 Michel Montalvo-Casimiro1†
Michel Montalvo-Casimiro1† Rodrigo González-Barrios1†
Rodrigo González-Barrios1† Marco Antonio Meraz-Rodriguez1†
Marco Antonio Meraz-Rodriguez1† Vasti Thamara Juárez-González2
Vasti Thamara Juárez-González2 Cristian Arriaga-Canon1Luis A. Herrera1,3*
Cristian Arriaga-Canon1Luis A. Herrera1,3*- 1Unidad de Investigación Biomédica en Cáncer, Instituto Nacional de Cancerología-Instituto de Investigaciones Biomédicas, UNAM, Mexico City, Mexico
- 2Departamento de Bioquímica, Facultad de Química, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 3Instituto Nacional de Medicina Genómica, Mexico City, Mexico
Gene mutations are strongly associated with tumor progression and are well known in cancer development. However, recently discovered epigenetic alterations have shown the potential to greatly influence tumoral response to therapy regimens. Such epigenetic alterations have proven to be dynamic, and thus could be restored. Due to their reversible nature, the promising opportunity to improve chemotherapy response using epigenetic therapy has arisen. Beyond helping to understand the biology of the disease, the use of modern clinical epigenetics is being incorporated into the management of the cancer patient. Potential epidrug candidates can be found through a process known as drug repositioning or repurposing, a promising strategy for the discovery of novel potential targets in already approved drugs. At present, novel epidrug candidates have been identified in preclinical studies and some others are currently being tested in clinical trials, ready to be repositioned. This epidrug repurposing could circumvent the classic paradigm where the main focus is the development of agents with one indication only, while giving patients lower cost therapies and a novel precision medical approach to optimize treatment efficacy and reduce toxicity. This review focuses on the main approved epidrugs, and their druggable targets, that are currently being used in cancer therapy. Also, we highlight the importance of epidrug repurposing by the rediscovery of known chemical entities that may enhance epigenetic therapy in cancer, contributing to the development of precision medicine in oncology.
Introduction
Since the turn of the century, epigenetics has become an important research area in human diseases study, where genetic mutations have been classically understood as the main cause in the development of human pathologies (1). The term epigenetics involves a wide variety of mechanisms that cells use to regulate the transcription of their DNA without changing its genetic material (2). Whether an epigenetic modification has a facilitating or inhibiting role in the gene expression depends on the chemical nature of the mark that is placed over the chromatin, and the type of modification that is set down on the proximal environment of these genes (3). Thus, epigenetics shapes a regulatory complex that bridges the gap between genetic sequences and actionable mutations. Due to current knowledge about these epigenetic mechanisms, the importance of this regulatory system has become more evident and it has led to the understanding that epigenetic alterations are some of the main mechanisms underlying many human diseases such as cancer, which arises through aberrant genetic and epigenetic alterations, both of which have a key role in malignant transformation, tumor progression and prognosis (4).
Nowadays, it is known that as cancer progresses, there are genetic aberrations that make tumors highly prone to developing resistance to therapies (5). Emerging data on cancer-associated epigenetic alterations have shown that epigenetic modifications leading to drug resistance may be the cue for individual variation in chemotherapy response, having the potential to be reversible using epigenetic therapy (6). The possibility to reprogram the cancer epigenome is becoming a promising target therapy for both, treatment development and reversibility of drug resistance. Which focuses on the development of pharmacological compounds that can reprogram the epigenetic landscape to enhance chemotherapy response (7).
For a few years, the design of therapeutic strategies has been a growing field of query for single-target epigenetic drugs (epidrugs); however, the traditional epidrug discovery pathway is time-consuming and expensive (8, 9). Hence, a promising strategy for epidrug development is based on tracing novel potential epi-targets in previously approved drugs through a process called drug repositioning or repurposing (10, 11). Epidrug repurposing allows exploring a wide diversity of molecular combinations in multifactorial diseases such as cancer, where combinational epigenetic therapies are likely to be more effective than monotherapy to overcome chemotherapy resistance (9). This review focuses on the emerging area of epidrug repurposing, highlighting strategies to enhance cancer therapy. To further understand this, we will discuss the main mechanisms and elements involved in epigenetic alterations in cancer and its relevance in cancer therapy response.
Background in Epigenetics
Epigenetics is the term coined by Conrad Hal Waddington seventy-six years ago, to refer to the molecular mechanisms that may exert their influence on gene expression that do not involve alterations in its gene code. Through these, an organism can develop and adapt its phenotype to environmental changes (12). Over time, many definitions of Epigenetics have arisen (13); however, we can understand epigenetics as reversible chemical modifications of DNA and histone proteins (epimarks) that regulate specific functions in chromatin remodeling without altering the DNA sequence (14). Epimarks are associated with the transcription and function of a gene, that may change the cellular phenotype or its functional patterns in response to a particular context, across different developmental stages, cellular differentiation, or maintenance of tissue-specific cell lineages (15).
At the molecular level, epigenetic machinery is composed mainly of three interconnected components working synergistically in the chromatin organization levels, which include DNA methylation, histone post-translational modifications, and regulatory non-coding RNAs (ncRNAs) (14, 16). In the nucleus, chromatin can exist in two physical and functional states: heterochromatin (condensed chromatin), which is associated with transcriptional repression; and euchromatin (relaxed chromatin), associated with transcriptional activation (17) (Figure 1). The organizational states of the chromatin are highly regulated by epigenetic mechanisms involving nucleosome, which is the basic packaging unit of chromatin, composed by an octamer of histone proteins (two dimers of H2A-H2B and a tetramer of H3-H4 histones) (Figure 1A), that constitutes a compact structure with 147 base pairs of DNA turned almost twice around it (17, 18). N-terminal tails of histone proteins can acquire post-translational modifications through multiple mechanisms including phosphorylation, ubiquitination, methylation/demethylation, and acetylation, the latter being the most studied. Histone and direct DNA modifications constitute “the epigenetic code”: an interplay between epigenetic factors and positive and negative feedback mechanisms that regulate it (18). Therefore, understanding the main mechanisms in the field of epigenetic research and their role in disease development is essential in its application in cancer therapy.
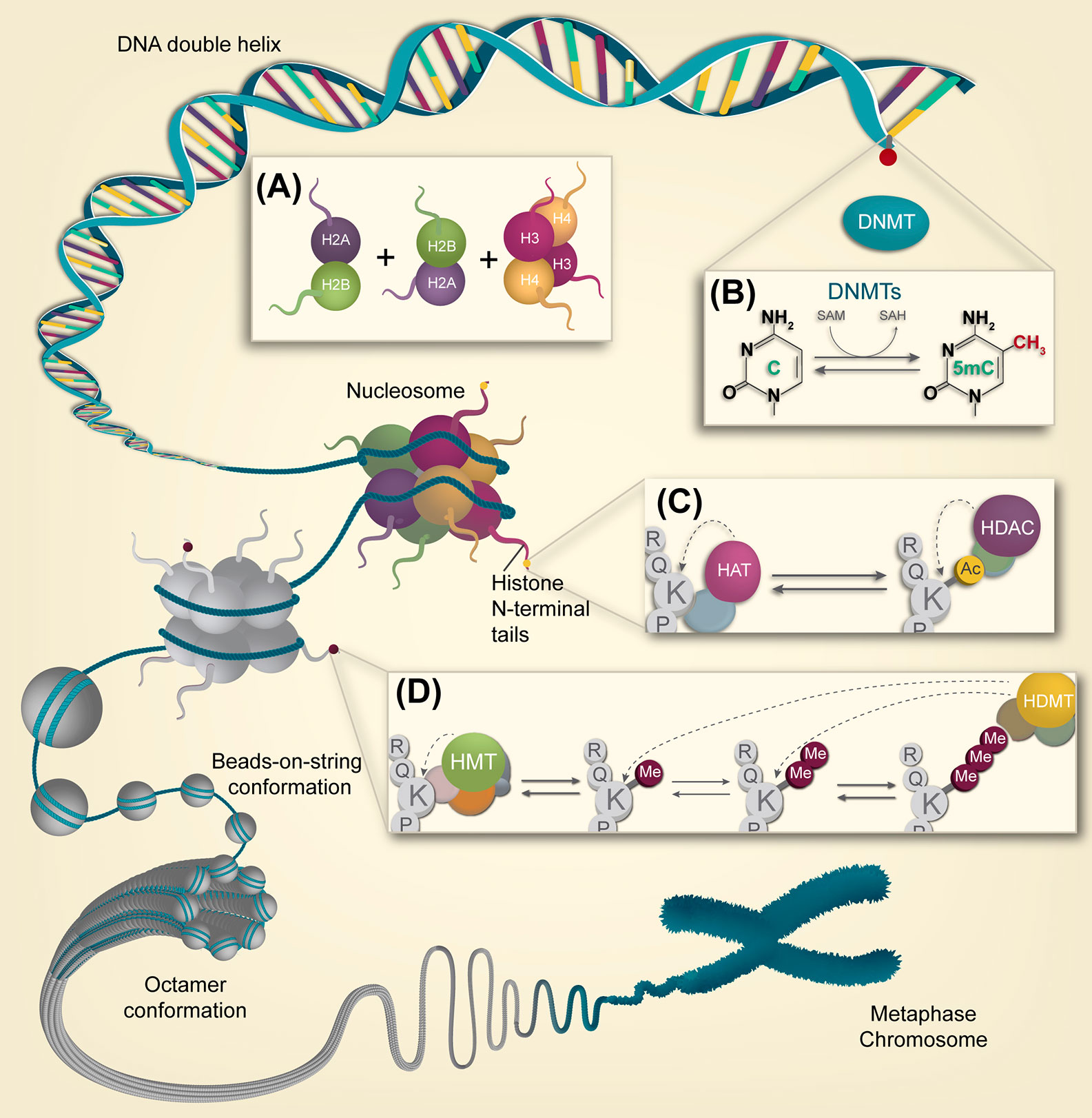
Figure 1 Overview of the epigenetic landscape. Different compaction levels of chromatin are depicted, from naked DNA to the metaphasic chromosome. (A) Two dimers of H2A-H2B and a tetramer of H3-H4 histones are required for nucleosome assembly, the chromatin’s basic packaging unit (B) DNA methylation is a process carried out by DMNTs in CpG dinucleotides, particularly on CpG islands. This dynamic epigenetic mark can be reversed by enzymatic conversion. (C) Histone acetylation is performed on lysine residues by HAT enzyme complexes. In contrast, histone lysine deacetylation is carried out by HDACs enzyme complexes. (D) Histone lysine methylation is carried out by HMT complexes. Lysines can be processively methylated from mono to di and trimethylation.
DNA Methylation
Methylation on DNA’s cytosine is the most broadly studied epigenetic modification in humans. It encompasses a reaction defined as “the covalent transfer of a methyl group to the C-5 position of a cytosine ring of DNA” (15, 19). Generally, in mammals, DNA methylation occurs predominantly—but not exclusively—in the context of genomic regions called CpG islands, which are formed by clusters of CpG dinucleotides, and it’s catalyzed by a group of enzymes called DNA methyltransferases (DNMTs). These enzymes transfer a methyl group from the donor molecule S-adenosylmethionine (SAM) to the fifth carbon of a cytosine residue to form 5-methylcytosine (5mC) (18, 19) (Figure 1B). This covalent modification is able to inhibit DNA transcription; either through the steric hindrance imposed by the methyl group which prevents transcription factors from binding DNA (18–20), or by the recruitment of proteins with methyl-CpG-binding domains (MBD). These proteins also contain domains able to recruit histone-modifying and chromatin-remodeling complexes to the methylated sites, forming repressor complexes that enhance the silencing state on that chromatin region (21). Three different DNMTs generate and maintain methylation patterns. DNMT1 is the methyltransferase enzyme specialized in the maintenance of previously placed methylation patterns, and DNMT3a & DNMT3b are instead involved in the establishment of de novo methylation patterns over DNA (18, 22, 23).
DNA methylation patterns occur in different regions of the genome. Alterations in these patterns lead to diseases (18). For instance, gene promoters which are mainly embedded in CpG islands (70%) are normally unmethylated, thus allowing transcription. Aberrant hypermethylation patterns of these gene regulatory elements lead to transcriptional inactivation and are tumor-type specific as well as a common hallmark of cancer (9). Alternatively, during diseases, other alterations occur, like the demethylation of the gene body. Such alteration allows transcription to be initiated at several incorrect sites. In consequence, DNA hypomethylation at specific regions can activate the aberrant expression of genes, some of which could behave as proto-oncogenes (18). Finally, as aforementioned, alterations of hypermethylated patterns in repetitive sequences can promote the activation of transposable elements and chromosomal instability, both phenomena being also correlated with carcinogenesis and metastasis (6).
However, the reactions that lead to altered patterns of DNA methylation can potentially be reversible and restored through DNMT inhibitors (DNMTi: see below) that contain nucleoside derivatives and non-nucleoside analogs, some of them have been highly researched and shown promise in cancer therapies (24).
Histone Post-Translational Modifications
Another axis of the epigenetic machinery, closely associated with DNA methylation, are the covalent post-translational modifications of nucleosomal histones. Through the addition of chemical groups at specific sites within the amino- or carboxy-terminus of each histone, different functional consequences influencing chromosome structure can be elicited. Chromatin is functionally divided into actively transcribed euchromatin and transcriptionally inactive heterochromatin, which finally regulates the accessibility to genomic DNA and has a role in the control of gene expression (18, 25). The principal histone proteins modifications include methylation, acetylation, phosphorylation, ubiquitylation, sumoylation, and ribosylation, from which methylation and acetylation are the most common and characterized, and generally occur in the proximity of promoter and enhancer genomic regions (26). Each histone residue can undergo one or more modifications, which have different effects depending on which residue is modified, giving rise to crosstalk between the different marks, constituting “the histone code” altogether (18).
Multiple enzymes catalyze histone post-translational modifications with specific catalytic activity based on each histone tail’s amino acids that can act as their substrates. Most of these modifications are reversible. There are specialized enzymes that can remove each type of covalent modification. Histone acetyltransferases (HATs) and deacetylases (HDACs) control acetylation, as well as histone methyltransferases (HMTs) and demethylases (HDMs) coordinate histone methylation. Acetylation and deacetylation of histones are among the most studied reversible, followed by methylation and demethylation of histone lysines (17, 27).
Due to the importance of histone epimarks in gene regulation and cellular function, aberrant histone post-translational modifications may change gene expression patterns and cause human pathologies (6). Thus, it is of great importance to understand the reversible nature of these marks as an advantageous alternative for the treatment of diseases where epigenome deregulation is one of the hallmarks.
Histone Acetylation and Deacetylation
Histone acetylation has a key role in many biological processes (cell cycle regulation, alternative splicing, nuclear transport, among others) (28). It can promote relaxed states of the chromatin (euchromatin) and favor gene transcription, while deacetylation exerts the opposite effect, generating heterochromatin domains that can inhibit transcription (2). Two families of enzymes with reverse functions control the feedback regulation between acetylation/deacetylation of histones: histones acetyltransferases (HATs or KATs) and histones deacetylases (HDACs) (2). HATs catalyze the transfer of acetyl groups to lysine-amino-terminal residues using acetyl-CoA as a donor; this reaction neutralizes the positive charge of the Lys (17, 29) (Figure 1C). As a result, the interaction between the histone and the DNA is weakened, forming an opening domain in chromatin, leading to exposure of DNA sequences and their transcription (2, 28). HATs are divided in three families based on their catalytic domain’s functional and structural identity, which bears the acetyltransferase activity for the recognition of acetyl-lysine residues (17). Several HATs associate with other protein complexes and subunits to selectively modify the different histones; however, p300/CBP is probably the most extensively studied HAT, since it is capable of acetylating all four histones along with many other coactivator or corepressor transcriptional complexes (30).
In contrast, HDACs remove acetyl groups from lysine residues through different reactions that reestablish the positive charges on histone tails, increasing its interaction with DNA and stabilizing the chromatin in place (2, 28) (Figure 1C). The histone deacetylase family includes 18 members (31), divided into two groups based on their enzymatic activity: Zn2+-dependent enzymes, which include classes I, II, and IV HDACs, exert their function through hydrolytic catalysis; and NAD+ cofactor-dependent enzymes, that include class III sirtuins (SIRTs), with a catalytic mechanism of nucleophilic substitution for histone deacetylation (28).
Both HATs and HDACs play a key role in the maintenance and regulation of chromatin accessibility, leading gene expression regulation, among other mechanisms. Histone acetylation global imbalance is one of the prominent alterations in the diseased state and a hallmark of many tumor types, where HDACs have been found overexpressed (32) or mutated (33). Additionally, abnormal genomic events such as translocations, mutations, or deletions in HAT- and acetylation readers-related genes may occur during cancer development (18). As a result, aberrant acetylation-related proteins contribute to the progression of the disease. For instance, germline mutations and overexpression of HDACs have been observed in various cancers, resulting in a global loss of histone acetylation and the consequent silencing of tumor suppressor genes (34). Also, it has been observed that reduced lysine 16 acetylation (H4K16ac), as well as the loss of acetylation of histone 3 (H3ac) are also hallmarks of human cancer (35, 36). Furthermore, HATs and HDACs are targeted to transcriptionally-active genes by phosphorylated RNA polymerase II through the recruitment of effector proteins with specialized reader domains (18), suggesting that the mechanistic switch between acetylation/deacetylation can be manipulated and restored by specific drugs inhibiting key enzymes by targeting their catalytic reaction (HATi and HDACi; see below).
Histone Methylation and Demethylation
Histone methylation occurs on arginine (R) and lysine (K) residues, and it is catalyzed by HMTs (or KMTs and RMTs) that use S-adenosyl-l-methionine (SAM) as a methyl donor group (Figure 1C). Lysine methyltransferases are divided into two broad groups based on the presence or the absence of a SET domain (Su(var)3-9, Enhancer-of-zeste, and Trithorax): SET-domain containing methyltransferase family and DOT1-domain lysine N-methyltransferase (37, 38).
KMTs can transfer three methyl groups onto lysine residues, prompting mono, di, and, trimethylation (me1, me2 and, me3 respectively) (17) (Figure 1D). The association of an active or repressive transcriptional state depends on the number of methyl groups and in the position of the lysine residue in the histone amino acid sequence. A repressed chromatin state (heterochromatin, constitutive, or facultative), correlates with methylation of H3K9me2,3, H3K27me2,3, and H4K20me3, while methylation of H3K4me2,3, H3K9me1, H3K27me1, H3K20me1, and H3K36me1 are associated with transcriptionally active chromatin (euchromatin) (17, 39). Besides, histone methylation also has an important role in DNA repair, DNA replication, alternative splicing, and chromosome condensation (18). Histone demethylases HDMs (or KDMs) can revert these modifications (Figure 1D), divided into two different families with distinct enzymatic mechanisms: KDM1A/LSD1 amine oxidase family, dependent on flavin adenine dinucleotide (FAD) as a cofactor; and the KDM2A/B dioxygenase family, which contain a Jumonji C (JmjC) domain and are iron Fe (II) and α-ketoglutarate-dependent to accomplish histone demethylation through methyl groups oxidation (40). The readers of methylated lysine residues consist of various proteins with specialized domains that can recognize these modifications (17).
Besides the global loss of acetylation and DNA hypomethylation, the deregulation of histone methylation/demethylation can lead to chromosome instability (18). It has been suggested that the aberrant expression of both histone methyltransferases and demethylases genes is the main cause of an altered distribution of histone methylation marks. Deregulation of histone methylation patterns can become a driver for mutations in many types of tumors (15). For instance, cancer cells have a global loss of activation marks, such as H4K20me3; along with a gain of methylation in repressive marks, such as H3K9me and H3K27me, as well as the monomethylation of H3K4me (35, 36) which are associated with DNA hypermethylation of silenced genes. The basal patterns of histone methylation are essential for establishing a permissive euchromatic state, allowing the expression of tumor suppressor genes. Therefore, its alteration results in the repression of some of these genes and oncogene aberrant expression (18, 35). Instead, instability of the methylation/demethylation mechanistic switch can promote proliferation and neoplastic transformation in several cancer types (41–43).
Epigenetic Alterations in Cancer and Cancer Therapy
As mentioned before, the cancer epigenome is characterized by global changes in DNA methylation, disruptions in histone posttranslational modification patterns, and alterations of normal chromatin-modifying enzymes expression (18, 36) (Figure 2) [see review (44)]. Accordingly, these changes can promote the disruption of cellular homeostasis in precancerous cells through the deregulation of genes implicated in cancer initiation and progression (4); for instance, those genes associated with apoptosis resistance, proliferation, invasive potential, and genomic instability, as well as genes correlated to therapeutic response (45, 46). Thus, the relationship between genetic disruptions and epigenetic abnormalities are mutually beneficial in order to drive cancer development and could be playing a key role in individual differences displayed by patients in the way they respond to therapies in both toxicity or treatment efficacy (15, 46, 47). Multiple studies demonstrate that reversing epigenetic patterns through de novo epidrugs and epidrug repurposing can resensitize cancer cells to chemotherapy (48–50).
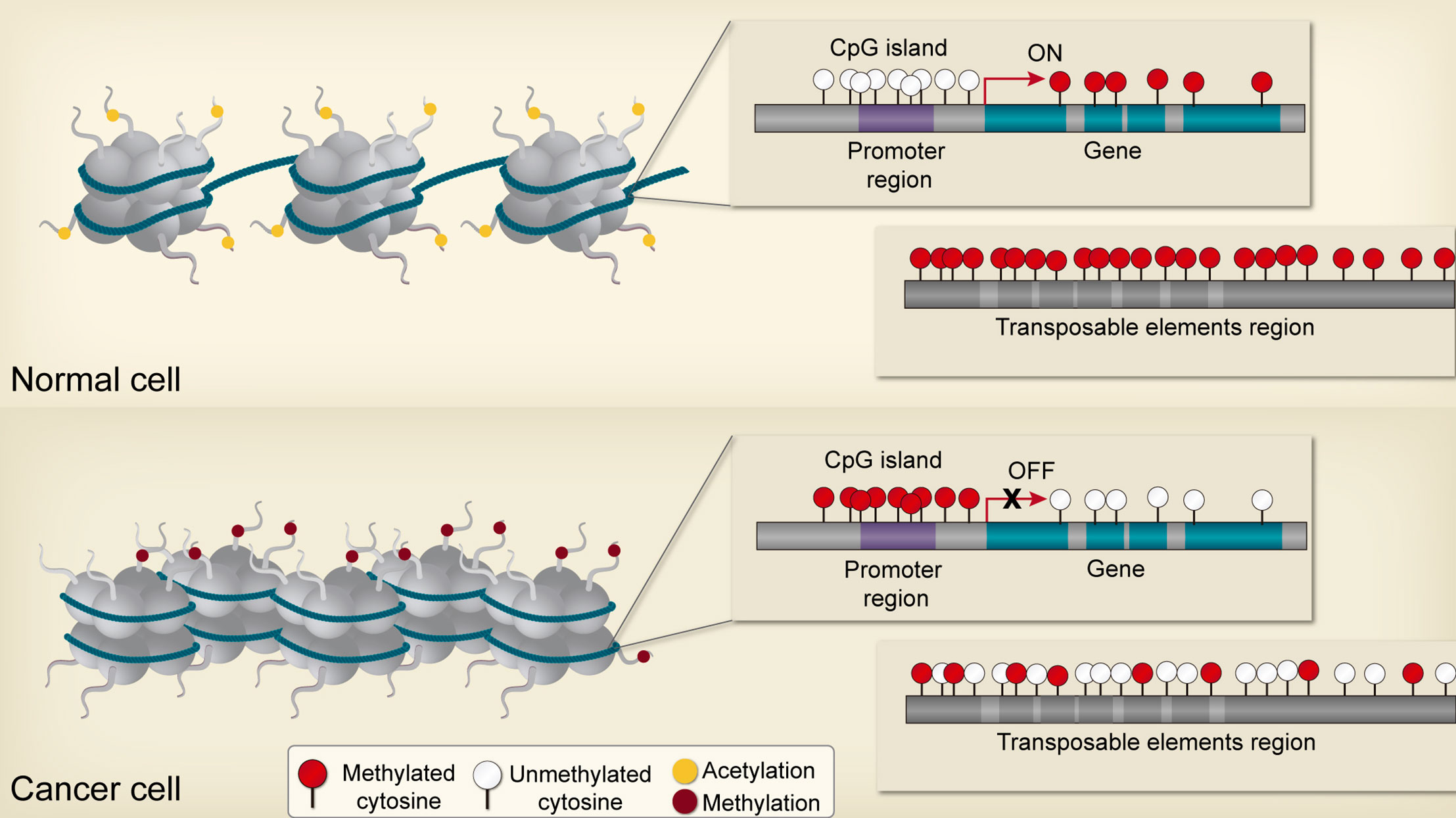
Figure 2 Epigenetic alterations in cancer cells. In non-neoplasic cells, CpG islands of tumor suppressor gene promoters are generally unmethylated and acetylated, resulting in transcriptional activation and expression. In contrast, non-coding regions and repetitive elements are hypermethylated, ensuring chromosome stability. Gene bodies are normally methylated, enhancing transcription. Neoplasic cells are characterized by global hypomethylation and local CpG island hypermethylation, especially at tumor suppressor gene promoters, resulting in aberrant transcription and genomic instability.
Principles of Epigenetic Therapy
Increasing understanding of epigenetic mechanisms and their importance in disease has led to the development of therapeutic interventions targeting epigenetic modulatory mechanisms. Due to the chemical reversibility nature of DNA methylation and histone posttranslational modifications, epigenetic proteins can be druggable targets by means of small-enzymatic inhibitors that aim for the restoration of the aberrant epigenetic machinery and hold the potential for reverting epigenetic signatures in cancer (14).
Epigenetic drugs (epidrugs) are chemical agents that modify the structure of DNA and chromatin, facilitating disruption of transcriptional and post-transcription changes, primarily by controlling the enzymes required for their establishment and maintenance, reactivating the tumor suppressor and DNA repair genes that are epigenetically silenced (51). Lately, epigenetic therapy has taken relevance in the field of oncology, where epidrugs have been successfully used in treatment, mostly in combination with standard chemotherapy (52).
Epidrugs (with one-target, as well as repurposed epidrugs; see below) that are designed based on these principles can exert direct cytotoxic effects over malignant cells (14, 46), function as sensitizers in complementary therapies (53, 54), or can be used to overcome epigenetically-acquired drug resistance against the limits of chemotherapy efficacy, as there are the dynamic associations between epigenetic pattern changes and resistance to therapeutic regimes for cancer (50, 52, 55). New epidrugs compounds are continually being tested for cytotoxicity, pharmacological parameters, and a better understanding of their mode of action; in both preclinical research (in vitro and in vivo) as well as in clinical trials. Epigenetics therapy is enhanced by a combination of laboratory and clinical data. The US Food and Drug Administration (FDA) has approved many epigenetic treatments and used them for treating cancer (6).
Epidrug Generations
Historically, molecules designed to inhibit the catalytic function of epigenetic factors have not only resulted in the reduction of the targeted enzymatic activity but also the appearance of indirect modifications of the transcription of large gene sets (56). Several epigenetic protein families have similar cofactors and co-substrates, similar epidrugs could target several epigenetic protein families. Some compounds can inhibit the functionality of a whole family of epigenetic proteins (Table 1).

Table 1 Current inhibition assays performed for different epigenetic factors.
The quest for finding epigenetic inhibitors led to the first generation of epidrugs, characterized by a meager degree of selectivity (57). Epidrugs of the first generation include DNMTi and HDACi, some of which have already been approved to treat hematological malignancies (58). DNMTi are pyrimidine analogs incorporated into DNA during replication and form covalent DNA adducts that cause DNA damage response activation and eventually lead to apoptosis. This was not without cytotoxic implications (3, 59). On the other hand, first generation HDACi are molecules that inhibit the Zn2+ dependent HDAC enzymes, except for sirtuin inhibitors, which inhibit a specific class of histone deacetylases that depend on NAD+ to perform their catalytic activity (59).
First-generation inhibitors represented many undesirable pharmacokinetic properties and poor target selectivity, resulting in the need for the creation of second-generation epidrugs, which included DNMTi (such as zebularine and guadecitabine), and HDACi (including hydroxamic acid, belinostat and panobinostat, tucidinostat and valproic acid) with improved physiological properties (59).
The second generation of epidrugs was characterized by strong academic research accompanied by industrial drug discovery to find molecules that resembled first generation epidrugs. The hypothesis was that molecules with more potent inhibitor action and fewer side-effects could be found. Another thing to consider was pharmacokinetics: first generation epidrugs had poor bioavailability, were more active within non pH physiological ranges, and were targets of cellular deaminases, which ultimately meant a short half-life for these compounds.
Ultimately, the third generation of epidrugs reflected that epigenetic factors could write, delete, or read epigenetic marks in the form of protein complexes. Therefore, a deeper understanding of epigenetic protein’s interactome is essential for the design of highly selective epidrugs (57). Epi-drugs of third generation includes, among others, histone methyltransferase inhibitors (HMTi), histone demethylase inhibitors (HDMi), and bromodomain and extra-terminal domain inhibitors (BETi) (59).
DNMT Inhibitors
DNA methylation inhibitors intercalate between DNA base pairs and suppress the CpG dinucleotide’s methylation, especially important at CpG islands. These inhibitors can be classified as DNMTi nucleoside analogs and non-nucleoside analogs (60) (Figure 3). DNMTi cytidine analogs are usually chemically unstable, and because of their similarity to cytidine, DNA and RNA polymerases identify both compounds and add them into growing nucleic acid chains, therefore hampering their selectivity (61).
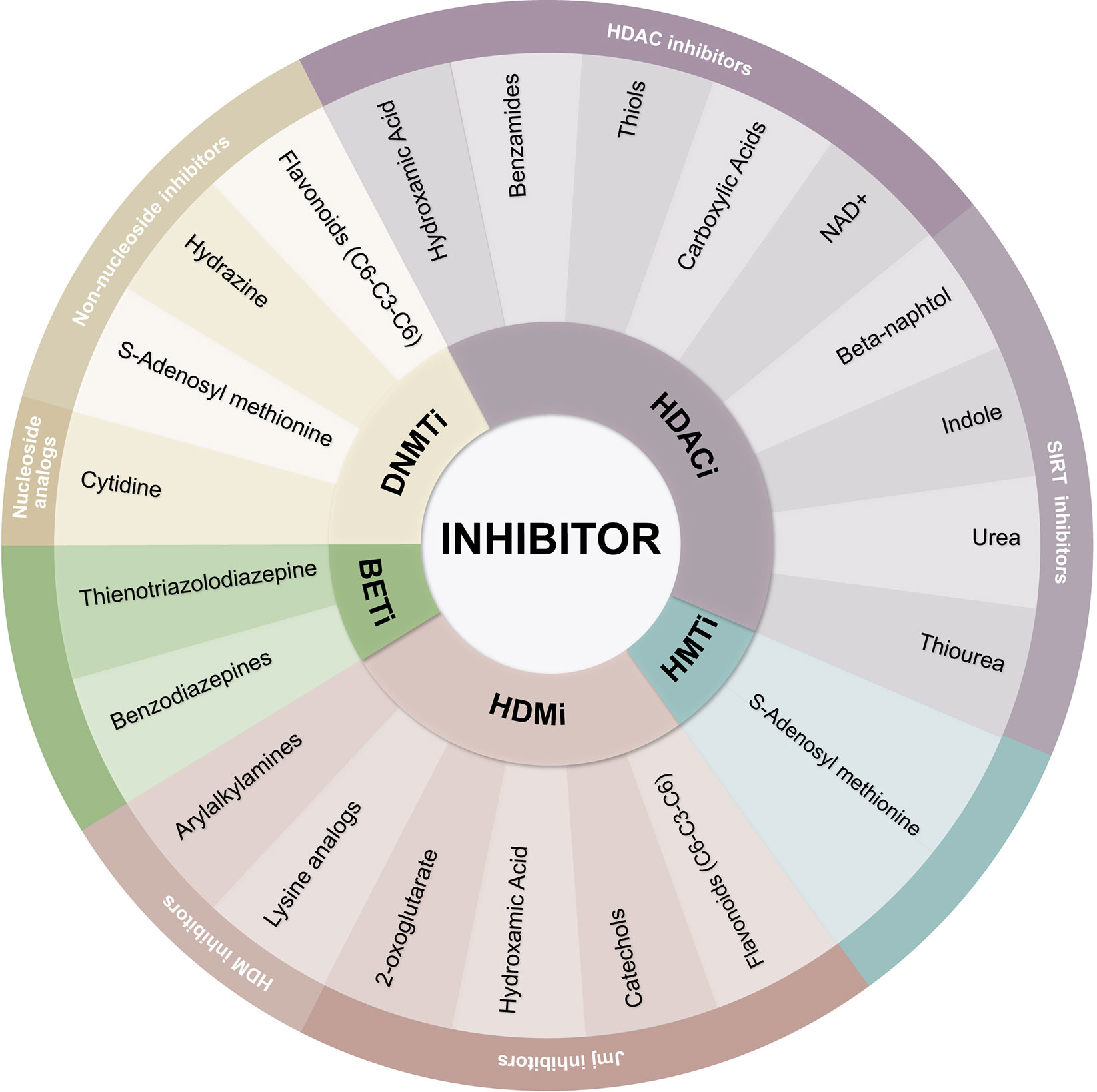
Figure 3 Classification of epigenetic inhibitors. Epigenetic inhibitors are classified as DNMTi, HDACi, HMTi, HDMI, and BETi. The chemical nature of each inhibitor defines the affinity of its targets.
Since the first DNMTi discovery (azacytidine), the number of inhibitors of DNMT has increased exponentially. The CHEMBL database reports 841 compounds tested for DNMT1 inhibition (CHEMBL1993), 258 compounds for DNMT3A (CHEMBL1992), and 80 compounds for DNMT3B (CHEMBL6095) (62) (Table 1, DNMTi section).
Among azacytidine derivatives, 5-aza-2’-deoxycytidine gained importance in the clinic, commonly known as “Decitabine”. Decitabine contains DNA sugar deoxyribose and is only integrated into DNA, while azacytidine allows for both RNA and DNA incorporation (14). Of note, Azacitidine and decitabine have both the same action mechanism. They both behave as a suicide substrate, trapping DNMTs after metabolic conversion and incorporation into DNA (3).
Guadecitabine is a hypomethylating agent of the second generation whose active metabolite is decitabine. Guadecitabine holds an amazing property: it is not a cytidine deaminase substrate, thus improving its selectivity. This drug has shown promise in treatments and recently tested in a Phase II clinical trial for treating non-intensive chemotherapy candidates with AML (63).
In 2004, azacytidine became the first medication approved by the FDA for all stages of myelodysplastic syndrome, a bone marrow disorder with a high risk of AML progression, characterized by irregular blood cell development, followed by decitabine in 2006 (64). These two drugs are currently used as first-line MDS therapy when other therapies are insufficient (14) (Table 2, DNMTi section).
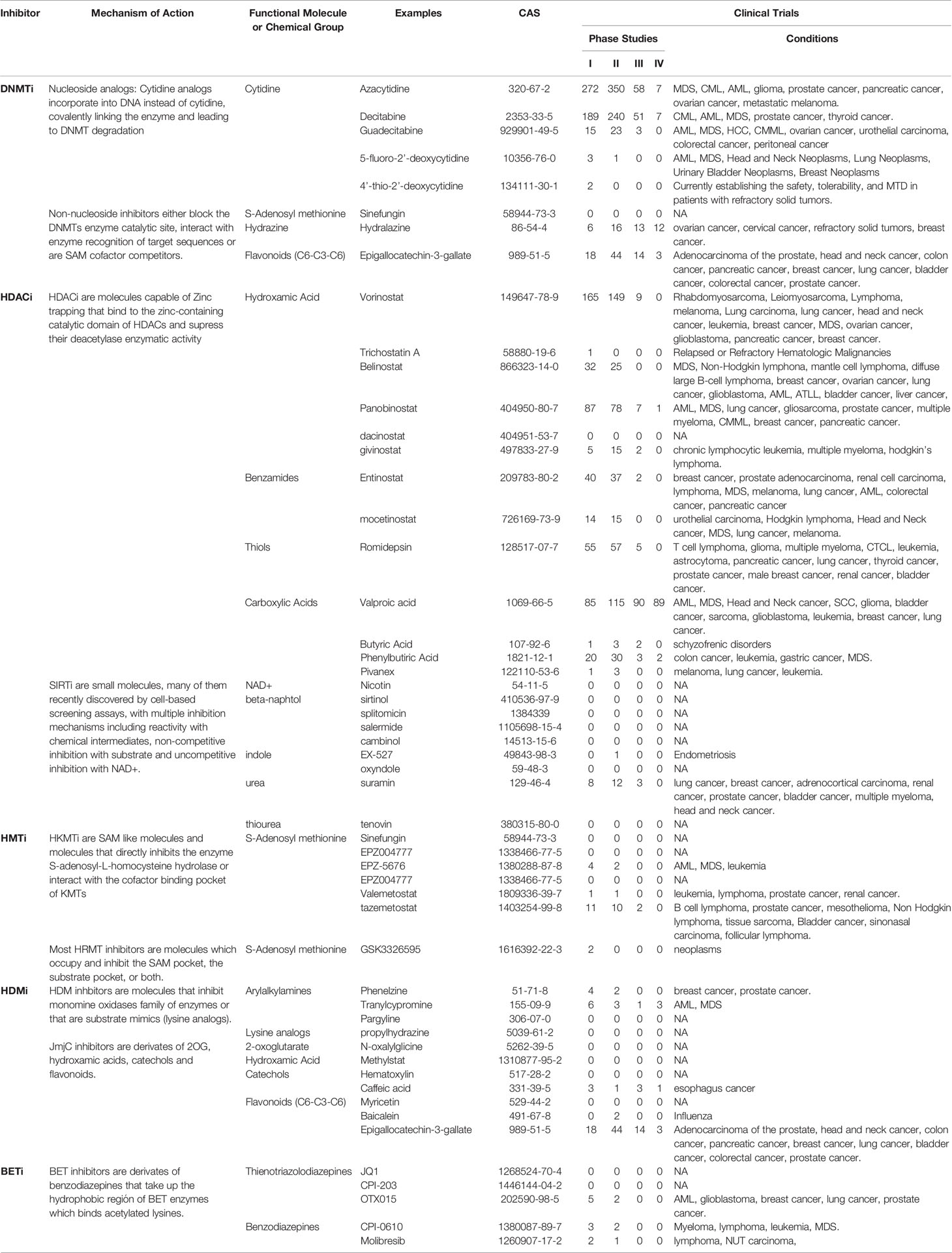
Table 2 Overview of epigenetic inhibitors currently in clinical trials for cancer therapies.
As mentioned before, DNMTs have two substrates, the methyl group donor cofactor SAM and the methylated cytosine. Non-nucleoside DNMTi includes analogs of the methyl donor S-adenosyl-L-methionine (SAM) and small molecules that interact with the active site of the enzyme DNMT (Figure 3). Indeed, it is possible to obtain potent DNMT inhibitors by designing substrate analogs and connecting them (65). This strategy has resulted in the most effective way to inhibit DNMTs and reactivate genes in cancer cells by promoting demethylation (60). Many forms of these derivatives have shown remarkable results in many models of cancer and other human diseases. These include hydralazine, EGCG, RG108, MG98, and disulfiram (66–71) (Table 2, DNMTi section). MG98 is a second-generation phosphorothioate antisense oligodeoxynucleotide that inhibits translation effects of DNMT1 mRNA but has no apparent impact on tumors (72).
Despite preclinical evidence indicating a potentiating chemotherapy cytotoxic activity of HDAC inhibitors and DNMT inhibitors, clinical outcomes have been discouraging: three of the five main combination randomized trials were stopped because of ineffectiveness or disadvantaged toxicity profiles compared to chemotherapy alone (59). The possible role of DNMT inhibitors remains unclear, but in conjunction with other therapies, these agents may theoretically still be of use.
There is a good scientific justification for combining DNMT inhibitors with HDAC inhibitors since both hypermethylated DNA and hypoacetylated histones are associated with closed chromatin states that repress gene expression by independent mechanisms. Further studies should be carried out into the efficacy of this combination at different dosages and durations of treatment. To date, hundreds of clinical trials have studied the effects of anti-DNA methylation therapy on different cancers.
HDAC Inhibitors
The development of the first HDACi commenced with the finding that erythroleukemia murine cells differentiated in the presence of dimethyl sulfoxide (DMSO). Later, chemical analogs that could make similar interactions as DMSO were studied (56). This was the case of vorinostat (SAHA), a molecule capable of metal coordination and hydrogen bonding. Interestingly, natural compounds inhibitors of HDACs (trichostatin A and trapoxin A) were found to chemically resemble vorinostat at the hydroxamic acid moiety. The mechanism of action of these compounds inhibits HDACs by reversibly binding to Zn2+ in the enzyme’s active site. Since the discovery of vorinostat, a lot of new activity assays are performed every day with inhibitor compounds (62) (Table 1, HDACi section).
Zinc binding is essential for the inactivation of most HDACs (56). As mentioned before, the Zn-binding hydroxamic moiety has proven to be one of the most successful inhibitors, and thousands of synthetic HDAC inhibitors with this moiety have been reported. Many of these inhibitors have focused primarily on optimizing the pharmacokinetics of vorinostat and trichostatin A (Figure 3; Table 2, HDACi section).
Currently, vorinostat therapy clinical applications have been applied to neurological conditions and, surprisingly, to reactivating chronic viral infection (73). Therapies for HIV-1 patients do not kill the virus entirely because it may be latent in reservoirs of CD4 + cells (74). Epigenetic mechanisms regulate viral latency, and so, clinical trials to test the effect of vorinostat therapy in reactivation of HIV-1 viral latency are currently being performed.
This optimizing focus led to the design of the hydroxamic acid containing HDACi, such as belinostat, dacinostat, givinostat, and panobinostat. The latter being the only HDACi with approval within the EU. As single agents, these molecules have shown limited efficacy, but when in combination with DNMTi, they have shown to be more effective, especially in patients with solid tumors (75, 76). Other metal-binding functional groups have been of great interest to this group. This is the case of thiols, benzamides, and carboxylic acids (56). Examples of these functional groups can be found in the drugs: romidepsin, entinostat, mocetinostat, and short-chain fatty acids, such as sodium butyrate, Pivanex, phenylbutyric acid, and valproic acid (Figure 3; Table 2, HDACi section).
Unlike hydroxamic acid analogs, short-chain fatty acids occupy an acetate escape tunnel, which may have a zinc-binding function or compete with an acetate group released in the deacetylation reaction. These are the least potent type of HDACi (77). The benzamide inhibitor class consists of a chemical moiety capable of contacting specific amino acids in the HDAC core tube active site, with or without zinc ion binding (78). These inhibitors are active at micromolar levels. The antiproliferative and cytotoxic activity has been shown by entinostat against several tumor cell lines in vitro. Entinostat is a clinical trial available orally active inhibitor (79) (Figure 3; Table 2, HDACi section).
Currently, the discovery of sirtuin inhibitors (SIRTi) is an ongoing quest in which most compounds are still under preclinical investigation (80). Most efforts have been driven toward the discovery of SIRT1 and SIRT2 inhibitors. SIRT1 inhibitors have been proposed for treating cancer, for they have shown to inhibit TNBC cell growth, survival, and tumorigenesis (56, 81). Nicotinamide is the only inhibitor of sirtuin currently used in solid tumor clinics (82). SIRTi can be categorized as β-naphthols (sirtinol, splitomicin, salermide, and cambinol), indoles (EX-527 and oxindole), and urea (suramin and tenovin) (83) (Figure 3; Table 2, SIRTi section).
HDACi have many biological effects due to changes in patterns of histone acetylation and many non-histone proteins, including proteins involved in gene expression control, extrinsic and intrinsic apoptosis pathways, the progression of the cell cycle, redox pathways, mitotic division, DNA repair, cell migration and angiogenesis (56). Whether selective inhibition of HDACs will be beneficial as anti-cancer agents over broader-acting HDACi is a question that remains unanswered (56).
Histone Methyltransferase Inhibitors
HMTs are enzymes that add up to three methyl groups to lysine (KMTs) or arginine (RMTs) residues in histone proteins (84). Lysine methylation may either activate or silence gene transcription depending on the lysine residue involved (85). Nearly 100 KMTs have been described which use the SAM molecule as the methyl donor (14). SAM-like molecules, such as sinefungin, compete with SAM for its binding site (Figure 3). These molecules are inhibitors of all SAM using enzymes, like HMTs (14). KMT drug discovery heavily relies on their cofactor binding pocket, which has structural characteristics convenient for inhibitor interaction and makes these enzymes appealing for the design of small molecular inhibitors for interference (80). Examples of HMTi can be found in drugs such as EPZ004777, EPZ-5676, DZNep, pinometostat, and tazemetostat. Pinometostat and tazemetostat are selective DOT1L and EZH2 inhibitors, respectively (Table 2, HMTi section).
Both inhibitors are of interest in some types of cancer because DOT1L is a KMT involved in abnormal methylation of H3K79 and expression of HOX genes that cause leukemia (Copeland et al., 2013), while elevated expression of the KMT, EZH2, is associated with many forms of cancer due to hypermethylation of H3K27 which facilitates transcriptional silencing (80). Also, in B-cell-lymphoma patients, EZH2 mutations occur with a frequency of approximately 15-20 percent in either tumor type, particularly in diffuse large-B cell-lymphomas and follicular lymphomas (86, 87). These modifications contribute to the more effective trimethylation of H3K27 by the mutant form of this protein (88). Preclinical studies showed that EZH2 inhibitors induced the arrest of proliferation, differentiation, and eventual apoptosis of DLBCL cells. These results were stronger in DLBCL cells that bear EZH2 mutations, but they also occurred in EZH2-wild-type DLBCL cells (89).
While several small molecule inhibitors have been developed for PRMTs with adequate potency, most PRMT inhibitors’ selectivity remains to be improved. Therefore, the detection of PRMT inhibitors involves further analysis of novel approaches (i.e., allosteric control) (90). Three PRMT inhibitors, including PRMT5 inhibitor GSK3326595 (Table 2, HRMTi section), and JNJ-64619178 as well as PRMT1 inhibitor GSK3368715 have entered clinical trials so far. PRMT inhibitors with novel action mechanisms and strong drug-like properties will shed new light on developments in drug discovery and development of PRMTi (87, 90). The number of inhibitor assays reported on CHEMBL database against the enzymatic activity of the HMTs increases everyday (62) (Table 1, HMTi section).
Histone Demethylase Inhibitors
Significant progress has been made in the development of JmjC-KDM inhibitors since the first inhibitors were identified in 2008 (91). The vast majority enter the catalytic domain and inhibit the enzyme’s activity by chelating the active site Fe (II), interfering with the 2OG binding. Because of the similarity between JmjC-KDMs’ active site pockets, it has proved difficult to achieve selectiveness in the broad superfamily of 2OG dioxygenases (92). The recent availability of JmjC-KDM crystal structures has encouraged medicinal chemistry efforts and has made it possible for the JmjC-KDMs to produce many chemical candidates. Examples of these inhibitors include hydroxamate derivatives, pyridinedicarboxylate derivatives, N-oxalyl amino acid derivatives, and agents which interfere with metal binding (71) (Figure 3; Table 2, HDMi section).
In 2004, Professor Yang Shi first described LSD1 and discovered that it had significant biological functions in a wide variety of biological processes, including cancer (93). During carcinogenesis, in AML and SCLC, elevated levels of LSD1 were observed (94). Pharmacological LSD1 inhibition with small molecules has shown that it suppresses the division, proliferation, invasion, and migration of cancer cells (95). LSD1 thus becomes an evolving clinical target for anticancer therapy. Many LSD1 inhibitors, including natural products, peptides, and synthetic compounds, have been identified.
The similarity of LSD demethylases with monoamine oxidases (MAOs) has started the quest for repurposing MAO inhibitors to find inhibitors for these types of enzymes. Initially approved by the FDA for the treatment of mood and anxiety disorders (96), the MAO inhibitor tranylcypromine (TCP) was found to be able to inhibit its homolog LSD1 moderately by forming covalent adducts (97). As a result, many MAO inhibitors (MAOi) such as pargyline, phenelzine, and tranylcypromine have been shown to inhibit HDM KDM1A (80) (Figure 3; Table 2, HDMi section). New studies are now ongoing in clinical trials with some TCP-based LSD1 inhibitors alone or combined therapy with other therapeutic agents for treating cancer (98).
Bromo and Extra Terminal Domain Inhibitors
Bromodomains are protein motifs present in several epigenetic readers including BET family, that recognize and bind to acetylated lysine residues located on histone tails. BETs consist of two bromodomains and an extra-terminal region. The BET family includes the Bromodomain testis-specific protein (BRDT), BRD2, BRD3, and BRD4 (99). BETs lead to malignancies production and progression by stimulating and enhancing the expression of main oncogenes such as MYC (100). Indeed, when treated with the inhibitor JQ1, BET inhibition resulted in MYC downregulation, which resulted in decreased levels of mRNA and protein in mouse MLL-fusion leukemia cells (101).
In various forms of cancers, including breast, neuroendocrine, ovarian, rhabdomyosarcoma, and glioma, preclinical studies of BET inhibitors have shown their efficacy (87). They disrupt the recognition by BET-containing reader proteins of acetylated lysine residues in histones, a mark associated with active transcription (102). The mechanism of BETi relies on the fact that the region that binds acetyl-lysine is hydrophobic and can be taken up by small hydrophobic molecules that specifically target this catalytic site. Examples of these inhibitors can be found in Thienotriazolodiazepines (JQ1, CPI-203, OTX015) and Benzodiazepines (CPI-0610 and molibresib) (Figure 3; Table 2, BETi section).
Preliminary clinical trials have demonstrated that BET inhibitors cannot induce long-lasting cytotoxic effects in human cancers when administered as single agents (103). Nevertheless, the potential of combinations with other epigenetic therapies is important (104). Although BET inhibitors’ toxicity may reduce such combinations, HDACi studies indicate that combinations with reduced doses may be effective, possibly reducing toxicity. This also reflects on the number of inhibitor assays for BRDs (62) (Table 1, BETi section).
The Basis for Drug Repurposing
Although epigenetic therapy has proven to be remarkably effective, epidrug discovery remains as a traditional “de novo” drug discovery pathway, which has significant disadvantages such as high costs, time consuming, and low success rate (105, 106) (Figure 4). An answer that addresses these problems and could speed up epidrugs in the clinic has arisen from the relatively recent idea of using known drugs for new targets, commonly known as drug repurposing (DR). This approach has gained considerable popularity, emerging as an interesting approach in cancer therapy research and many fields within medicine (107).
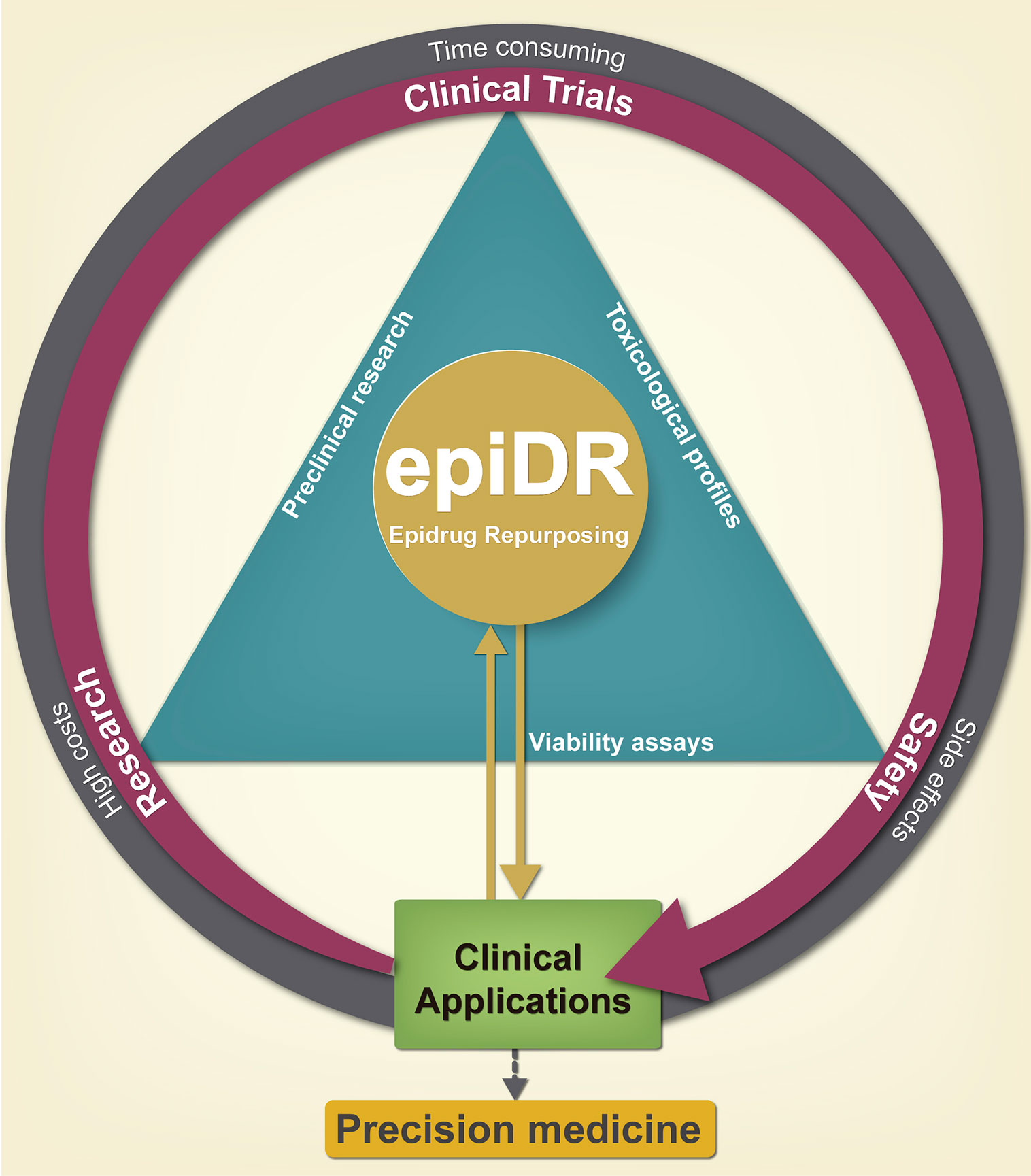
Figure 4 Advantages of pharmacological epi-drug repurposing in clinical applications. Drug repurposing serves as a shortcut reducing the time of incorporating a drug into the clinic; since the preclinical phase has already been carried out previously, giving a second chance to old drugs. Initially, it reduces the cost of development and toxicity research, which leads to greater cost-benefit efficiency for the pharmaceutical industry by generating a new cancer therapy. The repositioning of epi-drugs is a promise for the generation of new drugs of precision medicine.
DR is the discovery process of finding new medical uses of a preexisting drug which was previously approved for another indication, withdrawn from the market due to adverse effects or disapproved for failing to prove its efficacy and safety (11, 107) (Figure 4).
This approach includes the selection of drugs with promising repurposing potential and it also has important advantages over the “de novo” drug discovery processes. Previously assessed drug safety significantly reduces both costs and time for making these drugs readily available for use in the clinic (108, 109).
Historically speaking, repurposing of medications was mainly fortuitous; if an off-target effect or newly discovered target was detected, it was sure for it to be targeted for commercial usage. Examples of this are shown in drugs like sildenafil citrate, whose repurposing for erectile dysfunction was not based on a systemic approach, nor was thalidomide repurposing for erythema nodosum leprosum (ENL) and multiple myeloma, which are still the most promising examples of DR (107). Sildenafil was first formulated as an antihypertensive medication. However, after Pfizer reprofiled it for erectile dysfunction therapy and sold it as Viagra, it held the lead market share in erectile dysfunction medications in 2012, with global sales totaling more than 2 billion (110). Thalidomide, an antiemetic first sold in 1957, was discontinued within four years due to its notorious association with teratogenic defects in infants born to mothers who took the drug during their first trimester of pregnancy (107). However, the efficacy of thalidomide, first in ENL and decades later in multiple myeloma has been successfully demonstrated. Ever since, thalidomide has achieved considerable market success for treating multiple myeloma and has also contributed to the production and authorization of many more effective formulations, such as lenalidomide, which had $8.2 billion in worldwide revenues in 2017 (111).
These achievements have led to the implementation of systematic approaches to detect repurposable substances (109). The field of DR is fascinating, and its importance reflects in the vast number of drug projects of pharmaceutical companies that already have several candidate molecules that, although successful in phase I, they did not prosper in Phase II or III clinical trials. This gives rise to the existence of several known molecules, which are relatively safe to use in the clinic. Hence, this large reservoir of molecules provides a vast niche for the search for repositionable drugs, which is much larger than the set of approved drugs (112).
A DR approach usually consists of three phases before the target drug is taken into further development: The selection of a target molecule for a specific indication, analysis of the drug impact in preclinical models, and the evaluation of the effectiveness in clinical trials in phase II, when enough adequate safety results are available from phase I tests. These methods can be classified into computational approaches and experimental approaches, which are now both being widely used synergistically. DR is encompassed within these two large fields, focused on clinical evidence (109).
Experimental approaches include binding assays for the identification of novel target interactions. These types of assays come from proteomic methods, like affinity chromatography and mass spectrometry are used to detect novel targets of existing drugs (113); and phenotypic screening, which are approaches based on in vitro or in vivo models of disease screening of compounds can indicate clinical potential (114). These approaches offer testing in a relevant biochemical context by performing in vitro assays with live cells (115, 116). The evolution of in vitro screening has led to systematize drug discovery, allowing ultra-high-throughput screening, analyzing up to 10,000 compounds per day (116, 117); however, the main limitation of these methodologies are the high costs of the required infrastructure, as well as nonspecific results (8).
Computational methods include the study of large sets of data (e.g., gene expression, chemical composition, genotype or proteomic data or electronic health records) that lead to the development of reprofiling hypotheses (118). Computational approaches include: signature matching, which results for comparing a drug signature such as its transcriptomic, structural or adverse effect profile to that of another pharmaceutical product or disease phenotype (119); molecular docking, a structural computational strategy focused to predict complementarity of the binding site between a drug and a receptor (120); genetic association, a high throughput analysis of genes associated with a disease which can turn out to be potential targets for drugs (121); pathway mapping, another approach that analyses biological pathways in order to develop networks of drugs or disorders based on patterns in gene expression, disease biology, protein interactions or GWAS data to better classify repurposable candidates (122); retrospective clinical analysis, a systematic review of electronic health records, data from clinical trials and surveillances post-marketing could be useful identifying repurposable drugs; and novel sources, which is the combination of large-scale in-vitro drug screens with genomic data, electronic health records and self-reported patient data represents new ways to repurpose drugs (123, 124).
In sum, these approaches allow multiple manners for conducting DR. However, these methodologies applications need to be taken with caution, as many of them seem to be reductionist (117, 125). Numerous strategies are now coupling drug networks with computational analysis to characterize different diseases’ metabolic pathways. These efforts aim to identify drugs acting not only on a single target but also on a whole network of proteins (126, 127). In every computational approach, experimental validation is compulsory since the actual methods are not 100% accurate.
HTS (High-Throughput Screening) is the most common approach in DR of epidrugs, and most of them are designed to inhibit catalytic sites of epigenetic writer enzymes (128). Computational methods, such as virtual screening, aim to efficiently discover novel active compounds against epigenetic factors (8). The increasing attention on epigenetic targets as an opportunity for DR provides high expectations. Next, we will summarize the current efforts in epidrug repurposing for cancer therapy.
Available Databases Focused on Exploration and Recompilation of DR Research
Nowadays, there is a large amount of information available focused on the search and annotation of drugs to be repurposed and the drugs that currently have research that supports their proposed new uses. Some public databases such as ChemBL, DrugBank, and DrugCentral are repositories of bioactivity data and drug chemical structures. These databases summarize multiple indications and chemical drug-target interactions. More specifically, the FDA-approved epidrugs are gathered in several databases focused on tested epidrugs and provides information about annotation tools (Table 3, Section Epidrugs). These databases are useful because they facilitate the integration of epidrug datasets obtained from experimental and computational approaches, reducing the manual search of information, and helping to increase collaboration on the field.
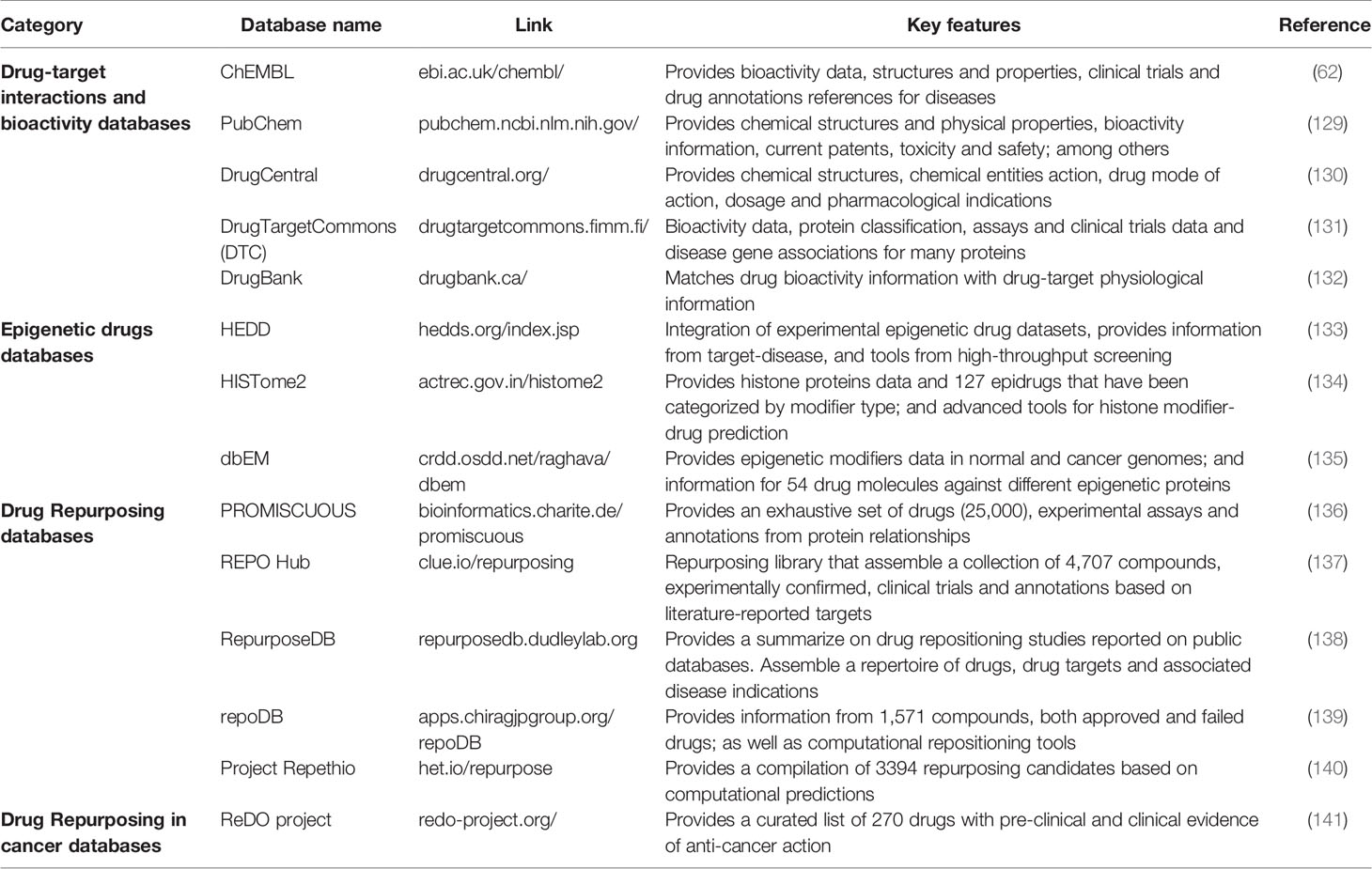
Table 3 Some databases and tools that summarize the current knowledge on DR.
Other databases that aim to summarize the current efforts and latest frontiers in DR research are the REPOHub, repoDB, and the Project Repethio; these include clinical trials, pre-clinical tools for annotations, and information resources. Unlike the previous ones, these databases focus on gathering and matching the results from both predictive tools and experimental or clinical trials, resulting in faster results on drugs that could be repurposed (Table 3, Section Drug Repurposing). Tanoli et al., 2020 summarize the types of data available through multi-database exploration focused on DR (142). Currently, the ReDO project (Repurposing Drugs in Oncology) is probably the only database focused on assembling DR for cancer targets. And it has played a crucial role in the development of research for new drugs to cancer therapy with the DR approach.
Epidrug Repurposing in Cancer (Epi-DR)
The interest in oncological DR has emerged as a response to the declining productivity of oncological drug development (143) and as a source of low-cost treatments to meet the increased demands for novel treatments, in efforts to overcome chemoresistance and reduce the development time of de novo drugs (144).
Some widely used and well-known drugs for cancer therapy are examples of epi-DR, with an effect on epigenetic targets, and are either currently FDA-approved or under clinical development (145). The first repurposed drugs as an anticancer epidrug in the field were the 5-azacytidine and 5‐aza‐2′‐deoxycytidine (decitabine) (146). At first, these drugs were both approved by the FDA to treat myelodysplastic syndromes due to their antimetabolic effects on in vitro assays in cancer cells (146). However, the toxicity shown by 5-azacytidine led to other chemotherapeutic regimens being preferred (146); later, it was found that azacytidine and decitabine could both inhibit DNA methylation and were incorporated by tumor cells and also in myelodysplastic syndromes (146–148).
DNMT Inhibitors
The natural compound Harmine downregulates the expression of DNMT1, which results in reactivation of the p15 tumor suppressor gene in AML. Future studies are expected to assess if Harmine can be considered a potential therapy for AML and if it can be used as a single agent or adjuvant (149). Chlorogenic acid is a polyphenol coffee that has been found to suppress DNMT1. Its inhibitory activity derives from a chemical change resulting in increased S-adenosyl-L-homocysteine (SAH) production. Chlorogenic acid has been shown to inhibit DNMT1, using breast cancer cell lines, which lowers DNA methylation (150).
Laccaic acid A is a direct, competing DNMT1 natural compound inhibitor that reactivates genes silenced by promoter DNA methylation synergistically with 5-azadC in breast cancer cells (151). Procaine is a promising treatment with growth-inhibiting and DNA-hypomethylation effects in cancer cells. Especially in gastric cancer where its antiproliferative and apoptotic effects have been proven (152). Its well-defined, safe use as a local anesthetic, with well-known pharmacology, should promote procaine to pre-clinical trials (153). Procainamide, a derivative of procaine, hinders the enzymatic activity of DNMT1 by directly reducing the enzyme affinity for both DNA and S-adenosyl-L-methionine. It would be important to analyze whether procainamide, a fairly stable non-nucleoside inhibitor of DNMT1, will prevent cancer from arising (154).
A computer-based search for similarities between a database of approved drugs and 5-aza-2’-deoxycytidine has recently been detected as an ideal candidate for DR. Mahanine, a plant derived alkaloid, was shown to induce DNMT1 and DNMT3B proteasomal degradation by inactivating Akt, which in turn restored RASSF1A expression in prostate cancer cells. Mahanine then represents a possible therapeutic agent for advanced prostate cancer when RASSF1A expression is inhibited (155).
Hydralazine, approved as an antihypertensive, is a non-nucleoside DNMTi that interacts with the binding domain of DNMTs, and can decrease DNMT1 and DNMT3A mRNA expression and protein levels in T cell leukemia cell lines (156). In advanced cervical cancer, bladder, and cervical cancer cell lines, respectively (157, 158), hydralazine induces DNA demethylation and decreases DNMT activity. Also, hydralazine, combined with magnesium valproate, is an opportunity to reverse imatinib resistance in patients with several malignancies, including lung (NCT00996060), cervical (NCT00404326), and locally advanced breast (NCT00395655) cancers, as well as different solid tumors which are refractory to current therapies (159–161) (NCT00404508). Olsalazine, an FDA approved anti-inflammatory agent, has proven its hypomethylating and very low cytotoxicity effects in cell-based screen tests (162).
Mithramycin A, an antibiotic with potent antitumor activity, binds to sequences of GC-rich or CG-rich DNA and upregulates tumor suppressor genes’ expression by reducing the methylation of their promoters through binding and depleting the DNMT1 protein in lung cancer cells (163). Nanaomycin A, an anthracycline antibiotic, has demonstrated selectivity to DNMT3B in biochemical assays. Dock modeling strategies suggest that nanaomycin A is capable of binding DNMT3B’s catalytic site. Treatment of the human tumor lines of the colon, lung, bone marrow with nanaomycin A demonstrated substantial genomic demethylation. While it is unclear if anthracyclines will be a successful choice for clinical DR due to certain long-term cardiotoxicity concerns, Nanaomycin A is the first non-SAH DNMT3B-selective compound that offers valuable biochemical properties for additional studies (164).
Disulfiram is an alcohol aversive drug that has been approved by the FDA for more than 60 years for treating alcohol abuse. It allows acetaldehyde to accumulate in the blood by inhibiting ALDH (165). Disulfiram’s anticancer activity is mediated by its ability to suppress DNMT1 and through the reactivation of epigenetically silenced genes such as APC and RARB in prostate cancer cell lines (70) (Table 4, Section 1).
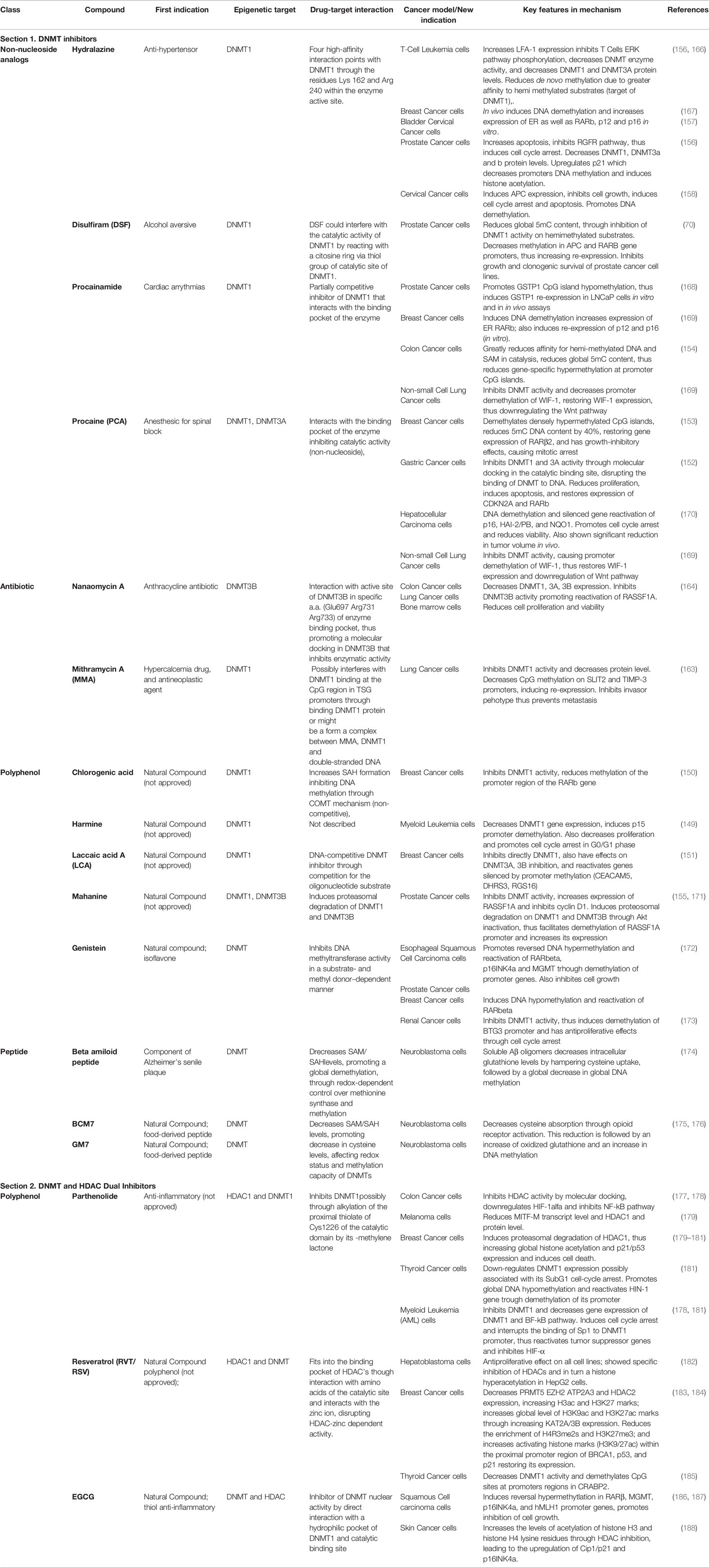
Table 4 Current DNMTi and DNMT-HDAC dual inhibitors repurposed drugs with applications in cancer therapy [*modified from Moreira-Silva et al. (9)].
Peptides are small proteins made up of fewer than 50 amino acids. Such compounds have several roles in the human body and can modulate epigenetic pathways, raising the exciting possibility of peptide-based therapy. Such peptides may be endogenous, or food derived. Amyloid beta (Aβ), the central component of Alzheimer’s senile plaque (AD), reduces global DNA methylation but increases DNA methylation in the Neprilysin gene promoting region, an Aβ-degrading enzyme (189). Soluble Aβ oligomers decrease intracellular glutathione levels by hampering cysteine uptake, followed by a global decrease in DNA methylation (174). BCM7 and GM7 are food derived peptides produced by hydrolytic casein and gliadin digestion. They decrease cysteine absorption through opioid receptor activation in neuronal and gastrointestinal cells. This reduction is followed by an increase of oxidized glutathione and an increase in DNA methylation (175, 176) (Table 4, Section 1).
Dual DNMT and HDAC Inhibitors
In most cancer types, altered DNMT and HDAC activity is observed (190). Therefore, some repurposed drugs that inhibit both DNMT and HDAC enzymes could improve efficacy over one-target agents (Table 4, Section 2).
Berberine, an isoquinoline alkaloid derived from Berberis vulgaris (191) and used to treat bacterial, parasitic, and fungal infections, has been repurposed as a DNMT and HDAC dual inhibitor (192). In multiple myeloma cell lines, berberine treatment showed downregulated DNMT1 and DNMT3A expression, restoring p53 expression through DNA hypomethylation (193). Berberine also inhibits Class I and II HDACs in lung cancer cell lines, down-regulates gene expression, and increases histone H3 and H4 acetylation (194). EGCG is a polyphenol found in green tea (Camellia sinensis) and is a known anti-inflammatory compound (195). It has recently been proposed as an inhibitor of DNMT by direct interaction with the catalytic site of DNMT (186–188). EGCG reduces cell growth and increases apoptosis in renal carcinoma cells through the upregulation of TFPI-2. In skin carcinoma cells, EGCG increases the levels of acetylation of histone H3 and histone H4 lysine residues through HDAC inhibition, leading to the upregulation of tumor-suppressor genes (188) (Table 4, Section 2). Resveratrol is a natural polyphenolic compound found in grapes and berries (196), and it has been proposed as a dual inhibitor of both DNMTs and HDACs. In breast cancer cell lines, resveratrol inhibits both HDAC and DNMT1 activity, decreases histone H3 lysine 27 methylation, and increases its acetylation (182–184). In thyroid cancer cell lines, treatment with resveratrol showed resensitization to therapy when in combination with retinoic acid through the demethylation of CpG sites at promoter regions of CRABP2 gene (185); the effect of resveratrol as a repurposed cancer drug was also investigated in clinical trials (NCT00256334, NCT01476592, NCT00433576). Finally, parthenolide is a terpenoid compound, isolated from Tanacetum parthenium, with anti‐inflammatory properties. Parthenolide downregulates HDAC1 gene expression (179) and increases histone acetylation (177, 180). It reverses drug resistance in some cancer cell lines (178) and restores silenced gene expression through a decrease in DNA methylation levels (181) (Table 4, Section 2).
HDAC Inhibitors
As previously mentioned, the use of HDACi among the chemotherapeutic agents is growing (Table 5, HDACi). Hydroxamic and carboxylic acids are being studied as potential HDACi; for instance, drugs like Vorinostat (SAHA), approved for psoriasis treatment, and Valproic acid (anticonvulsant) are currently included in several clinical trials against different types of cancers (236). A complete overview about clinical trials in some of the most studied HDACi repurposed, such as Vorinostat, Valproate, Belinostat, Panobinostat, and cyclic peptide Romidepsin is available (236) (Table 5, HDACi).
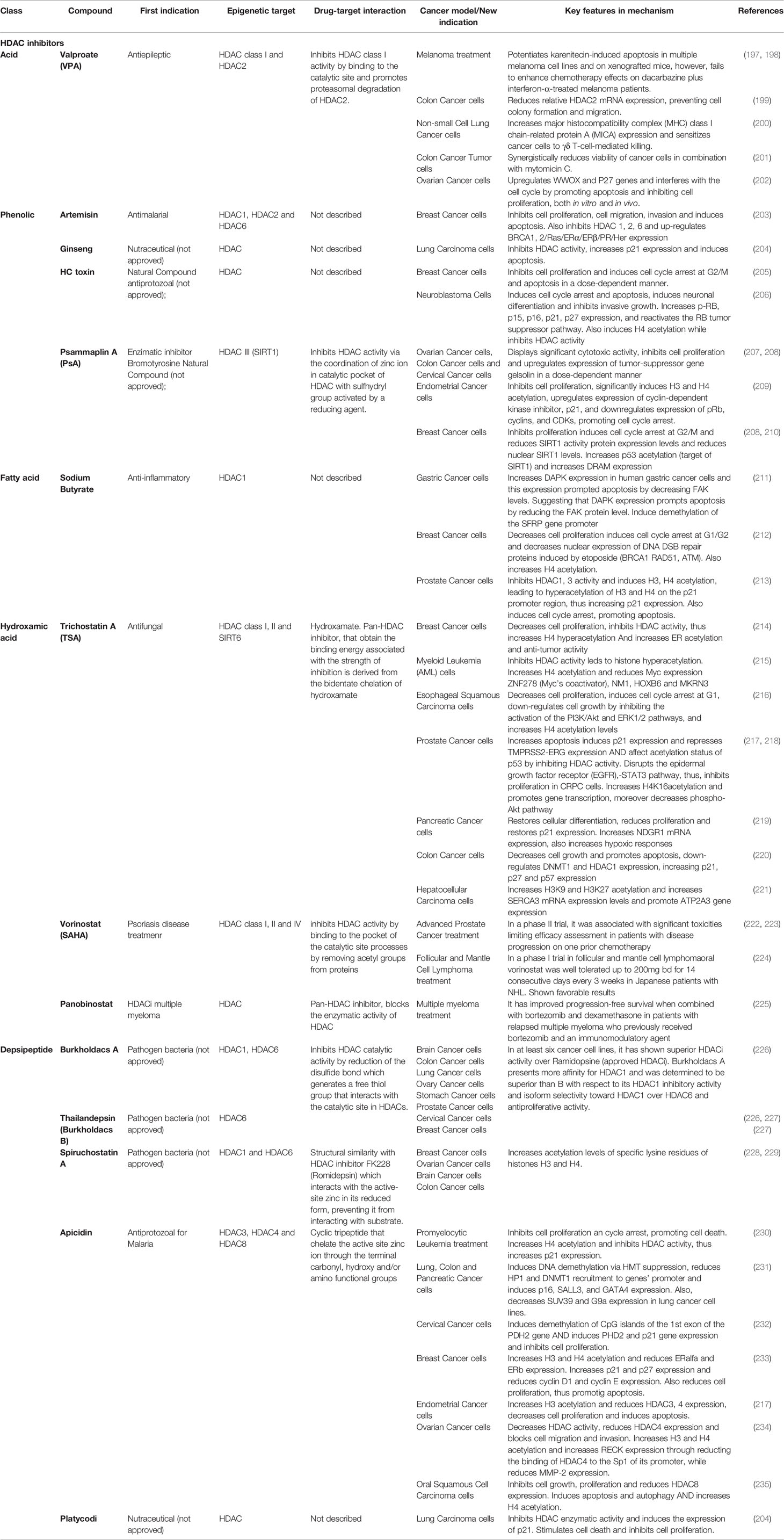
Table 5 Current HDACi repurposed drugs with applications in cancer therapy [*modified from Moreira-Silva et al. (9)].
Compounds with HDACi potential have been found in plants. Ginseng (Panax ginseng) is a popular plant extract commonly used in South Korea and traditional Chinese medicine, which contains several compounds (ginsenosides) with pharmacological properties (144). Platycodi radix (Platycodon grandiflorum), commonly known as balloon flower, is used to treat many diseases related to obesity in East Asia (237). Recently, Byun and cols. demonstrated that ginseng and platycodi have significant HDACi activity in Lung Carcinoma cell lines, thus upregulating p21 gene expression and promoting cell death (204). HC toxin is a cyclic tetrapeptide derived from a plant-fungal parasitic-association between Helminthosporium carbonum (ascomycetes) and its host, (commonly Poaceae plants family). It was reported as a Maize Histone Deacetylase inhibitor (238) and proposed as an analog of Apicidin and Artemisin, a fungal metabolite (239), and antimalarial drug, respectively; with antiprotozoal HDACi activity proved for Malaria (Plasmodium berghei) in mice. However, recently HC-toxin has been rediscovered and identified as HDACi in different cancer cell lines (205). In breast cancer and neuroblastoma cell lines, HC toxin inhibited HDAC activity and promoted cell proliferation inhibition, cellular death, and induced H4 acetylation (205, 206). Artemisin has been repurposed as an HDAC1, HDAC2, and HDAC6 inhibitor in the breast cancer cell line MCF‐7 (203) (Table 5, HDACi).
Psammaplin A (PsA) is a phenolic compound that derives from the marine sponge-association, Poecillastra sp. and Jaspis sp., (Pseudoceratina purpurea) whose active substances are monomers of thiol groups with enzymatic inhibition activity (210, 240). These monomers play a key role for both HDACi and DNMTi activity (241). In endometrial cancer cells, PsA showed HDAC1 and HDAC6 inhibition, reduction of HDAC1 expression the elevation of histone H3 and H4 acetylation, induction of cell cycle arrest, and apoptosis (208, 209). Burkholdacs A and B, with a structure similar to Thailandepsin A, was identified as a novel HDACi through the systematic overexpression of transcription factors associated with Burkholderia thilandensis (227). They are bicyclic depsipeptide compounds, proposed as potent HDACi in brain cancer cells, but also in other cancer cell lines (226). Using a panel of 39 human cancer cell lines, burkholdacs have shown superior HDACi activity over Ramidopsine (approved HDACi) in at least six cancer cell lines (226). Burkholdacs’ affinity for HDAC1 is greater than that for HDAC6. Structural changes in burkholdacs A and B structures may increase their activity and selectivity, giving rise to isoform selective inhibition of HDACs therapeutical potential (226) (Table 5, HDACi). Other depsipeptides have also been studied for repurposing. Spiruchostatin A, and Plitidepsin (Aplidin) are natural depsipeptides derived from Pseudomonas sp. (228) and Aplidium albicans (242), respectively. In cancer cell lines, reduced spiruchostatin A effectively inhibited HDAC1, an effect not observed when oxidized, and it showed an increase in the acetylation levels of specific lysine residues of histones H3 and H4 (228). Plitidepsin is currently in clinical trials to treat multiple myeloma (243, 244) but it has also displayed interesting properties against hematological malignancies (245). Some depsipeptides display a greater affinity for HDAC1 than HDAC6 and class II HDACs, but this does not appear to limit their activity as anti-cancer agents judging by in vitro effects in cancer cells (208, 226, 228). Structure-function studies on depsipeptides can lead to the generation of chemical analogs with enhanced selectivity as HDACi drugs (Table 5, HDACi).
HAT, HMT, HDM, and BET Inhibitors
Recently, HATi, HMTi, HDMi, and BETi have become of great interest for personalized cancer treatment. Multiple studies have consistently shown the enormous potential of known drugs and compounds for DR as epigenetic modulators (Table 6, HATi, HMTi, HDMi, and BETi).
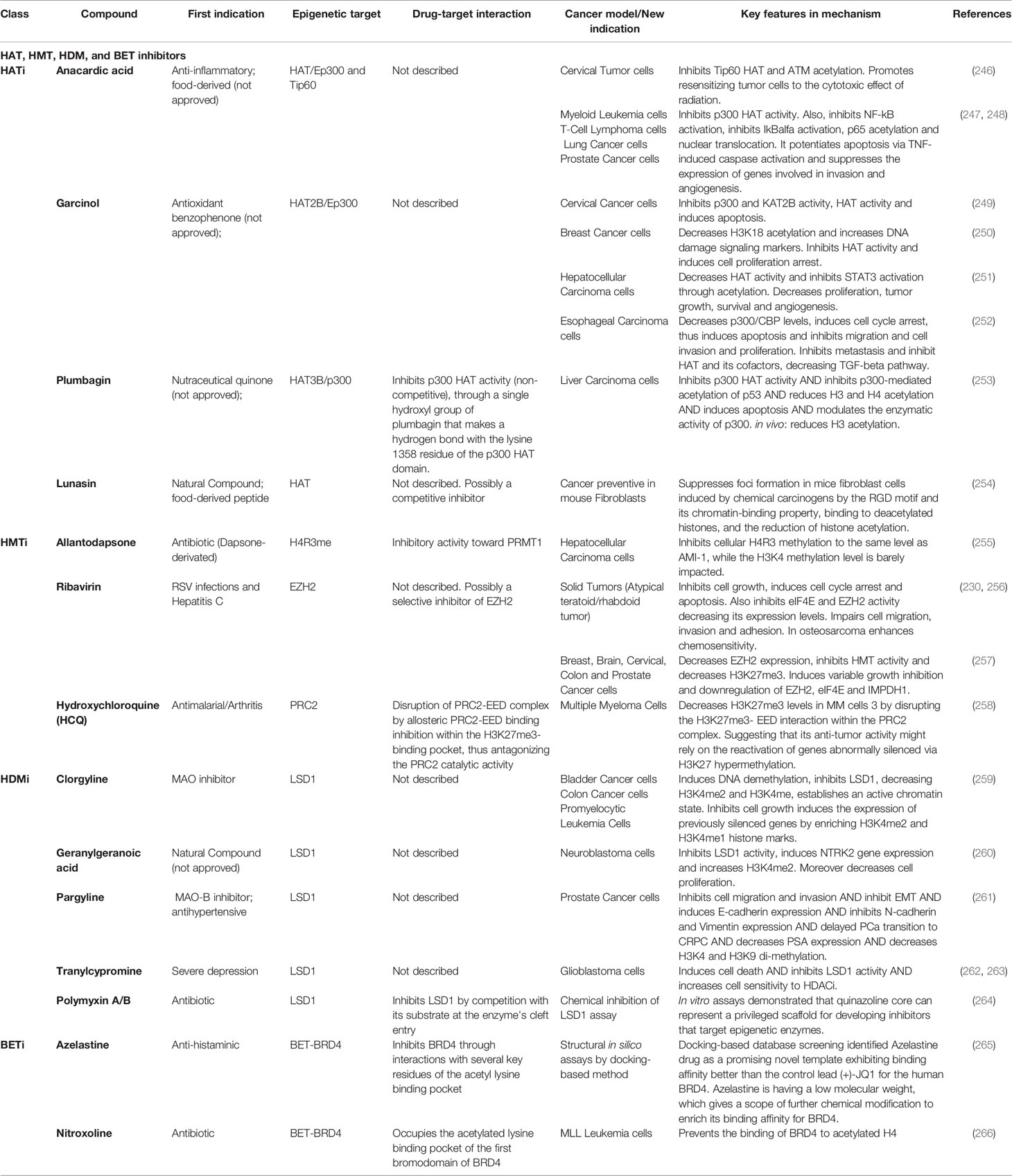
Table 6 Current HAT HMT, HDM, and BET inhibitors repurposed with epigenetic applications in cancer therapy [*modified from Moreira-Silva et al., (9)].
HAT Inhibitors
Anacardic acid, a small molecule obtained from cashew nutshell liquid with known antitumor activity, inhibits the p300’s and PCAF’s HAT activity. Anacardic Acid is not specific to any particular HAT group, but it can be used to synthesize other specific HAT activity modulators based on this molecule (246). Plumbagin is an in vivo, potent acetyltransferase inhibitor, hydroxynaphthoquinone isolated from the roots of Plumbago Rosea. A single hydroxyl group in Plumbagin confers its HATi properties. Replacing this group with other chemical moieties results in complete loss of its inhibitory activity. Plumbagin has also been reported to suppress the activation of NFκ-B, leading to apoptosis potentiation. Plumbagin may be a potential anticancer agent, but its cell toxicity properties could be the main limitation of its use as a therapeutic molecule (253). Garcinol is a potent inhibitor of the p300 and PCAF HATs. It inhibits in vivo histone acetylation in HeLa cells but does not affect histone deacetylation. Garcinol suppresses chromatin transcription dependent on HAT p300 but does not affected transcription of DNA (249). Lunasin is a 43 amino acid peptide found in soybean, barley, wheat, and rye. Previous studies have shown that lunasin can suppress the proliferation and migration of cancer cells with no effect on wild-type cells. Lunasin is a competitive inhibitor of HATs. It inhibits histone acetylation and regulates the cell cycle. This binding is probably achieved through its helical structure, similar to chromatin-binding protein structures (267) (Table 6, HATi).
HMT Inhibitors
Allantodapsone was recovered from a virtual screening based on the PRMT1 structure. Allantodapsone inhibits H4R3 methylation in the hepatocellular carcinoma cell line HepG2 while leaving H3K4 methylation unaffected (255). Ribavirin is an antiviral drug that has become of interest as a therapeutic agent in cancer. Ribavirine selectively inhibits pediatric osteosarcoma and improves chemosensitivity (256). It also possesses in vitro growth inhibitory effects against various malignant cell lines at clinically reasonable concentrations; also, ribavirin treatment results in the reduction of EZH2 at RNA and protein levels, inhibition of EZH2 enzyme activity, and reduction of H3K27 methylation (257). The anti-malarial drug, hydroxychloroquine, has also been effective in treating rheumatoid lupus, arthritis, and porphyria cutanea tarda. Structural experiments have shown that hydroxychloroquine inhibits the allosteric binding of PRC2 to EED within the H3K27me3-binding region, thereby antagonizing the catalytic function of the PRC2. These findings suggest a new epigenetic function of hydroxychloroquine with possible therapeutic repositioning (258) (Table 6, HMTi).
HDM Inhibitors
Clorgyline is a selective MAO A inhibitor- used as an antidepressant until severe dietary adverse effects are commonly known as the “cheese effect” were reported for this drug (268). As a member of MAO inhibitors, clorgyline can also inhibit LSD1, and it has been demonstrated to have cell-type dependent synergic effects when combined with DNMTi (259). Geranylgeranoic acid, an acyclic diterpenoid present in medicinal plants, has recently been found to be a potent inhibitor of recombinant LSD1. Geranylgeranoic acid inhibits the proliferation and induces a neuronal phenotype through increasing the abundance of H3K4me2 of NTRK2 gene promoter in human SH-SY5Y-derived neuroblastoma cells (260). Pargyline, a MAO B selective inhibitor with antidepressant activity, affects the transition from androgen-dependent to androgen-independent in prostate cancer. Inhibition of LSD1 with a concomitant reduction of H3K4me2 and H3K9me2 levels have been reported for pargyline. Pargyline, in combination with androgen deprivation therapy, could be an effective adjunctive treatment for advanced prostate cancer (261). Unlike selective MAO inhibitors such as pargyline, non-selective MAO inhibitors strongly repress the nucleosomal demethylation of histone H3K4. Tranylcypromine, a drug used in treating severe depression, has demonstrated strong LSD1 inhibitory effects with an IC50 of less than 2 mM (262). Tranylcypromine contributes to GBM cell synergistic apoptosis in association with other HDAC inhibitors (263). Recently, molecular docking studies have highlighted the potential of approved drugs such as decitabine, entecavir, abacavir, penciclovir, and DZNep as KDM5B inhibitors. Their role as HDMi could be of great importance in lung cancer, melanoma, hepatocellular carcinoma, gastric cancer, and prostate cancer, among others. Decitabine is a DNMTi used in myelodysplastic syndrome (MDS), abacavir, entecavir, and penciclovir are antivirals used in the treatment of HIV, hepatitis B, and herpes infections, respectively. DZNep is a specific HMTi with promising results in cancer immunotherapy (269). Finally, Polymyxin B and polymyxin E are antibiotics used in multidrug resistant bacterial infections. These compounds were shown to inhibit LSD1 by competition with its substrate at the enzyme’s cleft entry. Polymyxins have significant side effects that limit their application to untreated infections, but they could still be the target of drug repurposing for other diseases, such as leukemia (264) (Table 6, HDMi).
BET Inhibitors
Azelastine, a selective H1 antagonist, was found to be a promising BETi, displaying a stronger binding affinity than BETi control JQ1 for human BRD4 by docking-based methodologies. These findings highlight the importance of computational methods for molecular drug design and will uncover new BRD4 inhibition candidates (265). The antibiotic approved by FDA, nitroxoline, disrupts the association of BRD4 bromodomain with acetylated H4. Nitroxoline has shown strong selectivity at inhibiting all BET family members compared with non-BET proteins. By causing cell cycle arrest and apoptosis, nitroxoline successfully prevents the proliferation of MLL leukemia cells. The possible use of nitroxoline and its derivatives as BET inhibitors in BET related diseases is now under investigation (266) (Table 6, BETi).
Concluding Remarks
Drug repositioning has emerged as a viable strategy to increase drug discovery’s overall productivity, resulting in a new and cheaper way to generate alternative therapies for various diseases, including cancer. The drug repositioning approach is growing due to a broad range of reposition candidate molecules that already have clinical and toxicity profiling developments. One factor that has strongly driven this approach is the increasing availability of biomedical data, including genomic data, which covers various aspects of cellular mechanisms, opening a search that is not restricted to biological factors involved in a disease. This omic perspective allows the deduction of complex interactions that can be inhibited or treated to cure or reverse a pathological condition. Advances in complementary bioinformatic analytical methods provide critical substrate candidates that enable their systematic evaluation. Therefore, a window of opportunity opens where the reuse of previously synthesized drugs can be investigated and given a new direction. Epi-DR has already shown a profit in epigenetics and cancer treatment, where it has proven its efficacy. Indeed, many epidrugs emerged this way, such as 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine) (146), Hydralazine (156), Vorinostat (SAHA), and Valproic acid (236).
Epigenetic alterations are considered to be among the earliest and most comprehensive genomic aberrations occurring during carcinogenesis, and therefore it has been classified as a hallmark of cancer (270). The impact of epigenetics in understanding cancer has been of great interest in recent years, and even more due to the advancement of the genomic era. Several works demonstrate the importance of epigenetic biomarkers that can predict the response or prognosis in various types of cancer. The promoter methylation of the MGMT gene in gliomas is a clear example, where it helps to indicate the use of precision medicine through the drug temozolomide (271). Another example is found in EHZ2 enzyme alterations, which indicate a poor prognosis in breast, prostate, and other types of cancers.
Epigenetic mechanisms have great flexibility to respond to environmental changes and modify gene expression. Consequently, search for artificial ways to induce epigenetic remodeling, which could improve therapy in the event of a disease as cancer. Therefore, the implementation of epigenetic therapies opens a new panorama for the fight against cancer. Epidrugs show enormous potential for clinical use, especially in cancer, because in these diseases, an epigenetic imbalance is a well-known characteristic that is both of origin, development, and severity of tumors.
Even though there are already some epidrugs approved by the FDA and the current knowledge about various mechanisms involved in gene regulation, promoted by the advancement of technologies that expand the information on specific epigenetic mechanisms, challenges remain in identifying epigenetic modifications of cancer and targeting them for therapeutic purposes. Among them stands out that epigenetic changes can be diverse in the types of cancer and between the different clinical phases and those that are dependent on environmental conditions. Therefore, we must distinguish between the dysregulation of driver genes and those whose changes are secondary to these. Also, the generation of epigenetic therapies as well as the molecular mechanisms that coordinate them is subject to understanding, and much research is still required of several of them to safely transport them to the clinic. However, identifying epigenetic alterations that affect the tumor’s fate and behavior finding drugs that target them are some of the promises of epigenetic therapy in cancer.
In this sense, the concept of reusing a medicine offers a broad scope to investigate the hidden potential behind the medicine and to recycle it. The reincorporation of a drug with the potential to remodel epigenetic characteristics, which are beneficial for cancer management, is of great interest to the field. Offering great advantages in drug development times could lead to precision medicine therapy with new and clearly encouraging prospects for the future (Figure 4).
Author Contributions
MM-C and MM-R wrote and designed the manuscript. RG-B coordinated, wrote, and designed the manuscript. RG-B and VJ-G revised the manuscript. VJ-G elaborated on the figures. CA-C wrote and revised the manuscript, and LH coordinated and directed the review development. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT) by the Fondo Sectorial de Investigación en Salud y Seguridad Social (FOSISS, grant no. SALUD-2017-2-290041). Marco Antonio Meraz-Rodriguez is a masters student in the “Programa de Maestría y Doctorado en Ciencias Bioquímicas, UNAM”, and received a fellowship from CONACyT (CVU 659273, no. 481908).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the National Cancer Institute of Mexico (INCan) for support to the present work. We thank Juan F. Duarte-Campos and Hugo R. Barajas for their critical comments and review.
References
1. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discovery (2012) 11(5):384–400. doi: 10.1038/nrd3674
2. Fardi M, Solali S, Farshdousti Hagh M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis (2018) 5(4):304–11. doi: 10.1016/j.gendis.2018.06.003
3. Kelly TK, De Carvalho DD, Jones PA. Epigenetic Modifications as Therapeutic Targets. Nat Biotechnol (2010) 28(10):1069–78. doi: 10.1038/nbt.1678
4. Jerónimo C, Bastian PJ, Bjartell A, Carbone GM, Catto JWF, Clark SJ, et al. Epigenetics in Prostate Cancer: Biologic and Clinical Relevance. Eur Urol (2011) 60(4):753–66. doi: 10.1016/j.eururo.2011.06.035
5. Strauss J, Figg WD. Using Epigenetic Therapy to Overcome Chemotherapy Resistance. Anticancer Res (2016) 36(1):1–4.
6. Miranda Furtado CL, Dos Santos Luciano MC, Da Silva Santos R, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics (2019) 14(12):1164–76. doi: 10.1080/15592294.2019.1640546
7. Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med (2011) 17(3):330–9. doi: 10.1038/nm.2305
8. Naveja JJ, Dueñas-González A, Medina-Franco JL. Drug Repurposing for Epigenetic Targets Guided by Computational Methods. In: Epi-Informatics. Elsevier (2016). p. 327–57. doi: 10.1016/B978-0-12-802808-7.00012-5
9. Moreira-Silva F, Camilo V, Gaspar V, Mano JF, Henrique R, Jerónimo C. Repurposing Old Drugs into New Epigenetic Inhibitors: Promising Candidates for Cancer Treatment? Pharmaceutics (2020) 12(5):410. doi: 10.3390/pharmaceutics12050410
10. Blatt J, Corey SJ. Drug repurposing in pediatrics and pediatric hematology oncology. Drug Discovery Today (2013) 18(1–2):4–10. doi: 10.1016/j.drudis.2012.07.009
11. Shim JS, Liu JO. Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs. Int J Biol Sci (2014) 10(7):654–63. doi: 10.7150/ijbs.9224
12. Waddington CH. The epigenotype. 1942. Int J Epidemiol (2012) 41(1):10–3. doi: 10.1093/ije/dyr184
13. Deans C, Maggert KA. What do you mean, ‘epigenetic’? Genetics (2015) 199(4):887–96. doi: 10.1534/genetics.114.173492
14. Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenet (2019) 11:1–2. doi: 10.1186/s13148-019-0776-0
15. Roberti A, Valdes AF, Torrecillas R, Fraga MF, Fernandez AF. Epigenetics in cancer therapy and nanomedicine. Clin Epigenet (2019) 11(1):81. doi: 10.1186/s13148-019-0675-4
16. el Bahhaj F, Dekker FJ, Martinet N, Bertrand P. Delivery of epidrugs. Drug Discovery Today (2014) 19(9):1337–52. doi: 10.1016/j.drudis.2014.03.017
17. Biswas S, Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol (2018) 837:8–24. doi: 10.1016/j.ejphar.2018.08.021
18. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol (2010) 28(10):1057–68. doi: 10.1038/nbt.1685
19. Moore LD, Le T, Fan G. DNA Methylation and Its Basic Function. Neuropsychopharmacology (2013) 38(1):23–38. doi: 10.1038/npp.2012.112
20. Kuroda A, Rauch TA, Todorov I, Ku HT, Al-Abdullah IH, Kandeel F, et al. Insulin Gene Expression Is Regulated by DNA Methylation. PloS One (2009) 4(9):e6953. doi: 10.1371/journal.pone.0006953 Maedler K (ed.).
21. Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet (2007) 16:R50–59. doi: 10.1093/hmg/ddm018
22. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet (2008) 9(6):465–76. doi: 10.1038/nrg2341
23. Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta (2009) 1789(1):45–57. doi: 10.1016/j.bbagrm.2008.06.005
24. Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire J-M, et al. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie (2012) 94(11):2280–96. doi: 10.1016/j.biochi.2012.07.025
25. Peterson CL, Laniel M-A. Histones and histone modifications. Curr Biol: CB (2004) 14(14):R546–551. doi: 10.1016/j.cub.2004.07.007
26. Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev (2009) 19(2):127–34. doi: 10.1016/j.gde.2009.02.001
27. Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Et Biophys Acta (2014) 1839(8):728–36. doi: 10.1016/j.bbagrm.2014.02.013
28. de Lera AR, Ganesan A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin Epigenet (2016) 8:4–9. doi: 10.1186/s13148-016-0271-9
29. Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene (2007) 26(37):5528–40. doi: 10.1038/sj.onc.1210619
30. Dancy BM, Cole PA. Protein Lysine Acetylation by p300/CBP. Chem Rev (2015) 115(6):2419–52. doi: 10.1021/cr500452k
31. Zhang Y, Fang H, Jiao J, Xu W. The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr Medicinal Chem (2008) 15(27):2840–9. doi: 10.2174/092986708786242796
32. Zhu P, Martin E, Mengwasser J, Schlag P, Janssen K-P, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell (2004) 5(5):455–63. doi: 10.1016/s1535-6108(04)00114-x
33. Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet (2006) 38(5):566–9. doi: 10.1038/ng1773
34. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell (2012) 22(1):9–20. doi: 10.1016/j.ccr.2012.06.008
35. Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet (2005) 37(4):391–400. doi: 10.1038/ng1531
36. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis (2010) 31(1):27–36. doi: 10.1093/carcin/bgp220
37. Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, et al. Methylation of H3-Lysine 79 Is Mediated by a New Family of HMTases without a SET Domain. Curr Biol (2002) 12(12):1052–8. doi: 10.1016/S0960-9822(02)00901-6
38. Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev (2011) 25(13):1345–58. doi: 10.1101/gad.2057811
39. Karlić R, Chung H-R, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A (2010) 107(7):2926–31. doi: 10.1073/pnas.0909344107
40. Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell (2007) 25(1):1–14. doi: 10.1016/j.molcel.2006.12.010
41. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature (2010) 463(7279):360–3. doi: 10.1038/nature08672
42. Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol (2004) 6(8):731–40. doi: 10.1038/ncb1151
43. Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res: Off J Japan Soc Hepatol (2007) 37(11):974–83. doi: 10.1111/j.1872-034X.2007.00141.x
44. Füllgrabe J, Kavanagh E, Joseph B. Histone onco-modifications. Oncogene (2011) 30(31):3391–403. doi: 10.1038/onc.2011.121
45. Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet (2019) 20(2):109–27. doi: 10.1038/s41576-018-0074-2
46. Lv J-F, Hu L, Zhuo W, Zhang C-M, Zhou H-H, Fan L. Epigenetic alternations and cancer chemotherapy response. Cancer Chemother Pharmacol (2016) 77(4):673–84. doi: 10.1007/s00280-015-2951-0
47. Evans WE, Johnson JA. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet (2001) 2:9–39. doi: 10.1146/annurev.genom.2.1.9
48. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell (2010) 141(1):69–80. doi: 10.1016/j.cell.2010.02.027
49. El-Khoury V, Breuzard G, Fourré N, Dufer J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br J Cancer (2007) 97(4):562–73. doi: 10.1038/sj.bjc.6603914
50. Kumar S, Kushwaha PP, Gupta S. Emerging targets in cancer drug resistance. Cancer Drug Resist (2019) 2(2):161–77. doi: 10.20517/cdr.2018.27
51. Salarinia R, Sahebkar A, Peyvandi M, Reza Mirzaei H, Reza Jaafari M, Matbou Riahi M, et al. Epi-Drugs and Epi-miRs: Moving Beyond Current Cancer Therapies. Curr Cancer Drug Targets (2016) 16(9):773–88. doi: 10.2174/1568009616666151207110143
52. Lauschke VM, Barragan I, Ingelman-Sundberg M. Pharmacoepigenetics and Toxicoepigenetics: Novel Mechanistic Insights and Therapeutic Opportunities. Annu Rev Pharmacol Toxicol (2018) 58(1):161–85. doi: 10.1146/annurev-pharmtox-010617-053021
53. Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer (2014) 14(11):747–53. doi: 10.1038/nrc3819
54. Kumar R, Harilal S, Gupta SV, Jose J, Thomas Parambi DG, Uddin M, et al. Exploring the new horizons of drug repurposing: A vital tool for turning hard work into smart work. Eur J Medicinal Chem (2019) 182:111602. doi: 10.1016/j.ejmech.2019.111602
55. Raynal NJ-M, Da Costa EM, Lee JT, Gharibyan V, Ahmed S, Zhang H, et al. Repositioning FDA-approved drugs in combination with epigenetic drugs to reprogram colon cancer epigenome. Mol Cancer Ther (2017) 16(2):397–407. doi: 10.1158/1535-7163.MCT-16-0588
56. Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol (2012) 6(6):657–82. doi: 10.1016/j.molonc.2012.09.004
57. Cartron P-F, Cheray M, Bretaudeau L. Epigenetic protein complexes: the adequate candidates for the use of a new generation of epidrugs in personalized and precision medicine in cancer. Epigenomics (2019) 12(2):171–7. doi: 10.2217/epi-2019-0169
58. Bennett RL, Licht JD. Targeting Epigenetics in Cancer. Annu Rev Pharmacol Toxicol (2018) 58:187–207. doi: 10.1146/annurev-pharmtox-010716-105106
59. Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol (2020) 17(2):91–107. doi: 10.1038/s41571-019-0267-4
60. Erdmann A, Halby L, Fahy J, Arimondo PB. Targeting DNA methylation with small molecules: what’s next? J Medicinal Chem (2015) 58(6):2569–83. doi: 10.1021/jm500843d
61. Leone G, Teofili L, Voso MT, Lübbert M. DNA methylation and demethylating drugs in myelodysplastic syndromes and secondary leukemias. Haematologica (2002) 87(12):1324–41.
62. Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res (2017) 45(D1):D945–54. doi: 10.1093/nar/gkw1074
63. Sébert M, Renneville A, Bally C, Peterlin P, Beyne-Rauzy O, Legros L, et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure. Haematologica (2019) 104(8):1565–71. doi: 10.3324/haematol.2018.207118
64. Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol (2013) 10(5):256–66. doi: 10.1038/nrclinonc.2013.42
65. Zhang J, Zheng YG. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem Biol (2016) 11(3):583–97. doi: 10.1021/acschembio.5b00812
66. Asgatay S, Champion C, Marloie G, Drujon T, Senamaud-Beaufort C, Ceccaldi A, et al. Synthesis and evaluation of analogues of N-phthaloyl-l-tryptophan (RG108) as inhibitors of DNA methyltransferase 1. J Medicinal Chem (2014) 57(2):421–34. doi: 10.1021/jm401419p
67. Candelaria M, de la Cruz-Hernandez E, Taja-Chayeb L, Perez-Cardenas E, Trejo-Becerril C, Gonzalez-Fierro A, et al. DNA Methylation-Independent Reversion of Gemcitabine Resistance by Hydralazine in Cervical Cancer Cells. PloS One (2012) 7(3):e29181. doi: 10.1371/journal.pone.0029181
68. Ceccaldi A, Rajavelu A, Champion C, Rampon C, Jurkowska R, Jankevicius G, et al. C5-DNA Methyltransferase Inhibitors: From Screening to Effects on Zebrafish Embryo Development. ChemBioChem (2011) 12(9):1337–45. doi: 10.1002/cbic.201100130
69. Davis AJ, Gelmon KA, Siu LL, Moore MJ, Britten CD, Mistry N, et al. and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Invest New Drugs (2003) 21(1):85–97. doi: 10.1023/a:1022976528441
70. Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate (2011) 71(4):333–43. doi: 10.1002/pros.21247
71. Lu Y, Chan Y-T, Tan H-Y, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer (2020) 19:6–8. doi: 10.1186/s12943-020-01197-3
72. Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci (2010) 31(11):536–46. doi: 10.1016/j.tips.2010.08.001
73. Archin NM, Kirchherr JL, Sung JAM, Clutton G, Sholtis K, Xu Y, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Invest (2020) 127(8):3126–35. doi: 10.1172/JCI92684
74. Jiang C, Lian X, Gao C, Sun X, Einkauf KB, Chevalier JM, et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature (2020) 585:1–7. doi: 10.1038/s41586-020-2651-8
75. Connolly RM, Li H, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC, et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin Cancer Res: Off J Am Assoc Cancer Res (2017) 23(11):2691–701. doi: 10.1158/1078-0432.CCR-16-1729
76. Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discovery (2011) 1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214
77. Mai A, Altucci L. Epi-drugs to fight cancer: from chemistry to cancer treatment, the road ahead. Int J Biochem Cell Biol (2009) 41(1):199–213. doi: 10.1016/j.biocel.2008.08.020
78. Pontiki E, Hadjipavlou-Litina D. Histone deacetylase inhibitors (HDACIs). Structure–activity relationships: history and new QSAR perspectives. Medicinal Res Rev (2012) 32(1):1–165. doi: 10.1002/med.20200
79. Jaboin J, Wild J, Hamidi H, Khanna C, Kim CJ, Robey R, et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res (2002) 62(21):6108–15.
80. Prachayasittikul V, Prathipati P, Pratiwi R, Phanus-Umporn C, Malik AA, Schaduangrat N, et al. Exploring the epigenetic drug discovery landscape. Expert Opin Drug Discovery (2017) 12(4):345–62. doi: 10.1080/17460441.2017.1295954
81. Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol: Off J Am Soc Clin Oncol (2013) 31(17):2128–35. doi: 10.1200/JCO.2012.43.7251
82. Tomaselli D, Lucidi A, Rotili D, Mai A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Medicinal Res Rev (2020) 40(1):190–244. doi: 10.1002/med.21600
83. Mahajan SS, Leko V, Simon JA, Bedalov A. Sirtuin modulators. Handb Exp Pharmacol (2011) 206:241–55. doi: 10.1007/978-3-642-21631-2_11
84. Copeland RA, Moyer MP, Richon VM. Targeting genetic alterations in protein methyltransferases for personalized cancer therapeutics. Oncogene (2013) 32(8):939–46. doi: 10.1038/onc.2012.552
85. Han D, Huang M, Wang T, Li Z, Chen Y, Liu C, et al. Lysine methylation of transcription factors in cancer. Cell Death Dis (2019) 10(4):1–11. doi: 10.1038/s41419-019-1524-2
86. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet (2010) 42(2):181–5. doi: 10.1038/ng.518
87. Sermer D, Pasqualucci L, Wendel H-G, Melnick A, Younes A. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol (2019) 16(8):494–507. doi: 10.1038/s41571-019-0190-8
88. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci (2010) 107(49):20980–5. doi: 10.1073/pnas.1012525107
89. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell (2013) 23(5):677–92. doi: 10.1016/j.ccr.2013.04.011
90. Li X, Wang C, Jiang H, Luo C. A patent review of arginine methyltransferase inhibitors (2010-2018). Expert Opin Ther Patents (2019) 29(2):97–114. doi: 10.1080/13543776.2019.1567711
91. Rose NR, Ng SS, Mecinović J, Liénard BMR, Bello SH, Sun Z, et al. Inhibitor Scaffolds for 2-Oxoglutarate-Dependent Histone Lysine Demethylases. J Medicinal Chem (2008) 51(22):7053–6. doi: 10.1021/jm800936s
92. Joberty G, Boesche M, Brown JA, Eberhard D, Garton NS, Humphreys PG, et al. Interrogating the Druggability of the 2-Oxoglutarate-Dependent Dioxygenase Target Class by Chemical Proteomics. ACS Chem Biol (2016) 11(7):2002–10. doi: 10.1021/acschembio.6b00080
93. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell (2004) 119(7):941–53. doi: 10.1016/j.cell.2004.12.012
94. Niebel D, Kirfel J, Janzen V, Höller T, Majores M, Gütgemann I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood (2014) 124(1):151–2. doi: 10.1182/blood-2014-04-569525
95. Yang G-J, Lei P-M, Wong S-Y, Ma D-L, Leung C-H. Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules (2018) 23(12):9–11. doi: 10.3390/molecules23123194
96. Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu Rev Neurosci (1999) 22:197–217. doi: 10.1146/annurev.neuro.22.1.197
97. Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, et al. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry (2007) 46(27):8058–65. doi: 10.1021/bi700664y
98. Zheng Y-C, Yu B, Chen Z-S, Liu Y, Liu H-M. TCPs: privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics (2016) 8(5):651–66. doi: 10.2217/epi-2015-0002
99. Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer (2017) 17(4):268. doi: 10.1038/nrc.2017.26
100. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol: Off J Eur Soc Med Oncol (2017) 28(8):1776–87. doi: 10.1093/annonc/mdx157
101. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature (2011) 478(7370):524–8. doi: 10.1038/nature10334
102. Morel D, Almouzni G, Soria J-C, Postel-Vinay S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann Oncol: Off J Eur Soc Med Oncol (2017) 28(2):254–69. doi: 10.1093/annonc/mdw552
103. Winkler J, Raina K, Altieri M, Dong H, Wang J, Chen X, et al. PROTAC BET degraders are more broadly effective than BET inhibitors. Eur J Cancer (2016) 69:S10. doi: 10.1016/S0959-8049(16)32621-1
104. Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, et al. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discovery (2010) 9(11):843–56. doi: 10.1038/nrd3216
105. Li YY, Jones SJ. Drug repositioning for personalized medicine. Genome Med (2012) 4(3):27. doi: 10.1186/gm326
106. Hopkins AL. Drug discovery: Predicting promiscuity. Nature (2009) 462(7270):167–8. doi: 10.1038/462167a
107. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discovery (2004) 3(8):673–83. doi: 10.1038/nrd1468
108. Pantziarka P. Scientific advice - is drug repurposing missing a trick? Nature Reviews. Clin Oncol (2017) 14(8):455–6. doi: 10.1038/nrclinonc.2017.69
109. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discovery (2019) 18(1):41–58. doi: 10.1038/nrd.2018.168
110. YCharts. Pfizer’s Expiring Viagra Patent Adversely Affects Other Drugmakers Too. Jersey City, NJ: Forbes (2020).
111. Urquhart L. Market watch: Top drugs and companies by sales in 2017. Nat Rev Drug Discovery (2018) 17(4):232. doi: 10.1038/nrd.2018.42
112. Tartaglia LA. Complementary new approaches enable repositioning of failed drug candidates. Expert Opin Invest Drugs (2006) 15(11):1295–8. doi: 10.1517/13543784.15.11.1295
113. Brehmer D, Greff Z, Godl K, Blencke S, Kurtenbach A, Weber M, et al. Cellular Targets of Gefitinib. Cancer Res (2005) 65(2):379–82.
114. Moffat JG, Vincent F, Lee JA, Eder J, Prunotto M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat Rev Drug Discovery (2017) 16(8):531–43. doi: 10.1038/nrd.2017.111
115. Chong CR, Xu J, Lu J, Bhat S, Sullivan DJ, Liu JO. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol (2007) 2(4):263–70. doi: 10.1021/cb600362d
116. Sundberg SA. High-throughput and ultra-high-throughput screening: solution- and cell-based approaches. Curr Opin Biotechnol (2000) 11(1):47–53. doi: 10.1016/s0958-1669(99)00051-8
117. Reaume AG. Drug repurposing through nonhypothesis driven phenotypic screening. Drug Discovery Today: Ther Strategies (2011) 8(3–4):85–8. doi: 10.1016/j.ddstr.2011.09.007
118. Hurle MR, Yang L, Xie Q, Rajpal DK, Sanseau P, Agarwal P. Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther (2013) 93(4):335–41. doi: 10.1038/clpt.2013.1
119. Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell (2006) 10(4):321–30. doi: 10.1016/j.ccr.2006.09.005
120. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discovery (2004) 3(11):935–49. doi: 10.1038/nrd1549
121. Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, Cardon LR, et al. Use of genome-wide association studies for drug repositioning. Nat Biotechnol (2012) 30(4):317–20. doi: 10.1038/nbt.2151
122. Greene CS, Voight BF. Pathway and network-based strategies to translate genetic discoveries into effective therapies. Hum Mol Genet (2016) 25(R2):R94–8. doi: 10.1093/hmg/ddw160
123. Wei W-Q, Denny JC. Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med (2015) 7(1):2–8. doi: 10.1186/s13073-015-0166-y
124. Wicks P, Vaughan TE, Massagli MP, Heywood J. Accelerated clinical discovery using self-reported patient data collected online and a patient-matching algorithm. Nat Biotechnol (2011) 29(5):411–4. doi: 10.1038/nbt.1837
125. Medina-Franco JL, Yoo J, Dueñas-González A. DNA Methyltransferase Inhibitors for Cancer Therapy. In: Epigenetic Technological Applications. Elsevier (2015). p. 265–90. doi: 10.1016/B978-0-12-801080-8.00013-2
126. Carrella D, Napolitano F, Rispoli R, Miglietta M, Carissimo A, Cutillo L, et al. Mantra 2.0: an online collaborative resource for drug mode of action and repurposing by network analysis. Bioinf (Oxford England) (2014) 30(12):1787–8. doi: 10.1093/bioinformatics/btu058
127. Chang RL, Xie L, Xie L, Bourne PE, Palsson BØ. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PloS Comput Biol (2010) 6(9):e1000938. doi: 10.1371/journal.pcbi.1000938
128. Knapp S, Weinmann H. Small-Molecule Modulators for Epigenetics Targets. ChemMedChem (2013) 8(11):1885–91. doi: 10.1002/cmdc.201300344
129. Wang Y, Bryant SH, Cheng T, Wang J, Gindulyte A, Shoemaker BA, et al. PubChem BioAssay: 2017 update. Nucleic Acids Res (2017) 45(D1):D955–63. doi: 10.1093/nar/gkw1118
130. Ursu O, Holmes J, Bologa CG, Yang JJ, Mathias SL, Stathias V, et al. DrugCentral 2018: an update. Nucleic Acids Res (2019) 47(D1):D963–70. doi: 10.1093/nar/gky963
131. Tanoli Z, Alam Z, Vähä-Koskela M, Ravikumar B, Malyutina A, Jaiswal A, et al. Drug Target Commons 2.0: a community platform for systematic analysis of drug-target interaction profiles. Database: J Biol Database Curation (2018) 2018:1–13. doi: 10.1093/database/bay083
132. Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res (2006) 34(Database issue):D668–672. doi: 10.1093/nar/gkj067
133. Qi Y, Wang D, Wang D, Jin T, Yang L, Wu H, et al. HEDD: the human epigenetic drug database(2016). (Accessed Accessed: 10th October 2020). doi: 10.1093/database/baw159
134. Shah SG, Mandloi T, Kunte P, Natu A, Rashid M, Reddy D, et al. HISTome2: a database of histone proteins, modifiers for multiple organisms and epidrugs. Epigenet Chromatin (2020) 13(1):31. doi: 10.1186/s13072-020-00354-8
135. Singh Nanda J, Kumar R, Raghava GPS. dbEM: A database of epigenetic modifiers curated from cancerous and normal genomes. Sci Rep (2016) 6(1):19340. doi: 10.1038/srep19340
136. von Eichborn J, Murgueitio MS, Dunkel M, Koerner S, Bourne PE, Preissner R. PROMISCUOUS: a database for network-based drug-repositioning. Nucleic Acids Res (2011) 39(Database issue):D1060–6. doi: 10.1093/nar/gkq1037
137. Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med (2017) 23(4):405–8. doi: 10.1038/nm.4306
138. Shameer K, Glicksberg BS, Hodos R, Johnson KW, Badgeley MA, Readhead B, et al. Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Briefings Bioinf (2018) 19(4):656–78. doi: 10.1093/bib/bbw136
139. Brown AS, Patel CJ. A standard database for drug repositioning. Sci Data (2017) 4(1):170029. doi: 10.1038/sdata.2017.29
140. Himmelstein DS, Lizee A, Hessler C, Brueggeman L, Chen SL, Hadley D, et al. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife (2017) 6:e26726. doi: 10.7554/eLife.26726
141. Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP. The Repurposing Drugs in Oncology (ReDO) Project (Accessed 10th October 2020).
142. Tanoli Z, Seemab U, Scherer A, Wennerberg K, Tang J, Vähä-Koskela M. Exploration of databases and methods supporting drug repurposing: a comprehensive survey. Briefings Bioinf (2020) 1–23. doi: 10.1093/bib/bbaa003
143. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol (2014) 32(1):40–51. doi: 10.1038/nbt.2786
144. Chen S, Wang Z, Huang Y, O’Barr SA, Wong RA, Yeung S, et al. Ginseng and Anticancer Drug Combination to Improve Cancer Chemotherapy: A Critical Review. Evidence-Based Complement Altern Med (2014) 2014:1–13. doi: 10.1155/2014/168940
145. Lötsch J, Schneider G, Reker D, Parnham MJ, Schneider P, Geisslinger G, et al. Common non-epigenetic drugs as epigenetic modulators. Trends Mol Med (2013) 19(12):742–53. doi: 10.1016/j.molmed.2013.08.006
146. Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene (2002) 21(35):5483–95. doi: 10.1038/sj.onc.1205699
147. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol (2009) 10(3):223–32. doi: 10.1016/S1470-2045(09)70003-8
148. Kantarjian H, Issa J-PJ, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer (2006) 106(8):1794–803. doi: 10.1002/cncr.21792
149. Oodi A, Norouzi H, Amirizadeh N, Nikougoftar M, Vafaie Z. Harmine, a Novel DNA Methyltransferase 1 Inhibitor in the Leukemia Cell Line. Indian J Hematol Blood Transfusion: Off J Indian Soc Hematol Blood Transfusion (2017) 33(4):509–15. doi: 10.1007/s12288-016-0770-z
150. Lee WJ, Zhu BT. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis (2006) 27(2):269–77. doi: 10.1093/carcin/bgi206
151. Fagan RL, Cryderman DE, Kopelovich L, Wallrath LL, Brenner C. Laccaic Acid A Is a Direct, DNA-competitive Inhibitor of DNA Methyltransferase 1. J Biol Chem (2013) 288(33):23858–67. doi: 10.1074/jbc.M113.480517
152. Li Y-C, Wang Y, Li D-D, Zhang Y, Zhao T-C, Li C-F. Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J Cell Biochem (2018) 119(2):2440–9. doi: 10.1002/jcb.26407
153. Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res (2003) 63(16):4984–9.
154. Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide Is a Specific Inhibitor of DNA Methyltransferase 1. J Biol Chem (2005) 280(49):40749–56. doi: 10.1074/jbc.M505593200
155. Agarwal S, Amin KS, Jagadeesh S, Baishay G, Rao PG, Barua NC, et al. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol Cancer (2013) 12(1):99. doi: 10.1186/1476-4598-12-99
156. Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum (2003) 48(3):746–56. doi: 10.1002/art.10833
157. Segura-Pacheco B, Perez-Cardenas E, Taja-Chayeb L, Chavez-Blanco A, Revilla-Vazquez A, Benitez-Bribiesca L, et al. Global DNA hypermethylation-associated cancer chemotherapy resistance and its reversion with the demethylating agent hydralazine. J Trans Med (2006) 4(1):32. doi: 10.1186/1479-5876-4-32
158. Song Y, Zhang C. Hydralazine inhibits human cervical cancer cell growth in vitro in association with APC demethylation and re-expression. Cancer Chemother Pharmacol (2009) 63(4):605–13. doi: 10.1007/s00280-008-0773-z
159. Candelaria M, Herrera A, Labardini J, González-Fierro A, Trejo-Becerril C, Taja-Chayeb L, et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann Hematol (2011) 90(4):379–87. doi: 10.1007/s00277-010-1090-2
160. Soto H, Sanchez K, Escobar JY, Constanzo A, Fernandez Z, Melendez C. Cost-Effectiveness Analysis of Hydralazine and Magnesium Valproate LP Associated With Treatment for Adult Patients with Metastatic Recurrent or Persistent Cervical Cancer in Mexico. Value Health: J Int Soc Pharmacoeconom Outcomes Res (2014) 17(7):A639. doi: 10.1016/j.jval.2014.08.2300
161. Cervera E, Candelaria M, López-Navarro O, Labardini J, Gonzalez-Fierro A, Taja-Chayeb L, et al. Epigenetic Therapy With Hydralazine and Magnesium Valproate Reverses Imatinib Resistance in Patients With Chronic Myeloid Leukemia. Clin Lymphoma Myeloma Leukemia (2012) 12(3):207–12. doi: 10.1016/j.clml.2012.01.005
162. Méndez-Lucio O, Tran J, Medina-Franco JL, Meurice N, Muller M. Toward Drug Repurposing in Epigenetics: Olsalazine as a Hypomethylating Compound Active in a Cellular Context. ChemMedChem (2014) 9(3):560–5. doi: 10.1002/cmdc.201300555
163. Lin R-K, Hsu C-H, Wang Y-C. Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anti Cancer Drugs (2007) 18(10):1157–64. doi: 10.1097/CAD.0b013e3282a215e9
164. Kuck D, Caulfield T, Lyko F, Medina-Franco JL. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther (2010) 9(11):3015–23. doi: 10.1158/1535-7163.MCT-10-0609
165. Kalra G, De Sousa A, Shrivastava A. Disulfiram in the management of alcohol dependence: A comprehensive clinical review. Open J Psychiatry (2014) 4(1):720–6. doi: 10.4236/ojpsych.2014.41007
166. Arce C, Segura-Pacheco B, Perez-Cardenas E, Taja-Chayeb L, Candelaria M, Dueñnas-Gonzalez A. Hydralazine target: From blood vessels to the epigenome. J Trans Med (2006) 4:10. doi: 10.1186/1479-5876-4-10
167. Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res: Off J Am Assoc Cancer Res (2003) 9(5):1596–603.
168. Lin X, Asgari K, Putzi MJ, Gage WR, Yu X, Cornblatt BS, et al. Reversal of GSTP1 CpG island hypermethylation and reactivation of pi-class glutathione S-transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide. Cancer Res (2001) 61(24):8611–6.
169. Gao Z, Xu Z, Hung M-S, Lin Y-C, Wang T, Gong M, et al. Procaine and procainamide inhibit the Wnt canonical pathway by promoter demethylation of WIF-1 in lung cancer cells. Oncol Rep (2009) 22(6):1479–84. doi: 10.3892/or_00000590
170. Tada M, Imazeki F, Fukai K, Sakamoto A, Arai M, Mikata R, et al. Procaine inhibits the proliferation and DNA methylation in human hepatoma cells. Hepatol Int (2007) 1(3):355–64. doi: 10.1007/s12072-007-9014-5
171. Jagadeesh S, Sinha S, Pal BC, Bhattacharya S, Banerjee PP. Mahanine reverses an epigenetically silenced tumor suppressor gene RASSF1A in human prostate cancer cells. Biochem Biophys Res Commun (2007) 362(1):212–7. doi: 10.1016/j.bbrc.2007.08.005
172. Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res: Off J Am Assoc Cancer Res (2005) 11(19 Pt 1):7033–41. doi: 10.1158/1078-0432.CCR-05-0406
173. Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, et al. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis (2009) 30(4):662–70. doi: 10.1093/carcin/bgp042
174. Hodgson N, Trivedi M, Muratore C, Li S, Deth R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J Alzheimer’s Disease: JAD (2013) 36(1):197–209. doi: 10.3233/JAD-130101
175. Trivedi MS, Shah JS, Al-Mughairy S, Hodgson NW, Simms B, Trooskens GA, et al. Food-derived opioid peptides inhibit cysteine uptake with redox and epigenetic consequences. J Nutr Biochem (2014) 25(10):1011–8. doi: 10.1016/j.jnutbio.2014.05.004
176. Trivedi MS, Hodgson NW, Walker SJ, Trooskens G, Nair V, Deth RC. Epigenetic effects of casein-derived opioid peptides in SH-SY5Y human neuroblastoma cells. Nutr Metab (2015) 12(1):54. doi: 10.1186/s12986-015-0050-1
177. Gopal YNV, Arora TS, Van Dyke MW. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem Biol (2007) 14(7):813–23. doi: 10.1016/j.chembiol.2007.06.007
178. Dawood M, Ooko E, Efferth T. Collateral Sensitivity of Parthenolide via NF-κB and HIF-α Inhibition and Epigenetic Changes in Drug-Resistant Cancer Cell Lines. Front Pharmacol (2019) 10:542. doi: 10.3389/fphar.2019.00542
179. Hartman ML, Talar B, Sztiller-Sikorska M, Nejc D, Czyz M. Parthenolide induces MITF-M downregulation and senescence in patient-derived MITF-Mhigh melanoma cell populations. Oncotarget (2016) 7(8):9026–40. doi: 10.18632/oncotarget.7030
180. Koprowska K, Czyz M. [Molecular mechanisms of parthenolide’s action: Old drug with a new face]. Postepy Higieny I Medycyny Doswiadczalnej (2010) 64:100–14.
181. Liu Z, Liu S, Xie Z, Pavlovicz RE, Wu J, Chen P, et al. Modulation of DNA Methylation by a Sesquiterpene Lactone Parthenolide. J Pharmacol Exp Ther (2009) 329(2):505–14. doi: 10.1124/jpet.108.147934
182. Izquierdo-Torres E, Hernández-Oliveras A, Meneses-Morales I, Rodríguez G, Fuentes-García G, Zarain-Herzberg Á. Resveratrol up-regulates ATP2A3 gene expression in breast cancer cell lines through epigenetic mechanisms. Int J Biochem Cell Biol (2019) 113:37–47. doi: 10.1016/j.biocel.2019.05.020
183. Chatterjee B, Ghosh K, Kanade SR. Resveratrol modulates epigenetic regulators of promoter histone methylation and acetylation that restores BRCA1, p53, p21CIP1 in human breast cancer cell lines. BioFactors (Oxford England) (2019) 45(5):818–29. doi: 10.1002/biof.1544
184. Venturelli S, Berger A, Böcker A, Busch C, Weiland T, Noor S, et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PloS One (2013) 8(8):e73097. doi: 10.1371/journal.pone.0073097
185. Liu X, Li H, Wu M-L, Wu J, Sun Y, Zhang K-L, et al. Resveratrol Reverses Retinoic Acid Resistance of Anaplastic Thyroid Cancer Cells via Demethylating CRABP2 Gene. Front Endocrinol (2019) 10:734. doi: 10.3389/fendo.2019.00734
186. Lee WJ, Shim J-Y, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol (2005) 68(4):1018–30. doi: 10.1124/mol.104.008367
187. Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res (2003) 63(22):7563–70.
188. Nandakumar V, Vaid M, Katiyar SK. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis (2011) 32(4):537–44. doi: 10.1093/carcin/bgq285
189. Chen K-L, Wang SS-S, Yang Y-Y, Yuan R-Y, Chen R-M, Hu C-J. The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Commun (2009) 378(1):57–61. doi: 10.1016/j.bbrc.2008.10.173
190. Yuan Z, Chen S, Gao C, Dai Q, Zhang C, Sun Q, et al. Development of a versatile DNMT and HDAC inhibitor C02S modulating multiple cancer hallmarks for breast cancer therapy. Bioorg Chem (2019) 87:200–8. doi: 10.1016/j.bioorg.2019.03.027
191. Cicero AFG, Baggioni A. Berberine and Its Role in Chronic Disease. Adv Exp Med Biol (2016) 928:27–45. doi: 10.1007/978-3-319-41334-1_2
192. Wang Z, Liu Y, Xue Y, Hu H, Ye J, Li X, et al. Berberine acts as a putative epigenetic modulator by affecting the histone code. Toxicol Vitro (2016) 36:10–7. doi: 10.1016/j.tiv.2016.06.004
193. Qing Y, Hu H, Liu Y, Feng T, Meng W, Jiang L, et al. Berberine induces apoptosis in human multiple myeloma cell line U266 through hypomethylation of p53 promoter. Cell Biol Int (2014) 38(5):563–70. doi: 10.1002/cbin.10206
194. Kalaiarasi A, Anusha C, Sankar R, Rajasekaran S, John Marshal J, Muthusamy K, et al. Plant Isoquinoline Alkaloid Berberine Exhibits Chromatin Remodeling by Modulation of Histone Deacetylase To Induce Growth Arrest and Apoptosis in the A549 Cell Line. J Agric Food Chem (2016) 64(50):9542–50. doi: 10.1021/acs.jafc.6b04453
195. Tipoe GL, Leung T-M, Hung M-W, Fung M-L. Green tea polyphenols as an anti-oxidant and anti-inflammatory agent for cardiovascular protection. Cardiovasc Hematol Disord Drug Targets (2007) 7(2):135–44. doi: 10.2174/187152907780830905
196. Fernandes GFS, Silva GDB, Pavan AR, Chiba DE, Chin CM, Dos Santos JL. Epigenetic Regulatory Mechanisms Induced by Resveratrol. Nutrients (2017) 9(11):1–2. doi: 10.3390/nu9111201
197. Daud AI, Dawson J, DeConti RC, Bicaku E, Marchion D, Bastien S, et al. Potentiation of a Topoisomerase I Inhibitor, Karenitecin, by the Histone Deacetylase Inhibitor Valproic Acid in Melanoma: Translational and Phase I/II Clinical Trial. Clin Cancer Res (2009) 15(7):2479–87. doi: 10.1158/1078-0432.CCR-08-1931
198. Rocca A, Minucci S, Tosti G, Croci D, Contegno F, Ballarini M, et al. A phase I–II study of the histone deacetylase inhibitor valproic acid plus chemoimmunotherapy in patients with advanced melanoma. Br J Cancer (2009) 100(1):28–36. doi: 10.1038/sj.bjc.6604817
199. Patel MM, Patel BM. Repurposing of sodium valproate in colon cancer associated with diabetes mellitus: Role of HDAC inhibition. Eur J Pharm Sci: Off J Eur Fed Pharm Sci (2018) 121:188–99. doi: 10.1016/j.ejps.2018.05.026
200. Du X, Li Q, Du F, He Z, Wang J. Sodium Valproate Sensitizes Non-Small Lung Cancer A549 Cells to γδ T-Cell-Mediated Killing through Upregulating the Expression of MICA. J Biochem Mol Toxicol (2013) 27(11):492–8. doi: 10.1002/jbt.21513
201. Friedmann I, Atmaca A, Chow KU, Jäger E, Weidmann E. Synergistic Effects of Valproic Acid and Mitomycin C in Adenocarcinoma Cell Lines and Fresh Tumor Cells of Patients with Colon Cancer. J Chemother (2006) 18(4):415–20. doi: 10.1179/joc.2006.18.4.415
202. Yan H-C, Zhang J. Effects of sodium valproate on the growth of human ovarian cancer cell line HO8910. Asian Pacific J Cancer Prevent: APJCP (2012) 13(12):6429–33. doi: 10.7314/apjcp.2012.13.12.6429
203. Kumari K, Keshari S, Sengupta D, Sabat SC, Mishra SK. Transcriptome analysis of genes associated with breast cancer cell motility in response to Artemisinin treatment. BMC Cancer (2017) 17(1):858. doi: 10.1186/s12885-017-3863-7
204. Byun MR, Lee DH, Jang YP, Lee HS, Choi JW, Lee SK. Repurposing natural products as novel HDAC inhibitors by comparative analysis of gene expression profiles. Phytomedicine (2019) 59:152900. doi: 10.1016/j.phymed.2019.152900
205. Joung KE, Kim D-K, Sheen YY. Antiproliferative effect of trichostatin a and hc-toxin in T47D Human breast cancer cells. Arch Pharmacal Res (2004) 27(6):640–5. doi: 10.1007/BF02980164
206. Deubzer HE, Ehemann V, Westermann F, Heinrich R, Mechtersheimer G, Kulozik AE, et al. Histone deacetylase inhibitor Helminthosporium carbonum (HC)-toxin suppresses the malignant phenotype of neuroblastoma cells. Int J Cancer (2008) 122(8):1891–900. doi: 10.1002/ijc.23295
207. Park Y, Liu Y, Hong J, Lee C-O, Cho H, Kim D-K, et al. New bromotyrosine derivatives from an association of two sponges, Jaspis wondoensis and Poecillastra wondoensis. J Natural Products (2003) 66(11):1495–8. doi: 10.1021/np030162j
208. Kim DH, Shin J, Kwon HJ. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp Mol Med (2007) 39(1):47–55. doi: 10.1038/emm.2007.6
209. Ahn MY, Jung JH, Na YJ, Kim HS. A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol Oncol (2008) 108(1):27–33. doi: 10.1016/j.ygyno.2007.08.098
210. Kim D, Lee IS, Jung JH, Lee CO, Choi SU. Psammaplin A, a natural phenolic compound, has inhibitory effect on human topoisomerase II and is cytotoxic to cancer cells. Anticancer Res (1999) 19(5B):4085–90.
211. Shin H, Lee YS, Lee YC. Sodium butyrate-induced DAPK-mediated apoptosis in human gastric cancer cells. Oncol Rep (2012) 27(4):1111–5. doi: 10.3892/or.2011.1585
212. Li L, Sun Y, Liu J, Wu X, Chen L, Ma L, et al. Histone deacetylase inhibitor sodium butyrate suppresses DNA double strand break repair induced by etoposide more effectively in MCF-7 cells than in HEK293 cells. BMC Biochem (2015) 16:3–8. doi: 10.1186/s12858-014-0030-5
213. Cang S, Xu X, Ma Y, Liu D, Chiao JW. Hypoacetylation, hypomethylation, and dephosphorylation of H2B histones and excessive histone deacetylase activity in DU-145 prostate cancer cells. J Hematol Oncol (2016) 9:2–3. doi: 10.1186/s13045-016-0233-x
214. Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res: Off J Am Assoc Cancer Res (2001) 7(4):971–6.
215. Chambers AE, Banerjee S, Chaplin T, Dunne J, Debernardi S, Joel SP, et al. Histone acetylation-mediated regulation of genes in leukaemic cells. Eur J Cancer (Oxford England: 1990) (2003) 39(8):1165–75. doi: 10.1016/s0959-8049(03)00072-8
216. Ma J, Guo X, Zhang S, Liu H, Lu J, Dong Z, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and promotes apoptosis of esophageal squamous cell lines. Mol Med Rep (2015) 11(6):4525–31. doi: 10.3892/mmr.2015.3268
217. Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, et al. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int J Oncol (2011) 39(1):111–9. doi: 10.3892/ijo.2011.1014
218. Zhang H, Zhao X, Liu H, Jin H, Ji Y. Trichostatin A inhibits proliferation of PC3 prostate cancer cells by disrupting the EGFR pathway. Oncol Lett (2019) 18(1):687–93. doi: 10.3892/ol.2019.10384
219. Tiffon C. Histone Deacetylase Inhibition Restores Expression of Hypoxia-Inducible Protein NDRG1 in Pancreatic Cancer. Pancreas (2018) 47(2):200–7. doi: 10.1097/MPA.0000000000000982
220. Sanaei M, Kavoosi F. Effect of 5-Aza-2’-Deoxycytidine in Comparison to Valproic Acid and Trichostatin A on Histone Deacetylase 1, DNA Methyltransferase 1, and CIP/KIP Family (p21, p27, and p57) Genes Expression, Cell Growth Inhibition, and Apoptosis Induction in Colon Cancer SW480 Cell Line. Adv Biomed Res (2019) 8:9–15. doi: 10.4103/abr.abr_91_19
221. Hernández-Oliveras A, Izquierdo-Torres E, Meneses-Morales I, Rodríguez G, Zarain-Herzberg Á, Santiago-García J. Histone deacetylase inhibitors promote ATP2A3 gene expression in hepatocellular carcinoma cells: p300 as a transcriptional regulator. Int J Biochem Cell Biol (2019) 113:8–16. doi: 10.1016/j.biocel.2019.05.014
222. San-Miguel JF, Hungria VTM, Yoon S-S, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol (2014) 15(11):1195–206. doi: 10.1016/S1470-2045(14)70440-1
223. Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer (2009) 115(23):5541–9. doi: 10.1002/cncr.24597
224. Watanabe T, Kato H, Kobayashi Y, Yamasaki S, Morita-Hoshi Y, Yokoyama H, et al. Potential efficacy of the oral histone deacetylase inhibitor vorinostat in a phase I trial in follicular and mantle cell lymphoma. Cancer Sci (2010) 101(1):196–200. doi: 10.1111/j.1349-7006.2009.01360.x
225. Raedler LA. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am Health Drug Benefits (2016) 9(Spec Feature):84–7.
226. Fukui Y, Narita K, Dan S, Yamori T, Ito A, Yoshida M, et al. Total synthesis of burkholdacs A and B and 5,6,20-tri-epi-burkholdac A: HDAC inhibition and antiproliferative activity. Eur J Medicinal Chem (2014) 76:301–13. doi: 10.1016/j.ejmech.2014.02.044
227. Benelkebir H, Donlevy AM, Packham G, Ganesan A. Total Synthesis and Stereochemical Assignment of Burkholdac B, a Depsipeptide HDAC Inhibitor. Organic Lett (2011) 13(24):6334–7. doi: 10.1021/ol202197q
228. Crabb SJ, Howell M, Rogers H, Ishfaq M, Yurek-George A, Carey K, et al. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem Pharmacol (2008) 76(4):463–75. doi: 10.1016/j.bcp.2008.06.004
229. Narita K, Fukui Y, Sano Y, Yamori T, Ito A, Yoshida M, et al. Total synthesis of bicyclic depsipeptides spiruchostatins C and D and investigation of their histone deacetylase inhibitory and antiproliferative activities. Eur J Medicinal Chem (2012) 60:295–304. doi: 10.1016/j.ejmech.2012.12.023
230. Hong J. Apicidin, a histone deacetylase inhibitor, induces differentiation of HL-60 cells. Cancer Lett (2003) 189(2):197–206. doi: 10.1016/S0304-3835(02)00500-1
231. Wu L-P, Wang X, Li L, Zhao Y, Lu S, Yu Y, et al. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol Cell Biol (2008) 28(10):3219–35. doi: 10.1128/MCB.01516-07
232. Durczak M, Jagodzinski P. Apicidin upregulates PHD2 prolyl hydroxylase gene expression in cervical cancer cells. Anti Cancer Drugs (2010) 21(6):619–24. doi: 10.1097/CAD.0b013e328339848b
233. Im JY, Park H, Kang KW, Choi WS, Kim HS. Modulation of cell cycles and apoptosis by apicidin in estrogen receptor (ER)-positive and-negative human breast cancer cells. Chemico Biol Interact (2008) 172(3):235–44. doi: 10.1016/j.cbi.2008.01.007
234. Ahn MY, Kang DO, Na YJ, Yoon S, Choi WS, Kang KW, et al. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer Lett (2012) 325(2):189–99. doi: 10.1016/j.canlet.2012.06.017
235. Ahn M-Y. HDAC inhibitor apicidin suppresses murine oral squamous cell carcinoma cell growth in vitro and in vivo via inhibiting HDAC8 expression. Oncol Lett (2018) 16(5):6552–60. doi: 10.3892/ol.2018.9468
236. Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenet (2010) 1(3–4):117–36. doi: 10.1007/s13148-010-0012-4
237. Zhao HL, Harding SV, Marinangeli CPF, Kim YS, Jones PJH. Hypocholesterolemic and anti-obesity effects of saponins from Platycodon grandiflorum in hamsters fed atherogenic diets. J Food Sci (2008) 73(8):H195–200. doi: 10.1111/j.1750-3841.2008.00915.x
238. Brosch G, Ransom R, Lechner T, Walton JD, Loidl P. Inhibition of maize histone deacetylases by HC toxin, the host-selective toxin of Cochliobolus carbonum. Plant Cell (1995) 7(11):1941–50. doi: 10.1105/tpc.7.11.1941
239. Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, et al. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci U S A (1996) 93(23):13143–7. doi: 10.1073/pnas.93.23.13143
240. Liu S, Fu X, Schmitz FJ, Kelly-Borges M. Psammaplysin F, a new bromotyrosine derivative from a sponge, Aplysinella sp. J Natural Products (1997) 60(6):614–5. doi: 10.1021/np970070s
241. Piña IC, Gautschi JT, Wang G-Y-S, Sanders ML, Schmitz FJ, France D, et al. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J Organic Chem (2003) 68(10):3866–73. doi: 10.1021/jo034248t
242. Rinehart KL, Holt TG, Fregeau NL, Keifer PA, Wilson GR, Perun TJ, et al. Bioactive compounds from aquatic and terrestrial sources. J Natural Products (1990) 53(4):771–92. doi: 10.1021/np50070a001
243. Mateos MV, Cibeira M, Richardson PG, Prosper F, Oriol A, de la Rubia J, et al. Phase II Clinical and Pharmacokinetic Study of Plitidepsin 3-Hour Infusion Every Two Weeks Alone or with Dexamethasone in Relapsed and Refractory Multiple Myeloma. Clin Cancer Res: Official J Am Assoc Cancer Res (2010) 16(12):3260–9. doi: 10.1158/1078-0432.CCR-10-0469
244. Spicka I, Ocio EM, Oakervee HE, Greil R, Banh RH, Huang S-Y, et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann Hematol (2019) 98(9):2139–50. doi: 10.1007/s00277-019-03739-2
245. Alonso-Álvarez S, Pardal E, Sánchez-Nieto D, Navarro M, Caballero MD, Mateos MV, et al. Plitidepsin: design, development, and potential place in therapy. Drug Design Dev Ther (2017) 11:253–64. doi: 10.2147/DDDT.S94165
246. Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small Molecule Modulators of Histone Acetyltransferase p300. J Biol Chem (2003) 278(21):19134–40. doi: 10.1074/jbc.M301580200
247. Collins HM, Abdelghany MK, Messmer M, Yue B, Deeves SE, Kindle KB, et al. Differential effects of garcinol and curcumin on histone and p53 modifications in tumour cells. BMC Cancer (2013) 13(1):37. doi: 10.1186/1471-2407-13-37
248. Sung B, Pandey MK, Ahn KS, Yi T, Chaturvedi MM, Liu M, et al. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood (2008) 111(10):4880–91. doi: 10.1182/blood-2007-10-117994
249. Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, et al. Polyisoprenylated Benzophenone, Garcinol, a Natural Histone Acetyltransferase Inhibitor, Represses Chromatin Transcription and Alters Global Gene Expression. J Biol Chem (2004) 279(32):33716–26. doi: 10.1074/jbc.M402839200
250. Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF, et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol Cancer (2014) 13:66. doi: 10.1186/1476-4598-13-66
251. Wang J, Wu M, Zheng D, Zhang H, Lv Y, Zhang L, et al. Garcinol inhibits esophageal cancer metastasis by suppressing the p300 and TGF-β1 signaling pathways. Acta Pharmacol Sin (2020) 41(1):82–92. doi: 10.1038/s41401-019-0271-3
252. Galvez AF, Chen N, Macasieb J, de Lumen BO. Chemopreventive property of a soybean peptide (lunasin) that binds to deacetylated histones and inhibits acetylation. Cancer Res (2001) 61(20):7473–8.
253. Ravindra KC, Selvi BR, Arif M, Reddy BAA, Thanuja GR, Agrawal S, et al. Inhibition of Lysine Acetyltransferase KAT3B/p300 Activity by a Naturally Occurring Hydroxynaphthoquinone, Plumbagin. J Biol Chem (2009) 284(36):24453–64. doi: 10.1074/jbc.M109.023861
254. Casaos J, Huq S, Lott T, Felder R, Choi J, Gorelick N, et al. Ribavirin as a potential therapeutic for atypical teratoid/rhabdoid tumors. Oncotarget (2018) 9(8):8054–67. doi: 10.18632/oncotarget.23883
255. Hu H, Qian K, Ho M-C, Zheng YG. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin Invest Drugs (2016) 25(3):335–58. doi: 10.1517/13543784.2016.1144747
256. Chen J, Xu X, Chen J. Clinically relevant concentration of anti-viral drug ribavirin selectively targets pediatric osteosarcoma and increases chemosensitivity. Biochem Biophys Res Commun (2018) 506(3):604–10. doi: 10.1016/j.bbrc.2018.10.124
257. De la Cruz-Hernandez E, Medina-Franco JL, Trujillo J, Chavez-Blanco A, Dominguez-Gomez G, Perez-Cardenas E, et al. Ribavirin as a tri-targeted antitumor repositioned drug. Oncol Rep (2015) 33(5):2384–92. doi: 10.3892/or.2015.3816
258. Catalano R, Rocca R, Juli G, Costa G, Maruca A, Artese A, et al. A drug repurposing screening reveals a novel epigenetic activity of hydroxychloroquine. Eur J Medicinal Chem (2019) 183:111715. doi: 10.1016/j.ejmech.2019.111715
259. Han H, Yang X, Pandiyan K, Liang G. Synergistic Re-Activation of Epigenetically Silenced Genes by Combinatorial Inhibition of DNMTs and LSD1 in Cancer Cells. PloS One (2013) 8(9):e75136. doi: 10.1371/journal.pone.0075136
260. Sakane C, Okitsu T, Wada A, Sagami H, Shidoji Y. Inhibition of lysine-specific demethylase 1 by the acyclic diterpenoid geranylgeranoic acid and its derivatives. Biochem Biophys Res Commun (2014) 444(1):24–9. doi: 10.1016/j.bbrc.2013.12.144
261. Wang M, Liu X, Guo J, Weng X, Jiang G, Wang Z, et al. Inhibition of LSD1 by Pargyline inhibited process of EMT and delayed progression of prostate cancer in vivo. Biochem Biophys Res Commun (2015) 467(2):310–5. doi: 10.1016/j.bbrc.2015.09.164
262. Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 Lysine 4 Demethylation Is a Target of Nonselective Antidepressive Medications. Chem Biol (2006) 13(6):563–7. doi: 10.1016/j.chembiol.2006.05.004
263. Singh MM, Manton CA, Bhat KP, Tsai W-W, Aldape K, Barton MC, et al. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro-Oncology (2011) 13(8):894–903. doi: 10.1093/neuonc/nor049
264. Speranzini V, Rotili D, Ciossani G, Pilotto S, Marrocco B, Forgione M, et al. Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci Adv (2016) 2(9):1–3. doi: 10.1126/sciadv.1601017
265. Wakchaure P, Velayutham R, Roy KK. Structure investigation, enrichment analysis and structure-based repurposing of FDA-approved drugs as inhibitors of BET-BRD4. J Biomol Struct Dynamics (2019) 37(12):3048–57. doi: 10.1080/07391102.2018.1507838
266. Jiang H, Xing J, Wang C, Zhang H, Yue L, Wan X, et al. Discovery of novel BET inhibitors by drug repurposing of nitroxoline and its analogues. Organic Biomol Chem (2017) 15(44):9352–61. doi: 10.1039/C7OB02369C
267. Wan X, Liu H, Sun Y, Zhang J, Chen X, Chen N. Lunasin: A promising polypeptide for the prevention and treatment of cancer. Oncol Lett (2017) 13(6):3997–4001. doi: 10.3892/ol.2017.6017
268. Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Delivery Rev (2008) 60(13–14):1527–33. doi: 10.1016/j.addr.2008.06.002
269. Jose A, Shenoy GG, Sunil Rodrigues G, Kumar NAN, Munisamy M, Thomas L, et al. Histone Demethylase KDM5B as a Therapeutic Target for Cancer Therapy. Cancers (2020) 12(8):4–7. doi: 10.3390/cancers12082121
270. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
Keywords: epidrugs, drug repurposing, cancer therapy, cancer, epigenetic inhibitors, epigenetics
Citation: Montalvo-Casimiro M, González-Barrios R, Meraz-Rodriguez MA, Juárez-González VT, Arriaga-Canon C and Herrera LA (2020) Epidrug Repurposing: Discovering New Faces of Old Acquaintances in Cancer Therapy. Front. Oncol. 10:605386. doi: 10.3389/fonc.2020.605386
Received: 12 September 2020; Accepted: 15 October 2020;
Published: 18 November 2020.
Edited by:
Teresita Padilla-Benavides, Wesleyan University, United StatesReviewed by:
Shameer Khader, AstraZeneca, United StatesFrancisco Cuellar-Perez, University of Texas Southwestern Medical Center, United States
Copyright © 2020 Montalvo-Casimiro, González-Barrios, Meraz-Rodriguez, Juárez-González, Arriaga-Canon and Herrera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis A. Herrera, bWV0aWxAaG90bWFpbC5jb20=
†These authors have contributed equally to this work