 Giorgia Nardo1
Giorgia Nardo1 Jessica Carlet2
Jessica Carlet2 Ludovica Marra2
Ludovica Marra2 Laura Bonanno2
Laura Bonanno2 Alice Boscolo3
Alice Boscolo3 Alessandro Dal Maso3
Alessandro Dal Maso3 Andrea Boscolo Bragadin3
Andrea Boscolo Bragadin3 Stefano Indraccolo1*†
Stefano Indraccolo1*† Elisabetta Zulato1†
Elisabetta Zulato1†- 1Immunology and Molecular Oncology Unit, Istituto Oncologico Veneto IOV - IRCCS, Padova, Italy
- 2Medical Oncology 2, Istituto Oncologico Veneto IOV - IRCCS, Padova, Italy
- 3Department of Surgery, Oncology and Gastroenterology, University of Padua, Padova, Italy
Background: Molecular profiling of advanced EGFR mutated NSCLC has recently demonstrated the co-existence of multiple genetic alterations. Specifically, co-existing KRAS-mutations in EGFR NSCLCs have been described, despite their prevalence at progression and their role in the response to EGFR tyrosine kinase inhibitors (TKIs) remain marginally explored. Aim of our study was to investigate the prevalence of co-existing KRAS mutations at the time of progressive disease and explore their impact on clinical outcome.
Materials and Methods: We retrospectively analyzed by digital droplet PCR prevalence of KRAS co-mutations in 106 plasma samples of EGFR mutated NSCLC patients, in progressive disease after EGFR TKI treatment as first-line therapy.
Results: KRAS co-mutations (codon 12 and 13) were identified in 3 patients (2.8% of analyzed samples), with low allelic frequency (<0.2%), and had a negative impact on clinical outcome to first-line EGFR TKI.
Conclusion: Detection of KRAS mutations in cell-free DNA of EGFR mutant NSCLC patients at progression after first or second generation EGFR TKI is a rare event. Due to their low abundance, the negative impact of KRAS mutations on the response to EGFR TKI remains to be confirmed in larger studies.
Introduction
Non-small-cell lung cancer (NSCLC) is the leading cause of cancer-related mortality worldwide with five-year survival rate less than 10% among patients with advanced disease (1). Activating mutations in the epidermal growth factor receptor (EGFR) gene occur as early cancer-driving clonal event (2) in a subset of NSCLC patients (approximately 15% of Caucasian patients) and predict sensitivity to EGFR tyrosine kinase inhibitors (TKIs) (3). Improvement in clinical outcome, in terms of objective response rate (ORR), progression-free survival (PFS) and overall survival (OS), compared with upfront platinum doublet chemotherapy, made TKIs standard of care for advanced stage EGFR mutant NSCLC (4–8). However, resistance invariably develops, with EGFR T790M mutation accounting for approximately 50–60% of the mechanisms of acquired resistance to first- or second-generation EGFR-TKI therapy (3). Other less common EGFR- independent mechanisms of resistance include activation of bypassing pathways and histologic transformation to small-cell lung cancer (10–15% of cases) (9–11). In addition, 20–30% of patients do not show response on EGFR TKI treatment, probably due to intrinsic mechanisms of resistance (12).
Recently, comprehensive molecular-pathological profiling of advanced EGFR mutated NSCLC prior to therapy demonstrated co-existence of multiple genetic alterations (13). Consequently, the question arises as to whether co-occurring genetic alterations cooperate with the primary driver EGFR gene in promoting tumor progression and limiting efficacy of target therapy. Recent studies showed that co-mutations in the TP53 gene are a negative predictive factor of response to EGFR-TKI and an independent prognostic factor of shorter survival in advanced EGFR mutant NSCLC (14–17). Moreover, co-existing KRAS-mutations in EGFR NSCLCs have been reported by several studies (18–23). However, their prevalence at progression and their role in the response to TKIs treatment has been investigated only in one study including a small number of patients (n=33) (24).
Here, we performed a retrospective analysis of KRAS co-genetic alterations in 106 EGFR mutated NSCLC patients with progressive disease after EGFR TKI first-line therapy. We quantitated KRAS mutation in plasma samples by droplet digital PCR (ddPCR), with the aim to investigate the prevalence of co-existing KRAS mutations at the time of progression and explore their impact on clinical outcome.
Material and Methods
Study Design and Patient Population
The primary aim of this study was to assess the prevalence of KRAS co-mutations in EGFR mutated NSCLC patients, in progressive disease after EGFR TKI treatment as first-line therapy. For this purpose, we retrospectively selected 122 consecutive patients with EGFR-mutated NSCLC with progressive disease after first-line TKI treatment, referring to our Institution from 2016 to 2019. Eligibility criteria were confirmed histological diagnosis of advanced NSCLC, presence of an EGFR exon 18 to 21 mutation at diagnosis, progression to front line systemic treatment with first- or second- generation EGFR TKIs (erlotinib, gefitinib, or afatinib), and available liquid biopsy material collected at progressive disease, and clinical follow-up. Patients who did not progress to first-line EGFR TKIs, or without available liquid biopsy material after progression were excluded.
At the time of diagnosis, tissue molecular analyses of EGFR gene exons 18 to 21 were performed according to standard clinical practice, and KRAS mutational status was not routinely examined because mutually exclusive with activating EGFR mutations in this patient population.
At progressive disease, plasma samples were collected for liquid biopsy to assess the T790M mutational status in cell-free (cf)-DNA. Molecular analyses were performed according to standard lab practice, using the CE IVD cobas® EGFR Mutation Test v2.
The studies involving human participants were reviewed and approved by IOV Institutional Review Board and Ethics Committee (CESC IOV 2020/57), and were performed in accordance with the declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study. For patients who were dead or lost to follow-up at the time of study enrolment, we used the Italian Data Protection Authority Authorisation 9/2016 on “privacy protective rules for recording clinical data for research and study purposes”.
Cell Free DNA (cfDNA) Extraction and Analysis
Residual plasma collected at the time of progression for routine diagnostic activity was used: cfDNA was extracted from 1–2 ml of plasma using the Maxwell® RSC ccfDNA Plasma Kit (Promega, Madison, Wisconsin, USA), and eluted into 60 µl of buffer, according to manufacturer’s instructions. cfDNA was quantified using the QuBit dsDNA HS Assay kit with QuBit 3.0 fluorimeter (Thermo Fisher Scientific, San Jose, CA), and stored at −20°C before use.
Detection of KRAS mutations in codons 12 and 13 in cfDNA was performed by droplet digital PCR (ddPCR), as previously described (25). The ddPCR assay was purchased from Bio-Rad (the ddPCR KRAS G12/G13 Screening Kit #186-3506), and it does not enable to distinguish among different mutations in KRAS codon G12/G13 (G12A, G12C, G12D, G12R, G12S, G12V, G13D). Each sample was analyzed in triplicate and in each test at least three control wells with a negative KRAS cfDNA, one negative control well without DNA and one positive control were included. In line with our previous study (25) and as reported in the manufacturer’s instructions, a cut-off of three droplets was used to call a sample mutant, according to the Poisson’s law of small numbers. The sensitivity of our assay to detect KRAS mutation in plasma samples was 48% (25).
Data Analysis
Progression free survival (PFS) was calculated as the time between the first day of treatment and the radiologic and/or clinical evidence of progression; time to treatment failure (TTF) was defined as the time from the first day of EGFR-TKI administration to the date of treatment failure; overall survival (OS) was measured as the time elapsed from diagnosis to death for any cause. Patients who did not develop an event during the study period were censored at the date of last observation. Median PFS, TTF and OS were estimated using the Kaplan–Meier method.
Chi-square test was used to evaluate whether the frequency of cases with single or double KRAS positive droplets differ among EGFR mutant and EGFR wild-type cfDNA samples.
Results
Patients
Study layout is summarized in Figure 1. From 2016 to 2019, 122 patients with advanced EGFR mutated NSCLC referring to our Institution received treatment with a first- or second -EGFR-TKI as first-line therapy and underwent cfDNA genotyping for assessment of EGFR mutations at progression. Residual plasma samples were available for 106 patients. Clinical characteristics of patients matching the eligibility criteria are shown in Table 1. At the time of diagnosis, median age was 68 years. Most patients were females (59%), with EGFR mutant stage III–IV lung adenocarcinoma (90.5%) and without smoking history (62%). Patients presented in an optimal or good Eastern Cooperative Oncology Group (ECOG) performance status (PS), with 46 (43%) and 55 (52%) having ECOG 0 and 1, respectively. EGFR exon 19 deletion was carried by 64 out of 106 patients (60%); 35 patients (33%) had an EGFR p.L858R point mutation and 7 (7%) had different EGFR mutations. The majority of patients (n=54) received gefitinib as first-line TKI treatment (51%), 26 out of 106 (24.5%) patients received erlotinib, and 26 (24.5%) afatinib (Table 1). Median Progression Free Survival (PFS) was 24.30 months (95%, CI: 19.29–29.31).
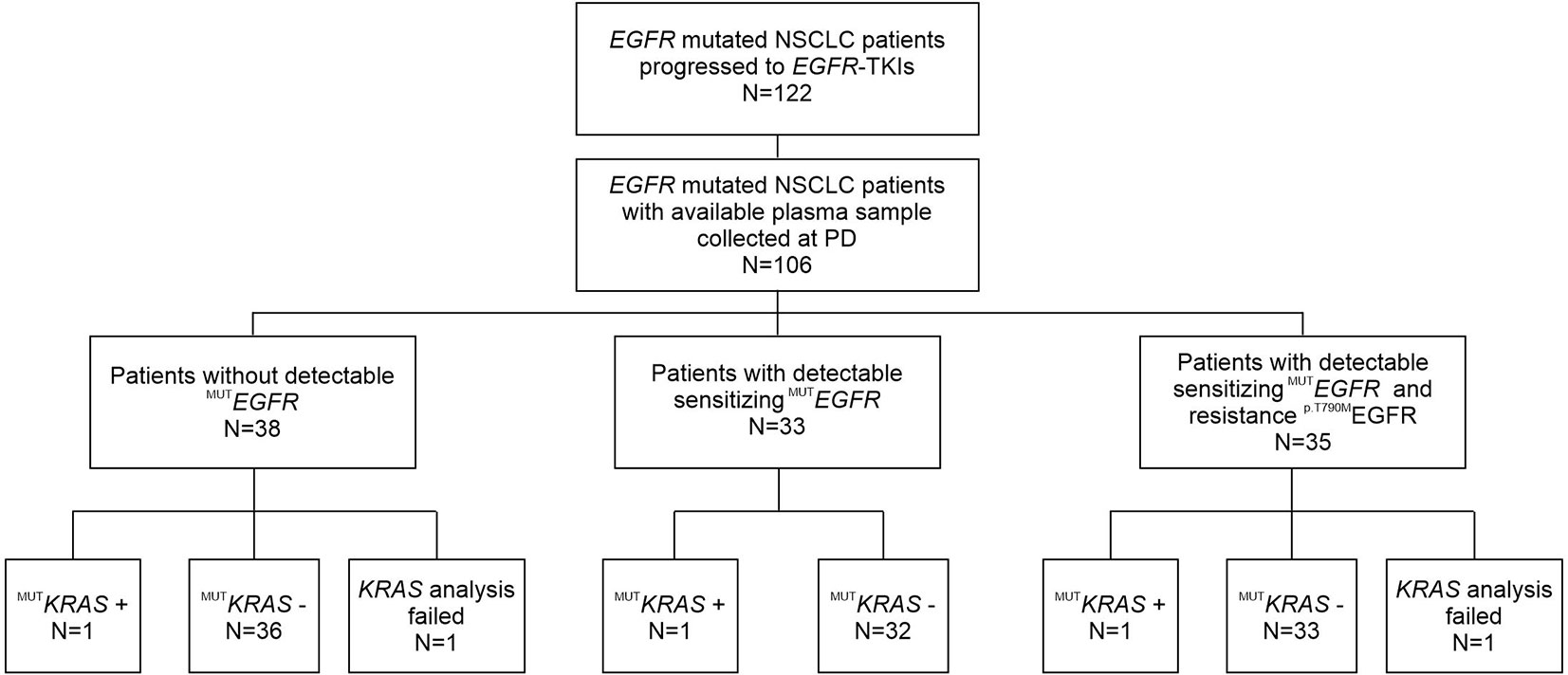
Figure 1 Flow chart of patients enrolled in this study, who progressed after front-line first- or second-generation EGFR-TKI treatment and underwent liquid biopsy to assess the T790M mutational status.
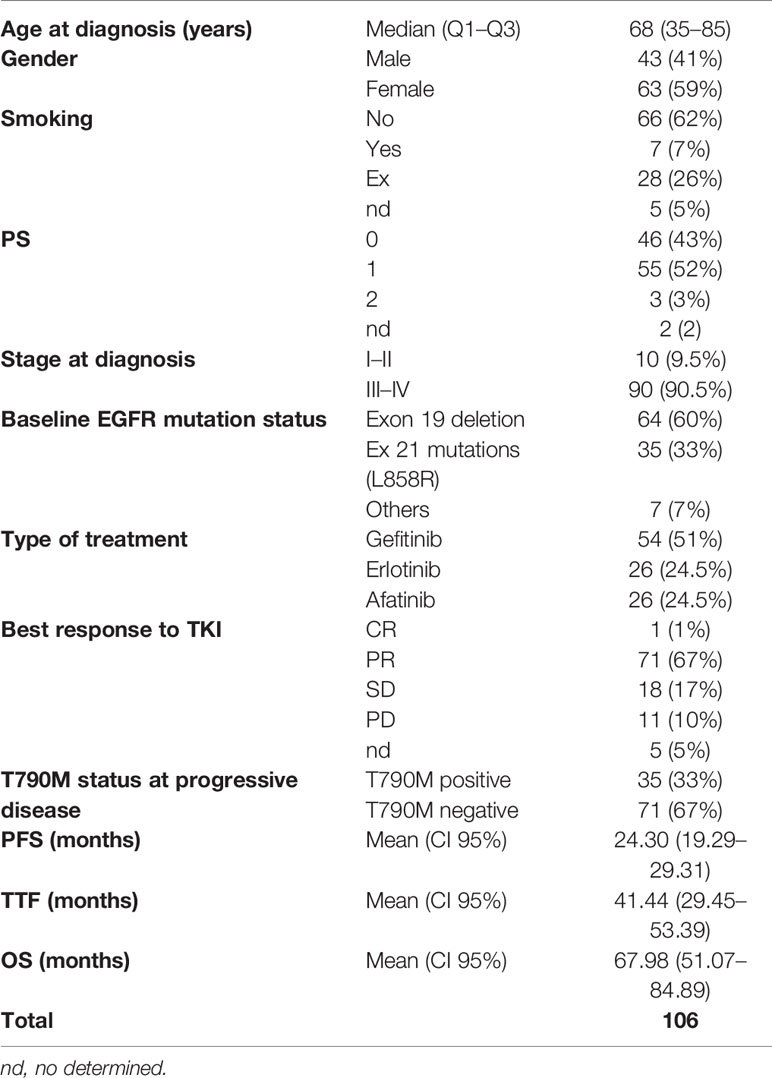
Table 1 Clinical features of patients at time of diagnosis.
At the time of progressive disease, EGFR sensitizing mutations were detected by liquid biopsy in 68 out of 106 plasma samples tested (64%), whereas the remaining 38 plasma samples (36%) did not bear EGFR mutations (Table 2). The T790M-resistance mutation was found in 35 out of 106 samples (33%), or 35 out of 68 plasma samples bearing EGFR sensitizing mutations (50.7%) (Table 2).
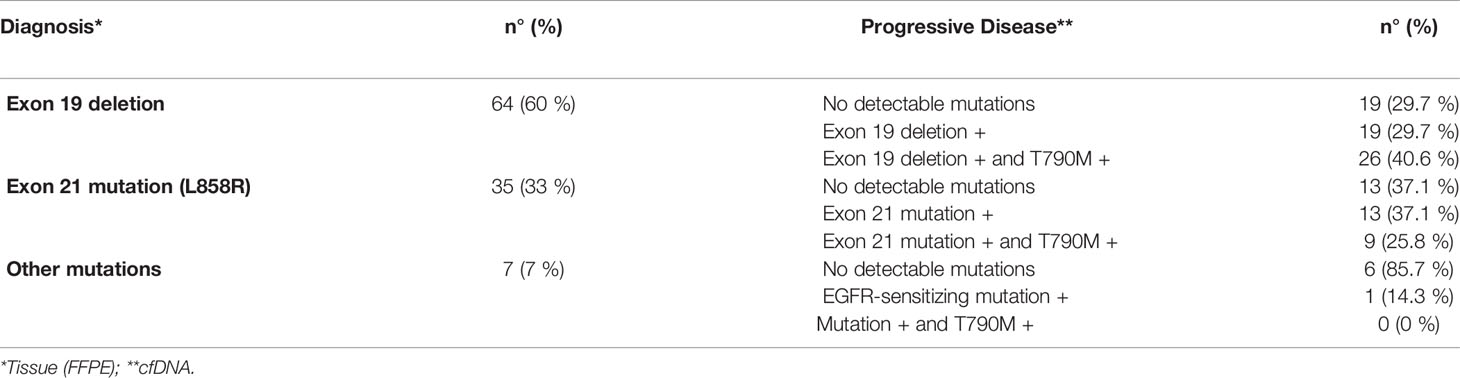
Table 2 EGFR mutational status.
Prevalence of KRAS Co-Mutations at Progressive Disease
Among 106 patients with plasma samples available, 104 were successfully screened by ddPCR for the presence of concomitant KRAS mutation in codon 12 and 13, whereas 2 samples were not evaluable (Figure 1).
Considering the standard cut-off value of three droplets, as detailed in the Materials and Methods section, KRAS mutations were detected in 3 patients (2.8%) (Figure 1). Tumor tissue collected at diagnosis was available only for one (ID#88) out of 3 KRAS positive patients, and its analysis confirmed the co-existence of EGFR and KRAS mutations (KRAS allelic frequency 13.8%). In all 3 positive cases, the allelic frequency of the KRAS mutations in the liquid biopsy samples was low (<0.2%) (Table 3). All KRAS positive patients (n=3) had poor clinical outcome to first-line EGFR TKI, in terms of TTF, PFS and OS (Figure 2; Table 3). Interestingly, these patients were current or former smoker and one of them had squamous cell carcinoma histology. At diagnosis they all presented with extra-thoracic disease, but they did not show any specific clinical negative prognostic marker (i.e. worse performance status; see Table 3). Two of them did not respond to first line EGFR TKI, while one of them achieved partial response with a PFS of about six months. At progression to first line TKI, only one of them carried T790M mutation (ID#39), but he did not respond to osimertinib.
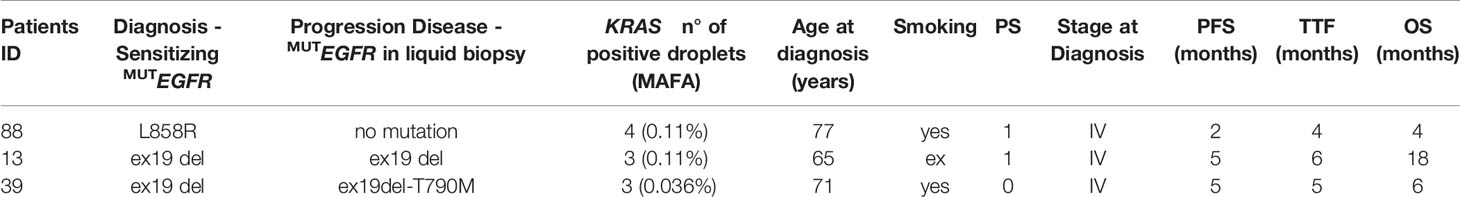
Table 3 EGFR and KRAS co-mutated cases.
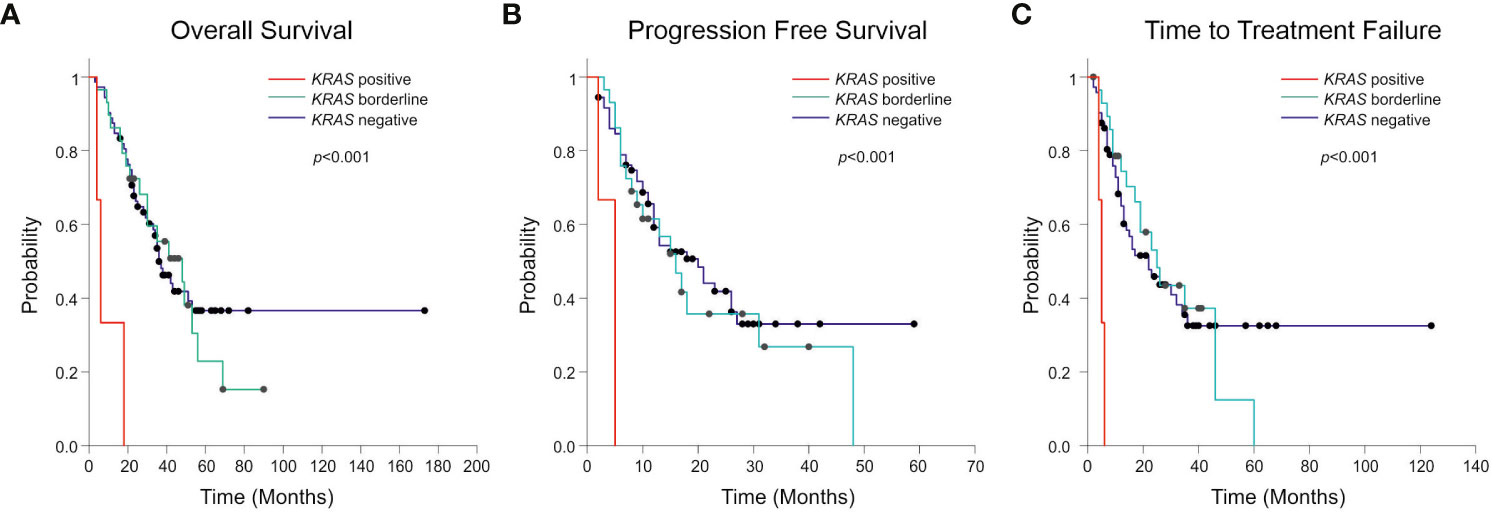
Figure 2 Kaplan-Meier curves showing Overall Survival (OS) (A), Progression Free Survival (PFS) (B) and Time to Treatment Failure (TTF) (C) according to the presence, the absence, or borderline positivity (1–2 positive droplets) of KRAS mutation. The p-value related to the presence or the absence of KRAS mutation is reported in figure.
Interestingly, one or two positive droplets for KRAS mutations were detected in additional 28 plasma samples out of 104 analyzed (27%), with allelic frequency of the KRAS variant very low (mean 0.15%; median 0.12%) and ranging between 0.016 and 0.32% (Table 4). These single or double KRAS positive droplets were similarly distributed among EGFR mutant and EGFR wild-type cfDNA samples (15 out of 68 EGFR mutant versus 13 out of 38 EGFR wild-type samples, respectively. Chi-square test, P=0.21). With regard to clinical correlates, patients with borderline KRAS positivity (n=28) behave similarly to KRAS negative patients (n=73) in terms of TTF, PFS, and OS (Figure 2). Details about median TTF, PFS and OS in KRAS positive, borderline and negative patients are reported in Table 5.
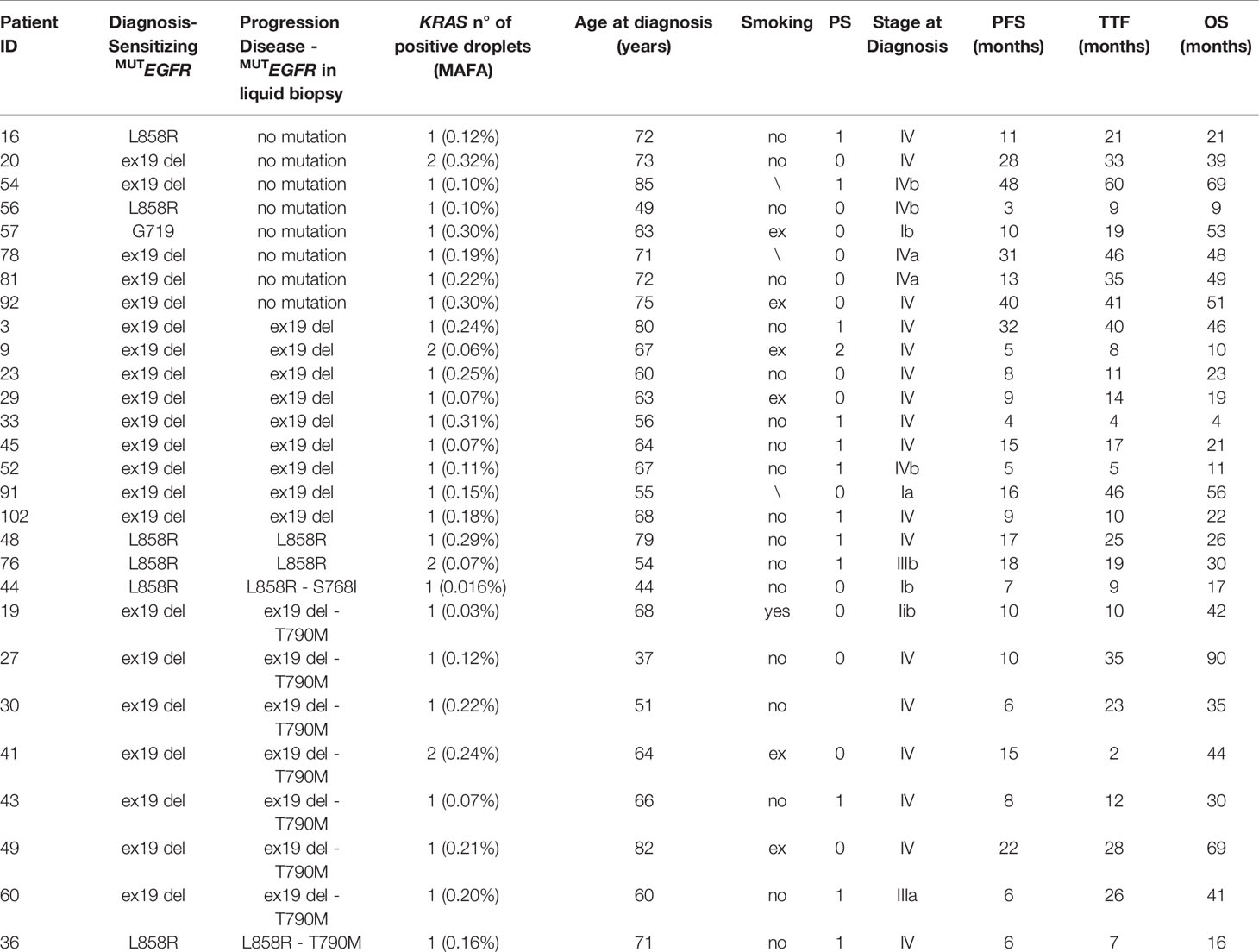
Table 4 KRAS borderline samples with one or two positive droplets in cfDNA.

Table 5 Overall Survival (OS), Progression Free Survival (PFS) and Time to Treatment Failure (TTF) in KRAS negative, borderline and positive patients at Progressive Disease (PD).
We conclude that frank positivity for codon 12 and 13 KRAS mutations in cfDNA of EGFR mutant NSCLC at progression after first or second generation EGFR TKI treatment is a rare event.
Discussion
We report a retrospective evaluation of the prevalence of codon 12 and 13 KRAS co-mutations in EGFR mutated NSCLC patients in progressive disease after EGFR TKI treatment as first-line therapy, with the aim to establish their prevalence and explore their impact on clinical outcome. Mutations in EGFR and KRAS are considered mutually exclusive in NSCLC (26) and this is also remarked by epidemiologic data, being KRAS mutations associated with smoke and EGFR mutations more common in non-smokers, respectively. On the other hand, genetic studies involving multi-region sequencing of tumors have clearly shown that genetic heterogeneity exists in lung adenocarcinoma and EGFR mutations generally occur in the genetic trunk of the tumor and are hence clonal, whereas KRAS mutations are often sub-clonal (2). This genetic model is also supported by studies which investigated EGFR and KRAS mutations in matched primary tumor and metastasis from the same patients and reported the occasional presence of KRAS mutations in metastatic lesions from EGFR mutant primary tumors (27). Moreover, up to 8–15% NSCLC are diagnosed with multiple lung nodules and can disclose extensive inter-tumor genetic variation in the same patient (28, 29).
In line with these arguments, previous studies investigated and found pathogenic KRAS mutations in EGFR mutant NSCLC at diagnosis (18–23, 30–32). Percentages of KRAS mutation vary widely among studies (range 1.2–10.5%), depending on the broadly different size of the study population (ranging from 58 to 6637 samples), the various analytical sensitivity of the techniques utilized (Sanger, RT-PCR, NGS, ddPCR) and the type of sample analyzed (tissue or cfDNA). Concomitant KRAS mutations often involve canonical codon 12 and 13 mutational hotspots and are well known pathogenic mutations which constitutively activate KRAS firing. These mutations could theoretically impact on the response to EGFR inhibitors, due to bypassing the inhibition of EGFR by TKIs. Consequences on clinical responses to EGFR TKIs have been investigated in some studies with variable results. In early studies, Takeda et al. and Pao et al. found that KRAS mutation is a negative predictor of response to EGFR-TKIs in EGFR mutation-positive NSCLC patients (33, 34). On the other hand, Benesoma et al. described 3 NSCLC patients with coexistence of EGFR and KRAS mutations uncoupled from negative response to EGFR TKIs (23). More recently, Hong et al. genotyped 58 EGFR mutant NSCLC patients before TKI treatment and found that concomitant KRAS mutations in cfDNA associated with shorter duration of PFS and OS (18). However, conclusions from these studies were based on small cohorts of patients and other groups reported overlapping clinical outcome in EGFR mutant NSCLC patients with or without concomitant KRAS mutations (32). These contrasting results could, among other factors, depend on the sub-clonal nature of KRAS mutations and their different abundance in the studied patients’ cohorts.
A field relatively less investigated so far involves the prevalence of KRAS mutations following treatment and onset of clinical resistance to EGFR TKI. Del Re et al. found that 16 out of 33 (48.5%) NSCLC samples studied at progression after EGFR TKI had concomitant codon 12 KRAS mutations in cfDNA, with percentages of mutated allele ranging from 1–98% (24). However, in this study it was not stated which cut-off has been used for interpretation of ddPCR results. Moreover, accurate assessment of the percentage of KRAS mutation in this patient population could be challenging, due to the small number of samples analyzed and the value reported (48.5%) was much greater than previously found by others (18–22, 30–32). In our study by using stringent criteria for interpretation of ddPCR data and analysing a large population of samples (n=104), KRAS mutations were rarely found in cfDNA from these patients (2.8%) and had a negative impact on response to TKI and clinical outcome (TTF, PFS, OS) (Figure 2). KRAS positivity was confirmed in one available matched tumor tissue biopsy at diagnosis. Although this is limited to one patient, results are in-line with a recent study suggesting that EGFR-mutated NSCLC patients with KRAS mutations detected in tumor before the start of treatment do not benefit from EGFR TKIs (22).
It is important to stress that technicalities, such as the cut-off values used to interpret ddPCR results are key to determine the result. In fact, if we lowered the cut-off and considered as KRAS mutant even samples with 1–2 positive droplets in cfDNA (n=28), the percentage of KRAS mutated samples was much higher (29%). In any case it should be considered that the abundance of KRAS mutations was very low, as indicated by the low MAFA values (mean 0.15%, median 0.12%, range 0.016–0.32%), compared with those found in cfDNA from NSCLC patients bearing KRAS mutant tumors (mean 8.87%, median 3%, range 0.46–53.7 %) (25). Of regard, we found no prognostic association of borderline KRAS mutations in cfDNA with PFS, nor with OS (Figure 2).
The main limitation of this study is the relatively limited number of frankly KRAS positive patients (n=3), compared with KRAS negative patients (n=73). However, our data suggest a potential negative prognostic impact, and confirmed recent reports indicating that EGFR-mutated NSCLC patients with additional driver alterations show reduced sensitivity to TKIs. Clearly, our findings should be confirmed in larger series to investigate the impact of KRAS mutation detection on clinical decisions, with particular regard to selection of patients for combination treatments, currently under investigation in lung cancer, such as EGFR inhibitor plus chemotherapy or plus antiangiogenic treatment (35).
On the other hand, 36% of plasma samples analyzed at progressive disease were negative for EGFR mutation, indicating the possible lack of circulating tumor DNA (ctDNA). This aspect could determine an underestimation of the patients with co-occurring KRAS mutations, even though the low presence of ctDNA could be associated with lower tumor burden and better prognosis (36).
Another limitation is represented by the fact that our study did not include systematic analysis of baseline KRAS mutation either in plasma or in tissue, which could unravel the multi-clonal character of the tumours. Therefore, we could not draw definitive conclusions on the role of sub-clones in the response to EGFR TKI.
We conclude that detection of KRAS mutations in cfDNA is rare in EGFR mutant patients treated with TKI and these mutations are more likely to be detected in smokers, possibly underlying broader genetic heterogeneity of these tumors compared with those on non-smokers.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by IOV Institutional Review Board and Ethics Committee (CESC IOV 2020/57). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
GN contributed to the design of the study, performed the analysis, and wrote sections of the manuscript. JC, LM, and ABB processed the samples and performed the experiments. EZ analyzed the data, and wrote the manuscript. LB, AB, and AM recruited patients, collected, and analyzed clinical data and performed statistical analysis. SI contributed conception and design of the study, wrote the advanced draft of the manuscript revising it critically for intellectual content, and provided approval for publication of the content. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by IOV intramural research grant 2017 – SINERGIA (to SI and LB). The QX200 ddPCR system (Bio-Rad Laboratories) was purchased through a grant provided by Università degli Studi di Padova, Padova, Italy (2015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cheng TY, Cramb SM, Baade PD, Youlden DR, Nwogu C, Reid ME. The International Epidemiology of Lung Cancer: Latest Trends, Disparities, and Tumor Characteristics. J Thorac Oncol (2016) 11(10):1653–71. doi: 10.1016/j.jtho.2016.05.021
2. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med (2017) 376(22):2109–21. doi: 10.1056/NEJMoa1616288
3. Piotrowska Z, Sequist LV. Epidermal Growth Factor Receptor-Mutant Lung Cancer: New Drugs, New Resistance Mechanisms, and Future Treatment Options. Cancer J (2015) 21(5):371–7. doi: 10.1097/PPO.0000000000000147
4. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med (2010) 362(25):2380–8. doi: 10.1056/NEJMoa0909530
5. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi: 10.1056/NEJMoa0810699
6. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13(3):239–46. doi: 10.1016/S1470-2045(11)70393-X
7. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol (2013) 31(27):3327–34. doi: 10.1200/JCO.2012.44.2806
8. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378(2):113–25. doi: 10.1056/NEJMoa1713137
9. Marcoux N, Gettinger SN, O’Kane G, Arbour KC, Neal JW, Husain H, et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J Clin Oncol (2019) 37(4):278–85. doi: 10.1200/JCO.18.01585
10. Van Der Steen N, Giovannetti E, Carbone D, Leonetti A, Rolfo CD, Peters GJ. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resistance (2018) 1(4):230–49. doi: 10.20517/cdr.2018.13
11. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer (2019) 121(9):725–37. doi: 10.1038/s41416-019-0573-8
12. Santoni-Rugiu E, Melchior LC, Urbanska EM, Jakobsen JN, de Stricker K, Grauslund M, et al. Intrinsic resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers (2019) 11(7):923. doi: 10.3390/cancers11070923
13. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet (2017) 49(12):1693–704. doi: 10.1038/ng.3990
14. Bria E, Pilotto S, Amato E, Fassan M, Novello S, Peretti U, et al. Molecular heterogeneity assessment by next-generation sequencing and response to gefitinib of EGFR mutant advanced lung adenocarcinoma. Oncotarget (2015) 6(14):12783–95. doi: 10.18632/oncotarget.3727
15. Canale M, Petracci E, Delmonte A, Chiadini E, Dazzi C, Papi M, et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res (2017) 23(9):2195–202. doi: 10.1158/1078-0432.CCR-16-0966
16. Labbe C, Cabanero M, Korpanty GJ, Tomasini P, Doherty MK, Mascaux C, et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer (2017) 111:23–9. doi: 10.1016/j.lungcan.2017.06.014
17. Molina-Vila MA, Bertran-Alamillo J, Gasco A, Mayo-de-las-Casas C, Sanchez-Ronco M, Pujantell-Pastor L, et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res (2014) 20(17):4647–59. doi: 10.1158/1078-0432.CCR-13-2391
18. Hong S, Gao F, Fu S, Wang Y, Fang W, Huang Y, et al. Concomitant Genetic Alterations With Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With EGFR-Mutant Advanced Non-Small Cell Lung Cancer. JAMA Oncol (2018) 4(5):739–42. doi: 10.1001/jamaoncol.2018.0049
19. Hu W, Liu Y, Chen J. Concurrent gene alterations with EGFR mutation and treatment efficacy of EGFR-TKIs in Chinese patients with non-small cell lung cancer. Oncotarget (2017) 8(15):25046–54. doi: 10.18632/oncotarget.15337
20. Jakobsen JN, Santoni-Rugiu E, Grauslund M, Melchior L, Sorensen JB. Concomitant driver mutations in advanced EGFR-mutated non-small-cell lung cancer and their impact on erlotinib treatment. Oncotarget (2018) 9(40):26195–208. doi: 10.18632/oncotarget.25490
21. Lee T, Lee B, Choi YL, Han J, Ahn MJ, Um SW. Non-small Cell Lung Cancer with Concomitant EGFR, KRAS, and ALK Mutation: Clinicopathologic Features of 12 Cases. J Pathol Transl Med (2016) 50(3):197–203. doi: 10.4132/jptm.2016.03.09
22. Rachiglio AM, Fenizia F, Piccirillo MC, Galetta D, Crino L, Vincenzi B, et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers (Basel) (2019) 11(3):341. doi: 10.3390/cancers11030341
23. Benesova L, Minarik M, Jancarikova D, Belsanova B, Pese M. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res (2010) 30(5):1667–71.
24. Del Re M, Tiseo M, Bordi P, D’Incecco A, Camerini A, Petrini I, et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: a study on circulating tumor DNA. Oncotarget (2017) 8(8):13611–9. doi: 10.18632/oncotarget.6957
25. Zulato E, Attili I, Pavan A, Nardo G, Del Bianco P, Boscolo Bragadin A, et al. Early assessment of KRAS mutation in cfDNA correlates with risk of progression and death in advanced non-small-cell lung cancer. Br J Cancer (2020) 123(1):81–91. doi: 10.1038/s41416-020-0833-7
26. Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol (2015) 10(5):768–77. doi: 10.1097/JTO.0000000000000516
27. Sun L, Zhang Q, Luan H, Zhan Z, Wang C, Sun B. Comparison of KRAS and EGFR gene status between primary non-small cell lung cancer and local lymph node metastases: implications for clinical practice. J Exp Clin Cancer Res (2011) 30:30. doi: 10.1186/1756-9966-30-30
28. Mansuet-Lupo A, Barritault M, Alifano M, Janet-Vendroux A, Zarmaev M, Biton J, et al. Proposal for a Combined Histomolecular Algorithm to Distinguish Multiple Primary Adenocarcinomas from Intrapulmonary Metastasis in Patients with Multiple Lung Tumors. J Thorac Oncol (2019) 14(5):844–56. doi: 10.1016/j.jtho.2019.01.017
29. Roepman P, Ten Heuvel A, Scheidel KC, Sprong T, Heideman DAM, Seldenrijk KA, et al. Added Value of 50-Gene Panel Sequencing to Distinguish Multiple Primary Lung Cancers from Pulmonary Metastases: A Systematic Investigation. J Mol Diagn (2018) 20(4):436–45. doi: 10.1016/j.jmoldx.2018.02.007
30. Li S, Li L, Zhu Y, Huang C, Qin Y, Liu H, et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. Br J Cancer (2014) 110(11):2812–20. doi: 10.1038/bjc.2014.210
31. Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, et al. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J Thorac Oncol (2019) 14(4):606–16. doi: 10.1016/j.jtho.2018.12.013
32. Zhuang X, Zhao C, Li J, Su C, Chen X, Ren S, et al. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med (2019) 8(6):2858–66. doi: 10.1002/cam4.2183
33. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PloS Med (2005) 2(1):e17. doi: 10.1371/journal.pmed.0020017
34. Takeda M, Okamoto I, Fujita Y, Arao T, Ito H, Fukuoka M, et al. De novo resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutation-positive patients with non-small cell lung cancer. J Thorac Oncol (2010) 5(3):399–400. doi: 10.1097/JTO.0b013e3181cee47e
35. Tam Z. Combination Strategies Using EGFR-TKi in NSCLC Therapy: Learning from the Gap between Pre-Clinical Results and Clinical Outcomes. Int J Biol Sci (2018) 14(2):204–16. doi: 10.7150/ijbs.22955
Keywords: KRAS, EGFR, liquid biopsy, non-small-cell lung cancer, cell free DNA, tyrosine kinase inhibitors
Citation: Nardo G, Carlet J, Marra L, Bonanno L, Boscolo A, Dal Maso A, Boscolo Bragadin A, Indraccolo S and Zulato E (2021) Detection of Low-Frequency KRAS Mutations in cfDNA From EGFR-Mutated NSCLC Patients After First-Line EGFR Tyrosine Kinase Inhibitors. Front. Oncol. 10:607840. doi: 10.3389/fonc.2020.607840
Received: 18 September 2020; Accepted: 30 November 2020;
Published: 15 January 2021.
Edited by:
Francesco Fabbri, Romagnolo Scientific Institute for the Study and Treatment of Tumors (IRCCS), ItalyReviewed by:
Vienna Ludovini, Hospital of Santa Maria della Misericordia in Perugia, ItalyMarcello Tiseo, University Hospital of Parma, Italy
Copyright © 2021 Nardo, Carlet, Marra, Bonanno, Boscolo, Dal Maso, Boscolo Bragadin, Indraccolo and Zulato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Indraccolo, c3RlZmFuby5pbmRyYWNjb2xvQHVuaXBkLml0
†These authors have contributed equally to this work