal gammopathy of undetermined significance and multiple myeloma. Cancer Sci (2011) 102:565–8. doi: 10.1111/j.1349-7006.2010.01819.x
206. Nair S, Sng J, Boddupalli CS, Seckinger A, Chesi M, Fulciniti M, et al. Antigen-mediated regulation in monoclonal gammopathies and myeloma. JCI Insight (2018) 3(8):e98259. doi: 10.1172/jci.insight.98259
207. Preuss KD, Hollak CEM, Fadle N, van Oers M, Regitz E, Pfreundschuh M. Saposin C is a frequent target of paraproteins in Gaucher disease-associated MGUS/multiple myeloma. Br J Haematol (2018) 184(3):384–91. doi: 10.1111/bjh.15659
208. Greenberg AJ, Rajkumar SV, Vachon CM. Familial monoclonal gammopathy of undetermined significance and multiple myeloma: Epidemiology, risk factors, and biological characteristics. Blood (2012) 119(23):5359–66. doi: 10.1182/blood-2011-11-387324
209. Neumann F, Pfreundschuh M, Preuss KD, Schormann C, Zwick C, Murawski N, et al. CD4(+) T cells in chronic autoantigenic stimulation in MGUS, multiple myeloma and Waldenstrom’s macroglobulinemia. Int J Cancer (2015) 137(5):1076–84. doi: 10.1002/ijc.29478
210. Siemer D, Kurth J, Lang S, Lehnerdt G, Stanelle J, Küppers R. EBV transformation overrides gene expression patterns of B cell differentiation stages. Mol Immunol (2008) 45(11):3133–41. doi: 10.1016/j.molimm.2008.03.002
211. Moller P, Lammler B, Herrmann B, Otto HF, Moldenhauer G, Momburg F. The primary mediastinal clear cell lymphoma of B-cell type has variable defects in MHC antigen expression. Immunology (1986) 59(3):411–7.
212. Möller P, Lämmler B, Eberlein-Gonska M, Feichter GE, Hofmann WJ, Schmitteckert H, et al. Primary mediastinal clear cell lymphoma. Virchows Arch A Pathol Anat Histopathol (1986) 409:79–92. doi: 10.1007/BF00705408
213. Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, LaCasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood (2010) 115(13):2578–85. doi: 10.1182/blood-2009-08-236471
214. Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med (2010) 363(14):1303–12. doi: 10.1056/NEJMoa1000500
215. Mullard A. FDA approves first-in-class SYK inhibitor. Nat Rev Drug Discovery (2018). doi: 10.1038/nrd.2018.96
216. Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, Barrientos J, et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med (2019) 381:432–43. doi: 10.1056/NEJMoa1817073
217. Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med (2014) 370:2286–94. doi: 10.1056/NEJMoa1400029
218. Liu TM, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood (2015) 126(1):61–8. doi: 10.1182/blood-2015-02-626846
219. Roschewski M, Lionakis MS, Melani C, Butman JA, Pittaluga S, Lucas AN, et al. Dose-Adjusted Teddi-R Induces Durable Complete Remissions in Relapsed and Refractory Primary CNS Lymphoma. Blood (2018) 132(supplement 1):4195. doi: 10.1182/blood-2018-99-112184
220. Sharman JP, Egyed M, Jurczak W, Skarbnik A, Pagel JM, Flinn IW, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet (2020) 18;395(10232):1278–91. doi: 10.1016/S0140-6736(20)30262-2
221. Ghia P, Pluta A, Wach M, Lysak D, Kozak T, Simkovic M, et al. Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol (2020) 38(25):2849–61. doi: 10.1200/jco.19.03355
222. Brown JR, Eichhorst BF, Ghia P, Kater AP, Li J, Khurana S, et al. A Phase 3 Trial Comparing the Efficacy and Safety of Acalabrutinib in Combination with Venetoclax with or without Obinutuzumab, Compared with Investigator’s Choice of Chemoimmunotherapy in Patients with Previously Untreated Chronic Lymphocytic Leukemia (CLL) without Del(17p) or TP53 Mutation. Blood (2019) 134(Supplement_1):4318. doi: 10.1182/blood-2019-123057
223. Naeem AS, Nguy WI, Tyekucheva S, Fernandes SM, Rai V, Ebata K, et al. LOXO-305: Targeting C481S Bruton Tyrosine Kinase in Patients with Ibrutinib-Resistant CLL. Blood (2019) 134(Supplement_1):478. doi: 10.1182/blood-2019-124362
224. Aslan B, Mahendra M, Peoples MD, Marszalek JR, Vellano CP, Zheng X, et al. Abstract 317: Vecabrutinib inhibits C481 mutated Bruton’s tyrosine kinase and its downstream signaling in vitro. 2019. doi: 10.1158/1538-7445.sabcs18-317
225. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The btk inhibitor arq 531 targets ibrutinib-resistant cll and richter transformation. Cancer Discovery (2018) 8(10):1–16. doi: 10.1158/2159-8290.CD-17-1409
226. Kuter DJ, Boccia RV, Lee E-J, Efraim M, Tzvetkov N, Mayer J, et al. Phase I/II, open-label adaptive study of oral bruton tyrosine kinase inhibitor PRN1008 in patients with relapsed/refractory primary or secondary immune thrombocytopenia. Blood (2019) 134(Supplement_1):87. doi: 10.1182/blood-2019-122336
227. Montalban X, Arnold DL, Weber M, Staikov I, Piasecka-Strycznska K, Willmer J, et al. Primary analysis of a randomized phase II study to evaluate the efficacy and safety of evobrutinib, a BTK inhibitor, in patients with relapsing MS. Mult Scler J (2019). doi: 10.1177/1352458519843084
228. Scholoff A, Obi G, Baker KR, Carrum G, Kamble RT. Complete Resolution of Extensive Chronic Graft-Versus-Host Disease with Ibrutinib. Blood (2015) 126(23):5474. doi: 10.1182/blood.v126.23.5474.5474
229. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med (2014) 370(11):997–1007. doi: 10.1056/NEJMoa1315226
230. Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol (2016) 27(10):1928–40. doi: 10.1093/ANNONC/MDW282
231. Flinn IW, Miller CB, Ardeshna KM, Tetreault S, Assouline SE, Zinzani PL, et al. Dynamo: A Phase 2 Study Demonstrating the Clinical Activity of Duvelisib in Patients with Relapsed Refractory Indolent Non-Hodgkin Lymphoma. Blood (2016) 128(22):1218. doi: 10.1182/blood.v128.22.1218.1218
232. Coutré SE, Barrientos JC, Brown JR, De Vos S, Furman RR, Keating MJ, et al. Management of adverse events associated with idelalisib treatment: Expert panel opinion. Leuk Lymphoma (2015) 2779–86. doi: 10.3109/10428194.2015.1022770
233. Philippar U, Lu T, Vloemans N, Bekkers M, van Nuffel L, Gaudiano M, et al. DISCOVERY OF A NOVEL, POTENTIAL FIRST-IN-CLASS MALT1 PROTEASE INHIBITOR FOR THE TREATMENT OF B CELL LYMPHOMAS. Hematol Oncol (2019). doi: 10.1002/hon.88_2629
234. Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: A phase 2 consortium trial. Blood (2018) 131(2):182–90. doi: 10.1182/blood-2017-09-804641
235. Torchia J, Weiskopf K, Levy R. Targeting lymphoma with precision using semisynthetic anti-idiotype peptibodies. Proc Natl Acad Sci U. S. A. (2016) 113(19):5376–81. doi: 10.1073/pnas.1603335113
236. Stepanov AV, Belogurov AA Jr., Ponomarenko NA, Stremovskiy OA, Kozlov LV, Bichucher AM, et al. Design of targeted B cell killing agents. PLoSOne (2011) 6:e20991. doi: 10.1371/journal.pone.0020991
237. Zocher M, Baeuerle PA, Dreier T, Iglesias A. Specific depletion of autoreactive B lymphocytes by a recombinant fusion protein in vitro and in vivo. IntImmunol (2003) 15:789–96. doi: 10.1093/intimm/dxg076
238. Nachreiner T, Kampmeier F, Thepen T, Fischer R, Barth S, Stocker M. Depletion of autoreactive B-lymphocytes by a recombinant myelin oligodendrocyte glycoprotein-based immunotoxin. JNeuroimmunol (2008) 195:28–35. doi: 10.1016/j.jneuroim.2008.01.001
Keywords: B-cell receptor, antigen, lymphoma, autoreactivity, posttransnational modification, antigens of infectious origin
Citation: Thurner L, Hartmann S, Neumann F, Hoth M, Stilgenbauer S, Küppers R, Preuss K-D and Bewarder M (2020) Role of Specific B-Cell Receptor Antigens in Lymphomagenesis. Front. Oncol. 10:604685. doi: 10.3389/fonc.2020.604685
Received: 10 September 2020; Accepted: 02 November 2020;
Published: 09 December 2020.
Edited by:
Jérôme Paggetti, Luxembourg Institute of Health, LuxembourgReviewed by:
Jan Burger, University of Texas MD Anderson Cancer Center, United StatesShihShih Chen, Feinstein Institute for Medical Research, United States
Copyright © 2020 Thurner, Hartmann, Neumann, Hoth, Stilgenbauer, Küppers, Preuss and Bewarder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenz Thurner, TG9yZW56LlRodXJuZXJAdWtzLmV1