 Yijia Guo
Yijia Guo Jun Song
Jun Song Letian Huang
Letian Huang Li Sun
Li Sun Jianzhu Zhao
Jianzhu Zhao Wei Jing
Wei Jing- Department of Clinical Oncology, Shengjing Hospital of China Medical University, Shenyang, China
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) greatly improve the survival and quality of life of non-small cell lung cancer (NSCLC) patients with EGFR mutations. However, many patients exhibit de novo or primary/early resistance. In addition, patients who initially respond to EGFR-TKIs exhibit marked diversity in clinical outcomes. With the development of comprehensive genomic profiling, various mutations and concurrent (i.e., coexisting) genetic alterations have been discovered. Many studies have revealed that concurrent genetic alterations play an important role in the response and resistance of EGFR-mutant NSCLC to EGFR-TKIs. To optimize clinical outcomes, a better understanding of specific concurrent gene alterations and their impact on EGFR-TKI treatment efficacy is necessary. Further exploration of other biomarkers that can predict EGFR-TKI efficacy will help clinicians identify patients who may not respond to TKIs and allow them to choose appropriate treatment strategies. Here, we review the literature on specific gene alterations that coexist with EGFR mutations, including common alterations (intra-EGFR [on target] co-mutation, TP53, PIK3CA, and PTEN) and driver gene alterations (ALK, KRAS, ROS1, and MET). We also summarize data for other biomarkers (e.g., PD-L1 expression and BIM polymorphisms) associated with EGFR-TKI efficacy.
Introduction
Lung cancer is the most prevalent cancer type and is one of the leading causes of cancer-related death (1). The discovery of epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) and the fact that most patients with EGFR-mutant non-small cell lung cancer (NSCLC) can benefit from TKI treatment have dramatically changed the therapeutic approach for NSCLC. EGFR mutations occur in approximately 10–35% of lung adenocarcinomas (2), with a higher prevalence of about 40–55% in East Asian patients (3). Because EGFR-TKIs, including first-generation gefitinib, erlotinib, and icotinib, second-generation afatinib and dacomitinib, and third-generation osimertinib, have demonstrated higher objective response rates (ORR) and prolonged progression-free survival (PFS) compared to standard chemotherapy (4–7), they have become the first choice for patients with advanced EGFR-mutated NSCLC. However, approximately 20–30% of patients exhibit primary resistance to EGFR-TKIs (4). Furthermore, even in patients with an initial response, significant heterogeneous outcomes have been observed. Some patients only respond for a few weeks, while others may benefit for years without progression.
Comprehensive genomic profiling has allowed us to understand various mutations and co-occurrence of genomic alterations in NSCLC and explore their impact on clinical outcomes. Many studies have demonstrated that concurrent genetic alterations potentially impair TKI efficacy and partly explain the heterogeneous patient outcomes (8–10). Hong et al. (8) analyzed 58 EGFR-mutant patients with metastatic NSCLC treated with first-line EGFR-TKIs. They demonstrated that concomitant mutations are widespread and significantly associated with reduced ORR and shorter overall survival (OS). Sato et al. (11) found that PFS for EGFR-TKIs in EGFR-mutant lung adenocarcinoma was associated with the number of concurrent genomic alterations. Significantly poorer PFS was observed in patients with four or more genomic alterations. The types of genomic alterations coexisting with EGFR mutations may also be associated with EGFR-TKI efficacy. For instance, Wang et al. (12) showed that patients with mutations in EGFR and oncogenes have shorter PFS compared to those with mutations in EGFR and tumor suppressor genes and patients with EGFR mutations only (4.7 vs. 9.3 vs. 13.2 months), which is consistent with other reports (13–15). In addition, EGFR mutations are thought to be mutually exclusive with other oncogenic drivers. However, recent studies have demonstrated that although at a small percentage (16–18), additional driver alterations coexist with EGFR mutations in TKI therapy-naïve NSCLC and may impact EGFR-TKI efficacy and partly explain the intrinsic resistance in some patients (19).
A greater understanding of the relationship between specific concurrent genes and TKI efficacy is urgently needed and will help predict clinical outcomes and guide clinicians in selecting the best treatment strategies for patients with these concurrent mutations. In this review, we describe the recent progress and future perspectives in this area, focusing on common concurrent genetic alterations [intra-EGFR (on target) co-mutation, TP53, PIK3CA, and PTEN] and concurrent driver gene alterations (ALK, KRAS, ROS1, and MET). Furthermore, we summarize the data on other biomarkers (e.g., PD-L1 expression and BIM polymorphisms) that appear to be associated with EGFR-TKI efficacy (Figure 1). The main characteristics of the studies described are summarized in Tables 1 and 2.
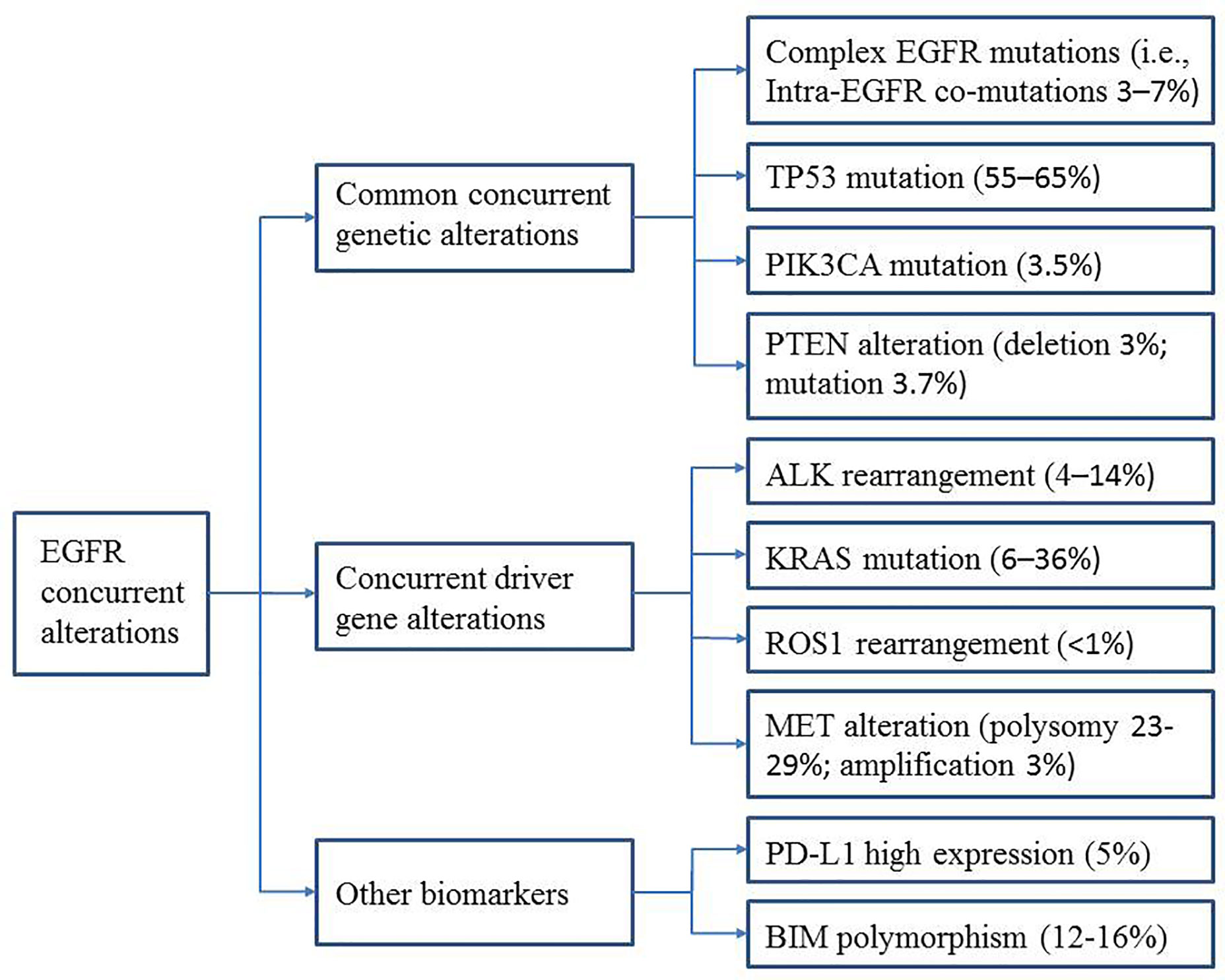
Figure 1 Overview of EGFR concurrent alterations.
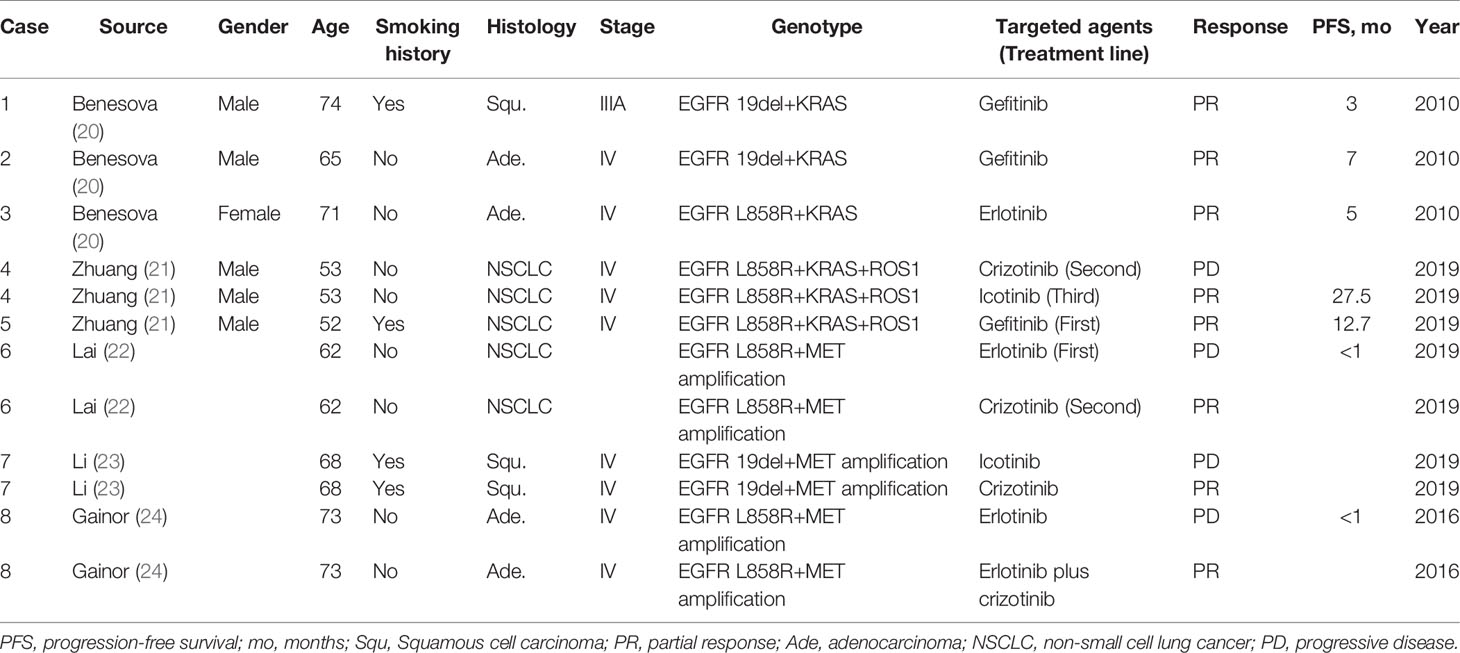
Table 1 Tyrosine kinase inhibitor responses of patients with concurrent alterations from literature review.

Table 2 Main characteristics of available studies.
Common Concurrent Genetic Alterations
Complex EGFR Mutations (Intra-EGFR Co-Mutations)
In-frame deletion mutations in exon 19 (19del) and the Leu858Arg (L858R) point mutation in exon 21 are two major types of EGFR activating mutations with an ORR of 70–80% and PFS of approximately 9–20 months for EGFR-TKIs. These mutations are sensitive or classical (i.e., typical or common) EGFR mutations (4–6, 44). Occasionally, complex mutations occur, meaning a single tumor sample has two or more different EGFR mutations (45–50). The frequency of complex mutations is 3–7% (25, 26, 50–54). Theoretically, the introduction of an additional mutation could change the molecular conformation of the EGFR tyrosine kinase domain, leading to increased or decreased TKI affinity that subsequently affects the clinical outcome (55). Zhang et al. (25) identified 187 patients with complex EGFR mutations out of 5898 EGFR-mutant NSCLC patients. Fifty-one of these patients had advanced lung adenocarcinoma and were treated with first-generation EGFR-TKIs as first-line therapy. The median PFS was 9.5 months. A total of 46 of the patients were evaluated for response. The ORR was 52.2%, and the disease control rate (DCR) was 71.7%. These patients were further divided into four groups: group A (n = 12), patients with double classic mutations; group B (n = 15), patients with classical mutations plus an atypical mutation; group C (n = 7), patients with double atypical mutations; group D (n = 12), patients with complex mutations harboring a primary T790M mutation or an exon 20 insertion. The ORRs for these four groups were 75, 60, 71, and 8.3%, respectively. The PFS values were 18.2, 9.7, 9.6, and 1.4 months, respectively. According to these results, patients with double classic mutations (group A) had the best ORR and PFS. Based on a literature review, the authors pooled 22 patients with concurrent 19del and L858R mutations treated with TKIs. Their ORR and PFS were 80% and 13.9 months, respectively. Consistent with other studies, they observed that patients harboring complex mutations with a classic mutation had similar clinical outcomes to those with single classical mutations (26, 27). In contrast, lower TKI efficacy was observed in patients with uncommon complex mutations, especially those with a primary T790M mutation or exon 20 insertions (27).
As the Zhang et al. (25) study showed, patients harboring complex mutations with a primary T790M or exon 20 insertion predominantly presented with primary EGFR-TKI resistance accompanied by the poorest ORR and PFS. Similar results were observed in other studies, which revealed that first and second-generation EGFR-TKIs were ineffective in patients with primary T790M mutations (PFS <3 months) (56, 57). Osimertinib showed better efficacy, with a PFS and OS of 17.0 months (95% CI, 14.0–20.0 months) and 29.9 months, respectively, for 18 patients with a primary T790M mutation (58). Another study by Zhang et al. (59) demonstrated that for 31 patients harboring compound EGFR mutations with primary T790M or exon 20 insertion and sensitive mutations, the median PFS for the patients treated with osimertinib (n = 15) was longer than that for patients treated with first-generation EGFR-TKIs (n = 16) (18.0 vs. 1.2 months; p < 0.001).
Previous studies showed that first-generation EGFR-TKIs had poor efficacy in patients with uncommon mutations (alone or as part of a compound mutation) (60, 61), while second-generation TKI afatinib had better activity (ORR 71.1%; median PFS 10.7 months) (57). Thus, afatinib has been approved by the United States Food and Drug Administration as first-line treatment for patients with uncommon EGFR mutations. A recent pooled analysis confirmed the efficacy of first-line afatinib in patients with uncommon compound mutations (n = 35), with an ORR of 77.1% and a median DOR of 16.6 months (62). In addition, osimertinib demonstrated moderate efficacy with an ORR of 50% and a median PFS of 8.2 months (62). Furthermore, some studies found that these patients had a significantly lower incidence of an acquired T790M mutation (63). Considering that patients with complex EGFR mutations and a secondary T790M mutation have a shorter PFS and OS when subsequently treated with osimertinib (64), first-line osimertinib is a choice for these patients. Additional studies on EGFR-TKIs efficacy focused specifically on this subgroup of patients are needed.
Collectively, it is difficult to analyze the efficacy of EGFR-TKIs in patients with uncommon complex EGFR mutations because almost 200 EGFR mutation types have been identified so far, and uncommon mutations have great heterogeneity (25, 65). It appears that afatinib and osimertinib are better options for these patients; however, more studies with larger populations are needed. Osimertinib provided a great survival benefit for patients harboring complex mutations with a primary T790M or an exon 20 insertion that had de novo resistance to first and second-generation TKIs and should be taken into consideration for treatment for this subset of patients.
TP53 Mutations
p53 can induce cell cycle arrest, senescence, and apoptosis and, thus, regulates the response to various cellular stress signals (66). Mutations in the tumor suppressor TP53 gene, which encodes p53, are found in 35–55% of NSCLC cases, more prevalent in squamous cell carcinoma than adenocarcinoma (67), and highly correlated with smoking habits (68). TP53 mutations may have a negative prognostic effect for NSCLC (69).
TP53 is the most prevalent co-alteration observed in EGFR-mutant NSCLC patients, with a frequency of 55–65% (8, 28, 70). Preclinical studies have shown a link between TP53 status and EGFR-TKI response (71–73). Apoptosis induced by gefitinib in NSCLC cell lines requires wild-type p53, which can induce Fas and caspase-dependent cell death, thus increasing TKI sensitivity. In contrast, gefitinib-induced apoptosis is reduced in mutated p53 cells (71). Some in vitro models of other tumor types also demonstrated a correlation between TKI response and TP53 mutation, especially in urothelial carcinoma (74, 75). Several studies revealed that patients with coexisting TP53 mutations treated with EGFR-TKIs showed a trend toward lower ORR, shorter PFS, and OS compared to patients with wild-type TP53 that did not reach statistical significance (28–31, 68). However, a recent meta-analysis indicated significantly poorer prognosis for patients with concurrent TP53 mutations treated with first-line EGFR-TKIs (pooled HRs for PFS and OS of 1.69 and 1.94, respectively) (76).
Interestingly, a study focused on TP53 exon 8 mutations demonstrated reduced responsiveness to EGFR-TKIs and worse prognosis, mainly in patients harboring EGFR 19del (29). The data showed that TP53 exon 8 mutations were associated with a significantly lower DCR (42 vs. 87%; p < 0.001) and a trend towards shorter PFS and OS compared to TP53 exon 8 wild-type patients in the whole cohort. These differences in PFS and OS became significant in the EGFR 19del subgroup (median PFS, 4.2 vs. 16.8 months; p < 0.001; median OS, 7.6 months vs. not reached; p = 0.006). In addition, patients with TP53 exon 8 mutations also showed a significantly higher risk of disease progression or death than TP53 exon 8 wild-type patients in this subgroup (HR for PFS, 6.99; 95% CI, 2.34–20.87; p = 0.006; HR for OS, 4.75; 95% CI, 1.38–16.29; p = 0.013).
Labbé et al. (31) found that the PFS for patients with TP53 missense mutations was significantly shorter than for TP53 wild-type patients. This study demonstrated that the patients with concurrent EGFR and TP53 mutations who progressed on EGFR-TKIs treatment (n = 24) were less likely to have a secondary EGFR T790M mutation [45% (5/11) vs. 69% (9/13); p = 0.41]. These data are similar to the recent work of VanderLaan et al. (30) who also showed a trend towards a decreased acquired T790M rate in tumors with concurrent TP53 mutations. Among the patients with secondary T790M, 12 received third-generation EGFR-TKIs (11 were evaluable). The ORR was not significantly different between TP53 mutant and wild-type patients in this subset [100% (3/3) vs. 88% (7/8)] (31). The influence of co-occurring TP53 mutations on the efficacy of third-generation EGFR-TKIs in patients with acquired T790M mutations following initial EGFR-TKI failure has also been explored (28). Surprisingly, patients with TP53 mutations had a significantly shorter PFS and worse OS compared to patients with wild-type TP53 (median PFS, 8.9 vs. 12.8 months; p = 0.029; median OS, 17.8 vs. 26.6 months; p = 0.007) in this cohort.
Coexisting of certain mutation sites (e.g., exon 8 mutations) or certain types of TP53 mutations (e.g., missense mutations) may be related to poor EGFR-TKI efficacy, and additional studies focusing on these different types of TP53 mutations are needed to confirm their prognostic impact. Nevertheless, it seems that EGFR-TKIs have lower efficacy in patients with concurrent TP53 mutations. A combination of EGFR-TKI with antiangiogenic therapy showed encouraging efficacy for these patients. In the RELAY study, ramucirumab plus erlotinib first-line therapy showed a superior PFS benefit in patients with EGFR-mutant metastatic NSCLC compared to erlotinib alone (19.4 vs. 12.4 months; HR, 0.591; p < 0.0001). Thus, this combination may be a better choice for these patients. Indeed, significant PFS improvement was observed in EGFR-mutant (19del/L858R) patients with concurrent TP53 mutations (77). In the ALTER-L004 study (NCT03736837), anlotinib plus icotinib showed encouraging efficacy and good tolerability for previously untreated, EGFR-mutant advanced NSCLC patients. Fourteen patients with TP53 mutations had an ORR of 78.5% and DCR of 100% (78). Moreover, in the ACTIVE phase III study, apatinib plus gefitinib demonstrated superior PFS as first-line therapy in patients with EGFR-activating mutations, and patients with TP53 exon 8 mutations significantly benefited from this combination (HR 0.24) (79).
PIK3CA Mutation
The PI3K/AKT pathway is a downstream signaling pathway of the HER family that is important in oncogenesis and lung cancer progression (80, 81). PIK3CA encodes the catalytic subunit, and PIK3CA mutations can activate the PI3K/AKT pathway (82). PIK3CA mutations are found in about 2–5% of NSCLC cases (83–85) and are considered rare oncogenic drivers in NSCLC. The majority of mutations occur in exon 9 (E545K, E545Q, E545G, E545A, Q546R, E542K, and T536I) and exon 20 (H1047R, H1047L, M1043L, G1007R, and Y1021C), with E545K and H1047R being the most frequent mutations (32, 33, 86, 87). In contrast to the mutual exclusivity of many oncogenic drivers in lung cancers, PIK3CA mutations frequently coexist with other oncogenic driver mutations, especially EGFR and KRAS (83, 88–90). Indeed, PI3KCA mutations have been found in approximately 3.5% of EGFR mutation-positive patients (91) and appear to be an indicator of resistance and poor survival for NSCLC patients treated with EGFR-TKIs (92). In a preclinical study, the introduction of an activated PIK3CA p.E545K mutation in exon 9 into the EGFR mutation-positive (19del) HCC827 cell line conferred resistance to gefitinib (93).
The role of PIK3CA mutations in predicting the efficacy of EGFR-TKIs was first investigated by Ludovini et al. (92, 94), who reported that six patients with a PIK3CA mutation had a shorter time to progression (TTP, median, 2.3 vs. 6.0 months; p = 0.01) and OS (median, 9.9 vs. 30.2 months; p < 0.001) after treatment with gefitinib or erlotinib. It should be mentioned that this study included patients with an unselected EGFR mutation status, with 75.3% (125/166) having a wild-type EGFR. Only two of the six PIK3CA-mutant patients had a concurrent EGFR mutation. One patient had an EGFR S784F mutation in exon 20 and experienced progressive disease (PD) on EGFR-TKI treatment. The second patient with EGFR 19del showed a partial response (PR) to erlotinib but experienced treatment failure after four months of therapy.
Three studies directly compared the efficacy of EGFR-TKIs in EGFR-mutant patients with or without concomitant PIK3CA mutations. Eng et al. (32) found that patients with concurrent PIK3CA mutations had a lower ORR (62% vs. 83%; p = 0.80) and shorter median TTP (7.8 vs. 11.1 months; p = 0.84) to EGFR-TKIs. However, these differences were not statistically significant, and these two groups had the same median duration of EGFR-TKI therapy (14.6 months; p = 0.65). A prospective study also explored the impact of PIK3CA mutations on the clinical characteristics and treatment response to EGFR-TKIs in lung adenocarcinoma (33). Of the 344 patients enrolled, six had coexisting PIK3CA mutations. These patients had a similar response to EGFR-TKIs as patients with wild-type PIK3CA (ORR, 66.7 vs. 78.7%; p = 0.476). Interestingly, these PIK3CA-mutant patients showed a tendency of longer PFS (median, 12.0 vs. 8.8 months) and OS (median, 25.1 vs. 21.4 months) compared to those with wild-type PIK3CA, although the differences were not significant (p = 0.401 and p = 0.247, respectively). These data are contrary to the Eng study described above (32). However, a recent study analyzing eight patients with coexisting PIK3CA mutations in a cohort of 54 EGFR-mutant advanced NSCLC patients treated with first-generation EGFR-TKIs found that concurrent PIK3CA mutations were significantly associated with a longer PFS compared to wild-type PIK3CA (95). Interestingly, further study revealed a domain-dependent effect of the PIK3CA mutations on PFS. Mutations in the p85 binding domain (R88Q, R108H, and K111E) were associated with an improved PFS, while mutations in the kinase (Y1021H and H1047R), helical (E542K), and C2 (N345K) domains were associated with a worse PFS (95). Although these findings need to be validated in larger cohorts, they provide a clue for understanding the controversial results of the described studies.
In summary, the available PIK3CA studies had limited sample sizes and inconsistent results. Therefore, additional studies on PIK3CA mutations in different domains and their impact on EGFR-TKI efficacy in a larger population are needed. Based on the present results, we believe that decision-making regarding whether to initiate EGFR-TKI therapy in the clinic should not be affected by the presence of a concurrent PIK3CA mutation.
PTEN Alterations
Gene of phosphate and tension homolog deleted on chromosome ten (PTEN) is a tumor suppressor gene and master negative regulator of the PI3K/AKT pathway (81, 96). PTEN inactivation, which can be caused by several mechanisms (e.g., decreased protein levels, mutations, loss of heterozygosity, and epigenetic silencing (97)), plays an important role in lung cancer oncogenesis and progression (81, 96). Indeed, it is a frequent event in NSCLC. Such a loss in PTEN function can constitutively stimulate the PI3K/AKT pathway and increase cellular proliferation (96) and may be associated with EGFR-TKI sensitivity (98–100).
Loss of PTEN occurs in more than 40% of NSCLC cases and is associated with poor clinical outcome (101–104). This association appears to be true for EGFR-mutant patients treated with EGFR-TKIs. Wang et al. (105) discovered that for 169 advanced NSCLC patients who harbored EGFR-sensitive mutations treated with EGFR-TKIs, patients with concurrent PTEN deletion had a shorter PFS and OS than those with intact PTEN (HR for PFS, 3.64; 95% CI, 1.47–9.00; HR for OS, 2.86; 95% CI, 1.04–7.89). In addition, both PTEN deletion (HR, 4.29; 95% CI, 1.72–10.70) and low PTEN protein expression (HR, 1.96; 95% CI, 1.22–3.13) were independent predictors of worse PFS for patients treated with EGFR-TKIs. In contrast, high PTEN expression has been reported to be a significant favorable prognostic marker (104). Endoh et al. (106) studied 78 patients with recurrent disease after surgical resection who were treated with gefitinib, and found that high PIK3CA and PTEN expression levels were associated with prolonged OS. In addition, the longest OS was observed in EGFR-mutant patients with concomitant high PTEN expression.
PTEN mutation is rare in NSCLC (2–5% of adenocarcinomas) but also a poor prognostic factor for EGFR-TKI treatment. Kim et al. (28) reported three patients with concurrent PTEN mutations out of 82 patients who acquired the T790M mutation following initial EGFR-TKI failure. They observed that the PTEN mutation was associated with significantly shorter PFS (median, 2.6 vs. 10.3 months; p = 0.001) and worse OS (median, 13.2 vs. 24.3 months; p = 0.005) for third-generation EGFR-TKIs.
These results provide some information about the efficacy of EGFR-TKIs in patients with concurrent PTEN alterations; however, further research is needed to confirm these findings to determine whether they can assist clinical selection for EGFR-TKI treatment.
Concurrent Driver Gene Alterations
ALK Rearrangement
The anaplastic lymphoma kinase (ALK) rearrangement is an oncogenic driver that occurs in approximately 5% of NSCLC patients (107). ALK-TKIs are recommended as first-line therapy for these patients. Early studies have suggested that ALK rearrangements are mutually exclusive with EGFR mutations (108). However, recent reports have described the incidence of concomitant EGFR mutations and ALK rearrangement at a rate of 0.45–1.6% in patients with NSCLC, accounting for 3.9–13.6% of EGFR-mutant and 15.4–18.8% of ALK-rearranged patients (17, 34, 36, 109). With the increased use of next-generation sequencing (NGS)-based mutational profiling, the detection of co-alterations is expected to increase.
EGFR-TKIs and ALK-TKIs play irreplaceable roles in treating NSCLC patients with single oncogenic driver alterations; however, their effects are controversial in double-positive patients. Yang et al. (34) revealed that 13 out of 977 NSCLC patients had co-altered EGFR and ALK. Ten patients received first-line EGFR-TKIs, with an ORR of 80% and a median PFS of 11.2 months (95% CI, 5.6–16.8). Four patients received crizotinib, with three of them first receiving first-line EGFR-TKIs. Of these four patients, two responded to EGFR-TKI but not to crizotinib, while one had de novo resistance to the EGFR-TKIs but was responsive to crizotinib. The fourth patient, who received first-line crizotinib, achieved a PR and 15.1 months of PFS but did not respond to subsequent EGFR-TKI treatment. The authors further found that the diverse responses to the ALK- and EGFR-TKIs observed in the ALK/EGFR co-altered patients were associated with phospho-EGFR and phospho-ALK levels. Overall, it seems that patients with EGFR/ALK co-alterations had more favorable responses to first-line EGFR-TKIs in this study.
Lou et al. (35) showed that the ORR (p = 0.57) and median PFS (HR, 0.95; 95% CI, 0.49–1.84; p = 0.87) were 80% and 11.2 months for EGFR/ALK-co-altered patients (n = 10) treated with first-line EGFR-TKIs and 65.5% and 13.2 months for single EGFR-mutant patients (n = 84). A less favorable result for second or further-line crizotinib therapy was found for double-positive patients (n = 5), with an ORR (p = 0.29) of 40% and a median PFS of 1.9 months (HR, 0.40; 95% CI, 0.15–1.10; p = 0.08) compared to 73.9% and 6.9 months for single ALK-rearranged patients (n = 23). The median OS for the single EGFR-mutant, single ALK-rearranged, and EGFR/ALK co-altered patients were 21.3, 23.7, and 18.5 months, respectively (p = 0.06). There was a statistically significant difference in OS between the single ALK-rearranged and EGFR/ALK co-altered patients (p = 0.03). Five EGFR/ALK co-altered patients received sequential treatment with both EGFR-TKIs and crizotinib. Of the four patients that received EGFR-TKI treatment followed by crizotinib, three had good responses and prolonged survival with the first-line EGFR-TKIs but had primary resistance to subsequent crizotinib. The patient that received crizotinib followed by EGFR-TKIs benefited from the crizotinib treatment, but not from the subsequent EGFR-TKI treatment. They also found that after developing resistance to the EGFR-TKIs, activation of ALK may be lower than the EGFR signaling pathway and the abundance of ALK rearrangement may be lower, which could account for the lack of crizotinib efficacy in EGFR/ALK co-altered patients. According to these two studies, first-line EGFR-TKI seems to be a reasonable therapy for EGFR/ALK co-altered patients and single ALK-TKI might be excluded after progressing on EGFR-TKIs. However, these studies contained a limited number of EGFR/ALK co-altered patients who used ALK-TKI as first-line therapy.
A recent study by Zhao et al. (36) presented conflicting results. They identified 26 cases (0.45%) of concomitant EGFR mutations and ALK rearrangement among 5816 NSCLC patients. There were no statistically significant differences in the ORR and PFS for EGFR-TKI treatment between patients with EGFR mutations alone and EGFR/ALK double-positive patients (ORR, 62.1% [59/95] vs. 63.2% [12/19]; p = 0.93; median PFS, 11.4 vs. 10.3 months; p = 0.87). Additionally, the ORR and median PFS for crizotinib were 65% (39/60) and 12.5 months for ALK-rearranged alone and 66.7% (8/12) and 11.1 months for double-positive patients, respectively. No statistically significant differences were found between these two groups (ORR, p = 1.00; HR for PFS, 1.39; 95% CI, 0.69–2.80; p = 0.28). Furthermore, nine patients were treated with both EGFR-TKIs and crizotinib, eight patients received crizotinib after progression on EGFR-TKI treatment, and the remaining patient received crizotinib before the first-generation EGFR-TKIs. In this subgroup, the ORR was 55.6% (5/9) for EGFR-TKIs and 66.7% (6/9) for crizotinib. A median PFS of 15.0 months was observed when crizotinib was used as a sequential therapy after failure with EGFR-TKI. Crizotinib was administered as first-line therapy in four patients (three received crizotinib monotherapy, and one received crizotinib combined with first-generation EGFR-TKI). The ORR in this subgroup was 75.0% (3/4). These results demonstrated that first-generation EGFR-TKIs and crizotinib in patients with concomitant EGFR and ALK alterations were as efficacious as in patients with single driver gene alterations. Furthermore, contrary to the first two studies mentioned above, crizotinib efficacy as a subsequent therapy after EGFR-TKI treatment failure was not influenced by the EGFR-TKIs. Thus, sequential treatment with EGFR-TKIs and crizotinib could be considered in EGFR and ALK double-positive patients. However, a study in Chinese patients found that EML4-ALK/EGFR and non-EML4-ALK/EGFR co-alterations displayed different clinical characteristics and responses to EGFR-TKIs. Non-EML4-ALK co-alterations are likely to occur as a resistance mechanism against the EGFR-TKIs, and dual-TKI therapy instead of single-TKI therapy might be a better choice for this subset of patients (110).
In summary, EGFR/ALK co-alterations can define a specific subgroup with tumor heterogeneity and diverse responses. Given the limited number of patients in these studies, it was not feasible to determine the best treatment strategy. There are different theories about the coexistence of EGFR mutations and ALK rearrangement in NSCLC—the two gene alterations coexist in different areas (i.e., different cells) of the tumor (111) or are present in the same tumor cells (112). These two scenarios can be distinguished by fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC). If the two alterations exist in different tumor cells, there might be a dominant driver clone. Thus, which agent becomes more effective might depend on the levels of the relevant gene alterations. Detection of the abundance of EGFR mutations and ALK rearrangements, levels of phosphorylation of EGFR and ALK and downstream proteins, their dynamic changes, and re-biopsy after progression might help guide the treatment selection and predict the efficacy of TKIs in clinical practice (34, 35). If tumor cells carry both EGFR and ALK alterations, a combination of both TKIs may be a potentially reasonable choice. Still, it is a challenging issue and requires detailed investigation.
KRAS Mutation
KRAS is one of the commonly detected gene mutations in NSCLC and present in approximately 20–30% of lung adenocarcinomas (113, 114). It rarely overlaps with other driver mutations and is thought to cause inherent resistance to EGFR-TKIs (114–117). However, studies have reported that 5.8–35.8% of EGFR-mutant patients have concurrent KRAS alterations (17, 118). Studies on the efficacy of EGFR-TKIs in these double-positive patients have yielded conflicting results.
Zhuang et al. (21) observed an ORR to EGFR-TKIs of 62.5% (5/8) for patients harboring EGFR/KRAS co-alterations and receiving first-line EGFR-TKI therapy. There were no significant differences in the PFS among patients harboring an EGFR/KRAS co-alteration and those harboring a single EGFR mutation (median, 8.2 vs. 9.6 months; p = 0.392). This result suggested that KRAS mutations do not influence the efficacy of EGFR-TKI therapy. However, Benesova et al. (20) identified three patients with concurrent EGFR and KRAS mutations. They all had an initial positive response to EGFR-TKI treatment; however, the efficacy did not last long, resulting in PFS of 3, 5, and 7 months. Rachiglio et al. (37) compared the response to EGFR-TKI treatment between 14 patients with concurrent EGFR and KRAS mutations, in which eight cases with dominant variant allelic frequency (VAF) of EGFR mutations relative to KRAS mutations. The patients with a dominant VAF of EGFR mutations had significantly improved PFS (11.09 vs. 2.42 months; p = 0.0081) and ORR (57.1 vs. 16.7%) compared to the remaining patients with a dominant VAF of KRAS mutations relative to EGFR mutations.
The limited number of patients harboring double EGFR and KRAS mutations does not allow us to draw definitive conclusions. It can be speculated that KRAS and EGFR mutations may be carried by two different clones, with the initial response due to the elimination of the TKI-sensitive clones by the targeted therapy. However, the growth of the remaining KRAS-resistant clones may lead to a shorter PFS (16). Overall, these studies suggested that EGFR-TKIs can be considered as therapy for treating patients with EGFR/KRAS co-mutations. Quantitative assessment of the allelic frequencies of both the EGFR and KRAS mutations might better identify patients who would benefit from EGFR-TKI treatment.
ROS1 Rearrangements
Oncogenic ROS1 is a well-recognized and targetable driver in NSCLC. ROS1 rearrangements occur in 1–2% of NSCLC patients (119, 120). The triple ALK, ROS1, and MET-TKI crizotinib is highly effective in ROS1-positive NSCLC patients (121, 122). Patients with ROS1 rearrangements concomitantly with EGFR are extremely rare, with a frequency of less than 1% (123). It would be almost impossible to conduct a clinical trial to compare the different therapeutic strategies in this subtype, although isolated cases can provide some information.
Lambros et al. (124) identified ten patients with ROS1 rearrangements and concomitant EGFR mutations that were treated with first-line EGFR-TKIs. Their responses included six PR, two stable diseases (SDs), and two PDs. Second-line crizotinib therapy was used in four patients with progressive disease during EGFR-TKI therapy, with two PR, one SD, and one PD observed. Interestingly, in the Zhuang study (21), two patients harbored triple EGFR/ROS1/KRAS co-alterations. One patient had PD after receiving second-line crizotinib and PR after third-line icotinib (PFS: 27.5 months), whereas the other patient had a PR after receiving gefitinib as first-line treatment (PFS: 12.7 months).
Despite the limited data, recommending EGFR-TKI as the first-line therapy for patients with dual EGFR and ROS1 alterations is reasonable, while crizotinib may be more useful as a second-line treatment after EGFR-TKI progression.
MET Alteration
Mesenchymal-epithelial transition (MET) receptor is a transmembrane tyrosine kinase. It can activate downstream signaling pathways (e.g., RAS/RAF/MAPK and PI3K/AKT/mTOR) by binding to the ligand hepatocyte growth factor. These pathways play important roles in cell proliferation, survival, migration, motility, and invasion (125–128). MET gene amplification has been recognized as a common mechanism of acquired resistance to EGFR-TKIs (129, 130), which encouraged researchers to pay more attention to the role of MET in the intrinsic resistance to these agents. In vitro studies have shown that MET amplification in HCC827 lung adenocarcinoma cells, which harbor EGFR 19del, mediates resistance to EGFR-TKIs (131). In addition, the coexistence of positive MET FISH status and EGFR mutations is associated with shorter DFS and OS after surgery in patients with lung adenocarcinoma (132). However, the relationship between MET FISH status and clinical outcomes for EGFR-TKI treatment is unclear.
Two distinct processes (i.e., polysomy and amplification) lead to MET copy-number gains (133). FISH, used to identify MET status in many clinical trials, can distinguish polysomy and true amplification. True MET amplification causes copy number increases without an increase in centromeric region of chromosome 7 (CEP7). Thus, the MET/CEP7 ratio increases. In polysomy, a MET copy increase is associated with an increase in the corresponding centromere. Therefore, polysomy has a preserved MET/CEP7 ratio (133). No standard criteria for MET positivity have been established. Two commonly used scoring systems are PathVysion (MET/CEP7 ratio ≥ 2), which only includes amplification, and the Cappuzzo scoring system (≥5 MET signals per cell), which includes both polysomy and amplification (132). MET amplification can be detected by FISH, IHC, and NGS. Lack of platform harmonization and thresholds for defining MET positivity has contributed to the conflicting results.
Noro et al. (38) explored the correlations between pre-treatment MET FISH status and OS and PFS in patients with EGFR-mutant lung adenocarcinoma treated with gefitinib. Eleven patients with MET FISH positivity (ten patients exhibited high polysomy [mean MET per cell ≥5 copies]; one patient exhibited amplification [MET gene/CEP7 ≥2 per cell]) had significantly shorter PFS and OS than 24 patients who were MET FISH-negative (PFS, 7.6 vs. 15.9 months; p = 0.001 and OS, 16.8 vs. 33 months; p = 0.03). Thus, pre-gefitinib MET FISH status may predict shortened PFS and OS.
Lai et al. (22) identified 52 patients with a high MET copy number gain (CNG) using FISH (MET-high: ≥5 copies per nucleus; polysomy: MET/CEP7 <2.0; amplification: MET/CEP7 ≥2.0) in 200 metastatic TKI-naive EGFR-mutant NSCLC patients. A total of 154 patients were treated with first-line EGFR-TKI monotherapy. The ORR was 74.4 and 53.9% in MET-high and MET-low patients, respectively (p = 0.033), while the median time-to-treatment failure (TTF) was similar in these two groups (12.2 vs. 13.1 months). However, all five patients with MET amplification had a poor or short-lived response to the EGFR-TKI (median TTF, 5 months; range, 1.0 to 6.4 months), suggesting that MET amplification concurrent with CNG ≥5 affects the response to EGFR-TKI. Furthermore, a patient harboring the EGFR L858R mutation and a coexisting MET amplification with a CNG of 7.3 and MET/CEP7 of 3.4 experienced disease progression within four weeks after starting erlotinib but showed significant regression of a pulmonary lesion when treated with crizotinib monotherapy. Li et al. (23) reported that a patient with coexisting EGFR 19del and de novo MET amplification had disease progression after initial treatment with icotinib, but the lung mass shrunk significantly after switching to crizotinib monotherapy. These data suggested that primary MET amplification could be a possible mechanism of intrinsic EGFR-TKI resistance, and patients harboring this genomic alteration may benefit from MET inhibitors.
Preliminary data from several small studies demonstrated crizotinib efficacy in patients with de novo MET amplification. In 15 patients with de novo MET amplification (A MET/centromere ratio [MET/CEN] ≥1.8), the ORR, PFS, and OS for crizotinib were 73.3%, 6.5 months (95% CI, 2.7–10.3), and 31 months, respectively (134). In addition, MET and EGFR inhibitor combination has had some effect in patients with EGFR-mutant NSCLC that developed resistance to prior EGFR-targeted therapies through MET gene amplification (127). A case report revealed that this combination might also have efficacy in patients with EGFR-mutant NSCLC and concomitant de novo MET amplification (24). While the patient had primary resistance to erlotinib, the tumor shrank after the addition of crizotinib to the treatment strategy.
In summary, it is still uncertain whether MET positivity is related to primary TKI resistance in patients with EGFR-mutant NSCLC, mainly due to the limited patient numbers and different evaluating systems. Further study in larger patient populations using uniform criteria to assess MET status is needed. MET inhibitor monotherapy and combinations with EGFR-TKIs appear to be potential treatment strategies for EGFR-mutant patients with primary MET alterations but require further validation.
Other Biomarkers
PD-L1 Expression
Programmed death-ligand 1 (PD-L1) is an immune checkpoint protein expressed on tumor and tumor-infiltrating immune cells. PD-L1 expression can be used as a predictive biomarker for PD-1 and PD-L1 blockade therapy (135–138). PD-1/PD-L1 antibodies, including nivolumab, pembrolizumab, and atezolizumab, have been approved for first- or second-line treatment of advanced NSCLC (139). However, PD-1/PD-L1 inhibitors lack efficacy in most NSCLC patients with EGFR mutations (140), even in those with high PD-L1 expression (tumor proportion score [TPS] ≥50%) (141). Positive PD-L1 expression (≥1%) occurs in about 50% of EGFR-mutant tumors, while high PD-L1 expression (≥50%) occurs in about 5%, which is less frequent compared to EGFR mutation-negative tumors (42). In addition, low tumor mutational burden and the lack of CD8+ tumor-infiltrating lymphocytes in the tumor microenvironment in lung cancer with EGFR mutations may be possible explanations for the lack of efficacy of PD-L1 inhibitors (142). A potential relationship may also exist between PD-L1 expression and EGFR-TKI efficacy in EGFR-mutant lung cancer patients.
Preclinical studies have demonstrated that EGFR activation can facilitate immune escape by inducing PD-L1 expression (143). Moreover, PD-L1 expression in EGFR-mutant NSCLC cell lines can be downregulated by EGFR inhibitors (144). A recent study revealed that EGFR-mutant NSCLC cell lines with higher PD-L1 expression were less sensitive to gefitinib. In addition, PD-L1 overexpression may induce epithelial-mesenchymal transition through the activation of the TGF-β/Smad canonical signaling pathway, leading to primary resistance to EGFR-TKIs (145).
Earlier clinical studies that examined PD-L1 expression generated conflicting results. Some studies showed that positive PD-L1 expression was significantly correlated with a greater DCR and longer PFS and OS after treatment with EGFR-TKIs, while another study found no significant correlation between PD-L1 expression and efficacy (146–148). Five recent studies in a larger Asian population showed that PD-L1 expression was associated with poor clinical outcomes in EGFR-mutant patients treated with EGFR-TKIs. Both Soo et al. (149) and Yoneshima et al. (150) (n = 90 and 71 EGFR-mutant NSCLC patients, respectively) found that PD-L1 expression was significantly associated with shorter PFS for EGFR-mutant NSCLC patients treated with EGFR-TKIs. Three additional studies revealed that high PD-L1 expression not only predicted a poor response to EGFR-TKIs but was also related to primary resistance to these agents. In a study of 101 patients with EGFR-mutant NSCLC, strong PD-L1 expression (TC3/IC3 ≥50% for tumor cells [TC] or ≥10% for immune cells [IC]) was significantly associated with decreased ORR and shortened PFS compared to weak (TC1-2/IC1-2: 5–49% for TC or 5–9% for IC) or negative (<5% for TC or IC) PD-L1 expression (ORR, 35.7 vs. 63.2 vs. 67.3%; p = 0.002; PFS, 3.8 vs. 6.0 vs. 9.5 months; p < 0.001), regardless of EGFR mutation (e.g., 19del or L858R) (39). Furthermore, patients with de novo resistance to EGFR-TKIs had a higher proportion of positive PD-L1 expression than those with acquired resistance (66.7 vs. 30.2%; p = 0.009).
The study performed by Hsu et al. (40) included 123 EGFR-mutant lung adenocarcinoma patients. The median PFS and OS for the EGFR-TKIs were 1.6 months (95% CI, 1.1–2.0) and 10.1 months (95% CI, 6.4–13.8) in patients with a PD-L1 ≥50%, which were clearly shorter than those for patients with a PD-L1 <1% (median PFS, 7.3 months; 95% CI, 2.7–12.0; median OS, 38.2 months; 95% CI, 26.1–50.3). Therefore, higher PD-L1 expression levels were associated with lower OS and PFS for patients with EGFR mutations treated with EGFR-TKIs. The patients were divided into two groups (primary resistance and disease control). In the primary resistance group, 22.7 and 30.3% of the patients had PD-L1 TPS ≥50 or ≥25%, respectively, while the frequencies were only 1.8 and 3.5%, respectively, in the disease control group (both p < 0.001). These results revealed that a higher PD-L1 expression level was associated with a higher incidence of primary EGFR-TKI resistance.
Yang et al. (41) performed a study with 153 EGFR-mutated lung adenocarcinoma patients. The ORR for EGFR-TKI and PFS was better in patients with PD-L1 expression <50% (ORR/PFS in PD-L1 0 vs. 1–49 vs. ≥50%: 65.6%/12.5 vs. 56.4%/12.8 vs. 38.9%/5.9 months, p < 0.05). The multivariate analysis showed that PD-L1 <50% was an independent prognostic factor for longer PFS (HR, 0.433; 95% CI, 0.250–0.751; p = 0.003). Furthermore, a significant proportion of patients with TPS ≥ 50% had primary resistance to EGFR-TKIs (44.4%). In addition, 91 patients were re-biopsied for T790M testing upon disease progression. Tumors with higher PD-L1 expression were less likely to develop an acquired T790M mutation (T790M+ in PD-L1 0 vs. 1–49 vs. ≥50%: 53.7% (36/67) vs. 35.7% (5/14) vs. 10% (1/10); p = 0.024).
Interestingly, a recent study by Brown et al. (42) revealed that unlike with erlotinib or gefitinib, the clinical benefit of first-line osimertinib treatment in NSCLC patients with EGFR mutations was unaffected by PD-L1 expression status (PFS: PD-L1-positive (TC ≥1%) vs. PD-L1-negative patients (TC <1%): 18.4 vs. 18.9 months). In addition, it can be inferred from the Yang et al. study (41) that patients with higher PD-L1 expression have less of a chance to use osimertinib as subsequent therapy because they are less likely to develop acquired T790M mutations, suggesting that first-line osimertinib may be a better choice for these patients.
Because immune checkpoint inhibitor (ICI) monotherapy showed poor efficacy in patients with EGFR mutations, different combinational strategies have been explored. Combination therapy consisting of EGFR-TKI with ICIs or sequential use of EGFR-TKI following ICI therapies are not proper choices because they showed increased grade three or higher toxicities and increased the risk of severe immune-related adverse events (142). Of note, in the IMpower-150 study (n = 124 EGFR-mutant NSCLC patients), atezolizumab plus bevacizumab and chemotherapy (ABCP; n = 34) yielded a higher ORR (71 vs. 42%), DOR (11.1 vs. 4.7 months), PFS (10.2 vs. 6.9 months; HR, 0.61; 95% CI, 0.36–1.03), and OS (not evaluable [NE] vs. 18.7 months; HR, 0.61; 95% CI, 0.29–1.28) compared to bevacizumab plus chemotherapy (BCP; n = 45) (151). In addition, toripalimab, a PD-1 inhibitor, in combination with chemotherapy, showed promising anti-tumor activity with a manageable safety profile in a phase II prospective clinical trial (152). These results suggest that chemotherapy and antiangiogenic therapy may be related to EGFR cancer immunity; however, additional studies are required to fully understand this relationship. Several Phase III prospective clinical trials are ongoing to explore the efficacy and safety of the combination of immunotherapy with chemotherapy and/or antiangiogenic therapy in EGFR-mutant NSCLC progressed on EGFR-TKIs, including KEYNOTE-789, KEYNOTE-722, ORIENT-31, and JS001 study (NCT03924050).
BIM Polymorphism
BIM, also known as B-cell chronic lymphocytic leukemia/lymphoma (Bcl-2)-like 11 (BCL2L11), is a BH3-only pro-apoptotic member of the Bcl-2 family. BIM gene products containing BH3 domains are required for the induction of apoptosis by EGFR-TKIs (153–157). The BIM deletion polymorphism is a 2,903-bp fragment deletion in intron 2 of the BIM gene. It results in splicing of exon 3 over exon 4 in the BIM pre-mRNA, generating an inactive BIM protein isoform lacking the crucial BH3 domain. This BIM deletion polymorphism occurs in 12–16% of lung cancer patients with EGFR mutations (158, 159). It impairs EGFR-TKI-related apoptosis and mediates intrinsic resistance to EGFR-TKIs in EGFR-mutant NSCLC cell lines (154).
The impact of the BIM deletion polymorphism on the clinical outcome of NSCLC patients with EGFR mutations treated with EGFR-TKIs has been evaluated in multiple studies with contradictory results. Some studies have shown that EGFR-mutant NSCLC patients with the BIM deletion polymorphism had inferior EGFR-TKI efficacy compared to those with wild-type BIM (154, 160–164). Others found that EGFR-mutant patients with and without the BIM deletion polymorphism had similar clinical outcomes in response to EGFR-TKI treatment (165–168). Several meta-analyses have indicated that the BIM deletion polymorphism is associated with poor ORR (168–172), consistent with other studies demonstrating that the BIM deletion polymorphism is associated with shorter PFS in patients with NSCLC harboring EGFR mutations who received EGFR-TKIs (168–175). Only one study showed no significant association between BIM status and the response to EGFR-TKIs (173).
These results suggested that BIM deletion polymorphism can be used as a predictive biomarker for EGFR-TKI treatment. Patients with the BIM deletion polymorphism may benefit less from EGFR-TKI therapy. EGFR-TKI combination therapy with histone deacetylase (HDAC) inhibitors may be one approach to overcome the inferior outcomes conferred by the BIM deletion. A recent phase I study with 12 patients treated with the combination of gefitinib and vorinostat (a small-molecule HDAC inhibitor) revealed a DCR of 83.3% (10/12) at six weeks, with a median PFS of 5.2 months (95% CI, 1.4–15.7) (43). In addition, retrospective analysis revealed that EGFR-TKIs plus chemotherapy conferred a significantly higher ORR (65.5 vs. 38.9%, p = 0.046), prolonged PFS (7.2 vs. 4.7 months; p = 0.008) and a longer OS (18.5 vs. 14.2 months; p = 0.107) compared to TKIs alone in advanced NSCLC patients with EGFR mutations and BIM deletion polymorphism (176). The latter difference did not reach statistical significance. Further studies are needed to determine the clinical efficacy of these combined therapies.
Conclusions
This report described the many studies on the efficacy of EGFR-TKIs in patients with EGFR-mutant NSCLC with coexisting intra-EGFR or other gene mutations and molecular markers that may predict EGFR-TKI efficacy. Most of these studies were retrospective studies with controversial results and inconsistencies between sample size, race, disease stage, and treatment line. In addition, tumor heterogeneity and a combination of several concurrent genes were not considered. Nevertheless, we presented additional information in this field and have provided clues for clinicians to identify which patients might be more effectively treated with EGFR-TKIs, and those for which EGFR-TKIs would be less effective, even though they have sensitive EGFR mutations. We also described some available treatment strategies for patients who might not benefit from EGFR-TKIs. To the best of our knowledge, this is the first review on EGFR-TKI efficacy encompassing so many specific concurrent gene mutations and other biomarkers. To initiate appropriate therapies for each individual patient in the future, the use of comprehensive genomic profiling should become routine. In addition, larger populations with advanced NSCLC stratified by uniform stage and therapy are needed to examine these concurrent alterations and validate their importance as prognostic factors for EGFR-TKI therapy in EGFR-mutant patients.
Author Contributions
YG wrote the original draft and did the largest amount of revising and editing. JS, YW, LH, LS, JZ, SZ, WJ, and JM collected related literature and did substantial editing of the manuscript. CH oversaw and coordinated the entire process and dramatically altered the orientation of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the 345 Talent Project of Shengjing Hospital.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Cheng L, Alexander RE, Maclennan GT, Cummings OW, Montironi R, Lopez-Beltran A, et al. Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol (2012) 25:347–69. doi: 10.1038/modpathol.2011.215
3. Yang CY, Yang JC, Yang PC. Precision Management of Advanced Non-Small Cell Lung Cancer. Annu Rev Med (2020) 71:117–36. doi: 10.1146/annurev-med-051718-013524
4. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med (2010) 362:2380–8. doi: 10.1056/NEJMoa0909530
5. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol (2010) 11:121–8. doi: 10.1016/S1470-2045(09)70364-X
6. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol (2011) 12:735–42. doi: 10.1016/S1470-2045(11)70184-X
7. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol (2013) 31:3327–34. doi: 10.1200/JCO.2012.44.2806
8. Hong S, Gao F, Fu S, Wang Y, Fang W, Huang Y, et al. Concomitant Genetic Alterations With Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With EGFR-Mutant Advanced Non-Small Cell Lung Cancer. JAMA Oncol (2018) 4:739–42. doi: 10.1001/jamaoncol.2018.0049
9. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet (2017) 49:1693–704. doi: 10.1038/ng.3990
10. Barnet MB, O’Toole S, Horvath LG, Selinger C, Yu B, Ng CC, et al. EGFR-Co-Mutated Advanced NSCLC and Response to EGFR Tyrosine Kinase Inhibitors. J Thorac Oncol (2017) 12:585–90. doi: 10.1016/j.jtho.2016.09.001
11. Sato S, Nagahashi M, Koike T, Ichikawa H, Shimada Y, Watanabe S, et al. Impact of Concurrent Genomic Alterations Detected by Comprehensive Genomic Sequencing on Clinical Outcomes in East-Asian Patients with EGFR-Mutated Lung Adenocarcinoma. Sci Rep (2018) 8:1005. doi: 10.1038/s41598-017-18560-y
12. Wang Z, Cheng Y, An T, Gao H, Wang K, Zhou Q, et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single-arm, multicentre clinical trial. Lancet Respir Med (2018) 6:681–90. doi: 10.1016/S2213-2600(18)30264-9
13. Chang SC, Lai YC, Chang CY, Huang LK, Chen SJ, Tan KT, et al. Concomitant Genetic Alterations are Associated with Worse Clinical Outcome in EGFR Mutant NSCLC Patients Treated with Tyrosine Kinase Inhibitors. Transl Oncol (2019) 12:1425–31. doi: 10.1016/j.tranon.2019.07.008
14. Chen M, Xu Y, Zhao J, Zhong W, Zhang L, Bi Y, et al. Concurrent Driver Gene Mutations as Negative Predictive Factors in Epidermal Growth Factor Receptor-Positive Non-Small Cell Lung Cancer. EBioMedicine (2019) 42:304–10. doi: 10.1016/j.ebiom.2019.03.023
15. Hu W, Liu Y, Chen J. Concurrent gene alterations with EGFR mutation and treatment efficacy of EGFR-TKIs in Chinese patients with non-small cell lung cancer. Oncotarget (2017) 8:25046–54. doi: 10.18632/oncotarget.15337
16. Guibert N, Barlesi F, Descourt R, Léna H, Besse B, Beau-Faller M, et al. Characteristics and Outcomes of Patients with Lung Cancer Harboring Multiple Molecular Alterations: Results from the IFCT Study Biomarkers France. J Thorac Oncol (2017) 12:963–73. doi: 10.1016/j.jtho.2017.02.001
17. Ulivi P, Chiadini E, Dazzi C, Dubini A, Costantini M, Medri L, et al. Nonsquamous, Non-Small-Cell Lung Cancer Patients Who Carry a Double Mutation of EGFR, EML4-ALK or KRAS: Frequency, Clinical-Pathological Characteristics, and Response to Therapy. Clin Lung Cancer (2016) 17:384–90. doi: 10.1016/j.cllc.2015.11.004
18. Lee JK, Kim TM, Koh Y, Lee SH, Kim DW, Jeon YK, et al. Differential sensitivities to tyrosine kinase inhibitors in NSCLC harboring EGFR mutation and ALK translocation. Lung Cancer (2012) 77:460–3. doi: 10.1016/j.lungcan.2012.04.012
19. Tetsu O, Hangauer MJ, Phuchareon J, Eisele DW, McCormick F. Drug Resistance to EGFR Inhibitors in Lung Cancer. Chemotherapy (2016) 61:223–35. doi: 10.1159/000443368
20. Benesova L, Minarik M, Jancarikova D, Belsanova B, Pesek M. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res (2010) 30:1667–71.
21. Zhuang X, Zhao C, Li J, Su C, Chen X, Ren S, et al. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med (2019) 8:2858–66. doi: 10.1002/cam4.2183
22. Lai GGY, Lim TH, Lim J, Liew PJR, Kwang XL, Nahar R, et al. Clonal MET Amplification as a Determinant of Tyrosine Kinase Inhibitor Resistance in Epidermal Growth Factor Receptor-Mutant Non-Small-Cell Lung Cancer. J Clin Oncol (2019) 37:876–84. doi: 10.1200/JCO.18.00177
23. Li JW, Cao SH, Xu JL, Zhong H. De novo MET amplification promotes intrinsic resistance to first-generation EGFR tyrosine kinase inhibitors. Cancer Biol Ther (2019) 20:1183–6. doi: 10.1080/15384047.2019.1617568
24. Gainor JF, Niederst MJ, Lennerz JK, Dagogo-Jack I, Stevens S, Shaw AT, et al. Dramatic Response to Combination Erlotinib and Crizotinib in a Patient with Advanced, EGFR-Mutant Lung Cancer Harboring De Novo MET Amplification. J Thorac Oncol (2016) 11:e83–85. doi: 10.1016/j.jtho.2016.02.021
25. Zhang B, Wang S, Qian J, Yang W, Qian F, Lu J, et al. Complex epidermal growth factor receptor mutations and their responses to tyrosine kinase inhibitors in previously untreated advanced lung adenocarcinomas. Cancer (2018) 124:2399–406. doi: 10.1002/cncr.31329
26. Keam B, Kim DW, Park JH, Lee JO, Kim TM, Lee SH, et al. Rare and complex mutations of epidermal growth factor receptor, and efficacy of tyrosine kinase inhibitor in patients with non-small cell lung cancer. Int J Clin Oncol (2014) 19:594–600. doi: 10.1007/s10147-013-0602-1
27. Wu SG, Chang YL, Hsu YC, Wu JY, Yang CH, Yu CJ, et al. Good response to gefitinib in lung adenocarcinoma of complex epidermal growth factor receptor (EGFR) mutations with the classical mutation pattern. Oncologist (2008) 13:1276–84. doi: 10.1634/theoncologist.2008-0093
28. Kim Y, Lee B, Shim JH, Lee SH, Park WY, Choi YL, et al. Concurrent Genetic Alterations Predict the Progression to Target Therapy in EGFR-Mutated Advanced NSCLC. J Thorac Oncol (2019) 14:193–202. doi: 10.1016/j.jtho.2018.10.150
29. Canale M, Petracci E, Delmonte A, Chiadini E, Dazzi C, Papi M, et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res (2017) 23:2195–202. doi: 10.1158/1078-0432.CCR-16-0966
30. VanderLaan PA, Rangachari D, Mockus SM, Spotlow V, Reddi HV, Malcolm J, et al. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer (2017) 106:17–21. doi: 10.1016/j.lungcan.2017.01.011
31. Labbé C, Cabanero M, Korpanty GJ, Tomasini P, Doherty MK, Mascaux C, et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer (2017) 111:23–9. doi: 10.1016/j.lungcan.2017.06.014
32. Eng J, Woo KM, Sima CS, Plodkowski A, Hellmann MD, Chaft JE, et al. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J Thorac Oncol (2015) 10:1713–9. doi: 10.1097/JTO.0000000000000671
33. Wu SG, Chang YL, Yu CJ, Yang PC, Shih JY. The Role of PIK3CA Mutations among Lung Adenocarcinoma Patients with Primary and Acquired Resistance to EGFR Tyrosine Kinase Inhibition. Sci Rep (2016) 6:35249. doi: 10.1038/srep35249
34. Yang JJ, Zhang XC, Su J, Xu CR, Zhou Q, Tian HX, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res (2014) 20:1383–92. doi: 10.1158/1078-0432.CCR-13-0699
35. Lou NN, Zhang XC, Chen HJ, Zhou Q, Yan LX, Xie Z, et al. Clinical outcomes of advanced non-small-cell lung cancer patients with EGFR mutation, ALK rearrangement and EGFR/ALK co-alterations. Oncotarget (2016) 7:65185–95. doi: 10.18632/oncotarget.11218
36. Zhao Y, Wang S, Zhang B, Qiao R, Xu J, Zhang L, et al. Clinical Management of Non-Small Cell Lung Cancer with Concomitant EGFR Mutations and ALK Rearrangements: Efficacy of EGFR Tyrosine Kinase Inhibitors and Crizotinib. Target Oncol (2019) 14:169–78. doi: 10.1007/s11523-019-00628-6
37. Rachiglio AM, Fenizia F, Piccirillo MC, Galetta D, Crinò L, Vincenzi B, et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers (Basel) (2019) 11(3):341. doi: 10.3390/cancers11030341
38. Noro R, Seike M, Zou F, Soeno C, Matsuda K, Sugano T, et al. MET FISH-positive status predicts short progression-free survival and overall survival after gefitinib treatment in lung adenocarcinoma with EGFR mutation. BMC Cancer (2015) 15:31. doi: 10.1186/s12885-015-1019-1
39. Su S, Dong ZY, Xie Z, Yan LX, Li YF, Su J, et al. Strong Programmed Death Ligand 1 Expression Predicts Poor Response and De Novo Resistance to EGFR Tyrosine Kinase Inhibitors Among NSCLC Patients With EGFR Mutation. J Thorac Oncol (2018) 13:1668–75. doi: 10.1016/j.jtho.2018.07.016
40. Hsu KH, Huang YH, Tseng JS, Chen KC, Ku WH, Su KY, et al. High PD-L1 expression correlates with primary resistance to EGFR-TKIs in treatment naïve advanced EGFR-mutant lung adenocarcinoma patients. Lung Cancer (2019) 127:37–43. doi: 10.1016/j.lungcan.2018.11.021
41. Yang CY, Liao WY, Ho CC, Chen KY, Tsai TH, Hsu CL, et al. Association between programmed death-ligand 1 expression, immune microenvironments, and clinical outcomes in epidermal growth factor receptor mutant lung adenocarcinoma patients treated with tyrosine kinase inhibitors. Eur J Cancer (2020) 124:110–22. doi: 10.1016/j.ejca.2019.10.019
42. Brown H, Vansteenkiste J, Nakagawa K, Cobo M, John T, Barker C, et al. Programmed Cell Death Ligand 1 Expression in Untreated EGFR Mutated Advanced NSCLC and Response to Osimertinib Versus Comparator in FLAURA. J Thorac Oncol (2020) 15:138–43. doi: 10.1016/j.jtho.2019.09.009
43. Takeuchi S, Hase T, Shimizu S, Ando M, Hata A, Murakami H, et al. Phase I study of vorinostat with gefitinib in BIM deletion polymorphism/epidermal growth factor receptor mutation double-positive lung cancer. Cancer Sci (2020) 111:561–70. doi: 10.1111/cas.14260
44. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361:947–57. doi: 10.1056/NEJMoa0810699
45. Hsieh MH, Fang YF, Chang WC, Kuo HP, Lin SY, Liu HP, et al. Complex mutation patterns of epidermal growth factor receptor gene associated with variable responses to gefitinib treatment in patients with non-small cell lung cancer. Lung Cancer (2006) 53:311–22. doi: 10.1016/j.lungcan.2006.06.005
46. Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res (2011) 17:3812–21. doi: 10.1158/1078-0432.CCR-10-3408
47. Zhang GC, Lin JY, Wang Z, Zhou Q, Xu CR, Zhu JQ, et al. Epidermal growth factor receptor double activating mutations involving both exons 19 and 21 exist in Chinese non-small cell lung cancer patients. Clin Oncol (R Coll Radiol) (2007) 19:499–506. doi: 10.1016/j.clon.2007.04.006
48. Huang SF, Liu HP, Li LH, Ku YC, Fu YN, Tsai HY, et al. High frequency of epidermal growth factor receptor mutations with complex patterns in non-small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res (2004) 10:8195–203. doi: 10.1158/1078-0432.CCR-04-1245
49. O’Kane GM, Bradbury PA, Feld R, Leighl NB, Liu G, Pisters KM, et al. Uncommon EGFR mutations in advanced non-small cell lung cancer. Lung Cancer (2017) 109:137–44. doi: 10.1016/j.lungcan.2017.04.016
50. Hata A, Yoshioka H, Fujita S, Kunimasa K, Kaji R, Imai Y, et al. Complex mutations in the epidermal growth factor receptor gene in non-small cell lung cancer. J Thorac Oncol (2010) 5:1524–8. doi: 10.1097/JTO.0b013e3181e8b3c5
51. Beau-Faller M, Prim N, Ruppert AM, Nanni-Metéllus I, Lacave R, Lacroix L, et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann Oncol (2014) 25:126–31. doi: 10.1093/annonc/mdt418
52. Kobayashi S, Canepa HM, Bailey AS, Nakayama S, Yamaguchi N, Goldstein MA, et al. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J Thorac Oncol (2013) 8:45–51. doi: 10.1097/JTO.0b013e3182781e35
53. Sriuranpong V, Chantranuwat C, Huapai N, Chalermchai T, Leungtaweeboon K, Lertsanguansinchai P, et al. High frequency of mutation of epidermal growth factor receptor in lung adenocarcinoma in Thailand. Cancer Lett (2006) 239:292–7. doi: 10.1016/j.canlet.2005.08.029
54. Stone E, Allen HA, Saghaie T, Abbott A, Daniel R, Mead RS, et al. High proportion of rare and compound epidermal growth factor receptor mutations in an Australian population of non-squamous non-small-cell lung cancer. Intern Med J (2014) 44:1188–92. doi: 10.1111/imj.12587
55. Shih JY, Gow CH, Yang PC. EGFR mutation conferring primary resistance to gefitinib in non-small-cell lung cancer. N Engl J Med (2005) 353:207–8. doi: 10.1056/NEJM200507143530217
56. Yu HA, Arcila ME, Hellmann MD, Kris MG, Ladanyi M, Riely GJ. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann Oncol (2014) 25:423–8. doi: 10.1093/annonc/mdt573
57. Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol (2015) 16:830–8. doi: 10.1016/S1470-2045(15)00026-1
58. Wang S, Yan B, Zhang Y, Xu J, Qiao R, Dong Y, et al. Different characteristics and survival in non-small cell lung cancer patients with primary and acquired EGFR T790M mutation. Int J Cancer (2019) 144:2880–6. doi: 10.1002/ijc.32015
59. Zhang B, Xu J, Zhang X, Gu P, Wang H, Wang S, et al. Coexistence of sensitive and resistant epidermal growth factor receptor (EGFR) mutations in pretreatment non-small cell lung cancer (NSCLC) patients: First or third generation tyrosine kinase inhibitors (TKIs)? Lung Cancer (2018) 117:27–31. doi: 10.1016/j.lungcan.2018.01.006
60. Watanabe S, Minegishi Y, Yoshizawa H, Maemondo M, Inoue A, Sugawara S, et al. Effectiveness of gefitinib against non-small-cell lung cancer with the uncommon EGFR mutations G719X and L861Q. J Thorac Oncol (2014) 9:189–94. doi: 10.1097/JTO.0000000000000048
61. Shen YC, Tseng GC, Tu CY, Chen WC, Liao WC, Chen WC, et al. Comparing the effects of afatinib with gefitinib or Erlotinib in patients with advanced-stage lung adenocarcinoma harboring non-classical epidermal growth factor receptor mutations. Lung Cancer (2017) 110:56–62. doi: 10.1016/j.lungcan.2017.06.007
62. Yang JC, Schuler M, Popat S, Miura S, Heeke S, Park K, et al. Afatinib for the Treatment of NSCLC Harboring Uncommon EGFR Mutations: A Database of 693 Cases. J Thorac Oncol (2020) 15:803–15. doi: 10.1016/j.jtho.2019.12.126
63. Yang S, Mao S, Li X, Zhao C, Liu Q, Yu X, et al. Uncommon EGFR mutations associate with lower incidence of T790M mutation after EGFR-TKI treatment in patients with advanced NSCLC. Lung Cancer (2020) 139:133–9. doi: 10.1016/j.lungcan.2019.11.018
64. Lin YT, Tsai TH, Wu SG, Liu YN, Yu CJ, Shih JY. Complex EGFR mutations with secondary T790M mutation confer shorter osimertinib progression-free survival and overall survival in advanced non-small cell lung cancer. Lung Cancer (2020) 145:1–9. doi: 10.1016/j.lungcan.2020.04.022
65. Yeh P, Chen H, Andrews J, Naser R, Pao W, Horn L. DNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clin Cancer Res (2013) 19:1894–901. doi: 10.1158/1078-0432.CCR-12-1894
66. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol (2009) 1:a001883. doi: 10.1101/cshperspect.a001883
67. Mogi A, Kuwano H. TP53 Mutations in Nonsmall Cell Lung Cancer. J Biomed Biotechnol (2011) 2011:583929. doi: 10.1155/2011/583929
68. Deben C, Deschoolmeester V, Lardon F, Rolfo C, Pauwels P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: Evaluating their prognostic and predictive value. Crit Rev Oncol Hematol (2016) 99:63–73. doi: 10.1016/j.critrevonc.2015.11.019
69. Ma X, Le Teuff G, Lacas B, Tsao MS, Graziano S, Pignon JP, et al. Prognostic and Predictive Effect of TP53 Mutations in Patients with Non-Small Cell Lung Cancer from Adjuvant Cisplatin-Based Therapy Randomized Trials: A LACE-Bio Pooled Analysis. J Thorac Oncol (2016) 11:850–61. doi: 10.1016/j.jtho.2016.02.002
70. Nahar R, Zhai W, Zhang T, Takano A, Khng AJ, Lee YY, et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat Commun (2018) 9:216. doi: 10.1038/s41467-017-02584-z
71. Rho JK, Choi YJ, Ryoo BY, Na II, Yang SH, Kim CH, et al. p53 enhances gefitinib-induced growth inhibition and apoptosis by regulation of Fas in non-small cell lung cancer. Cancer Res (2007) 67:1163–9. doi: 10.1158/0008-5472.CAN-06-2037
72. Munsch D, Watanabe-Fukunaga R, Bourdon JC, Nagata S, May E, Yonish-Rouach E, et al. Human and mouse Fas (APO-1/CD95) death receptor genes each contain a p53-responsive element that is activated by p53 mutants unable to induce apoptosis. J Biol Chem (2000) 275:3867–72. doi: 10.1074/jbc.275.6.3867
73. Chang GC, Hsu SL, Tsai JR, Liang FP, Lin SY, Sheu GT, et al. Molecular mechanisms of ZD1839-induced G1-cell cycle arrest and apoptosis in human lung adenocarcinoma A549 cells. Biochem Pharmacol (2004) 68:1453–64. doi: 10.1016/j.bcp.2004.06.006
74. Li J, Lin B, Li X, Tang X, He Z, Zhou K. Biomarkers for predicting response to tyrosine kinase inhibitors in drug-sensitive and drug-resistant human bladder cancer cells. Oncol Rep (2015) 33:951–7. doi: 10.3892/or.2014.3639
75. MacLaine NJ, Wood MD, Holder JC, Rees RW, Southgate J. Sensitivity of normal, paramalignant, and malignant human urothelial cells to inhibitors of the epidermal growth factor receptor signaling pathway. Mol Cancer Res (2008) 6:53–63. doi: 10.1158/1541-7786.MCR-07-0134
76. Qin K, Hou H, Liang Y, Zhang X. Prognostic value of TP53 concurrent mutations for EGFR- TKIs and ALK-TKIs based targeted therapy in advanced non-small cell lung cancer: a meta-analysis. BMC Cancer (2020) 20:328. doi: 10.1186/s12885-020-06805-5
77. Nakagawa K NE, Garon EB, Nishio M, Seto T, Yamamoto N, Park K, et al. RELAY, erlotinib plus ramucirumab or placebo in untreated EGFR-mutated metastatic NSCLC: Outcomes by EGFR mutation type. Ann Oncol (2020) 31(suppl_4):S754–840. doi: 10.1016/annonc/annonc283
78. Zhong D ZC, Zhang Y, Shang Y, Wang L, Huang D. Anlotinib combined with icotinib provides a promising first-line treatment option for EGFR positive NSCLC patients harboring concomitant mutations: Exploratory analysis of the ALTER-L004 study. Ann Oncol (2020) 31(suppl_4):S754–840. doi: 10.1016/annonc/annonc283
79. Zhang L ZH, Zhang Z, Yao W, Min X, Gu K, Yu G, et al. ACTIVE: Apatinib plus gefitinib versus placebo plus gefitinib as first-line treatment for advanced epidermal growth factor receptor-mutant (EGFRm) non-small-cell lung cancer (NSCLC): A multicentered, randomized, double-blind, placebo-controlled phase III trial (CTONG1706). Ann Oncol (2020) 31(suppl_4):S1142–215. doi: 10.1016/annonc/annonc325
80. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer (2006) 94:455–9. doi: 10.1038/sj.bjc.6602970
81. Balsara BR, Pei J, Mitsuuchi Y, Page R, Klein-Szanto A, Wang H, et al. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis (2004) 25:2053–9. doi: 10.1093/carcin/bgh226
82. Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA (2005) 102:802–7. doi: 10.1073/pnas.0408864102
83. Kawano O, Sasaki H, Endo K, Suzuki E, Haneda H, Yukiue H, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer (2006) 54:209–15. doi: 10.1016/j.lungcan.2006.07.006
84. Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res (2008) 68:6913–21. doi: 10.1158/0008-5472.CAN-07-5084
85. Spoerke JM, O’Brien C, Huw L, Koeppen H, Fridlyand J, Brachmann RK, et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res (2012) 18:6771–83. doi: 10.1158/1078-0432.CCR-12-2347
86. Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol (2005) 6:279–86. doi: 10.1016/S1470-2045(05)70102-9
87. Fiala O, Pesek M, Finek J, Benesova L, Bortlicek Z, Minarik M. Gene mutations in squamous cell NSCLC: insignificance of EGFR, KRAS and PIK3CA mutations in prediction of EGFR-TKI treatment efficacy. Anticancer Res (2013) 33:1705–11. doi: 10.1016/S0169-5002(13)70293-9
88. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther (2012) 11:485–91. doi: 10.1158/1535-7163.MCT-11-0692
89. Scheffler M, Bos M, Gardizi M, König K, Michels S, Fassunke J, et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget (2015) 6:1315–26. doi: 10.18632/oncotarget.2834
90. Wang L, Hu H, Pan Y, Wang R, Li Y, Shen L, et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PloS One (2014) 9:e88291. doi: 10.1371/journal.pone.0088291
91. Li S, Li L, Zhu Y, Huang C, Qin Y, Liu H, et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. Br J Cancer (2014) 110:2812–20. doi: 10.1038/bjc.2014.210
92. Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol (2011) 6:707–15. doi: 10.1097/JTO.0b013e31820a3a6b
93. Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borrás AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest (2006) 116:2695–706. doi: 10.1172/JCI28656
94. Ludovini V, Bianconi F, Pistola L, Pistola V, Chiari R, Colella R, et al. Optimization of patient selection for EGFR-TKIs in advanced non-small cell lung cancer by combined analysis of KRAS, PIK3CA, MET, and non-sensitizing EGFR mutations. Cancer Chemother Pharmacol (2012) 69:1289–99. doi: 10.1007/s00280-012-1829-7
95. Jin Y, Bao H, Le X, Fan X, Tang M, Shi X, et al. Distinct co-acquired alterations and genomic evolution during TKI treatment in non-small-cell lung cancer patients with or without acquired T790M mutation. Oncogene (2020) 39:1846–59. doi: 10.1038/s41388-019-1104-z
96. Vazquez F, Sellers WR. The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling. Biochim Biophys Acta (2000) 1470:M21–35. doi: 10.1016/S0304-419X(99)00032-3
97. Pérez-Ramírez C, Cañadas-Garre M, Molina M, Faus-Dáder MJ, Calleja-Hernández M. PTEN and PI3K/AKT in non-small-cell lung cancer. Pharmacogenomics (2015) 16:1843–62. doi: 10.2217/pgs.15.122
98. Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, et al. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res (2010) 70:8715–25. doi: 10.1158/0008-5472.CAN-10-0043
99. Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res (2009) 69:3256–61. doi: 10.1158/0008-5472.CAN-08-4055
100. Bidkhori G, Moeini A, Masoudi-Nejad A. Modeling of tumor progression in NSCLC and intrinsic resistance to TKI in loss of PTEN expression. PloS One (2012) 7:e48004. doi: 10.1371/journal.pone.0048004
101. Xiao J, Hu CP, He BX, Chen X, Lu XX, Xie MX, et al. PTEN expression is a prognostic marker for patients with non-small cell lung cancer: a systematic review and meta-analysis of the literature. Oncotarget (2016) 7:57832–40. doi: 10.18632/oncotarget.11068
102. Yanagawa N, Leduc C, Kohler D, Saieg MA, John T, Sykes J, et al. Loss of phosphatase and tensin homolog protein expression is an independent poor prognostic marker in lung adenocarcinoma. J Thorac Oncol (2012) 7:1513–21. doi: 10.1097/JTO.0b013e3182641d4f
103. Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer (2006) 51:181–91. doi: 10.1016/j.lungcan.2005.10.00
104. Bepler G, Sharma S, Cantor A, Gautam A, Haura E, Simon G, et al. RRM1 and PTEN as prognostic parameters for overall and disease-free survival in patients with non-small-cell lung cancer. J Clin Oncol (2004) 22:1878–85. doi: 10.1200/JCO.2004.12.002
105. Wang F, Diao XY, Zhang X, Shao Q, Feng YF, An X, et al. Identification of genetic alterations associated with primary resistance to EGFR-TKIs in advanced non-small-cell lung cancer patients with EGFR sensitive mutations. Cancer Commun (Lond) (2019) 39:7. doi: 10.1186/s40880-019-0354-z
106. Endoh H, Yatabe Y, Kosaka T, Kuwano H, Mitsudomi T. PTEN and PIK3CA expression is associated with prolonged survival after gefitinib treatment in EGFR-mutated lung cancer patients. J Thorac Oncol (2006) 1:629–34. doi: 10.1016/S1556-0864(15)30374-9
107. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med (2010) 363:1693–703. doi: 10.1056/NEJMoa1006448
108. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res (2013) 19:4273–81. doi: 10.1158/1078-0432.CCR-13-0318
109. Won JK, Keam B, Koh J, Cho HJ, Jeon YK, Kim TM, et al. Concomitant ALK translocation and EGFR mutation in lung cancer: a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann Oncol (2015) 26:348–54. doi: 10.1093/annonc/mdu530
110. Liu J, Mu Z, Liu L, Li K, Jiang R, Chen P, et al. Frequency, clinical features and differential response to therapy of concurrent ALK/EGFR alterations in Chinese lung cancer patients. Drug Des Devel Ther (2019) 13:1809–17. doi: 10.2147/DDDT.S196189
111. Cai W, Lin D, Wu C, Li X, Zhao C, Zheng L, et al. Intratumoral Heterogeneity of ALK-Rearranged and ALK/EGFR Coaltered Lung Adenocarcinoma. J Clin Oncol (2015) 33:3701–9. doi: 10.1200/JCO.2014.58.8293
112. Baldi L, Mengoli MC, Bisagni A, Banzi MC, Boni C, Rossi G. Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung Cancer (2014) 86:291–5. doi: 10.1016/j.lungcan.2014.09.011
113. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol (2015) 16:e342–351. doi: 10.1016/S1470-2045(15)00077-7
114. Cardarella S, Ortiz TM, Joshi VA, Butaney M, Jackman DM, Kwiatkowski DJ, et al. The introduction of systematic genomic testing for patients with non-small-cell lung cancer. J Thorac Oncol (2012) 7:1767–74. doi: 10.1097/JTO.0b013e3182745bcb
115. Wen YS, Cai L, Zhang XW, Zhu JF, Zhang ZC, Shao JY, et al. Concurrent oncogene mutation profile in Chinese patients with stage Ib lung adenocarcinoma. Med (Baltimore) (2014) 93:e296. doi: 10.1097/MD.0000000000000296
116. Fiala O, Pesek M, Finek J, Benesova L, Belsanova B, Minarik M. The dominant role of G12C over other KRAS mutation types in the negative prediction of efficacy of epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Genet (2013) 206:26–31. doi: 10.1016/j.cancergen.2012.12.003
117. Choughule A, Sharma R, Trivedi V, Thavamani A, Noronha V, Joshi A, et al. Coexistence of KRAS mutation with mutant but not wild-type EGFR predicts response to tyrosine-kinase inhibitors in human lung cancer. Br J Cancer (2014) 111:2203–4. doi: 10.1038/bjc.2014.401
118. Zhu YC, Liao XH, Wang WX, Xu CW, Zhuang W, Wei JG, et al. Dual drive coexistence of EML4-ALK and TPM3-ROS1 fusion in advanced lung adenocarcinoma. Thorac Cancer (2018) 9:324–7. doi: 10.1111/1759-7714.12578
119. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol (2012) 30:863–70. doi: 10.1016/j.yonc.2012.06.023
120. Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res (2012) 18:4449–57. doi: 10.1158/1078-0432.CCR-11-3351
121. Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res (2012) 18:4570–9. doi: 10.1158/1078-0432.CCR-12-0550
122. Arnaoutakis K. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med (2015) 372:683. doi: 10.1056/NEJMc1415359
123. Mao Y, Wu S. ALK and ROS1 concurrent with EGFR mutation in patients with lung adenocarcinoma. Onco Targets Ther (2017) 10:3399–404. doi: 10.2147/OTT.S133349
124. Lambros L, Guibourg B, Uguen A. ROS1-rearranged Non-Small Cell Lung Cancers With Concomitant Oncogenic Driver Alterations: About Some Rare Therapeutic Dilemmas. Clin Lung Cancer (2018) 19:e73–4. doi: 10.1016/j.cllc.2017.08.005
125. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol (2011) 3:S7–s19. doi: 10.1177/1758834011422556
126. Cipriani NA, Abidoye OO, Vokes E, Salgia R. MET as a target for treatment of chest tumors. Lung Cancer (2009) 63:169–79. doi: 10.1016/j.lungcan.2008.06.011
127. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol (2017) 12:15–26. doi: 10.1016/j.jtho.2016.10.014
128. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discovery (2015) 5:850–9. doi: 10.1158/1538-7445.AM2015-1118
129. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316:1039–43. doi: 10.1126/science.1141478
130. Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett (2016) 380:494–504. doi: 10.1016/j.canlet.2016.07.021
131. Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res (2008) 14:2895–9. doi: 10.1158/1078-0432.CCR-07-2248
132. Tanaka A, Sueoka-Aragane N, Nakamura T, Takeda Y, Mitsuoka M, Yamasaki F, et al. Co-existence of positive MET FISH status with EGFR mutations signifies poor prognosis in lung adenocarcinoma patients. Lung Cancer (2012) 75:89–94. doi: 10.1016/j.lungcan.2011.06.004
133. Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, Nishio K. Targeting MET Amplification as a New Oncogenic Driver. Cancers (Basel) (2014) 6:1540–52. doi: 10.3390/cancers6031540
134. Song Z, Wang H, Yu Z, Lu P, Xu C, Chen G, et al. De Novo MET Amplification in Chinese Patients With Non-Small-Cell Lung Cancer and Treatment Efficacy With Crizotinib: A Multicenter Retrospective Study. Clin Lung Cancer (2019) 20:e171–6. doi: 10.1016/j.cllc.2018.11.007
135. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
136. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
137. Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (2016) 387:1540–50. doi: 10.1016/S0140-6736(15)01281-7
138. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
139. Pu X, Wu L, Su D, Mao W, Fang B. Immunotherapy for non-small cell lung cancers: biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer (2018) 18:1082. doi: 10.1186/s12885-018-4990-5
140. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin Cancer Res (2016) 22:4585–93. doi: 10.1158/1078-0432.CCR-15-3101
141. Lisberg A, Cummings A, Goldman JW, Bornazyan K, Reese N, Wang T, et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J Thorac Oncol (2018) 13:1138–45. doi: 10.1016/j.jtho.2018.03.035
142. Calles A, Riess JW, Brahmer JR. Checkpoint Blockade in Lung Cancer With Driver Mutation: Choose the Road Wisely. Am Soc Clin Oncol Educ Book (2020) 40:372–84. doi: 10.1200/EDBK_280795
143. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discovery (2013) 3:1355–63. doi: 10.1158/1535-7163.TARG-13-B290
144. Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol (2014) 25:1935–40. doi: 10.1093/annonc/mdu242
145. Zhang Y, Zeng Y, Liu T, Du W, Zhu J, Liu Z, et al. The canonical TGF-β/Smad signalling pathway is involved in PD-L1-induced primary resistance to EGFR-TKIs in EGFR-mutant non-small-cell lung cancer. Respir Res (2019) 20:164. doi: 10.1186/s12931-019-1137-4
146. D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer (2015) 112:95–102. doi: 10.1038/bjc.2014.555
147. Lin C, Chen X, Li M, Liu J, Qi X, Yang W, et al. Programmed Death-Ligand 1 Expression Predicts Tyrosine Kinase Inhibitor Response and Better Prognosis in a Cohort of Patients With Epidermal Growth Factor Receptor Mutation-Positive Lung Adenocarcinoma. Clin Lung Cancer (2015) 16:e25–35. doi: 10.1016/j.cllc.2015.02.002
148. Tang Y, Fang W, Zhang Y, Hong S, Kang S, Yan Y, et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget (2015) 6:14209–19. doi: 10.18632/oncotarget.3694
149. Soo RA, Kim HR, Asuncion BR, Fazreen Z, Omar MFM, Herrera MC, et al. Significance of immune checkpoint proteins in EGFR-mutant non-small cell lung cancer. Lung Cancer (2017) 105:17–22. doi: 10.1016/j.lungcan.2017.01.008
150. Yoneshima Y, Ijichi K, Anai S, Ota K, Otsubo K, Iwama E, et al. PD-L1 expression in lung adenocarcinoma harboring EGFR mutations or ALK rearrangements. Lung Cancer (2018) 118:36–40. doi: 10.1016/j.lungcan.2018.01.024
151. Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med (2019) 7:387–401. doi: 10.1016/S2213-2600(19)30084-0
152. Ren S, Zhang J, Zhao Y, Mu X, Zhou J, Bao Z, et al. A multi-center phase II study of toripalimab with chemotherapy in patients with EGFR mutant advanced NSCLC patients resistant to EGFR TKIs: Efficacy and biomarker analysis. J Clin Oncol (2020) 38:e21618–8. doi: 10.1200/JCO.2020.38.15_suppl.e21618
153. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol (2008) 9:47–59. doi: 10.1038/nrm2308
154. Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med (2012) 18:521–8. doi: 10.1038/nm.2713
155. Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PloS Med (2007) 4:1669–1679; discussion 1680. doi: 10.1371/journal.pmed.0040315
156. Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PloS Med (2007) 4:1681–1689; discussion 1690. doi: 10.1371/journal.pmed.0040316
157. Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PloS Med (2007) 4:e294. doi: 10.1371/journal.pmed.0040294
158. Ebi H, Oze I, Nakagawa T, Ito H, Hosono S, Matsuda F, et al. Lack of association between the BIM deletion polymorphism and the risk of lung cancer with and without EGFR mutations. J Thorac Oncol (2015) 10:59–66. doi: 10.1097/JTO.0000000000000371
159. Cho EN, Kim EY, Jung JY, Kim A, Oh IJ, Kim YC, et al. BCL2-like 11 intron 2 deletion polymorphism is not associated with non-small cell lung cancer risk and prognosis. Lung Cancer (2015) 90:106–10. doi: 10.1016/j.lungcan.2015.07.017
160. Isobe K, Hata Y, Tochigi N, Kaburaki K, Kobayashi H, Makino T, et al. Clinical significance of BIM deletion polymorphism in non-small-cell lung cancer with epidermal growth factor receptor mutation. J Thorac Oncol (2014) 9:483–7. doi: 10.1097/JTO.0000000000000125
161. Zhao M, Zhang Y, Cai W, Li J, Zhou F, Cheng N, et al. The Bim deletion polymorphism clinical profile and its relation with tyrosine kinase inhibitor resistance in Chinese patients with non-small cell lung cancer. Cancer (2014) 120:2299–307. doi: 10.1002/cncr.28725
162. Zhong J, Li ZX, Zhao J, Duan JC, Bai H, An TT, et al. Analysis of BIM (BCL-2 like 11 gene) deletion polymorphism in Chinese non-small cell lung cancer patients. Thorac Cancer (2014) 5:509–16. doi: 10.1111/1759-7714.12119
163. Cardona AF, Rojas L, Wills B, Arrieta O, Carranza H, Vargas C, et al. BIM deletion polymorphisms in Hispanic patients with non-small cell lung cancer carriers of EGFR mutations. Oncotarget (2016) 7:68933–42. doi: 10.18632/oncotarget.12112
164. Qian K, Zhang Y, Zhi X. Retrospective Study of Efficacy in BIM Gene Polymorphism on First-line EGFR-TKIs Treatment for Advanced Lung Adenocarcinoma]. Zhongguo Fei Ai Za Zhi (2017) 20:543–8. doi: 10.3779/j.issn.1009-3419.2017.08.08
165. Lee JK, Shin JY, Kim S, Lee S, Park C, Kim JY, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol (2013) 24:2080–7. doi: 10.1093/annonc/mdt127
166. Lee JY, Ku BM, Lim SH, Lee MY, Kim H, Kim M, et al. The BIM Deletion Polymorphism and its Clinical Implication in Patients with EGFR-Mutant Non-Small-Cell Lung Cancer Treated with EGFR Tyrosine Kinase Inhibitors. J Thorac Oncol (2015) 10:903–9. doi: 10.1097/JTO.0000000000000535
167. Wu SG, Liu YN, Yu CJ, Yang PC, Shih JY. Association of BIM Deletion Polymorphism With Intrinsic Resistance to EGFR Tyrosine Kinase Inhibitors in Patients With Lung Adenocarcinoma. JAMA Oncol (2016) 2:826–8. doi: 10.1001/jamaoncol.2016.0016
168. Sun S, Yu H, Wang H, Zhao X, Zhao X, Wu X, et al. Exploratory cohort study and meta-analysis of BIM deletion polymorphism in patients with epidermal growth factor receptor-mutant non-small-cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Onco Targets Ther (2017) 10:1955–67. doi: 10.2147/OTT.S126075
169. Ying HQ, Chen J, He BS, Pan YQ, Wang F, Deng QW, et al. The effect of BIM deletion polymorphism on intrinsic resistance and clinical outcome of cancer patient with kinase inhibitor therapy. Sci Rep (2015) 5:11348. doi: 10.1038/srep11348
170. Ma JY, Yan HJ, Gu W. Association between BIM deletion polymorphism and clinical outcome of EGFR-mutated NSCLC patient with EGFR-TKI therapy: A meta-analysis. J Cancer Res Ther (2015) 11:397–402. doi: 10.4103/2152-7806.157308
171. Zou Q, Zhan P, Lv T, Song Y. The relationship between BIM deletion polymorphism and clinical significance of epidermal growth factor receptor-mutated non-small cell lung cancer patients with epidermal growth factor receptor-tyrosine kinase inhibitor therapy: a meta-analysis. Transl Lung Cancer Res (2015) 4:792–6. doi: 10.3978/j.issn.2218-6751.2015.12.09
172. Su W, Zhang X, Cai X, Peng M, Wang F, Wang Y. BIM deletion polymorphism predicts poor response to EGFR-TKIs in nonsmall cell lung cancer: An updated meta-analysis. Med (Baltimore) (2019) 98:e14568. doi: 10.1097/MD.0000000000014568
173. Huang WF, Liu AH, Zhao HJ, Dong HM, Liu LY, Cai SX. BIM Gene Polymorphism Lowers the Efficacy of EGFR-TKIs in Advanced Nonsmall Cell Lung Cancer With Sensitive EGFR Mutations: A Systematic Review and Meta-Analysis. Med (Baltimore) (2015) 94:e1263. doi: 10.1097/MD.0000000000001263
174. Nie W, Tao X, Wei H, Chen WS, Li B. The BIM deletion polymorphism is a prognostic biomarker of EGFR-TKIs response in NSCLC: A systematic review and meta-analysis. Oncotarget (2015) 6:25696–700. doi: 10.18632/oncotarget.4678
175. Soh SX, Siddiqui FJ, Allen JC, Kim GW, Lee JC, Yatabe Y, et al. A systematic review and meta-analysis of individual patient data on the impact of the BIM deletion polymorphism on treatment outcomes in epidermal growth factor receptor mutant lung cancer. Oncotarget (2017) 8:41474–86. doi: 10.18632/oncotarget.17102
Keywords: non-small cell lung cancer, concurrent genetic alteration, epidermal growth factor receptor tyrosine kinase inhibitors, PD-L1 expression, BIM polymorphism
Citation: Guo Y, Song J, Wang Y, Huang L, Sun L, Zhao J, Zhang S, Jing W, Ma J and Han C (2020) Concurrent Genetic Alterations and Other Biomarkers Predict Treatment Efficacy of EGFR-TKIs in EGFR-Mutant Non-Small Cell Lung Cancer: A Review. Front. Oncol. 10:610923. doi: 10.3389/fonc.2020.610923
Received: 27 September 2020; Accepted: 10 November 2020;
Published: 10 December 2020.
Edited by:
Pasquale Pisapia, University of Naples Federico II, ItalyReviewed by:
Valerio Gristina, University of Palermo, ItalyPaola Ulivi, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRCCS), Italy
Copyright © 2020 Guo, Song, Wang, Huang, Sun, Zhao, Zhang, Jing, Ma and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengbo Han, aGFuY2hlbmdib0Bzai1ob3NwaXRhbC5vcmc=