 Matteo Molica
Matteo Molica Carla Mazzone1
Carla Mazzone1 Paolo de Fabritiis
Paolo de Fabritiis- 1Haematology Unit, S. Eugenio Hospital, ASL Roma 2, Rome, Italy
- 2Department of Biomedicina and Prevenzione, Tor Vergata University, Rome, Italy
TP53 is a key tumor suppressor gene with protean functions associated with preservation of genomic balance, including regulation of cellular senescence, apoptotic pathways, metabolism functions, and DNA repair. The vast majority of de novo acute myeloid leukemia (AML) present unaltered TP53 alleles. However, TP53 mutations are frequently detected in AML related to an increased genomic instability, such as therapy‐related (t-AML) or AML with myelodysplasia-related changes. Of note, TP53 mutations are associated with complex cytogenetic abnormalities, advanced age, chemoresistance, and poor outcomes. Recent breakthroughs in AML research and the development of targeted drugs directed at specific mutations have led to an explosion of novel treatments with different mechanisms. However, optimal treatment strategy for patients harboring TP53 mutations remains a critical area of unmet need. In this review, we focus on the incidence and clinical significance of TP53 mutations in de novo and t-AML. The influence of these alterations on response and clinical outcomes as well as the current and future therapeutic perspectives for this hardly treatable setting are discussed.
Introduction
The tumor suppressor gene TP53 encodes a transcription factor, which operates after the stimulation of various forms of cellular stress and, employs multiple anti-proliferative functions (Figure 1). The transcription factor includes a DNA binding domain, a transcription activation domain, a tetramerization domain and a proline-rich domain (1). The most well‐known property of TP53 gene is its capability in containing cellular proliferation/differentiation associated with aberrant and uncontrolled oncogene expression. Therefore, TP53 inactivation inducing by gene mutation or deletion favors the activities of oncogenes, thereby promoting uncontrolled proliferation of cancer cells (2). Conversely to the vast majority of tumor suppressor genes that are predominantly characterized by truncating mutations, a substantial proportion of TP53 alterations include missense substitutions (75%); other mutations are frameshift insertions and deletions (9%), non-sense mutations (7%), silent mutations (5%), and other rare aberrations (2%). While wild-type (WT) p53 tends to prevent a tumorigenic phenotype, either loss-of-function or oncogenic gain-of-function (GOF) TP53 mutations favor tumorigenesis events. The majority of the gene alterations detected in patients with hematological diseases include GOF mutations. For instance, IDH1/2 gene acquires a novel function to convert α-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG) determining the inhibition of TET2 as well as some histone demethylase and promoting tumorigenesis (3). TP53 gene represents the most commonly altered gene in somatic cells of human malignancies, with mutations in TP53 gene detected in more than 50% of solid tumors (4–6). In some tumors, such as colorectal cancer, TP53 alteration is frequently a belated occurrence in a multi-stage carcinogenic pathway that proceeds from hyper-proliferative cells in colonic epithelium to colorectal adenomas and ultimately to metastatic colorectal cancer (7, 8). Contrariwise, in high-grade serous ovarian tumors, TP53 mutations are a relatively precocious event, likely arising in forerunner lesions (9, 10). Germline TP53 mutations may cause Li-Fraumeni disease; carriers tend to develop several cancers including early onset brain and adrenocortical tumors, breast cancer, soft tissue sarcoma, and leukemia (11). In the last decade, robust discoveries have been made exploring the mutational landscape of hematological diseases mainly thanks to developments in sequencing techniques. The recent progress of next-generation sequence (NGS) has increasingly included it in clinical research allowing the cytogenetic analysis in several hematological malignancies, including acute myeloid leukemia (AML) (12). Therefore, to date, NGS is the most common and reliable method used to detect TP53 mutations in AML, holding higher sensitivity than other techniques such as immunohistochemistry (IHC), fluorescent in situ hybridization (FISH), or real-time qualitative reverse transcription PCR (RT-qPCR). Despite the frequency of TP53 mutations detected in human cancers, they are observed in only 5–10% of patients with AML. Although all classes of TP53 variants have been observed in AML, the vast majority of mutations include missense alterations usually arising in the DNA binding domain (encoded by exons 5–8) with a predilection for arginine residues and noted mutational “hot spots” such as R175H, Y220C, R248Q, and R273C (13). Interestingly, despite variants in TP53 usually determined a loss-of-function of the tumor-suppressor activity of the protein, the R282 site was found to belong to the gain-of function (GOF) mutations (14). R282 represents a structural mutation (as also e.g. R175, G245, R248, und R249) inducing conformational instability of the p53 protein, by contrast to the contact mutations (R273 and R248) that are sited in the p53-DNA binding surface (15). Mutations in R282 were only observed in the acute diseases AML and acute lymphoblastic leukemia (ALL) and might accelerate the rapid proliferation of the malignant cells in these diseases (14, 16). The single p53 deletion caused by loss of 17p chromosome in also included among TP53 mutations. A deletion of 17p commonly involves the tumor suppressor gene p53 on band 17p13.1 with allelic loss of the gene (17). It was widely demonstrated that p53 deletion without multiple cytogenetic aberrations is an independent negative prognostic factor for disease-free survival (DFS), relapse risk, and overall survival (OS) in AML (17). Therefore, the single p53 deletion should be always considered as a high-risk aberration for new risk-adapted treatment strategies in AML. Quintás-Cardama et al. have recently reported that, in addition to somatic mutations, p53 dysfunction can occur via aberrant expression of proteins that regulate p53 stability and function (e.g. overexpression of its canonical negative regulators Mdm2 and/or Mdm4) (18). They demonstrated that reactivation of p53 functions via Mdm2-antagonists in the context of Mdm2 overexpression and wild-type p53 restored p53’s anti-tumor effects. These data suggest that specific recommendations could be considered in patients with AML expressing p53 protein stabilization and to those exhibiting Mdm2 protein overexpression. In the latter case, a strong recommendation to enroll in trials assessing experimental agents should include those with Mdm2 blocking activity. Mutations in other functional domains hold specific effects on p53 protein activities; however, to date, their clinical implications in AML are not well understood (19). TP53 is the sole mutated gene detected in up to 75% of patients, while patients who harbor co-occurring mutations show a lower incidence of mutations in several AML-related genes such as NPM1, FLT3, IDH1, IDH2, WT1, DNMT3A, RUNX1, and RAS (20, 21). The median variant allele frequency (VAF) has been reported to be the highest of all recurrently mutated genes in AML at nearly 50%, with a higher VAF potentially predictive of inferior survival in MDS and AML patients (22). TP53 alterations are associated with higher age in AML and ALL, while no such difference was found in chronic lymphocytic leukemia (CLL) and myelodysplastic syndromes (MDS) patients. Notably, this effect was specifically detected for TP53 mutations but not for TP53 deletions. It can be suggested that TP53 alterations seem to have a higher impact in acute diseases (AML, ALL) compared to MDS and CLL patients, as they are more frequent and show a clear increase with age (23).
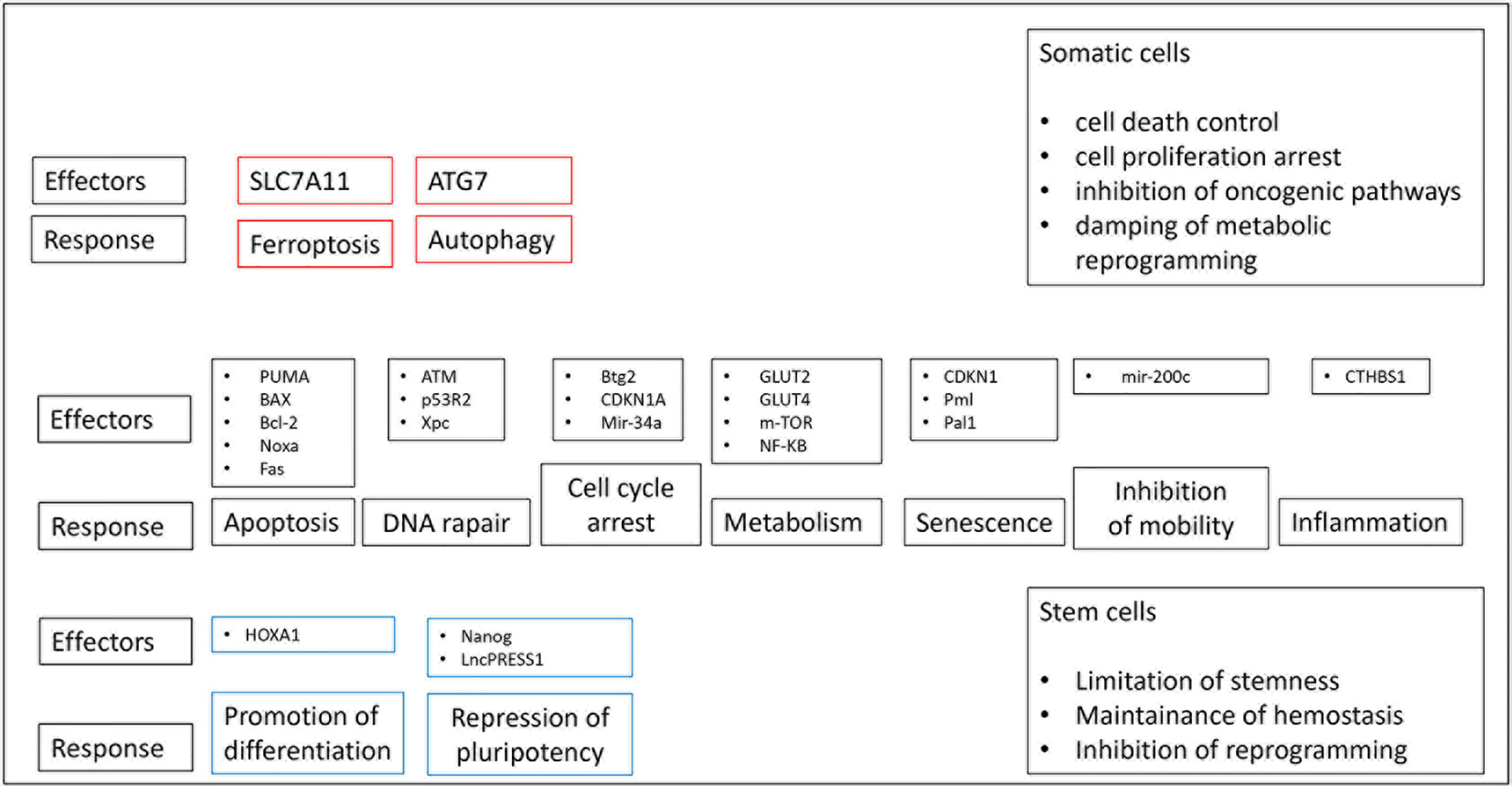
Figure 1 Several p53-mediated pathways potentially affect upon a common result of tumor suppression. P53 has the capability to control common and different biological pathways in somatic and stem cells. P53-mediated biological pathways in red boxes are specific to somatic cells, those in the blue boxes are specific to stem cells and those in black boxes are common to both cell types.
Significance and Incidence of TP53 Mutations in De Novo Acute Myeloid Leukemia (AML)
The vast majority of cases of de novo AML have unaltered TP53 alleles; data from The Cancer Gene Atlas (TCGA) including adult patients with AML documented that ~8% of AML cases harbor TP53 mutations (24, 25) (Figure 2). A consistent proportion (70–80%) of TP53 mutations in de novo AML includes missense substitutions causing amino-acid changes; the remaining mutations are truncating alterations. Missense changes determine a prolonged half‐life of the altered proteins, compared with its wild‐type, short‐lived counterpart. The molecular mechanisms leading to the prolonged half-life of mutant TP53 protein are not still completely clarified. It could in part be associated with the incomplete degradation produced by the E3 ubiquitin ligase Mdm2, whose gene is considered as a selected transcriptional target of WT-TP53. Consequently, its activation is impaired in cells carrying mutant TP53 proteins (26). The vast majority of mutations recurrently earmark “hot-spot” codons. Some “hot-spot” codon positions such as Y220C, R248Q, P72R, R273C, and R175H often overlap between AML-related gene mutations and TP53 alterations detected in solid malignancies (2, 4). These alterations either directly produce the degradation of the DNA-binding domain of TP53 or induce conformational variations of the TP53 protein, thus determining a sever impairment of TP53 functions (27). Analysis of the respective frequency of TP53 mutations across the TCGA datasets shows that AML is one of the tumors with the lowest TP53 alteration rate among all human cancers (24). This may mean that both TP53 inactivation is not necessary for the occurrence or maintenance of AML and AML may influence TP53 activities due to alternate mechanisms. In AML, the loss of one allele of chromosome 17p is frequently associated with TP53 mutation resulting in a loss of heterozygosity (25). In contrast, TP53 mutations unrelated to cytogenetic alterations represent a rare event (28). In addition, 10–30% of de novo AML cases harboring TP53 mutations hold a cytogenetically detectable TP53 deletion with wild-type conformation of the remaining allele (29). Results from studies conducted on mice show that the loss of one TP53 allele might be sufficient for promoting multistep leukemogenesis mechanisms (30). This may be significant for the occurrence of leukemia in patients harboring a single TP53 deletion. An alternative hypothesis consists in the inhibition of downstream mediators involving in the TP53 intracellular pathways, which affect both the cell-cycle arrest and the repair of DNA damages and mechanism of apoptosis. Alternatively, up-regulation of genes, which inhibit TP53 functions and favor inactivation of TP53 may be contemplated, for instance MDM2 gene amplifications have been identified in CLL (31). Aberrant overexpression of the p53 protein is usually associated with TP53 alterations and a complex karyotype, but the prevalence and impact of p53 overexpression in AML with diploid cytogenetics is unknown. Assi et al. analyzed survival outcomes according to p53 expression level quantified in bone marrow core biopsy samples using immunohistochemistry and computer‐assisted image analysis, in patients with diploid karyotype. They observed that the median leukemia free survival was significantly shorter in the subset of patients with p53 overexpression (9 vs 55 months; p = 0.026), including those who were consolidated with allogeneic stem-cell transplantation (ASCT) (32). These data suggested that p53 expression level could be helpful to stratify patients with AML and a diploid karyotype, currently classified as intermediate‐risk disease and that p53 expression level could help to identify patients who could possibly benefit from post-ASCT maintenance therapy. Several reports have also described the association between erythroid/myeloid leukemia or AML-M6a (acute erythroid leukemia [AEL]) with high-risk biological features, including high frequency of TP53 mutations in up to 53% of patients with AEL (33, 34). Recently, Montalbano-Bravo et al. showed that also in pure erythroid leukemia (PEL) which represents 14% of cases of all erythroid leukemia, there is a high prevalence of chromosome 17 abnormalities and mutations in TP53; interestingly, in several patients, there was co-occurrence of both, or double TP53 mutations. Therefore, these data demonstrated that the loss of TP53 function was a strong influential factor for PEL pathogenesis consisting in dismal prognosis irrespective of currently available therapies and that the enrolment in clinical trials targeting or circumventing mutant p53, and less intensive approaches is the current optimal strategy for this subset of patients (35).
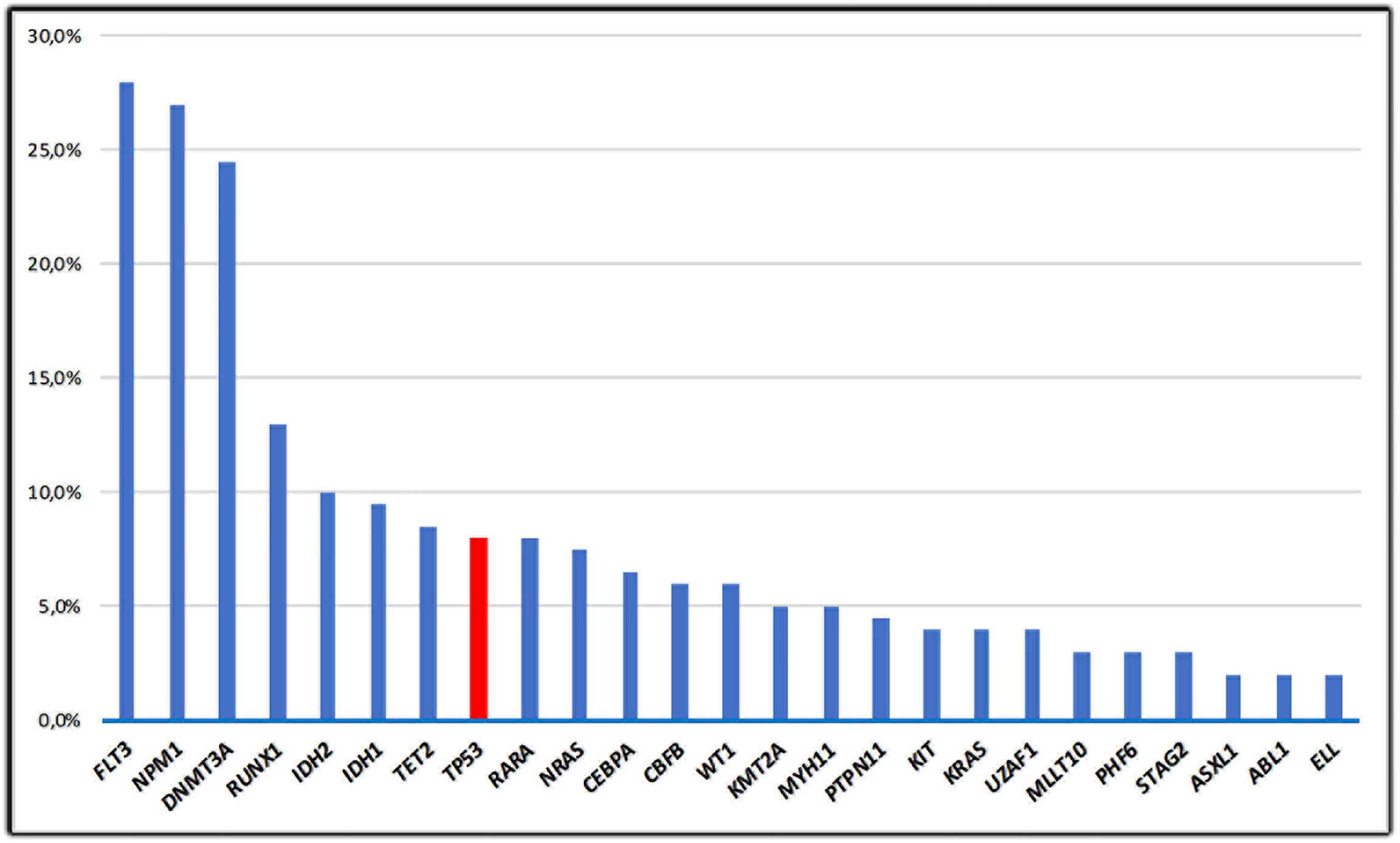
Figure 2 The frequency of TP53 mutations in de novo AML; mutations in AML from the TCGA study (11) are shown in this figure. Only the 25 most frequent mutations are represented.
TP53 Co-occurring Mutations in AML
Assessment of co-occurring gene mutations has identified TP53-altered AML cluster as a unique disease subgroup; nevertheless, AML with TP53 mutations are not specifically correlated with a unique transcriptional signature by standard RNA sequencing. In fact, TP53 alterations may co-exist with some other frequent AML-related single-nucleotide mutations such as TET2, NPM1, FLT3, DNMT3A, IDH1, and IDH2; in addition, TP53-mutant AML are often characterized by recurrent co-occurring karyotypic structural aberrations, especially genomic abnormalities detected in certain chromosomes (chromosome 5, 7, and 17), and with events involving chromothripsis (36–38). Interestingly, although karyotypic instability has been associated with TP53 inactivation, only distinct forms of increased mutagenesis have been identified among TP53-mutated AML; TP53-mutations tend to co-exist with augmented numbers of major karyotipic abnormalities, marker chromosomes and chromothripsis (39), but not with a potentially rise in single-nucleotide mutations (40). Mutations can co-occur within founding clones or sub-clones or can be found in alternative clones, which are not correlated with the neoplastic hematopoietic cell. Two patterns of TP53 co-mutations often emerged. First, mutations in TP53 and epigenetic genes (i.e. IDH1, IDH2, DNMT3A, TET2) and transcription factors (i.e. CEBPA, RUNX1, NPM1) commonly co-occur in the founding clone. Contrariwise, mutations involving triggered signaling pathways (i.e. FLT3, RAS, PTPN11, BCOR, JAK2, NF1) and polycomb mechanisms (i.e. SF3B1, KDM6A, SRSF2) frequently occurred in sub-clones, indicating that these alterations may productively stimulate TP53 AML throughout clonal evolution. Mutations involving ASXL1, CBL, and U2AF1 genes arise almost identically in sub-clones of TP53, and with TP53 as a variant in a novel incoming sub-clone (36). Kadia et al. analyzed peripheral blood or bone marrow samples from 293 de novo AML patients using targeted amplicon-based NGS-based mutation analysis. They found TP53 mutations in 53 patients (18%); 45 patients harbored a missense mutations with the most frequent amino acid replacement being a substitution of arginine for histidine in various codons. There was a significant lower presence of mutations involving FLT3 (6 vs. 19%; p = 0.02), RAS (4 vs. 14%; p = 0.04), and NPM1 (8 vs. 20%; p = 0.03) genes in mutant-TP53 AML in comparison with WT-TP53 AML (41). Mutations of TP53 were detected by NGS in 105 out of 2,272 (5%) AML patients enrolled in the Study Alliance Leukemia AML96 and AML2003 multicenter studies. Among these patients, none showed additional low-risk mutations, such as t(8;21) or inv (16), while a significant direct correlation between TP53 deletion and other high-risk features was observed for del(5q) (P < 0.001), −5 (P < 0.001) and −7 (P < 0.05). The well-known molecular risk factors FLT3-ITD (internal tandem duplication) and NPM1 mutation showed a lower incidence then TP53 deletion in patients with complex karyotype (P < 0.001) (17). An Italian study analyzed 40 AML patients for copy number alterations (CNAs) (detected by Affymetrix SNP arrays) potentially associated with TP53 and found several CNAs ranging from loss to gain of complete chromosome arms. Specifically, gains located on chromosome 8 were statistically associated with TP53 mutations. In addition to trisomy of chromosome 8, others CNAs related to other chromosomes are significantly associated with TP53 mutations: loss of chromosome 5q, deletion of chromosome 3(p22.3), deletion of chromosome 12(p12.3), and gain of chromosome 17(p11.2), chromosome 16p(11.2-11.3), and chromosome 14(q32.33) (42). Recently, the International Working Group for Prognosis in MDS (IWG-PM) assessed 3,324 MDS patients for TP53 mutations and allelic imbalances of the TP53 locus observing that one in three patients harboring TP53 mutations displayed mono-allelic targeting of the gene whereas two in three showed multiple hits reflecting bi-allelic targeting. The noted correlation between TP53 and high-risk presentation, complex karyotype, rapid leukemic transformation, and poor survival were directly associated with patients displaying the multi-hit state only. TP53 multi-hit state resulted in combination with high-risk occurrence, complex karyotype, and dismal survival and predicted risk of leukemic transformation and death regardless of the Revised International Prognostic Scoring System, while patients presenting mono-allelic alterations did not show differences when compared with those with WT-TP53 (43). In AML, TP53 mutations correlate with low-rate of response to standard chemotherapy regimens and dismal OS in patients presenting or not a complex karyotype. By using a multivariate analysis, the presence of TP53 mutations without a cytogenetic aberration predicted for inferior OS and lower response to chemotherapy (26). In fact, all patients with TP53 deletion showed significantly poorer 2-year DFS as well as shorter 2-year OS compared to patients with complex aberrancy without TP53 deletion (29). Furthermore, 17p-loss of heterozygosity (LOH) (44), chromosome 17 abnormalities (45), and up-regulation of full-length protein isoform 22 (46) additionally predicted for unfavorable prognosis in AML. Detection of the TP53 allelic state is crucial for defining diagnosis, risk assessment and precise prognostication in MDS, and further studies should include the evaluation of TP53 allelic state in AML. TP53 mutations were assessed not only as a dichotomous variable. With the advent of NGS technologies, mutational sub-clones can now be detected with high sensitivity. A recent study investigated the clinical characteristics associated with sub-clonal TP53 mutations and their prognostic impact in a large cohort of AML patients prospectively treated within studies of the “German-Austrian AML Study Group” (AMLSG). A total of 108 TP53 mutations were found in 98 patients (6.4%) and patients with TP53 mutations were categorized by their VAF into groups with frequencies >40, 20–40, and <20%. Authors demonstrated that even TP53-mutated sub-clones, defined by a VAF <20%, have a statistically significant negative prognostic impact with respect to complete remission rate, OS, and EEFS. However, compared to patients who harbored clonal TP53 mutations, those with sub-clonal alterations showed significantly fewer complex karyotypes and chromosomal losses (47). These data suggest that sub-clonal TP53 mutations represent a novel significant prognostic factor in AML and may have implications for incorporating NGS among TP53 routinely screening methods and for re-defining future risk stratification in AML.
Role of TP53 Mutations in Therapy-Related AML (t-AML)
Mutations involving TP53 gene are more commonly found in AML characterized by augmented genomic instability, such as relapsed AML, therapy-related (t-AML), and older AML patients (48, 49). In t-AML, TP53 mutations are present in about 30% of patients (Table 1) (44), while in elderly patients with complex‐karyotype AML, TP53 lesions can be present in up to 70% of cases (57). Over the last years, it was hypothesized that the higher rate of TP53 mutations in t-AML was directly associated with the cytotoxic effect of chemotherapy and radiation on hematopoietic stem/progenitor cell (HSPC) DNA. However, recently Wong et al. reported that the cytotoxic effects of standard chemotherapies do not directly produce TP53 mutations. They postulated a model in which a low number of hematopoietic stem/progenitor cells (HSPC) (<0.7%) harboring age-related TP53 mutations present resistance to conventional therapies and expand preferentially after chemotherapy (54). The precocious occurrence of TP53 genetic alterations in the founding HSPC clones likely determines the increased rate of cytogenetic aberrations and scarce responses to treatments that are typically observed in t-AML/t-MDS patients. It is thus challenging to predict which cases may develop a t-AML and to find ways to prevent it. An MD Anderson Cancer Center (MDACC) study, which collected 108 consecutive patients with t-MDS/t-AML showed that TP53 mutations in this setting were predominantly located in DNA-binding domains presenting an allelic frequency of 37% (range between 7.1 and 98.8). The majority of TP53 gene mutations included single nucleotide variants, among which transitions (65.9%) were more frequently observed than transversions (34.1%). Missense mutations appeared the most commonly detected, followed by non-sense and frameshift alterations and the TP53 mutation pattern was notably similar to that observed in newly diagnosed MDS/AML (29). Peculiar subsets of altered genes detected in t-AML and in newly diagnosed AML have been reported. According to prior studies (41, 44, 45), TP53 mutations were remarkably higher in t-AML compared with de novo AML but presented lower rate of driver mutations such as NPM1 and DNMT3A. Furthermore, TP53 mutations seem to be mutually exclusive with other gene mutations such as NPM1, FLT3, ARF, and MDM2 (58).
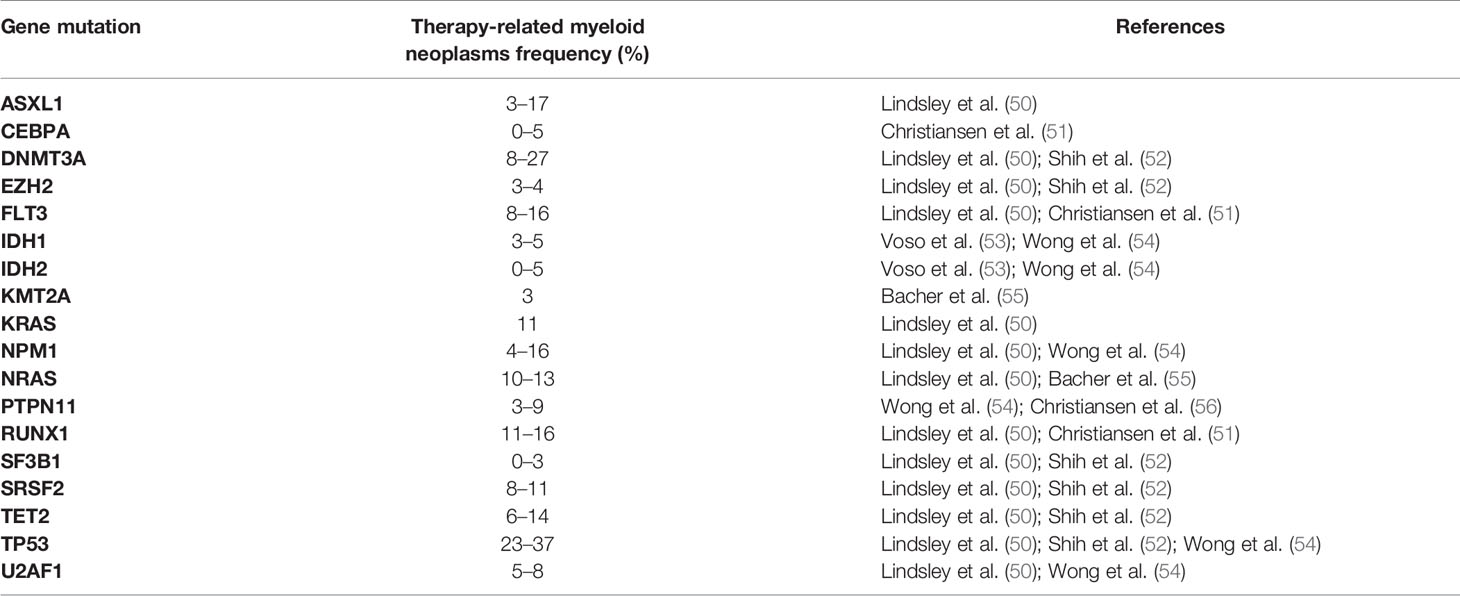
Table 1 Recurrent mutations in therapy-related neoplasms.
Prognostic Impact of TP53 Mutations in AML
The European Leukemia net (ELN) guidelines represent probably the most established source for evaluating risk of resistance and classifying patients into “favorable,” “intermediate,” and “unfavorable” groups according to cytogenetic and molecular mutations detected in leukemic cells. The 2017-updated version considers TP53 mutated cases as an independent group to be included in the adverse prognostic risk category (59). In 2012, the German group identified five distinct prognostic subgroups according to molecular characterization through a univariate and multivariate Cox regression analyses (60). TP53 mutated cases treated with intensive standard chemotherapy [“3+7” scheme, cytarabine (ARA-C)-daunorubucin (DNR) based regimen] were a very unfavorable prognostic group presenting the worse outcome (OS at 3 years: 0%). Recently, Papaemmanuil et al. analyzed 1,540 patients with AML using targeted sequencing of 111 myeloid cancer genes (13). Patterns of co-mutations distinguished AML patients into 11 categories, each with a specific clinical behavior and outcome. In addition to noted disease classes, three more heterogeneous AML categories were identified: AML presenting mutations of chromatin and RNA-splicing regulators, AML harboring TP53 mutations and/or chromosomal aneuploidies, and AML with IDH2R172 abnormalities. Patients with chromatin–spliceosome and TP53–aneuploidy AML presented dismal prognosis, with the different class-defining mutations independently and additively affecting outcomes. Multiple studies have reported that the presence of TP53 mutations produce low response to treatments, significant rate of relapses, and poor prognosis (60, 61). Using a multivariate analysis adjusted for age, performance status, cytogenetic risk group, de novo/t-AML leukemia, and white blood cell count, Bowen et al. demonstrated that having a TP53 mutation was independently correlated with inferior survival in adult AML patients treated with ARA-C-anthracycline based treatments (DAT = DNR, ARA-C, thioguanine and ADE = DNR, ARA-C, etoposide regimens) (62). Outcome results of patients presenting high-risk cytogenetic score at diagnosis showed that the presence of TP53 mutations contributed to an inferior rate of complete response (CR) (28% mutated versus 50% un-mutated), and a lower DFS and OS (59). Although ASCT represents the unique potentially curative treatment for TP53-mutated AML/MDS patients, the risk of death after the procedure remains significantly high, with the majority of deaths mostly related to the occurrence of disease relapse. In a recently published analysis of the European Society for Blood and Marrow Transplantation (EBMT), authors reported that patients with a 17p abnormality who achieved a CR1 and underwent ASCT had a 2-year OS and leukemia-free survival rate of 28 and 24%, respectively. The 2-year non-relapse mortality (NRM) was 15%, and the 2-year rate of relapse was 61% (63). Among a large cohort of 289 patients with TP53-mutated MDS assessed in a Center for International Blood and Marrow Transplant Research study, the 3-year OS was 20%, while the median OS was 0.7 years (64). The MDACC group postulated that the outcome of TP53-mutated AML/MDS patients are not uniformly poor and focused on identify potential prognostic factors predicting survival in those patients who underwent ASCT after achieving a CR (65). Investigators stratified patients with TP53-mutated AML/MDS according to the hematopoietic cell transplantation specific comorbidity index (HCT-CI), performance status (PS), and disease status at the time of transplant and individualized a distinctive group of patients who showed significantly better outcomes after the procedure. Patients presenting more than one risk factor in the analysis showed inferior long-term survivals and should be enrolled in new trials including novel therapies before and/or after ASCT in order to reduce the potential risk of relapse and prolong survival after the procedure.
Functional Classification of TP53 Mutation in AML
It is not yet elucidated whether distinct types of TP53 mutations (non-sense, missense, deletions, insertions, slice site mutations) produce a uniformly dismal outcome in AML. Several studies have demonstrated that there are no differences in terms of OS and EFS between AML patients with missense TP53 mutations and those with truncating aberrations and that GOF activities elated for some missense TP53 variants do not represent the crucial mechanism of treatment resistance (66–69). A statistically significant impact on survival was identified between non-disruptive and disruptive TP53 mutations in a large study including patients with head and neck cancers (70). “Non-disruptive mutations” are identified as mutations arising inside the L2 or L3 binding domains that determine a substitution of an aminoacid from the same polarity/charge category, or mutations outside the L2 or L3 binding domain (except stop mutations). “Disruptive mutations” are categorized as DNA hot-spot aberrations that produce a STOP-sequence determining a desegregation of TP53 protein codification or DNA sequence alterations that arise inside the L2 or L3 binding domains (codons 163–195 or 236–251) and cause the substitution of an aminoacid with another of a different polarity/charge category. Conversely to what has been shown for solid tumors, discrimination into disruptive and non-disruptive TP53 mutations does not indicate a different outcome in patients with AML (69). The Evolutionary Trace (ET) method is an extensively standardized approach to assess key functional or structural residues in proteins (71). A scoring system (EAp53) was developed in solid tumors (72) to categorize missense mutations of TP53 gene into high- and low-risk alterations. Consequently, the same evolutionary action score (EAp53) has been also applied in AML patients to predict which types of TP53 mutations are more deleterious. This algorithm includes mutations scoring between 0 and 100 with WT-TP53 sequences holding a score of 0. In a recent study including a large cohort of 1,537 patients (AMLSG cohort) with newly diagnosed AML who received intensive treatments (69), the EAp53 score was applied to identify missense mutations and distinguish between low-risk (<75 mutations) and high-risk (≥75 mutations) groups (49 and 35 patients in the high-risk and in the low-risk group, respectively). However, utilizing a multivariate Cox proportional analysis a significant difference on OS and/or EFS was not detected (69). Recently, the Israeli group created a synthetic TP53 mutation dataset and calculated the functional impact of more than 10,000 DNA-binding domain (DBD) TP53 alterations in human cells in culture and in vivo (73). They proposed an algorithm based on the average of the relative enhancement or depletion of a specific TP53 mutation detected at three different time points called “relative fitness score (RFS).” A high RFS identifies advantageous expansion in culture showing higher fitness of the variant, while a low RFS indicates advantageous depletion. In the AMLSG cohort a statistically significant superior OS (median 12.9 versus 5.5 months, p = 0.017) and a trend in longer EFS (7.3 vs 5.2 months, p = 0.054) were found for patients with a low-risk RFS comparing with those with a high-risk RFS (69). Therefore, the AML-specific RFS algorithm seems to hold a prognostic significance in TP53-mutant AML and to be a functional source for treatment decision-making.
Treatment Options Beyond Standard Chemotherapy
Patients with mutant TP53 AML who are treated with standard anthracycline-based and ARA-C-based induction regimens show poor outcomes, with post-induction response rates to standard therapeutic approaches amounting to 20–30% (37, 61). Furthermore, median survival in this setting range between 4 and 6 months, with an estimated OS after ASCT of roughly 10 months (37, 61). These unsatisfactory results clearly indicate that the dismal prognosis in patients with an unfavorable-risk cytogenetic profile, the presence of TP53 mutations, or both may be crucial for the therapeutic approaches and may be mitigated with other potentially effective treatment modalities.
Decitabine
Decitabine (DAC) (5-aza-2′-deoxycytidine) is routinely employed as monotherapy for treating patients with MDS and older patients with AML (74). Several studies have sought to identify biomarkers such as mutations and DNA methylation alterations in IDH1, IDH2, TET2, and DNMT3A that might predict responses to DAC (75, 76). Interestingly, recent clinical trials have demonstrated that patients with AML harboring TP53 mutations displayed favorable responses to treatment with DAC (77, 78), despite the molecular mechanisms associated with DAC responses being currently unclear. Moreover, clonal assessments of patients treated with DAC showed the prominent, but not durable, clearance of sub-clones with TP53 mutations (79). In a study, including 116 patients (26 with MDS) treated with a 10-day regimen of DAC in monthly cycles, Welch et al. reported high rates of complete morphologic response among TP53 mutant patients (77). They observed a higher response rate in AML patients presenting an unfavorable-risk cytogenetic score compared with those stratified as intermediate-risk or favorable-risk according to cytogenetic analysis (67 vs. 34%). A higher response rate was also seen comparing patients with TP53 mutations and patients with WT-TP53 (100 vs. 41%). However, this study does not contribute to elucidate the exact mechanisms causing the marked sensitivity of patients with TP53-mutant AML or MDS to DAC. Further studies including 5-day regimens of DAC in monthly cycles reported 62 and 66% response rates in TP53-mutant AML or MDS patients, respectively (17, 80) and cell-line analysis also suggested a specific sensitivity of TP53-altered cells to hypomethylating treatment (81). Later, Chang et al. demonstrated in univariate and multivariate analyses that TP53 mutations were the unique molecular abnormalities, which predicted a CR achievement after DAC in patients with MDS (78). Using routine immunohistochemistry (IHC), Ruzinova et al. also found that expression levels of TP53 is associated with either TP53 gene mutation profile or clearance in patients treated with DAC (82). Therefore, the authors postulated that TP53 IHC may be used as a valid surrogate for assessing TP53 mutation profile in patients with AML and may be specifically functional in cases for whom molecular testing cannot be applicable. Four studies (83–86), although they primarily included patients with MDS (532 of 550 patients, 97%), showed that patients with TP53 mutations treated with various regimens of hypomethylating agents were characterized by response rates that were similar to, but not higher than, the rates observed among patients with WT-TP53 (Table 2). In a single-center, open-label, randomized phase II trial, Short et al. assessed the efficacy of DAC given in either 5-day or 10-day schedules. There was no difference in response rates and outcomes between the two DAC schedules overall or when patients were stratified by cytogenetic risk, de novo versus t- or s-AML, or TP53 mutation status. Authors observed several preliminary findings in this study that should be further evaluated in larger cohorts. First, they did not observe a correlation between baseline TP53 VAF and response. In addition, they observed that the median TP53 VAF at the time of remission was 8.4%, with four patients with a VAF of ≥20% at the time of remission, suggesting that some of these TP53 mutations may be present in pre-leukemic clones and not exclusively in myeloblasts. Finally, they reported that the two patients in whom a TP53 mutation could no longer be detected in the remission bone marrow had the longest durations of remission (87). Therefore, despite the current fervor for DAC, DAC as monotherapy does not seem to produce deep and durable responses in a TP53-mutated setting, and additional consolidation therapies appear to be necessary.
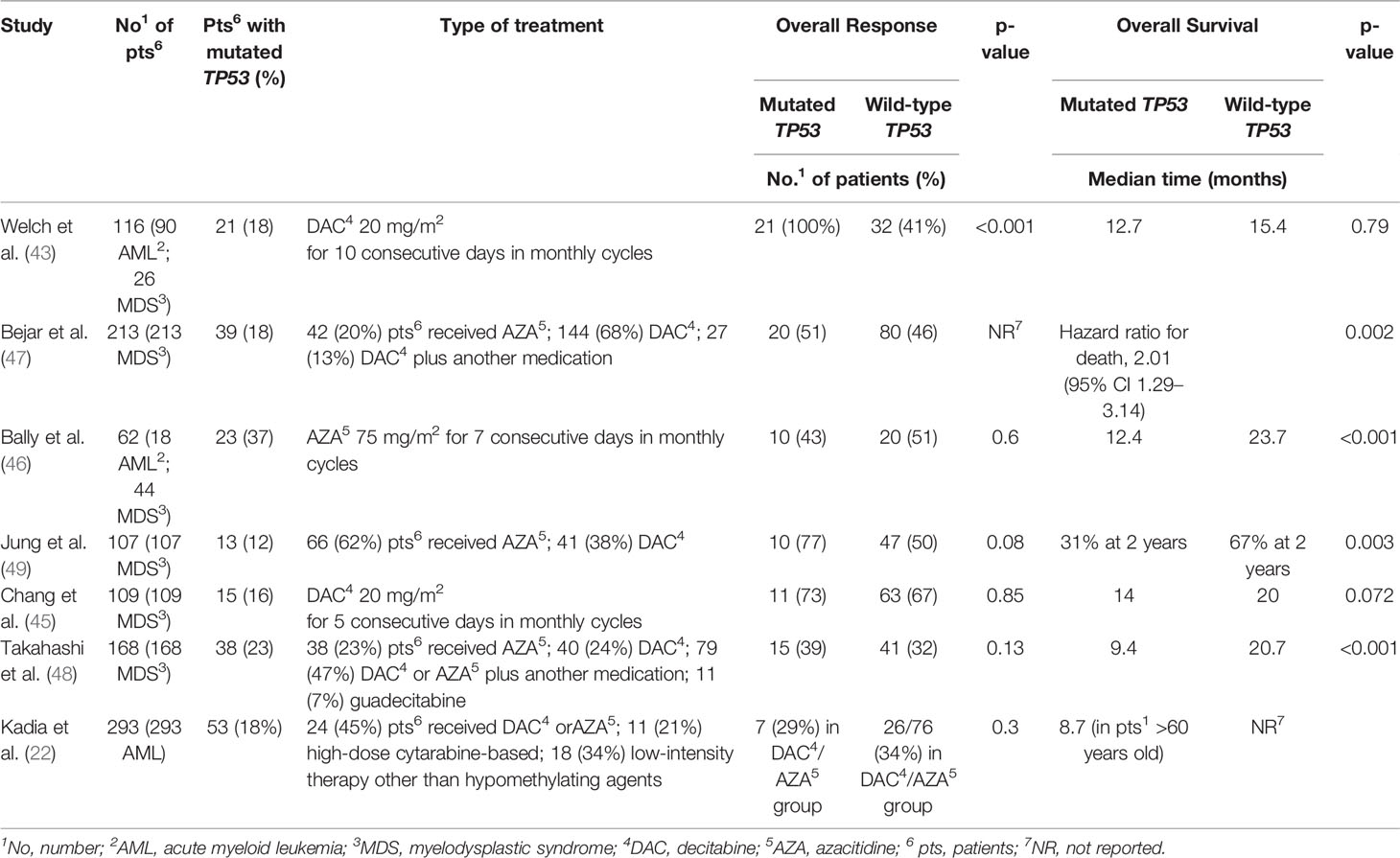
Table 2 Outcomes of patients with AML and MDS treated with hypomethylating treatments as single agent according to TP53 mutation status.
Venetoclax
Venetoclax (VEN), a bcl-2 inhibitor, has displayed promising efficacy in combination with hypomethylating agents (HMAs) either in de novo or in relapsed/refractory (R/R) AML (81, 88, 89). Apoptosis mediated by VEN appears to be TP53‐independent (90, 91), and VEN/HMAs combination activity was reported in several high-risk leukemia genetic groups (81, 88, 89). In the trial that led to VEN approval by the FDA, encouraging remission rates were reported in patients harboring TP53 mutations; in this setting, CR + complete remission with incomplete blood count recovery (CRi) rates was 47%, median duration of CR + CRi and median OS was 5.6 and 7.2 months, respectively (88). In a post hoc exploratory analysis assessing the baseline prognostic factors, TP53 status figured as a significantly positive predictor for CR + CRi using univariate logistic regression models (92). These data suggested improvement compared with historical results that documented CR rates as low as 28%, although duration of response was short, consistent with those reported elsewhere (93, 94). Also, Aldoss et al. have reported that responses to VEN/HMAs combination strategies were encouraging not only in de novo AML, but also in the group of patients with R/R TP53 mutated AML (CR/CRi rate 38%), which included also patients relapsing after prior ASCT (95). Furthermore, in this cohort, response rates were similar comparing patients who received either a 5‐ or a 10‐day schedule of DAC plus VEN and the VAF of TP53-mutated cases was comparable in responders and non‐responders, conversely to an MDS trial in which TP53 VAF affected outcomes of patients treated with HMAs (96). The MDACC group reviewed 69 patients with AML and TP53 mutations who received VEN-based regimens between 2014 and 2018 (52% in frontline and 48% in the salvage setting). The overall response rate (ORR) was 47 and 24% in de novo and R/R patients with AML, respectively; all six patients with negative MRD obtained a complete cytogenetic response after receiving VEN and maintained a CR for a median of 3.4 months (97) (Table 3). Recently, DiNardo et al. analyzed 81 patients with AML treated with VEN-based combinations in order to identify molecular correlates of durable remission, response followed by relapse (adaptive resistance), or refractory disease (primary resistance) (92). High response rates and durable remissions were typically associated with NPM1 or IDH2 mutations, with prolonged molecular remissions prevalent for NPM1 mutations. Primary and adaptive resistance to VEN-based combinations were mainly correlated with two major mechanisms of resistance: activated kinase signaling and bi-allelic TP53 perturbation. Using CRISPR/Cas9 to disrupt TP53 function in AML cell lines, authors demonstrated that TP53 loss favored therapeutic resistance to VEN, HMAs, and ARA-C alone as well as in combination. Notably, these data are strongly sustained by further preclinical reports which, using a genome-wide CRISPR/Cas9 screen in leukemic cell lines, showed VEN resistance associated with defects in apoptotic pathway members (TP53, BAX, and PMAIP1) (98, 99). Therefore, these findings suggest the need of surveying genetic TP53 integrity both at diagnosis and in remission in order to identify patients likely to develop resistance to VEN-based therapy. The combination of VEN/HMAs seems to produce better outcomes compared with the poor prognosis reported in TP53-mutated AML patients who received combination treatments based on conventional chemotherapy (93, 94), indicating that improving response rates and remission duration may represent a safe bridge to transplantation for more patients.
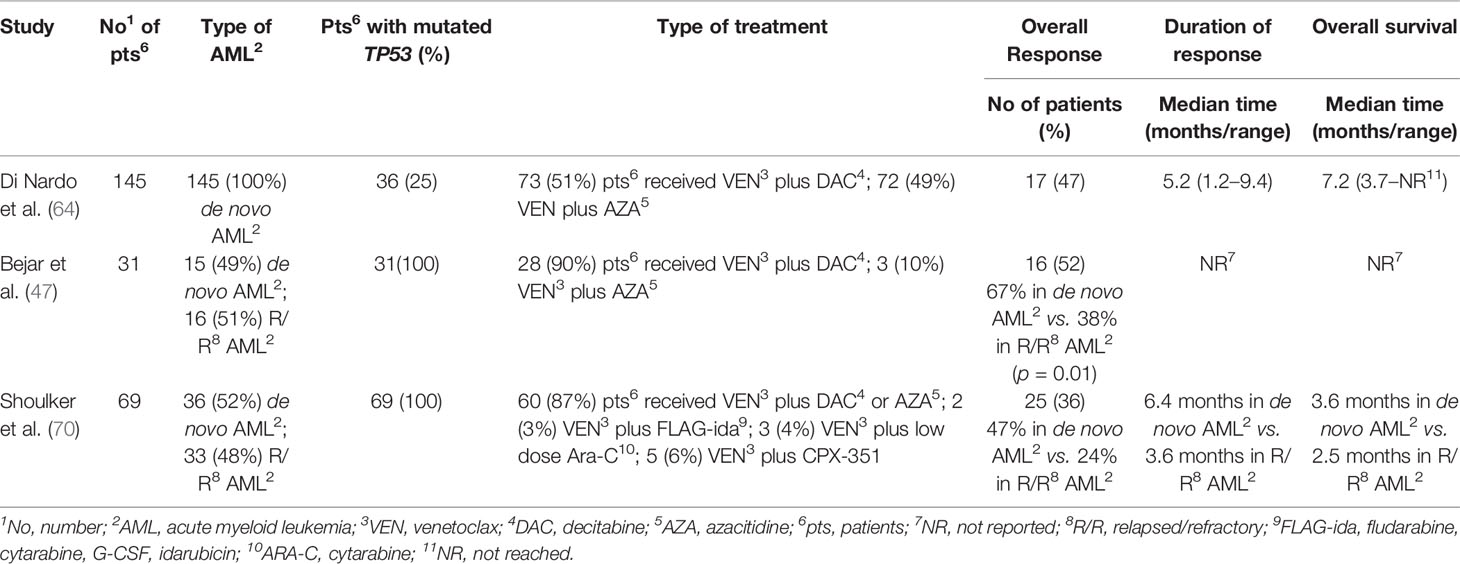
Table 3 Outcomes of patients with AML treated with venetoclax combination strategies according to TP53 mutation status.
Other Therapeutic Perspectives
Alternative approaches are ongoing to treat the ultra-high-risk class of patients with TP53-mutated AML. To date, AML with TP53 mutations lacks a clinically standardized therapeutic strategy aimed at targeting these alterations. The primary therapeutic end-point in this setting should be to restore normal TP53 functions, reactivating intact TP53 activities. However, multiple clinical trials are ongoing with the aim of selectively targeting mutant TP53. Recently, small molecules have been developed with the specific abilities of destabilizing individual TP53 point mutants (100). These are exciting, targeted therapies although their effectiveness might be challenging to evaluate in AML clinical trials due to the small number of patients with target mutations (Table 4).
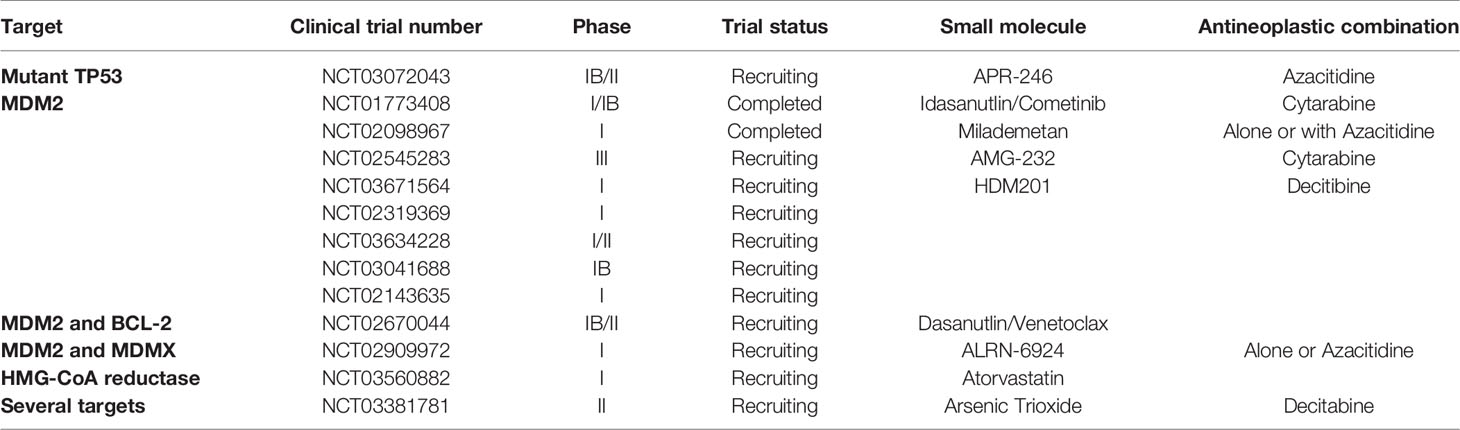
Table 4 Ongoing clinical trials targeting TP53 mutant and wild-type AML.
Potential Molecularly Targeted Therapies for p53-Mutant Cases
Several approaches, including inactivation of mutant p53, degradation of mutant p53, and restoration of the wild type function of p53, have been studied. Overall, two crucial approaches have been used to target mutant TP53 in treatment of human cancers. The first approach includes small agents able to directly target mutant p53 by induction of its degradation or reactivation of its tumor-suppressive transcriptional activity (101). Among compounds that induce degradation of mutant p53 there are agents such as Hsp90 inhibitors [reverse the Hsp90’s function to inactivate MDM2 and Hsc70‐interacting protein (CHIP)] (102), statins (induce CHIP-dependent degradation of p53 with conformational changes) (103), or HDAC inhibitors (inhibit HDAC6 and disrupt the HDAC/Hsp90/mutant p53 complex) (104). Among compounds that restore wild-type function of p53 there are agents such as CP-31398 (stabilize the DNA-binding core domain and induce conformational changes) (105), PETIC (sensitize the p53 mutant to proteasome-mediated degradation and restore p53 WT conformation) (106), RITA (reactivate p53 in mutant p53 cancers by inhibiting the p53-HDM2 interaction) (107), and COTI-2 (restore WT p53 activity by targeting and binding to misfolded p53 mutant) (108). The alternative approach is to targeting pathways that are critical for the survival and growth of p53 mutant cancers. Specific molecular targets include G2/M regulators (CHK1/2, MK2, PLK2) (109–111) kinases (SGK2, MPS1) (112, 113) and growth pathways (p38, DAPK1) (114, 115). This latter approach is justified by the fact that, over the last years, several molecules was identified to be critical for survival or growth of cells with p53 mutations that can be targeted for the selective treatment of p53-mutant cancers (116, 117). Ongoing efforts continue to identify such critical mutant p53-specific survival and growth regulatory pathways. Therefore, novel drugs that target mutant p53 or the critical pathways activated by p53 mutation are highly promising for effective treatment of many cancers, including AML and probably these agents represent the future landscape in AML scenario.
APR 246
Pharmacologic strategies able to stabilize both WT and mutant TP53 promoting the reactivation of tumor-suppressor activities are under study in AML. The most encouraging is APR-246, a pro-drug converted to methylene quinuclidinone (MQ), which links through a covalent bond the mutant TP53 core domain, promoting the increased regulation of transcriptional factors involving in apoptotic pathways (116, 117). Therefore, cells acquire functions promoting apoptotic mechanisms and cell cycle arrest pathways. The first human trial including the use of this agent showed a reduction of blast percentage from 46 to 26% in the bone marrow of the unique patient enrolled in the study with AML harboring a TP53 core domain mutation (118). In a recent phase Ib/II trial, APR-246 was associated with 5-azacitidine (5-AZA) for the treatment of TP53-mutated MDS and AML patients. Interestingly in 11 evaluable patients, the combination of these agents provided an ORR of 100% and a CR rate of 82%. Moreover, the majority of remissions were characterized by deep molecular responses showing a median VAF of 0.3% in NGS-negative patients (119). The efficacy of APR-246 as maintenance therapy after ASCT in AML and MDS patients is currently being assessed in phase II trials. A phase I dose-finding and cohort expansion trial is also ongoing to assess the safety and preliminary efficacy of APR-246 combined with VEN and 5-AZA in patients with myeloid malignancies (NCT04214860).
Immunotherapeutic Approaches
Checkpoint inhibitors function inhibit co-receptors on T-cells, such as PD-1 and CLTA-4, which play a role in immune evasion by cancer cells. Augmented expression of these receptors and their ligands have been detected on leukemic blasts, with further upregulation observed after treatment with HMAs (120). This suggests a possible resistance mechanism to HMA therapy and provides rationale for combination therapy. Notably, increased expression of PD-L1 has been identified in blasts and hematopoietic stem cells in TP53 mutant patients in comparison to wild-type patients (121). A recent phase 2 study of nivolumab in combination with 5-AZA in R/R AML showed tolerable toxicity and an ORR of 33%, although higher response rates were observed in HMAs-naïve patients and those with increased bone marrow and peripheral blood T-cells by flow cytometry (122). TP53 mutations were found in 23% of patients and mutation status did not appear predictive of response. Preliminary results from trials assessing checkpoint inhibitors in combination with HMAs, in combination with induction chemotherapy and as post-remission maintenance treatment in MDS and AML patients have recently been presented demonstrating tolerability and promising early efficacy data, with numerous trials ongoing (123–129). Macrophages represent a crucial cell type involving in the innate immune response with CD47 consisted in a dominant macrophage checkpoint. CD47 has a key role in signaling pathways and appears overexpressed in myeloid malignancies leading to tumor evasion of phagocytosis by macrophages. Inhibition of CD47 induces engulfment of leukemic cells and their potential therapeutic elimination (130). Pre-clinical data has showed encouraging anti-cancer activity in several hematologic diseases including AML and MDS. Magrolimab, a CD47 inhibitor, was assessed in a phase 1b study either as single agent in R/R AML/MDS or in association with 5-AZA in untreated AML patients non-eligible for intensive chemotherapy and untreated MDS patients who are stratified as intermediate, high, or very high risk according to the Revised International Prognostic Scoring System (IPSS-R) (131). Forty-six AML/MDS patients were efficacy evaluable. In AML patients, 14/22 (64%) showed an objective response with 55% achieving a CR/CRi. In MDS 22/24 (92%) patients responded with 50% achieving a CR. The MRD negativity was seen in 57 and 23% in AML and MDS responders, respectively. Magrolimab + 5-AZA efficacy was also investigated in AML patients with TP53 mutations. The CR/CRi rate in 9 TP53 mutant AML patients was 78% with 44% achieving CR and 33% achieving CRi. In addition, MRD negativity was observed in 57% of responders and median duration or median survival was not reached with a median follow-up of 6.9 months. TP53 mutant VAF appeared substantially decreased or eliminated in all patients (131).
MDM2 Inhibitors
Recent results demonstrate that several AML subgroups can develop TP53 dysfunction due to the occurrence of different events, such as NPM1 and FLT3 mutations, MDM2/MDMX upregulation, fusion proteins promoting chromosomal reciprocal translocations, and aberrant expression of specific miRNAs (132). MDM2 inhibition is a promising target in the treatments of AML, serving as a negative regulator of TP53; the activation of TP53 functions induced by any stimuli or DNA damages up-regulates transcription of MDM2 mRNA and codification of protein that in turn links TP53 and directly decreases or inhibits its activity through multiple mechanisms (132). The first MDM2 inhibitor tested into human clinical trials was RG7112 (133). In AML scenario, RG7112 was assessed either as single agent (133) or combined with low-doses of ARA-C (134). Some patients even reached CR after having RG7112 and received transplant. RG7388 (idasanutlin) is a second-generation MDM2 inhibitor that resulted more selective and potent, and hold a better pharmacodynamics/pharmacokinetic profile when it was compared with RG7112. Furthermore, this agent produced dose-dependent TP53 stabilization, apoptosis, and cell cycle arrest (135). In a multicenter phase I/Ib trial, idasanutlin was assessed in AML patients either as single agent (5-day schedule of idasanutlin in monthly cycles) or in association with ARA-C (1 g/m2 IV for 5 days every 28 days) at escalation doses (136). Seventy-six patients were enrolled in the combination arm; specifically, 23 and 21 patients in the dose escalation and dose expansion cohorts, respectively, while 32 patients in a bridging cohort (137). The CR rate was 25% (n = 19); the composite CR rate [composite CR (cCR), CR + CR with incomplete platelet recovery (CRp) + Cri] was 29%. In order to define biomarkers able to predict response, MDM2 protein expression on peripheral blood leukemic blasts and HSPC was measured by flow cytometry. A grater MDM2 expression both on leukemic blasts and on HSPC was strictly correlated with CR; TP53 mutational status alone was not. These data highlight the significance of MDM2 expression in leukemic blasts identifying it as a positive predictive factor for disease remission. Preclinical studies in TP53 wild-type AML tumor models have also shown synergism between idasanutlin and bcl2-inhibitors, such as VEN and ABT-263 (navitoclax) (138, 139). The bcl-2 inhibitors may induce apoptosis in G1 compartments while idasanutlin may determine G1 arrest causing nuclear fragmentation in the G1 phase of the second cycle (140). A phase I/Ib study including >60 years old patients with R/R AML who are not eligible for intensive chemotherapy is now ongoing to evaluate the efficacy of idasanutlin and VEN combination (NCT02670044).
Other Agents
Arsenic trioxide (ATO) in association with ATRA is the standard combination therapeutic approach for low and intermediate-risk acute promyelocytic leukemia (APL) patients (141). It has been reported that ATO is able to inactivate TP53 functions via the 26S proteasome pathway (142). Furthermore, ATO amplifies WT-TP53 functions and indices the upregulation of its target genes, thereby promoting apoptosis and inactivating the proliferation of neoplastic cells (143). In a recent in vitro study, leukemic blast cells deriving from patients with AML and APL were tested with graded doses of ascorbate (ASC), alone or in combination with standard concentration (1 μM) of ATO. The ASC/ATO combination was enabled to induce myeloid blasts degradation, including leukemic CD34+ cells, sparing CD34+ progenitors deriving from bone marrow and normal cord blood (144). The pro-apoptotic effect of ASC/ATO treatment seemed to be associated with augmented oxidative stress and reactive oxygen species (ROS) overexpression. Furthermore, recent studies identify a statin (atorvastatin), a cholesterol-lowering drug, as a degradation inducer for conformational or misfolded TP53 mutants with minimal effects on WT TP53 and DNA contact mutants (103).
Conclusions
TP53 mutations characterize a distinctive setting of AML associated with recurrent karyotypic aberrations, the lacking of recurrent single nucleotide mutations, dismal outcomes, and scarce responses to intensive therapeutic regimens. However, the role of TP53 mutations in AML is enigmatic. Conversely to several other human cancers, a great majority of AML display no genomic TP53 alterations. There is now growing appreciation of the fact that the unaltered TP53 status of tumor cells can be exploited therapeutically. Therefore, the use of pharmacological activators of the TP53 pathway may provide clinical benefit in AML. The importance of detecting TP53 mutations in all AML age categories is justified by the fact that the presence of TP53 mutations contribute to a very high risk of treatment failure with standard chemotherapy approaches and thus TP53 status is an important consideration for designing novel therapeutic strategies for AML. In patients with TP53 mutations, these alterations may lead to novel, selective vulnerabilities, creating opportunities for therapeutic targeting of TP53 mutant AML. DAC has been reported as a potential efficient approach, being characterized by a TP53-independent mechanism of action. Some studies showed that the survival rate among AML patients with unfavorable-risk cytogenetic profiles, TP53 mutations, or both and are treated with DAC is comparable to that of patients who have an intermediate-risk cytogenetic profile and receive DAC. However, further studies are required to verify whether these differences in survival are truly due to improved responses associated with DAC or to decreased survival rate of patients with unfavorable-risk cytogenetic profiles treated with conventional chemotherapy including anthracycline and ARA-C. The combination of VEN/HMAs seems also to give some benefit in this setting, improving response rates and remission duration and potentially representing a safe bridge to transplantation for selected more patients. Unconventional strategies and TP53-targeted therapeutics are now being tested as monotherapy or in combination with conventional drugs in order to further improve the response rate especially in R/R AML patients and to increase the number of patients potentially eligible for transplantation, which remains the only curative option for these patients. A wide armamentarium of small-molecule activators of the TP53 pathway, and an increasing understanding of molecular pathways triggered by mutant TP53 have accelerated efforts aimed at targeting TP53 function in AML. In combination with standard AML chemotherapy or emerging targeted therapies, pharmacological targeting of the TP53 pathway may provide therapeutic benefit in AML. Further therapeutic efforts and novel targeted therapeutic options will be necessary in order to overcome the unfavorable risk related to TP53 mutations.
Author Contributions
MM, CM, and PN wrote the manuscript. PF critically revised the paper and approved the final version. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
No funding or sponsorship for this article. Authors wish to thank Prof. Barbara Bain for help in revising the Manuscript.
Abbreviations
AEL, acute erythroid leukemia; AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; ASC, ascorbate; ASCT, allogenic stem cell transplantation; ATO, Arsenic trioxide; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; CR, complete response; cCR, composite complete remission; Cri, complete responses with incomplete blood count recovery; CRp, complete remission with incomplete platelet recovery; DAC, decitabine; DBD, DNA-binding domain; DFS, disease free survival; DNR, daunorubicin; EBMT, European Society for Blood and Marrow Transplantation; EFS, event free survival; ELN, European Leukemia net; ET, evolutionary trace; GOF, gain of function; HCT-CI, cell transplantation specific comorbidity index; HMAs, hypomethylating agents; HSPC, hematopoietic stem/progenitor cells; IHC, immunohistochemistry; IWG-PM, International Working Group for Prognosis in MDS; LOH, loss of heterozygosity; MDACC, MD Anderson Cancer Center; MDS, myelodysplastic syndromes; MQ, methylene quinuclidinone; MRC-AML, myelodysplasia-related changes acute myeloid leukemia; MRD, minimal residual disease; NGS, next generation sequencing; NRM, non relapse mortality; OS, overall survival; PEL, pure erythroid leukemia; PS, performance status; R/R AML, relapsed/refractory acute myeloid leukemia; ROS, reactive oxygen species; t-AML, therapy related acute myeloid leukemia; TCGA, the Cancer Gene Atlas; t-MDS, therapy related myelodysplastic syndromes; VAF, variant allele frequency; VEN, venetoclax; WT-TP53, wild-type TP53; 5-Azacitidine, 5-AZA
References
1. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature (2000) 408:307–10. doi: 10.1038/35042675
2. Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat (2002) 19:607–14. doi: 10.1002/humu.10081
3. Kitamura T, Watanabe-Okochi N, Enomoto Y, Nakahara F, Oki T, Komeno Y, et al. Novel working hypothesis for pathogenesis of hematological malignancies: combination of mutations-induced cellular phenotypes determines the disease (cMIP-DD). J Biochem (2016) 159(1):17–25. doi: 10.1093/jb/mvv114
4. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol (2010) 2:a001107. doi: 10.1101/cshperspect.a001107
5. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer (2014) 14:359–70. doi: 10.1038/nrc3711
6. Kandoth C, Mc Lellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature (2013) 502:333–9. doi: 10.1038/nature12634
7. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell (1990) 61:759–67. doi: 10.1016/0092-8674(90)90186-I
8. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer (2011) 2:466–74. doi: 10.1177/1947601911408889
9. Bernardini MQ, Baba T, Lee PS, Barnett JC, Sfakianos GP, Secord AA, et al. Expression signatures of TP53 mutations in serous ovarian cancers. BMC Cancer (2010) 10:237. doi: 10.1186/1471-2407-10-237
10. Piek JM, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol (2001) 195:451–6. doi: 10.1002/path.1000
11. Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature (1990) 348:747–9. doi: 10.1038/348747a0
12. Ross L, Peter JM. Valk Next-Generation Sequencing In The Diagnosis And Minimal Residual Disease Assessment Of Acute Myeloid Leukemia. Haematologica (2019) 104:868–71. doi: 10.3324/haematol.2018.205955
13. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
14. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Canc (2004) 4(10):793–805. doi: 10.1038/nrc1455
15. Zhao D, Tahaney WM, Mazumdar A, Savage MI, Brown PH. Molecularly targeted therapies for p53-mutant cancers. Cell Mol Life Sci (2017) 74(22):4171–87. doi: 10.1007/s00018-017-2575-0
16. Hof J, Krentz S, van Schewick C, Körner G, Shalapour S, Rhein P, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol (2011) 29:3185–93. doi: 10.1200/JCO.2011.34.8144
17. Seifert H, Mohr B, Thiede C, Oelschlägel O, Schäkel U, Illmer T, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia (2009) 23:656–63. doi: 10.1038/leu.2008.375
18. Quintás-Cardama A, Hu C, Qutub A, Qiu YH, Zhang X, Post SM, et al. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia (2017) 31(6):1296–305. doi: 10.1038/leu.2016.350
19. Kim MP, Zhang Y, Lozano G. Mutant p53: multiple mechanisms define biologic activity in cancer. Front Oncol (2015) 5:249. doi: 10.3389/fonc.2015.00249
20. Haase D, Stevenson KE, Neuberg D, Maciejewski JP, Nazha A, Sekeres MA, et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia (2019) 33:1747–58. doi: 10.1038/s41375-018-0351-2
21. Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute myeloid leukemia: the good, the bad, and the ugly. Am Soc Clin Oncol Educ Book (2018) 38:555–73. doi: 10.1200/EDBK_199519
22. Dohner H, Dolnik A, Tang L, Seymour JF, Minden MD, Stone RM, et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia (2018) 32(12):2546–57. doi: 10.1038/s41375-018-0257-z
23. Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia (2016) 31(3):705–11. doi: 10.1038/leu.2016.263
24. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
25. Ley TJ, Ding L, Raphael BJ, Raphael BJ, Mungall AJ, Robertson AG, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia the cancer genome atlas research network. N Engl J Med (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
26. Soenen V, Preudhomme C, Roumier C, Daudignon A, Laï JL, Fenaux P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood (1998) 91:1008–15. doi: 10.1182/blood.V91.3.1008
27. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387:296–9. doi: 10.1038/387296a0
28. Fenaux P, Jonveaux P, Quiquandon I, Lai JL, Pignon JM, Loucheux-Lefebvre MH, et al. P53 gene mutations in acute myeloid leukemia with 17p monosomy. Blood (1991) 78:1652–7. doi: 10.1182/blood.V78.7.1652.bloodjournal7871652
29. Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia (2008) 22:1539–41. doi: 10.1038/leu.2008.143
30. Lai JL, Preudhomme C, Zandecki M, Flactif M, Vanrumbeke M, Lepelley P, et al. Myelodysplastic syndromes and acute myeloid leukemia with 17p deletion. An entity characterized by specific dysgranulopoiesis and a high incidence of P53 mutations. Leukemia (1995) 9:370–81. doi: 10.1016/0145-2126(94)90142-2
31. Zenz T, Eichherst B, Busch R, Denzel T, Häbe S, Winkler D, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol (2010) 28(29):4473–9. doi: 10.1200/JCO.2009.27.8762
32. Assi R, Gur HR, Loghavi S, Konoplev SN, Konopleva M, Dever N, et al. P53 protein overexpression in de novo acute myeloid leukemia patients with normal diploid karyotype correlates with FLT3 internal tandem duplication and worse relapse-free survival. Am J Hematol (2018) 93(11):1376–83. doi: 10.1002/ajh.25255
33. Santos FP, Faderl S, Garcia-Manero G, Koller C, Beran M, O’Brien S, et al. Adult acute erythroleukemia: an analysis of 91 patients treated at a single institution. Leukemia (2009) 23(12):2275–80. doi: 10.1038/leu.2009.181
34. Liu W, Hasserjian RP, Hu Y, Zhang L, Miranda RN, Medeiros LJ, et al. Pure erythroid leukemia: a reassessment of the entity using the 2008 World Health Organization classification. Mod Pathol (2011) 24(3):375–83. doi: 10.1038/modpathol.2010.194
35. Montalbano-Bravo G, Benton CB, Wang SA, Ravandi F, Kadia T, Cortes J, et al. More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia. Blood (2017) 129(18):2584–7. doi: 10.1182/blood-2016-11-749903
36. Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53-mutated AML is associated with recurrent co-occurring karyotypic structural alterations. Blood (2012) 119(9):2114–21. doi: 10.1182/blood-2011-08-375758
37. Rücker FG, Dolnik A, Blätte TJ, Teleanu V, Ernst A, Thol F, et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica (2018) 103(1):e17–20. doi: 10.3324/haematol.2017.180497
38. Metzeler KH, Herold T, Thurley M, Amler S, Sauerland MC, Görlich D, et al. AMLCG Group. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood (2016) 128:686–98. doi: 10.1182/blood-2016-01-693879
39. Fontana MC, Marconi G, Feenstra JDM, Fonzi E, Papayannidis C, Ghelli Luserna di Rorà A, et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia (2018) 32(7):1609–20. doi: 10.1038/s41375-018-0035-y
40. Cumbo C, Tota G, Anelli L, Zagaria A, Specchia G, Albano F. TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings. Int J Mol Sci (2020) 21(10):3432.
41. Kadia TM, Jain P, Ravandi F, Garcia-Manero G, Andreef M, Takahashi K, et al. TP53 mutations in newly diagnosed Acute Myeloid Leukemia –Clinico-molecular characteristics, response to therapy, and outcomes. Cancer (2016) 122(22):3484–91. doi: 10.1002/cncr.30203
42. Ferrari A, Papayannidis C, Bladazzi C, Iacobucci I, Paolini S, Padella S, et al. Leukemia Associated TP53 Mutations in AML Patients ARE Strongly Associated with Complex Karyotype and Poor Outcome. Blood (2014) 124(21):2379. doi: 10.1182/blood.V124.21.2379.2379
43. Bernard E, Nannya Y, Yoshizat T, Hasserjian RP, Saiki R, Shiozawa Y, et al. TP53 State Dictates Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Blood (2019) 134:675. doi: 10.1182/blood-2019-129392
44. Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol (2001) 19:1405–13. doi: 10.1200/JCO.2001.19.5.1405
45. Nahi H, Lehmann S, Bengtzen S, Jansson M, Möllgård L, Paul C. Merup M.Chromosomal aberrations in 17p predict in vitro drug resistance and short overall survival in acute myeloid leukemia. Leuk Lymphoma (2008) 49:508–16. doi: 10.1080/10428190701861645
46. Anensen N, Hjelle SM, Van Belle W, Haaland I, Silden E, Bourdon JC, et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene (2012) 31:1533–45. doi: 10.1038/onc.2011.348
47. Prochazka KT, Pregartner G, Rücker FG, Heitzer E, Pabst G, Wölfler A, et al. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Hematologica (2019) 104:3. doi: 10.3324/haematol.2018.205013
48. Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res (2015) 39:348–54. doi: 10.1016/j.leukres.2014.12.006
49. Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia (2008) 22:240–8. doi: 10.1038/sj.leu.2405078
50. Lindsley RC, Brenton GM, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood (2015) 125(9):1367–76.
51. Christiansen DH, Andersen MK, Desta F, Pedersen-Bjergaard J. Mutations of genes in the receptor tyrosine kinase (RTK)/RAS-BRAF signal transduction pathway in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia (2005) 19(12):2232–40.
52. Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica (2013) 98:6.
53. Voso MT, Fabiani E, Fianchi L, Falconi G, Criscuolo M, Santangelo R, et al. Mutations of epigenetic regulators and of the spliceosome machinery in therapy-related myeloid neoplasms and in acute leukemias evolved from chronic myeloproliferative diseases. Leukemia (2013) 27(4):982–5.
54. Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature (2015) 518:552–5. doi: 10.1038/nature13968
55. Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica (2007) 92:6.
56. Christiansen DH, Desta F, Andersen MK. Pedersen-Bjergaard J.Mutations of the PTPN11 gene in therapy-related MDS and AML with rare balanced chromosome translocations. Genes Chromosomes Cancer (2007) 46(6):517–21.
57. Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer (2005) 43:227–38. doi: 10.1002/gcc.20193
58. Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Peng J, Tang J, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol (2015) 8:45. doi: 10.1186/s13045-015-0139-z
59. Herold T, Rothenberg-Thurley M, Grunwald VV, Janke H, Goerlich D, Sauerland MC, et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia (2020) 34(12):3161–72. doi: 10.1038/s41375-020-0806-0
60. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FP, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
61. Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood (2012) 120(15):2963–72. doi: 10.1182/blood-2012-03-419622
62. Bowen D, Groves MJ, Burnett AK, Patel Y, Allen C, Green E, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia (2009) 23(1):203–6. doi: 10.1038/leu.2008.173
63. Poiré X, Labopin M, Maertens J, Yakoub-Agha I, Blaise D, Ifrah N, et al. Allogeneic stem cell transplantation in adult patients with acute myeloid leukaemia and 17p abnormalities in first complete remission: a study from the Acute Leukemia Working Party (ALWP) of the European Society for Blood and Marrow Transplantation (EBMT). J Hematol Oncol (2017) 10(1):20. doi: 10.1186/s13045-017-0393-3
64. Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med (2017) 376(6):536–47. doi: 10.1056/NEJMoa1611604
65. Ciurea SO, Chilkulwar A, Saliba RM, Chen J, Rondon G, Patel KP, et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood (2018) 131(26):2989–92. doi: 10.1182/blood-2018-02-832360
66. Brosh R, Rotter V. When Mutants Gain New Powers: News from the Mutant p53 Field. Nat Rev Cancer (2009) 9:701–13. doi: 10.1038/nrc2693
67. Loizou E, Banito A, Livshits G, Ho YJ, Koche RP, Sánchez-Rivera FJ, et al. A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1. Cancer Discovery (2019) 9:962–79. doi: 10.1158/2159-8290.CD-18-1391
68. Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, et al. A Dominant-Negative Effect Drives Selection of TP53 Missense Mutations in Myeloid Malignancies. Science (2019) 365:599–604. doi: 10.1126/science.aax3649
69. Dutta S, Pregartner G, Rucker FG, Heitzer E, Zebisch A, Bullinger L, et al. Functional Classification of TP53 Mutations in Acute Myeloid Leukemia. Cancers (Basel) (2020) 12(3):637. doi: 10.3390/cancers12030637
70. Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2007) 357:2552–61. doi: 10.1056/NEJMoa073770
71. Lichtarge O, Wilkins A. Evolution: a guide to perturb protein function and networks. Curr Opin Struct Biol (2010) 20(3):351–9. doi: 10.1016/j.sbi.2010.04.002
72. Neskey DM, Osman AA, Ow TJ, Katsonis P, McDonald T, Hicks SC, et al. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res (2015) 75:1527–36. doi: 10.1158/0008-5472.CAN-14-2735
73. Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, et al. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol Cell (2018) 71:873. doi: 10.1016/j.molcel.2018.08.013
74. Malik P, ashen AF. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res (2014) 6:53–61. doi: 10.2147/CMAR.S40600
75. Metzeler KH, Walker A, Geyer S, Garzon R Klisovic RB, Bloomfield CD, Blum W, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia (2012) 26:1106–7. doi: 10.1038/leu.2011.342
76. DiNardo CD, Patel KP, Garcia-Manero G, Luthra R, Pierce S, Borthakur G, et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk Lymphoma (2014) 55:1925–9. doi: 10.3109/10428194.2013.855309
77. Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med (2016) 375:2023–36. doi: 10.1056/NEJMoa1605949
78. Chang CK, Zhao YS, Xu F. TP53 mutations predict decitabine-induced complete responses in patients with myelodysplastic syndromes. Br J Haematol (2017) 176(4):600–8. doi: 10.1111/bjh.14455
79. Yi L, Sun Y, Levine A. Selected drugs that inhibit DNA methylation can preferentially kill p53 deficient cells. Oncotarget (2014) 5:8924–36. doi: 10.18632/oncotarget.2441
80. Bally C, Adès L, Renneville A, Sebert M, Eclache Y, Preudhomme C, et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res (2014) 38:751–5. doi: 10.1016/j.leukres.2014.03.012
81. Aldoss I, Yang D, Aribi A, Ali H, Sandhu K, Malki MM, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica (2018) 103:e404– e407. doi: 10.3324/haematol.2018.188094
82. Ruzinova MB, Lee YS, Duncavage EJ, Welch JS. TP53 immunohistochemistry correlates with TP53 mutation status and clearance in decitabine treated patients with myeloid malignancies. Haematologica (2019) 104(8):e345–8. doi: 10.3324/haematol.2018.205302
83. Bejar R, Lord A, Stevenson K, Bar-Natan M, Pérez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood (2014) 124:2705–12. doi: 10.1182/blood-2014-06-582809
84. Takahashi K, Patel K, Bueso-Ramos C, Zhang J, Gumbs C, Jabbour E, et al. Clinical implications of TP53 mutations in myelodysplastic syndromes treated with hypomethylating agents. Oncotarget (2016) 7:14172–87. doi: 10.18632/oncotarget.7290
85. Jung SH, Kim YJ, Yim SH, Kim YH, Kwon YR, Hur EH, et al. Somatic mutations predict outcomes of hypomethylating therapy in patients with myelodysplastic syndrome. Oncotarget (2016) 7:55264–75. doi: 10.18632/oncotarget.10526
86. Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol (2017) 18:112–21. doi: 10.1016/S1470-2045(16)30627-1
87. Short NJ, Kantarjian H, Loghavi S, Huang X, Qiao W, Borthakur G, et al. Treatment with a 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukemia: a randomized phase 2 trial. Lancet Haematol (2019) 6(1):e29–37. doi: 10.1016/S2352-3026(18)30182-0
88. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker MS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood (2019) 133:7– 17. doi: 10.1182/blood-2018-08-868752
89. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. New Engl J Med (2020) 383(7):617–29. doi: 10.1056/NEJMoa2012971
90. Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood (2016) 127:3215– 3224. doi: 10.1182/blood-2016-01-688796
91. Schiffer CA. Promoting Apoptosis with Venetoclax - A Benefit for Older Patients with AML. N Engl J Med (2020) 383(7):677–9. doi: 10.1056/NEJMe2023326
92. DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood (2020) 135:11. doi: 10.1182/blood.2019003988
93. Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. AMLCG Study Group. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood (2016) 128(5):686–98. doi: 10.1182/blood-2016-01-693879
94. Patel JP, Gönen M, Figueroa ME, Fernandez, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med (2012) 366(12):1079–89. doi: 10.1056/NEJMoa1112304
95. Aldoss I, Zhang J, Pillai R, Shouse G, Sanchez JF, Mei M, et al. Venetoclax and ypomethylating agents in TP53-muatated acute myeloid leukaemia. Br J Hematol (2019) 187(2):e45–8. doi: 10.1111/bjh.16166
96. Sallman DA, Komrokji R, Vaupel C, Cluzeau T, Geyer SM, McGraw KL, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia (2016) 30:666– 673. doi: 10.1038/leu.2015.304
97. Shoukier M, Konopleva M, Dinardo CD, Ravandi F, Andreeff M, Garcia-Manero G, et al. Activity of venetoclax-based therapy in TP53-mutated acute myeloid leukemia. JCO (2019) 37(15):7034–4. doi: 10.1200/JCO.2019.37.15_suppl.7034
98. Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro C, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discovery (2019) 9(7):910–25. doi: 10.1158/2159-8290.CD-19-0125
99. Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discovery (2019) 9(7):890–909. doi: 10.1158/2159-8290.CD-19-0117
100. Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis (2018) 9(5):439. doi: 10.1038/s41419-018-0463-7
101. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell (2014) 25:304–17. doi: 10.1016/j.ccr.2014.01.021
102. Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ (2011) 18(12):1904–13. doi: 10.1038/cdd.2011.71
103. Parrales A, Ranjan A, Iyer SV, Padhye S, Weir SJ, Roy A, et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol (2016) 18:1233–43. doi: 10.1038/ncb3427
104. Jhaveri K, Modi S. Ganetespib: research and clinical development. Onco Targets Ther (2015) 8:1849–58. doi: 10.2147/OTT.S65804
105. Tang X, Zhu Y, Han L, Kim AL, Kopelovich L, Bickers DR, et al. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J Clin Invest (2007) 117(12):3753–64. doi: 10.1172/JCI32481
106. Gupta P, Srivastava SK. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med (2012) 10:80. doi: 10.1186/1741-7015-10-80
107. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med (2004) 10(12):1321–8. doi: 10.1038/nm1146
108. Salim KY, Vareki SM, Danter WR, Koropatnick J. COTI-2, a new anticancer drug currently under clinical investigation, targets mutant p53 and negatively modulates the PI3K/AKT/mTOR pathway. Eur J Cancer (2016) 69:S19–S. doi: 10.1016/S0959-8049(16)32638-7
110. Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell (2005) 17(1):37–48. doi: 10.1016/j.molcel.2004.11.021
111. Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol (2014) 15(7):433–52. doi: 10.1038/nrm3819
112. Baldwin A, Grueneberg DA, Hellner K, Sawyer J, Grace M, Li WL, et al. Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. P Natl Acad Sci USA (2010) 107(28):12463–8. doi: 10.1073/pnas.1007462107
113. Winey M, Huneycutt BJ. Centrosomes and checkpoints: the MPS1 family of kinases. Oncogene (2002) 21(40):6161–9. doi: 10.1038/sj.onc.1205712
114. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer (2009) 9(8):537–49. doi: 10.1038/nrc2694
115. Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, et al. DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol (1999) 146(1):141–8. doi: 10.1083/jcb.146.1.141
116. Rangel LP, Ferretti GDS, Costa CL, Andrade S, Carvalho RS, Costa D. Silva JL. P53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J Biol Chem (2019) 294:3670–82. doi: 10.1074/jbc.RA118.004671
117. Lehmann S, Bykov VJN, Ali D, Andrén O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol (2012) 30(29):3633–9. doi: 10.1200/JCO.2011.40.7783
118. Sallman D, Dezern A, Sweet K, Sweet KL, Cluzeau T, Sekeres MA, et al. Phase 1B/2 combination study of APR-246 and azacitidine (AZA) in patients with TP53 mutant myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Blood (2018) 132(Supplement 1):3091. doi: 10.1182/blood-2018-99-119990
119. Prokocimer M, Molchadsky A, Rotter V. Perspective dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood (2017) 130:699– 712. doi: 10.1182/blood-2017-02-763086
120. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia (2014) 28(6):1280–8. doi: 10.1038/leu.2013.355
121. Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer (2018) 125(9):1470–81. doi: 10.1002/cncr.31896
122. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discovery (2018) 9(3):370–83. doi: 10.1158/2159-8290.CD-18-0774
123. Garcia-Manero G, Montalban-Bravo G, Sasaki K, Daver NG, Jabbour EJ, Alvarado Y, et al. Double immune checkpoint inhibitor blockade with nivolumab and ipilimumab with or without azacitidine in patients with myelodysplastic syndrome (MDS). In: American society of hematology annual meeting. San Diego, CA.
124. Assi R, Kantarjian HM, Daver NG, Garcia-Manero G, Benton CB, Thompson PA, et al. Results of a phase 2, open-label study of idarubicin (I), cytarabine (A) and nivolumab (nivo) in patients with newly diagnosed acute myeloid leukemia and highrisk myelodysplastic syndrome (MDS). In: American society of hematology annual meeting. San Diego, CA.
125. Garcia-Manero G, Sasaki K, Montalban-Bravo G, Daver NG, Jabbour EJ, Alvarado Y, et al. A phase II study of nivolumab or ipilimumab with or without azacitidine for patients with myelodysplastic syndrome (MDS). In: American society of hematology annual meeting. San Diego, CA.
126. Wong E, Dawson E, Davis J, Koldej R, Ludford-Menting M, Lansdown M, et al. Nivolumab for relapsed or residual haematological malignancies after allogeneic haematopoietic stem cell transplantation (NIVALLO). In: American society of hematology annual meeting. San Diego, CA.
127. Daver NG, Garcia-Manero G, Basu S, Cortes JE, Ravandi F, Kadia TM, et al. Safety, efficacy, and biomarkers of response to azacitidine (aza) with nivolumab (nivo) and aza with nivo and ipilimumab (ipi) in relapsed/refractory acute myeloid leukemia: a non-randomized, phase 2 study. In: American society of hematology annual meeting. San Diego, CA.
128. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med (2016) 375(2):143–53. doi: 10.1056/NEJMoa1601202
129. Kadia TM, Cortes JE, Ghorab A, Ravandi F, Jabbour E, Daver NG, et al. Nivolumab (Nivo) maintenance (maint) in high-risk (HR) acute myeloid leukemia (AML) patients. JCO (2018) 36(15):7014. doi: 10.1200/JCO.2018.36.15_suppl.7014
130. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138:271–85. doi: 10.1016/j.cell.2009.05.046
131. Sallman DA, Asch AS, Al Malki MM, Lee DJ, Donnellan WB, Marcucci G, et al. The first-in-class anti-CD47 antibody magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: ongoing phase 1b results. In: American Society of Hematology Annual Meeting Oral Presentation. (2019). doi: 10.1182/blood-2019-126271
132. Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev (1993) 7:1126–32. doi: 10.1101/gad.7.7a.1126
133. Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang D, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett (2013) 4:466–9. doi: 10.1021/ml4000657
134. Martinelli G, Assouline S, Kasner M, Kasner M, Vey N, Kelly KR, et al. Phase 1b study of the MDM2 antagonist RG7112 in combination with 2 doses/schedules of cytarabine. Blood (2013) 122:498. doi: 10.1182/blood.V122.21.498.498
135. Ding Q, Zhang Z, Liu J-J, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53–MDM2 inhibitor in clinical development. J Med Chem (2013) 56:5979–83. doi: 10.1021/jm400487c
136. Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S, et al. Phase 1/1b study of RG7388, a potent MDM2 antagonist, in Acute Myelogenous Leukemia (AML) patients (Pts). Blood (2014) 124:116. doi: 10.1182/blood.V124.21.116.116
137. Martinelli G, Pappayannidis C, Yee K, Vey N, Drummond M, Kelly K, et al. Phase 1b results of idasanutlin + cytarabine (ARA-C) in acute myeloid leukemia patients. (2016). Denmark: European Hematology Association (2016).
138. Dangl M, Chien Y, Lehmann C, Friess T. Synergistic anticancer activity of clinical stage, non-genotoxic apoptosis inducing agents RG7388 (MDM2 antagonist) and ABT-199 (GDC-0199, BCL2 inhibitor) in p53 wild-type AML tumor models. Cancer Res (2014) 74:5505. doi: 10.1158/1538-7445.AM2014-5505
139. Hoffman-Luca CG, Ziazadeh D, McEachern D, Zhao Y, Sun W, Debussche L, et al. Elucidation of acquired resistance to Bcl-2 and MDM2 inhibitors in acute leukemia in vitro and in vivo. Clin Cancer Res (2015) 21:2558–68. doi: 10.1158/1078-0432.CCR-14-2506
140. Pan R, Kojima K, Zheng Z, Ruvolo VR, Gwen N, Leverson JD, et al. Activation of p53 by novel MDM2 antagonist RG7388 overcomes AML inherent and acquired resistance to Bcl-2 inhibitor ABT-199 (GDC-0199). Blood (2014) 124:2162. doi: 10.1182/blood.V124.21.2162.2162
141. Cicconi L, Platzbecker U, Avvisati G, Paoloni F, Thiede C, Vignetti M, et al. Long-term results of all-trans retinoic acid and arsenic trioxide in non-high-risk acute promyelocytic leukemia: update of the APL0406 Italian-German randomized trial. Leukemia (2020) 34(3):914–8. doi: 10.1038/s41375-019-0589-3
142. Yan W, Jung YS, Zhang Y, Chen X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PloS One (2014) 9:e103497. doi: 10.1371/journal.pone.0103497
143. Yan W, Zhang Y, Zhang J, Liu S, Cho SJ, Chen X, et al. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J Biol Chem (2011) 286:17478– 17486. doi: 10.1074/jbc.M111.231639
144. Noguera NI, Pelosi E, Angelini DF, Piredda ML, Guerrera G, Piras E, et al. High-dose ascorbate and arsenic trioxide selectively kill acute myeloid leukemia and acute promyelocytic leukemia blasts in vitro. Oncotarget (2017) 8:32550–65. doi: 10.18632/oncotarget.15925
Keywords: acute myeloid leukemia, TP53 mutations, poor outcome, decitabine, venetoclax (BCL-2 inhibitor)
Citation: Molica M, Mazzone C, Niscola P and de Fabritiis P (2021) TP53 Mutations in Acute Myeloid Leukemia: Still a Daunting Challenge? Front. Oncol. 10:610820. doi: 10.3389/fonc.2020.610820
Received: 27 September 2020; Accepted: 21 December 2020;
Published: 08 February 2021.
Edited by:
Alessandro Isidori, AORMN Hospital, ItalyReviewed by:
Guru Prasad Maiti, Oklahoma Medical Research Foundation, United StatesLucia Masarova, University of Texas MD Anderson Cancer Center, United States
Copyright © 2021 Molica, Mazzone, Niscola and de Fabritiis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Molica, bW9saWNhQGJjZS51bmlyb21hMS5pdA==