 Hyun Jung Kim
Hyun Jung Kim Jung Won Park
Jung Won Park Jeong Ho Lee
Jeong Ho Lee- 1Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, South Korea
- 2SoVarGen, Inc., Daejeon, South Korea
An aggressive primary brain cancer, glioblastoma (GBM) is the most common cancer of the central nervous system in adults. However, an inability to identify its cell-of-origin has been a fundamental issue hindering further understanding of the nature and pathogenesis of GBM, as well as the development of novel therapeutic targets. Researchers have hypothesized that GBM arises from an accumulation of somatic mutations in neural stem cells (NSCs) and glial precursor cells that confer selective growth advantages, resulting in uncontrolled proliferation. In this review, we outline genomic perspectives on IDH-wildtype and IDH-mutant GBMs pathogenesis and the cell-of-origin harboring GBM driver mutations proposed by various GBM animal models. Additionally, we discuss the distinct neurodevelopmental programs observed in either IDH-wildtype or IDH-mutant GBMs. Further research into the cellular origin and lineage hierarchy of GBM will help with understanding the evolution of GBMs and with developing effective targets for treating GBM cancer cells.
Introduction
Glioblastoma (GBM) is a common, but aggressive, primary brain cancer of the central nervous system in adults and is associated with poor prognosis due to its invasiveness and resistance to therapy. According to 2016 WHO classification of glioma, GBMs are divided into: 1) IDH-wildtype (about 90% of cases), 2) IDH-mutant (about 10% of cases), and 3) IDH not otherwise specified (1). Molecular genetic features have emerged as fundamental factors contributing to its prognosis, particularly isocitrate dehydrogenase (IDH) mutation, which is considered a favorable factor. Whereas patients with IDH-wildtype GBM show a low median rate of survival of 14 to 16 months, patients with IDH-mutant GBM exhibit prolonged survival (median survival up to 31 months) and slower progression (1, 2). Over the past two decades, extensive and comprehensive genetic analysis of GBM has improved our understanding of GBM pathogenesis, and researchers have hypothesized that GBM arises from an accumulation of somatic mutations (3, 4). However, redundant signaling pathways and intratumoral heterogeneity underlie treatment failure and tumor recurrence (5–7). Thus, identifying the cellular origin of GBM would help with further understanding of tumor initiation/propagation and effective targets of use in treating GBM cancer cells. Regarding the cellular origin of cancer, cell-of-origin refers to normal cells in which oncogenic mutations first occur and accumulate to initiate tumor formation, while cancer stem cells (CSCs) refers to a subset of proliferating cancer cells that sustain tumor growth (8). The CSCs in GBM have been well-reviewed in many other papers (9–11). In this mini review, we mainly focus on the cell-of-origin in GBM and discuss the recent genomic analyses of GBM and genetically engineered mouse models (GEMMs) investigating tumorigenesis of GBM.
Genetic Alterations in Glioblastoma
Recent large-scale sequencing analyses have uncovered molecular alterations in somatic single nucleotide variants, copy number variations, gene expression profiles, and epigenetic signatures in GBM (3, 4, 12, 13). In addition, longitudinal genetic characterization of GBM has supported predictions of the order of mutation events and patterns of tumor evolution (14–18). Reviewing these studies, we summarize in the following paragraphs key somatic mutations, known as driver mutations, frequently occurring in IDH-wildtype and IDH-mutant GBM, respectively (Figure 1).
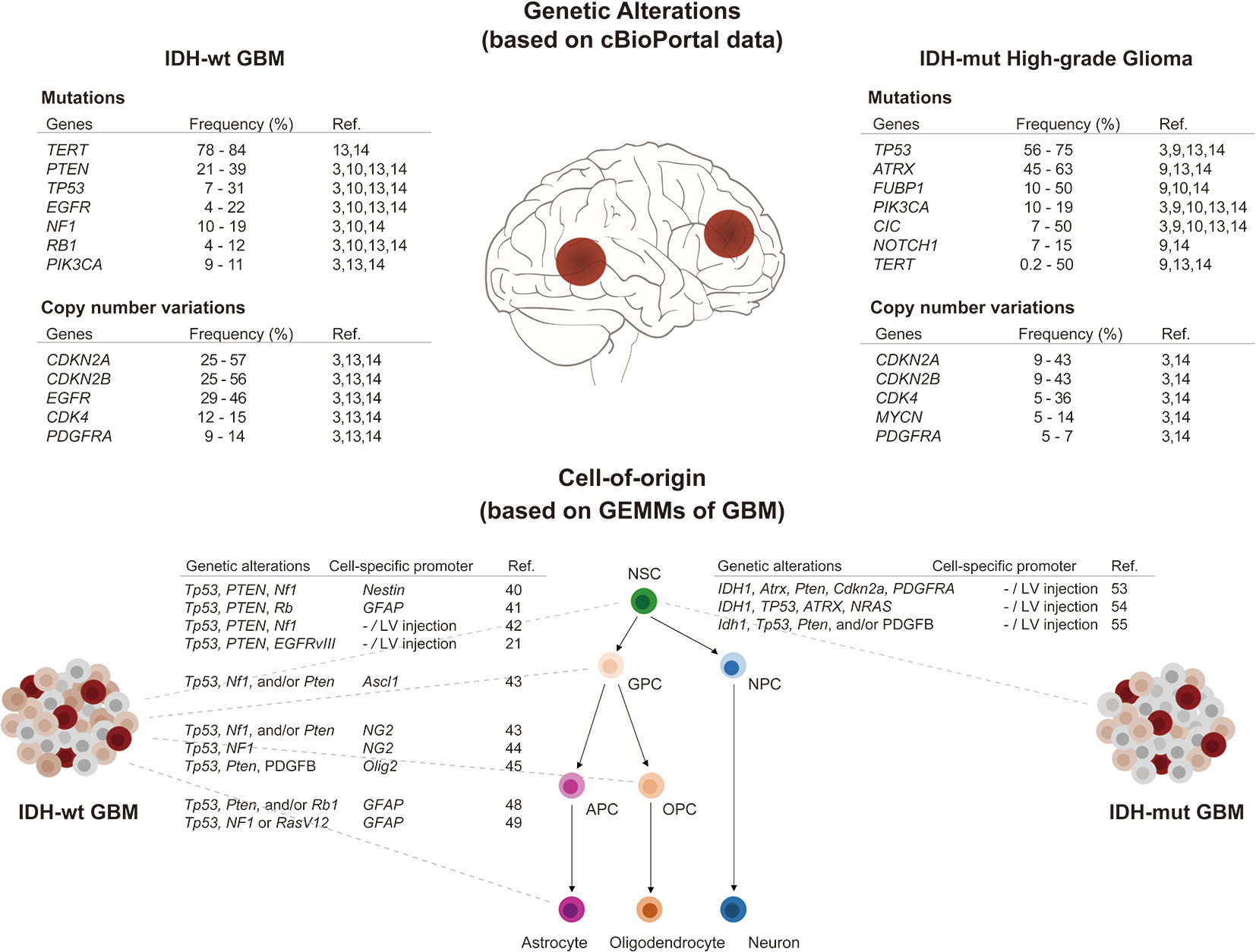
Figure 1 Overview of genetic alterations and cell-of-origin in IDH-wildtype and IDH-mutant GBMs. (Upper panel) Frequently occurring driver mutations and CNVs in IDH-wildtype and IDH-mutant high-grade gliomas (WHO grade 3 & 4) were listed as above. The frequencies were obtained from published data using cBioPortal (19, 20). (Lower panel) Multipotent neural stem cells (NSCs) have capability to self-renewal and differentiate into progenitors with restricted potential including glial precursor cells (GPCs), oligodendrocyte precursor cells (OPCs), astrocyte precursor cells (APCs), and neural progenitor cells (NPCs). Using specific genetic alterations and cell-specific promoters, NSCs and progenitor cells can be transformed to generate either IDH-wildtype or IDH-mutant GBMs in GEMMs.
IDH-Wildtype Glioblastoma
Although GBM is genetically and transcriptionally heterogeneous, previous studies have demonstrated concordant genetic alterations, including those in TP53, PTEN, EGFR, PIK3CA, and PIK3R1, NF1, and RB1, in human GBM samples (3, 4, 12). These mutations represent a set of deregulated signaling pathways, including growth factor (receptor tyrosine kinase [RTK]/phosphatidylinositide 3-kinase [PI3K]/Ras), p53, and retinoblastoma (Rb) signaling pathways. In the growth factor signaling pathway, EGFR is frequently activated with variant III deletion of the extracellular domain in GBM. Additionally, activating mutations in PI(3)K complex and inactivating mutations or deletions in tumor suppressor genes, such as PTEN and NF1, lead to uncontrolled proliferation. In the p53 pathway, inactivating mutations in TP53, along with CDKN2A (ARF) deletion, have been reported. Finally, deletions in CDKN2A/CDKN3B and amplifications of CDK4 have been found to result in Rb pathway inactivation, along with mutation or deletion of RB1 itself. The majority of GBMs harbor genetic alterations in multiple signaling pathways, suggesting that these pathways are required for GBM pathogenesis.
Interestingly, up to 83% of IDH-wildtype GBMs exhibit telomerase reverse transcriptase promoter (TERTp) mutations (3, 21). The TERTp mutations, at positions 124 bp (C228T) and 146 bp (C250T) upstream of the TERT ATG site, generate de novo transcriptional factor binding sites leading to increased expression of TERT and subsequent telomere activation (21, 22). A recent study has demonstrated that IDH-wildtype GBM patients carry a high frequency of TERTp mutations in the astrocytic ribbon, the neurogenic niche of the postnatal human brain (23). This suggests that mutation of TERTp is an early shared event through which NSCs in the SVZ avoid replicative senescence, thereby increasing the possibility that these cells acquire GBM driver mutations (24). On the other hand, Körber and colleagues argued that TERTp mutations are subsequent mutations following copy number changes in EGFR, PTEN, or CDKN2A (14). These studies imply that among many GBM driver mutations, TERTp, EGFR, PTEN, or CDKN2A mutations seem to play a key role in the early stage of IDH-wildtype GBM formation.
IDH-Mutant Glioblastoma
IDH-mutant GBM accounts for about 12% of all GBMs, with an occurrence rate of IDH1/2 mutations of approximately 73% to 83% in secondary GBMs (12, 25). IDH1/2 mutations have been observed in the vast majority of astrocytomas and oligodendrogliomas, and have been described as early molecular events during gliomagenesis (25, 26). Mutated IDH1 elicits altered catalytic functions in metabolic, epigenetic, and reactive oxygen species managing pathways (27). GBMs with IDH1 mutations show a higher frequency of loss-of-function mutations in TP53 (3, 28). Based on a recent longitudinal study on IDH-mutant glioma, mutations in IDH1 and/or TP53 occur prior to ATRX alteration on the evolutionary trajectories of IDH-mutant gliomagenesis (24, 29). In addition, IDH-mutant GBMs exhibit alternating lengthening of telomeres (ALT) due to concurrent ATRX mutations, which are mutually exclusive with TERTp mutations (30, 31). Thus, genetic alteration enabling telomere maintenance are likely to be critical steps in GBM tumorigenesis.
Researchers have attempted to classify GBMs with similar molecular genetic characteristics into proneural, classical, and mesenchymal subtypes (32, 33). Each of these subtypes show an enrichment of lineage-specific gene signatures from distinct neural-glial lineages; for example, proneural GBMs show enrichment in oligodendrocyte precursor cell (OPC) genes (34). This implies that gene expression patterns in different subtypes may reflect the phenotype of their specific cell-of-origin.
Cell-of-origin in Glioblastoma
To identify the cell-of-origin in GBM, understanding of normal cellular hierarchy is required. NSCs are ubiquitously found in all regions of the central nervous system during embryonic development and are capable of initiating cell lineages, leading to the formation of differentiated neurons and glial cells (35). NSCs give rises to intermediate progenitor cells with more restricted potential, which can proliferate and further differentiate into the three major cell types of the central nervous system. A subset of NSCs and lineage-restricted progenitor cells continue to reside in restricted regions of the postnatal and adult brain: the subventricular zone (SVZ) of the lateral ventricle and the subgranular zone (SGZ) of the dentate gyrus in the hippocampus (36, 37).
Considering that multiple oncogenic mutations are necessary for gliomagenesis, the self-renewal and proliferative properties of NSCs ensure appropriate conditions for endogenous accumulation of somatic mutations. Moreover, research has indicated that most driver mutations in cancer are attributable to DNA replicative errors, which are correlated with the total number of divisions of stem cells (38). Based on this notion, it has been hypothesized that the NSCs in the ventricular-subventricular zone is the main source of de novo somatic mutations throughout one’s lifetime. A recent study indeed showed that 55.5% of tumor-free SVZ tissue contains low-level mutations, such as TP53, EGFR, RB1, PDGFR, or TERT variations shared by matching tumor tissue in IDH-wildtype GBM patients, but not in IDH-mutant GBM patients (23). However, this study did not show any evidence of which cell type is the cell-of-origin in IDH-mutant GBM. Knowing now that human genetic studies provide the evidence of the cellular origin of IDH-wildtype GBM, we can recapitulate human GBM in mouse models, which are an invaluable tool with which to study the processes of tumorigenesis from originating cells (39–41). Below, we give an overview of GEMMs reflective of specific cell lineages and different combinations of GBM driver mutations, with or without IDH mutation (Figure 1).
Animal Modeling of IDH-Wildtype Glioblastoma
To target NSCs in the adult brain, researchers utilized Cre recombinase-expressing adenovirus injected into the SVZ of mutant mice with conditional Tp53, Pten, and Nf1 or Rb knockout, which resulted in the development of GBM (42, 43). Induction of the same tumor suppressor mutations in mice with Nestin-CreER transgenes also led to GBM formation (42). In addition, GBM has been successfully generated from NSCs harboring somatic mutations in NF1, TP53, and PTEN using in utero electroporation of CRISPR/Cas9 system (44). Similarly, TP53 and PTEN mutations were introduced into the SVZ of conditional EGFRvIII transgenic mouse to generate a GBM (23).
Another model has suggested that GBM arises from committed precursor cells, such as glial precursor cells (GPCs), OPCs, and astrocytes. Researchers have used mice with an Ascl1-CreER transgene to target bipotential progenitors expressed in both adult neural and oligodendrocyte lineage progenitors (45). Bipotential progenitors carrying NF1, TP53, and/or PTEN mutations give rise to GBM, as do NG2-expressing OPCs (45–47). Several studies have suggested OPCs as the prominent cell-of-origin in GBMs, because of their aberrant growth prior to malignancy (23, 34, 47, 48). In contrast to glial lineage, susceptibility to malignant transformation declines with neural lineage restriction (49). Researchers utilized cell-specific promoters such as Dlx1, Neurod1, and Camk2a to introduce oncogenic mutations at specific time points during neural lineage specification.
There were several studies showing that mature astrocytes are also capable of tumor formation through de-differentiation. Loss-of-function mutations in TP53, PTEN, and/or RB1 in GFAP-CreER mice (50) and injection of shNF1-shp53- or H-RasV12-shp53 lentivirus in the cortex of GFAP-cre resulted in glioma formation (51). The oncogenic virus induced astrocytes to de-differentiate into NS/PC-like state, by expressing the transcriptional factors Sox2, c-myc, and Nanog. The manipulation of pluripotency regulators are capable of inducing de-differentiation or cellular reprogramming (52, 53); however, the above studies have a limitation that GFAP-cre does not discriminate GFAP+ astrocytes from GFAP+ NSCs.
Animal Modeling of IDH-Mutant Glioblastoma
The expression of IDH1R132H mutation in SVZ NSCs led to a proliferating phenotype, but it was insufficient to generate glioma (54, 55). Therefore, researchers have examined tumor-forming capacity by induction of additional oncogenic mutations in conjunction with IDH1 mutation. Researchers utilized the RCAS-TVA system to express IDH1R132H and PDGFA in Cdkn2a, Pten, Atrx conditional knockout mice, thereby showing high-grade IDH-mutant glioma formation (55). Similarly, IDH-mutant glioma also was successfully generated by IDH1R132H and NRAS knock-in and shp53 and shATRX knockout in neonatal mice lateral ventricle using the Sleeping Beauty transposon system (56). Induction of Idh1R132H mutation with the loss of p53 and Pten led to GBM formation using retrovirus expressing PDGFB-IRES-Cre recombinase and adenovirus expressing Cre recombinase (57). To date, all of IDH-mutant animal models mainly target NSCs in the SVZ; thus, additional animal studies need to be done to carefully examine the tumorigenic potential of other lineage-restricted cell populations following IDH1R132H and co-occurring oncogenic mutations.
Collectively, the cell-of-origin and subsequent mutant cell behavior appear to underlie different biological and genomic phenotypes in GBM. A recent study demonstrated that distinct characteristics in transcriptome profiles, obtained from GBM animal models targeting either NSCs or oligodendrocyte lineage cells, can be used to classify IDH-wildtype GBMs into two subtypes based on the cellular origin (58). However, individual cells from the same tumor harbor different mutations and exhibit diverse transcriptional patterns and phenotypes (59), making it difficult to completely unravel cellular origins and tumor evolution processes.
Dissecting Cellular Hierarchy in Glioblastoma
With advances in single-cell sequencing, brain tumors have been examined at the single-cell level in an attempt to document developmental programs in GBM. Using single-cell whole-genome sequencing, researchers noted intratumoral clonal evolution based on EGFR aberrations (60). Patel and colleagues also showed the mosaic pattern of EGFR and other RTK signaling molecules (59). Despite the observed clonal heterogeneity in GBM, researchers have attempted to identify key neurodevelopmental programs from transcriptional profiles. Hierarchical clustering revealed that a subset of genes regulating oligodendrocyte function are important in primary GBM, along with genes related to the cell cycle, hypoxia, and complement/immune responses (59). Müller and colleagues also demonstrated that PDGF-driven GBMs exhibit a progressive induction of OPC-like cells (61). Additionally, several studies have recently indicated that IDH-wildtype GBM recapitulates a normal neurodevelopmental hierarchy (62, 63): malignant cells exist in four cellular states of distinct neural cell types, including NPC-like, OPC-like, astrocyte-like, and mesenchymal-like cells (62). Meanwhile, Couturier and colleagues demonstrated that putative originating cell populations share similar expression profiles of glial progenitors and that tumor cells are organized into the normal neural lineage hierarchy observed in fetal brain (63).
Although single-cell RNA sequencing of IDH-mutant GBM has not been conducted due to a small number of patients, several studies of IDH-mutant glioma have shed some light on the cellular hierarchy of IDH-mutant GBM. Therein, most malignant cells are differentiated into and are reminiscent of glial lineages (oligodendrocyte-like and astrocyte-like), while a small subset of cells remain undifferentiated, exhibiting features of NSCs (64, 65). Overall, aberrant differentiation toward glial lineage cells and developmental programs appears to dominate the cellular diversity in IDH-mutant glioma. These studies suggest that IDH-mutant GBM might originate from progenitor cells with more restricted potential.
Discussion
A number of studies have described the cellular origin and hierarchy of IDH-wildtype GBMs in humans, and accumulating evidence from genome, transcriptome, and animal studies suggests that IDH-mutant GBMs have characteristics distinct from those in IDH-wildtype GBMs. This raises the hypothesis that IDH-mutant GBMs may arise from a different cell-of-origin that undergoes malignant transformation. Based on the hypothesis, we may consider another possible candidates for the cell-of-origin of brain tumor such as glial progenitor cells (66). Accordingly, additional genetic analysis and animal modeling of IDH-mutant GBM should be performed to identify the cell-of-origin. Furthermore, future research should seek to carefully characterize the underlying mechanisms of which cells initially acquire mutations and how mutation-harboring cells evolve and undergo lineage specification during gliomagenesis. Such research may benefit from focusing on influences from the tumor microenvironment (e.g., immune cell infiltration) on the fate of tumor initiating cells and subsequent expression-based subtypes in GBM.
Author Contributions
HJK, JWP, and JHL conceived the topic for the mini review and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Suh Kyungbae Foundation (to JHL), from the Sovargen, Inc. (to JHL), from the National Research Foundation of Korea (NRF) funded by the Korea government, Ministry of Science and ICT (No. 2019R1A3B2066619 to JHL), and from the Daewoong Foundation (to HJK).
Conflict of Interest
JHL is a co-founder and CTO of SoVarGen, which seeks to develop new diagnostics and therapeutics for intractable brain disorders.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
GBM, Glioblastoma; NSCs, Neural stem cells; IDH, Isocitrate dehydrogenase; GEMM, genetically engineered mouse model; RTK, receptor tyrosine kinase; PI3K, Phosphatidylinositide 3-kinase; TERTp, telomerase reverse transcriptase promoter; SVZ, Subventricular zone; ALT, alternating lengthening of telomeres; OPC, Oligodendrocyte precursor cell; SGZ, subgranular zone; GPC, glial precursor cell; APC, astrocyte precursor cell; NPC, neural progenitor cell.
References
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
3. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
4. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi: 10.1038/nature07385
5. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science (2007) 318(5848):287–90. doi: 10.1126/science.1142946
6. Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science (2014) (6166)343:72–6. doi: 10.1126/science.1241328
7. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med (2014) 370(8):699–708. doi: 10.1056/NEJMoa1308573
9. Batista CM, Mariano ED, Barbosa BJ, Morgalla M, Marie SK, Teixeira MJ, et al. Adult neurogenesis and glial oncogenesis: when the process fails. BioMed Res Int (2014) 2014:438639. doi: 10.1155/2014/438639
10. Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front Surg (2016) 3:21. doi: 10.3389/fsurg.2016.00021
11. Gimple RC, Bhargava S, Dixit D, Rich JN. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev (2019) 33(11-12):591–609. doi: 10.1101/gad.324301.119
12. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321(5897):1807–12. doi: 10.1126/science.1164382
13. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell (2016) 164(3):550–63. doi: 10.1016/j.cell.2015.12.028
14. Korber V, Yang J, Barah P, Wu Y, Stichel D, Gu Z, et al. Evolutionary Trajectories of IDH(WT) Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell (2019) 35(4):692–704.e12. doi: 10.1016/j.ccell.2019.02.007
15. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy i glioblastoma. Nat Med (2019) 25(3):462–9. doi: 10.1038/s41591-019-0349-y
16. Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, et al. Clonal evolution of glioblastoma under therapy. Nat Genet (2016) 48(7):768–76. doi: 10.1038/ng.3590
17. Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature (2019) 565(7741):654–8. doi: 10.1038/s41586-019-0882-3
18. Jonsson P, Lin AL, Young RJ, DiStefano NM, Hyman DM, Li BT, et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin Cancer Res (2019) 25(18):5537–47. doi: 10.1158/1078-0432.CCR-19-0032
19. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
20. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6(269):pl1. doi: 10.1126/scisignal.2004088
21. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr., et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA (2013) 110(15):6021–6. doi: 10.1073/pnas.1303607110
22. Bell RJ, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science (2015) 348(6238):1036–9. doi: 10.1126/science.aab0015
23. Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature (2018) 560(7717):243–7. doi: 10.1038/s41586-018-0389-3
24. Barthel FP, Wesseling P, Verhaak RGW. Reconstructing the molecular life history of gliomas. Acta Neuropathol (2018) 135(5):649–70. doi: 10.1007/s00401-018-1842-y
25. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res (2009) 15(19):6002–7. doi: 10.1158/1078-0432.CCR-09-0715
26. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol (2009) 11(4):341–7. doi: 10.1215/15228517-2009-025
27. Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ. Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol (2011) 12(1):83–91. doi: 10.1016/S1470-2045(10)70053-X
28. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360(8):765–73. doi: 10.1056/NEJMoa0808710
29. Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature (2019) 576(7785):112–20. doi: 10.1038/s41586-019-1775-1
30. Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol (2012) 124(5):615–25. doi: 10.1007/s00401-012-1031-3
31. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget (2012) 3(7):709–22. doi: 10.18632/oncotarget.588
32. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
33. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell (2017) 32(1):42–56.e6. doi: 10.1016/j.ccell.2017.06.003
34. Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, et al. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS One (2011) 6(5):e20041. doi: 10.1371/journal.pone.0020041
35. Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci (2009) 32:149–84. doi: 10.1146/annurev.neuro.051508.135600
36. Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature (2004) 427(6976):740–4. doi: 10.1038/nature02301
37. Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell (2018) 22(4):589–99.e5. doi: 10.1016/j.stem.2018.03.015
38. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science (2017) 355(6331):1330–4. doi: 10.1126/science.aaf9011
39. Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell (2012) 149(1):36–47. doi: 10.1016/j.cell.2012.03.009
40. Zong H, Parada LF, Baker SJ. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb Perspect Biol (2015) 7(5):a020610. doi: 10.1101/cshperspect.a020610
41. Alcantara Llaguno SR, Parada LF. Cell of origin of glioma: biological and clinical implications. Br J Cancer (2016) 115(12):1445–50. doi: 10.1038/bjc.2016.354
42. Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell (2009) 15(1):45–56. doi: 10.1016/j.ccr.2008.12.006
43. Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J (2010) 29(1):222–35. doi: 10.1038/emboj.2009.327
44. Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun (2015) 6:7391. doi: 10.1038/ncomms8391
45. Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell (2015) 28(4):429–40. doi: 10.1016/j.ccell.2015.09.007
46. Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RG, et al. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc Natl Acad Sci USA (2014) 111(40):E4214–23. doi: 10.1073/pnas.1414389111
47. Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell (2011) 146(2):209–21. doi: 10.1016/j.cell.2011.06.014
48. Lu F, Chen Y, Zhao C, Wang H, He D, Xu L, et al. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell (2016) 29(5):669–83. doi: 10.1016/j.ccell.2016.03.027
49. Alcantara Llaguno S, Sun D, Pedraza AM, Vera E, Wang Z, Burns DK, et al. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat Neurosci (2019) 22(4):545–55. doi: 10.1038/s41593-018-0333-8
50. Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell (2011) 19(3):305–16. doi: 10.1016/j.ccr.2011.01.039
51. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science (2012) 338(6110):1080–4. doi: 10.1126/science.1226929
52. Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep (2014) 15(3):244–53. doi: 10.1002/embr.201338254
53. Xiong S, Feng Y, Cheng L. Cellular Reprogramming as a Therapeutic Target in Cancer. Trends Cell Biol (2019) 29(8):623–34. doi: 10.1016/j.tcb.2019.05.001
54. Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell (2016) 30(4):578–94. doi: 10.1016/j.ccell.2016.08.017
55. Philip B, Yu DX, Silvis MR, Shin CH, Robinson JP, Robinson GL, et al. Mutant IDH1 Promotes Glioma Formation In Vivo. Cell Rep (2018) 23(5):1553–64. doi: 10.1016/j.celrep.2018.03.133
56. Nunez FJ, Mendez FM, Kadiyala P, Alghamri MS, Savelieff MG, Garcia-Fabiani MB, et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci Transl Med (2019) 11(479):eaaq1427. doi: 10.1126/scitranslmed.aaq1427
57. Zhang Y, Pusch S, Innes J, Sidlauskas K, Ellis M, Lau J, et al. Mutant IDH Sensitizes Gliomas to Endoplasmic Reticulum Stress and Triggers Apoptosis via miR-183-Mediated Inhibition of Semaphorin 3E. Cancer Res (2019) 79(19):4994–5007. doi: 10.1158/0008-5472.CAN-19-0054
58. Wang Z, Sun D, Chen YJ, Xie X, Shi Y, Tabar V, et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell (2020) 38(3):366–79.e8. doi: 10.1016/j.ccell.2020.06.003
59. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (2014) 344(6190):1396–401. doi: 10.1126/science.1254257
60. Francis JM, Zhang CZ, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discovery (2014) 4(8):956–71. doi: 10.1158/2159-8290.CD-13-0879
61. Muller S, Liu SJ, Di Lullo E, Malatesta M, Pollen AA, Nowakowski TJ, et al. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF- and EGF-driven gliomas. Mol Syst Biol (2016) 12(11):889. doi: 10.15252/msb.20166969
62. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell (2019) 178(4):835–49.e21. doi: 10.1016/j.cell.2019.06.024
63. Couturier CP, Ayyadhury S, Le PU, Nadaf J, Monlong J, Riva G, et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun (2020) 11(1):3406. doi: 10.1038/s41467-020-17186-5
64. Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature (2016) 539(7628):309–13. doi: 10.1038/nature20123
65. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (2017) 355(6332):eaai8478. doi: 10.1126/science.aai8478
Keywords: glioblastoma, somatic mutation, neural stem cells, subventricular zone, genetically engineered mouse model
Citation: Kim HJ, Park JW and Lee JH (2021) Genetic Architectures and Cell-of-Origin in Glioblastoma. Front. Oncol. 10:615400. doi: 10.3389/fonc.2020.615400
Received: 09 October 2020; Accepted: 01 December 2020;
Published: 21 January 2021.
Edited by:
Carmen Castro, University of Cádiz, SpainReviewed by:
Christian Badr, Massachusetts General Hospital and Harvard Medical School, United StatesMaya Srikanth Graham, Cornell University, United States
Copyright © 2021 Kim, Park and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeong Ho Lee, amhsZWU0MjQ2QGthaXN0LmFjLmty