 Astha Malik1†
Astha Malik1† Unmesha Thanekar1†
Unmesha Thanekar1† Surya Amarachintha1
Surya Amarachintha1 Reena Mourya1
Reena Mourya1 Shreya Nalluri1
Shreya Nalluri1 Alexander Bondoc2,3
Alexander Bondoc2,3 Pranavkumar Shivakumar1,2*
Pranavkumar Shivakumar1,2*- 1Division of Gastroenterology, Hepatology and Nutrition, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 2Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 3Division of Pediatric General and Thoracic Surgery, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver and a leading cause of death in the US and worldwide. HCC remains a global health problem and is highly aggressive with unfavorable prognosis. Even with surgical interventions and newer medical treatment regimens, patients with HCC have poor survival rates. These limited therapeutic strategies and mechanistic understandings of HCC immunopathogenesis urgently warrant non-palliative treatment measures. Irrespective of the multitude etiologies, the liver microenvironment in HCC is intricately associated with chronic necroinflammation, progressive fibrosis, and cirrhosis as precedent events along with dysregulated innate and adaptive immune responses. Central to these immunological networks is the complement cascade (CC), a fundamental defense system inherent to the liver which tightly regulates humoral and cellular responses to noxious stimuli. Importantly, the liver is the primary source for biosynthesis of >80% of complement components and expresses a variety of complement receptors. Recent studies implicate the complement system in liver inflammation, abnormal regenerative responses, fibrosis, carcinogenesis, and development of HCC. Although complement activation differentially promotes immunosuppressive, stimulant, and angiogenic microenvironments conducive to HCC development, it remains under-investigated. Here, we review derangement of specific complement proteins in HCC in the context of altered complement regulatory factors, immune-activating components, and their implications in disease pathogenesis. We also summarize how complement molecules regulate cancer stem cells (CSCs), interact with complement-coagulation cascades, and provide therapeutic opportunities for targeted intervention in HCC.
HCC: Incidence, Etiology, and Treatment
Hepatocellular Carcinoma (HCC) is the major form of primary malignancy of the liver, derived mostly from hepatocytes in more than 80% of the cases. HCC ranks as the fifth most common cancer in men and the seventh in women, representing a third of all cancer-related deaths (1) and centralizing mostly in developing countries. Globally, the incidence of HCC continues to rise, with rates increasing from 2.7/100,000 in 1997 to 8.8/100,000 in 2016 in men and from 0.8/100,000 to 2.2/100,000 in women. HCC is associated with unfavorable trends in North America, Northern and Central Europe, and Latin America. Development of HCC with enhanced tumor burden is highly prevalent in patients with liver cirrhosis as the single-most important etiology (2, 3). While HCC uniformly results in high mortality, the etiology and epidemiology differ widely in their geographical distributions. In western countries, including USA, and in Japan, chronic hepatitis C virus (HCV) infection is the primary risk factor (4, 5) and hepatitis B virus (HBV) infection is more prevalent in Southeast Asia, China, and sub-Saharan Africa (6). Since liver cirrhosis underpins the fundamental cause of HCC, patients with chronic liver diseases and a predisposition to cirrhosis are at substantial risk (7). However, contributions from nonalcoholic steatohepatitis (NASH), diabetes mellitus, obesity, and autoimmune and cholestatic diseases as predisposing factors in the onset of HCC are relatively minor (8). In contrast, an alarming rise in HCV, alcohol-related, and post-NASH HCC has been found in the United States, Canada, areas of Europe, Australia, and New Zealand (9, 10).
As an aggressive disease typified frequently by late diagnosis, the prognosis for HCC remains very poor (7), with median survival following diagnosis ranging from 6 to 20 months (11) and a 5-year relative survival rate of 18.4%. Cirrhosis and portal vein occlusion define the length of survival and severely limit therapeutic options, resulting in liver failure, tumor progression, and death. The existence of underlying advanced chronic liver disease, tumor stage, and portal hypertension in most of patients with HCC dictates and complicates the adoption of treatment strategies and prognosis. Treatment options including medical and transplantation for large non-resectable HCC patients, unfortunately, share high tumor recurrence rates due to persistent cirrhosis that confers a preneoplastic environment (12). The only curative treatment strategies involve orthotopic liver transplantation (OLT) and surgical liver resections (LR). OLT, however, is limited by organ shortage, resulting in increased utilization of extended-criteria donor (ECD) allografts (13). Other surgical interventions include but are not limited to the locoregional tumor ablation therapies including TACE (14), trans-arterial radioembolization with Yttrium-90 (Y-90) (15), stereotactic body radiotherapy (SBRT) (16), percutaneous ethanol injection (PEI) (17), high-intensity focused ultrasound (HIFU) (18), irreversible electroporation (IRE) (19), and radiofrequency-, microwave-, and cryo- ablations (19, 20). While surgical therapy remains the mainstay of treating HCC, systemic treatments targeting the molecular signaling pathways are frequently implemented for patients with unresectable and/or advanced HCC. Taking advantage of the molecular signaling pathways, systemic therapies involving Sorafenib (21), Lenvatinib (22), Nivolumab (23), Regorafenib (24), and Cabozantinib (25) have shown survival benefits in HCC patient cohorts. However, the surgical, systemic, and locoregional therapies currently advocated and in practice for treating HCC are associated with several adverse events summarized in Table 1. The ability to systemically treat, albeit partially, the highly chemotherapy resistant HCC tumors and increased understanding of disease pathogenesis are expected to pave way for future therapeutics.

Table 1 Adverse events associated with systemic and surgical hepatocellular carcinoma (HCC) treatment approaches.
Since systemic therapies target proliferative and angiogenic pathways involving tyrosine kinases, vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR)-β tyrosine kinases, fibrosarcoma kinases, etc (26), and HCC is characterized as an immunogenic cancer, greater opportunities can be envisaged for specific and more effective treatment strategies. In general, cancer-associated inflammation, present at different stages of tumorigenesis, contributes to genomic instability, stimulation of angiogenesis, epigenetic modifications, aggressive cancer cell proliferation, enhanced anti-apoptotic pathways, and cancer dissemination (27). Studies in the last two decades have implicated inflammatory pathways in cancer with emphasis on understanding how immune cells impact tumor fate in different stages of disease: early neoplastic transformation, clinically detected tumors, metastatic dissemination, and therapeutic intervention. Despite the significant advances in our understandings of the immunological basis of cancer (28), the immunopathogenesis of HCC remains underexplored.
Immunopathogenesis of HCC
While the liver is highly tolerogenic and prevents hostile immune responses, organ homeostasis is maintained by natural killer (NK), natural killer T (NKT) cells, γδT cells, Kupffer cells (KCs), etc (29). However, breakdown of this tight regulation by virus infection, alcohol abuse, and lipid accumulation results in chronic inflammation and destruction of hepatocyte and cholangiocyte epithelial cells, leading to cirrhosis (30). Inflammation-associated cellular proliferation, genomic DNA mutations, and reactive oxygen species (ROS) production further enhance malignant transformation (31). In this environment, cancer cells evade immune surveillance and are associated with increased tumor infiltration by immune cells and, amplification of pro-tumorigenic cytokines, etc (32). Several cell-death pathways linked to TNFα, IL-6, NF-κB, STAT3, and JNK, and innate and adaptive immunity are activated in HCC, attesting to the dominant roles of immune mechanisms in hepatocarcinogenesis (30, 33). In particular, innate immune responses involving NK, NKT cells, dendritic cells (DCs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), myeloid derived suppressor cells (MDSCs), regulatory T cells (Tregs), and cytokines/chemokines derived from these cells form the first-line events in either dampening or promoting tumor initiation and progression within the tumor microenvironment (TME) (34).
In HCC, NK cells are activated by NKT, DC, and KCs, and suppressed by Tregs and hepatic stellate cells (HSCs) (35). However, NK cell numbers are reduced in HCC lesions, with reduced levels of IFNγ and cytotoxic potentials (36), possibly due to hypoxic stress and/or transitory behavior of activating/inhibitory NK receptors. In addition, α-fetoprotein (AFP), MDSCs, and TAMs dampen activating NKG2D receptors and block NK cell cytotoxicities (37). The role of NKT cells, however, remains less understood, with Th2 cytokine-producing tumor-promoting and anti-tumor CD4+ NKT cells that accumulate in the TME. Another important component of innate immunity involves DCs that serve as professional antigen-presenting cells (APCs), priming T cells against tumor associated antigens (TAAs) in HCC. However, DCs in patients with HCC remain refractory to high inflammatory cytokine maturation stimuli and show defective antigen presentations due to decreased HLA class-I expressions and a weakened T cell response (38). Furthermore, the frequencies of activated CD83+ DCs are lower in HCC livers and absent in tumor nodules, denoting impaired cytotoxic responses.
In parallel with the DC phenotypes, the alternatively activated CD163+ M2 TAMs promote tumor initiation, progression, and metastatic malignancy, and are considered as negative prognostic markers associated with low survival rates (39). In HCC, this M2 polarization is sustained by high levels of colony stimulating factor-1 (CSF-1) and reduced innate and adaptive immunity via IL4 (40). TAM-derived IL-10 and interactions with MDSCs result in decreased IL-6, IL-12, and MHCII, and increased anti-inflammatory IL-10, TGF-β1, and Foxp3+ Treg frequencies to facilitate tumor growth and immune tolerance (41, 42). Similar to TAMs, the recently described TANs recruit macrophages and Tregs to the TME, promoting tumorigenesis and resistance to sorafenib in preclinical studies (43). In patients with HCC, CD66B+ neutrophils colocalized with CCL2 and CCL17, infiltrating the liver stroma (44). In experimental models, TANs secrete BMP2 and TGF-β2, trigger miR-301-3p expression in HCC cells, suppress LSAMP and CYLD expressions, and enhance HCC stemness (44). In patient specimens, increased TANs were associated with increased CXCL5 expression and miR-301b-3p levels, decreased LSAMP and CYLD expressions, and nuclear p65 accumulation, collectively contributing to immunosuppression and HCC patient prognosis (45).
The immunosuppressive TME is further elevated by MDSCs, a heterogeneous inhibitory cell population with increased arginase-1, nitric oxide, ROS, and TGF-β activities that promote induction of Tregs (46). While CD14+/HLA-DR–/low MDSCs populate HCC livers and block T-cell responses, circulating MDSCs have been negatively correlated with reduced HCC recurrence-free survival (47). Furthermore, MDSCs in the TME suppress IFN-γ production by NKT cells, express Galectin-9 to interact with and induce T-cell apoptosis, and inhibit NK cell cytotoxicity via interactions with Nkp30 receptor (48, 49). In HCC, increased intratumoral Treg activity is always associated with defective anti-tumor responses and poor prognosis. Higher frequencies of Tregs were found to be intricately associated with lower CD8+ T cell responses, absent tumor encapsulation, and increased tumor vascular invasion (50). A concerted interaction between Amphiregulin (AR)-expressing HCC cells and Tregs triggered mTORC1 expression in Tregs, suppressing CD8+ T cell mediated anti-tumor responses. Similarly, inhibiting mTORC1 via rapamycin or blocking AR/EGFR signaling using Gefitinib enhanced anti-tumor CD8+ T-cell functions, highlighting the importance of Treg-driven processes in HCC TME (51). Similarly, increased accumulation of Tregs in HCC tumors correlated with reduced CD8+ T-cell infiltrations and reduced Granzyme A, Granzyme B, and Perforin expressions. Importantly, these events are associated with significantly reduced survival times and increased mortality of HCC patients. Such intratumoral inverse correlations of Tregs and CD8+ T-cells also contribute to the prognostic value of HCC patients by facilitating angiogenesis and substantially modulating anti-tumor CD8+ T-cell functions (52). An immunosuppressive functional role has also been identified for IL-35 in HCC, a cytokine expressed primarily by Foxp3+ Tregs. IL-35 induces conversion of naïve T cells and B cells into Tregs and Bregs, respectively, and is involved in negative regulation of autoimmune diseases (53, 54). Patients with elevated IL-35 were at a higher risk of postoperative recurrence after curative HCC resection and correlated with increased infiltration of a new CD39+Foxp3+ Treg subset (55). Meta-analysis of 23 studies with a total of 1,279 patients with HCC and 547 healthy controls revealed that a) the frequency of circulating Tregs was 87% higher than in healthy controls and b) intratumoral Treg levels were higher than the surrounding tissue and healthy controls (56).
Cytokines and Chemokines in HCC
In conjunction with the suppressive functions and escape mechanisms of the immune-cell compartments, several proinflammatory and immunomodulatory Th1 and Th2 cytokines and chemokines define the outcomes of tumorigenesis in HCC (57, 58). The sustained and permissive cytokine and chemokine synthesis in the TME promotes a maladaptive immune response, amplifying dysplastic cellular responses. Immune and epithelial cells within the hepatobiliary system elaborate a range of cytokines with simultaneous expression of receptors.
Pro- and Anti-Inflammatory Cytokines
In patients with cirrhotic livers, high levels of Kupffer cell derived IL-6 are associated with poor disease prognosis (59). TAMs also utilize IL-6/STAT3 axis to promote expansion of liver cancer stem cells (CSCs) via autocrine IL-6 signaling (60). High levels of IL-4 and IL-5 in the TME are also associated with increased HCC metastasis and a polarized Th2 phenotype (61). IL-22, a member of the immunosuppressive IL-10 family, is also elevated in the TME, promoting HCC tumorigenesis, metastasis, and inhibition of apoptosis via activation of STAT3 (62). IL-10 itself is upregulated in HCC TME, defining risk of progression after tumor resection (63).
IL-1, IL-18, and IL-36, members of the IL-1 family of the cytokines, are pro-inflammatory and mostly associated with tumor growth. IL-1 induces synthesis of DC-derived CCL22 to recruit immunosuppressive Tregs and further enhance HCC. However, antitumor activity is shown by the presence of IL-36a in HCC, decreased levels of which predict poor prognosis and survival (64). Similarly, IL-37 inhibits HCC growth via CD57+ NK cells (65), limiting G2/M cell cycle arrest and decreasing cell proliferation (66). The pro-inflammatory cytokines TNF-α and IL-1β are robustly involved in HCC tumor invasion, angiogenesis, and metastasis. IL-1β has been found to increase soluble MHC Class I Polypeptide-Related Sequence A (MICA) thereby blocking NK activity and enhancing HCC (67). TNF-α suppresses anti-tumor CD8+ T-cell responses by upregulating macrophage cell surface expressions of the negative co-stimulatory molecules B7H1 or PDL1 (68). IL-1β, while promoting increased synthesis of IL-2, IL-6, and TNF-α, also acts as a tumor growth promoting molecule in conditions of chronic inflammation. TAM-derived IL-1β in the TME is known to drive metastatic potentials of HCC (69). A recent study showed that HCC patients with necrotic tumors harbored significantly higher levels of CD68+ TAMs and were associated with elevated levels of serum IL-1β and poor prognosis. Importantly, areas with TAMs showed high expressions of IL-1 receptor, HIF-1α and Vimentin suggesting epithelial mesenchymal transition (EMT). In a Huh-7 xenograft mouse model, the authors showed that IL-1β-induced EMT was mediated through HIF-1α resulting in metastatic lesions (70). Higher levels of TNF-α and IL-1β are also found in tumor-independent areas of tissue metastases (71). In association with TNF-α and IL-1β, increased levels of IL-17A predict poor prognosis (72). IL-17A also induces EMT via AKT signaling, promotes invasion/metastasis and HCC cell colonization (73), and increases cell motility by upregulating MMP-2 and MMP-9 and activating NF-κβ (74). IL-17 acts directly on HCC cells, inducing AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression (75). In contrast, increased infiltration of IL-33+ cells derived mostly from CD8+ T-cells was associated with better prognosis in patients undergoing surgical resection (76). Key cytokines of the IL-2 family, including IL-2 and IL-15, potently stimulate lymphocyte activity and proliferation of cytotoxic T lymphocytes (CTLs) and NK cells. IL-2 enhances CTL activity and IFN-γ production and modulates HCC progression in mice (71). Similarly, an increase in Th1 IL-2 expression is associated with enhanced CD8+ T-cell activity, increased IFN-γ production, and improved prognosis (77). IL-15, which positively upregulates proliferation and activation of NK, NK-T, and CD8+ T-cells, corrects NK cell dysfunction (78), controls HCC tumorigenesis (79), and promotes tumor-specific CD8+ T-cell responses (80).
This cytokine milieu not only regulates developmental and regenerative responses in the liver but also contributes to pathogenesis of hepatic cirrhosis, fibro-inflammation, and HCC. In particular, altered levels of proinflammatory IL-1α, IL-1β, IL-2, TNF-α, and Th2-like IL-4, IL-5, IL-8, and IL-10 cytokines have been associated with HCC phenotypes. In general, the cytokine milieu in HCC is skewed towards an anti-inflammatory over pro-inflammatory environment.
Role of Chemokines in HCC
Aligned with the pro-tumorigenic roles of cytokines, the chemokines and their receptors promote extravasation of immune cells and migration along a chemotactic gradient towards areas of fibroinflammation. The most relevant chemokine-dependent immunoregulatory pathways in HCC include the CXCL12–CXCR4, CXCL5/8–CXCR2, CCL2–CCR2, and CCL3/5–CCR1/5 axes. The CXCL12-CXCR4 axis represents the most extensively investigated system in HCC, which regulates angiogenesis and promotes tumorigenesis. In liver specimens from HCC patients, CXCL12–CXCR4 signals are more selectively localized to tumors than the adjacent normal or cirrhotic areas (81). In HCC cell lines, this chemokine axis promoted and enhanced the growth, invasion and metastatic potentials (82), and migration of tumor cells (83). Associations of the CXCL12–CXCR4 pathway in supporting metastasis and disease severity have also been demonstrated using HCC cell lines, showing increased MMP2 and MMP9 secretion (84) and decreased 3-year-survival rates in patients (85). Importantly, the CXCL12–CXCR4 axis interacts with MMP10 (86), further supporting tumor development, angiogenesis, and metastasis. The importance of MMPs in early invasion of HCC is further exemplified by the interactions of CXCL12 and CXCR4 with MMP2, MMP7, and MMP9. In this context, the CXCL12–CXCR4 axis provides avenues for development of novel therapeutics (87). In the aforementioned pathway, TGFβ interactions with CXCR4 shift HCC cells towards a mesenchymal phenotype (88) and increase invasiveness when treated with exogenous CXCL12 (89). High levels of CXCR4-expressing OV6+ tumor-initiating cells in HCC patient livers are associated with aggressive pathobiology, increased invasion, metastasis, and poor prognosis (90). Signaling pathways linked to EGF-EGFR in concert with CXCL5 regulate development of HCC (91), while the CXCL5-CXCR2 axis contributes to EMT of HCC cells via PI3K/Akt/GSK-3β/Snail signaling (92). CXCL5 also influences the development of an inflammatory TME by regulating the infiltration of MDSCs in HCC tumor sites via elaboration of IL-17A in γδ T cells (93). In conjunction with CXCL5, high serum levels of CXCL8 in HCC have been associated with increased tumor burden, aggressiveness, and poor patient prognosis (94). Additionally, epithelial cell derived CXCL8 chemoattracts peritumoral neutrophils and regulates disease progression by stimulating angiogenesis via secretion of MMP9 (95) and via VEGF–VEGFR2 axis in endothelial cells (96).
Similar to the CXC chemokines, the CC chemokines CCL2 and CCL5 interact with their receptors CCR2 and CCR1/5 respectively and are primarily involved in driving pro-tumorigenic and pro-fibrogenic responses. HSC, hepatocyte, macrophage, and endothelial cell derived CCL2 drives hepatic macrophage infiltrations (97) and provides pro-angiogenic signals via VEGF and MMP9 (98). Activation of CCL2–CCR2 also promotes migration, invasion, epithelial-mesenchymal transition, and metastasis of HCC via endothelial progenitor cells (99). Correspondingly, CCL5 promotes fibrogenic responses via resident Kupffer cells, bone marrow-derived macrophages, and HSCs (100) necessary for development of HCC. Investigations into other CCL chemokines CCL19, CCL20, and CCL21 showed specific upregulation of CCL20 in HCC tissues, together with increased expressions of the cognate receptor CCR6 (101). The authors demonstrated that CCL20-CCR6 axis regulates tumorigenicity in HCC, with increased CCL20 and CCR6 expressions in grade III tumors. Elevated expressions of CCR6 also correlate with formation of pseudopodia in HCC cell lines, increased metastasis, and poor survival in patients (102). Recent studies have also identified Fractalkine–CX3CR1 interactions in HCC cell cycle and CX3CL1 dependent cytotoxic T cell, IL-2, and IFN-γ responses that block tumor development (103).
In summary, the immune cells, soluble effector molecules and the chemokine receptors have been a subject of intense research and investigations as potential therapeutic targets to treat the chronic inflammatory states in HCC.
Targeting Immunity in HCC: Current Strategies, Limitations, and New Mechanisms
As discussed above, the complex interplay of the immune cells with soluble effector molecules in chronic inflammatory states of HCC alters the immune system, either suppressing or facilitating tumor growth. Harnessing these multimodal mechanisms via immunotherapeutics is therefore expected to be beneficial in early and advanced stages of HCC. Utilizing the differential responses, systemic therapies to perturb VEGF-dependent angiogenesis, WNT, PI3K/AKT/mTOR, AMPK, and c-MET pathways in the TME are either approved or in clinical trials (104). However, Sorafenib, an oral tyrosine kinase inhibitor (TKI), remains the only FDA-approved treatment with survival benefits for HCC. It inhibits VEGFR, Raf-1, B-Raf, platelet-derived growth factor receptor (PDGFR), c-KIT receptor, and p38 signaling pathways involved in angiogenesis and tumor proliferation (105). Notwithstanding the survival benefits, therapeutic efficacy of Sorafenib is limited, with patients experiencing severe adverse effects and disease progression; prognosis is poor in patients discontinuing Sorafenib, with no additional available therapies (106). Similarly, Lenvatinib is a first-line TKI for unresectable HCC currently in clinical trials (107). Second-line therapies for advanced HCC that are intolerant to first-line treatment include Regorafenib, Cabozantinib, Sunitinib, Linifanib, Brivanib, Tivantinib, Donafenib, etc. which target tyrosine kinases, HGF-MET axis, and related pathways (108). Though many of these newer therapies show improved survival with robust and durable responses, development of drug resistance, severe adverse events, and cytostatic properties limit therapeutic benefits and patient acceptability. In this context, several indirect and direct immunotherapies that target adaptive and innate immune cells have been developed. The immune checkpoint inhibitors (ICIs) block T cell activation and promote T cell exhaustion by primarily targeting either CTLA-4 (cytotoxic T lymphocyte antigen-4; CD152) or PD-1/PD-L1, some currently approved or under clinical trials for HCC (109). Anti-PD-1 ICIs (Nivolumab, Pembrolizumab, Tislelizumab, Camrelizumab, Cemiplimab, Sintilimab) block the co-inhibitory receptor PD-1 on T cells, activating antitumor T cell responses, durable response, and improved survival. Alternatively, anti-PD-L1 ICIs (Durvalumab, Atezolizumab, Avelumab) target increased PD-L1 expression on DCs, macrophages, T and B cells, and tumor and endothelial cells. Mono or combination therapies have demonstrated reasonable response rates, improving progression-free survival (110). Similarly, in an immunosuppressive environment, CTLA-4 inhibits T cell activation, promotes Treg differentiation, and deregulates antigen-presenting functions of DCs (111). The anti-CTLA-4 ICIs (Tremelimumab, Ipilimumab) enhance anti-tumor immunity by reducing Treg frequencies, increasing activation threshold, and preventing anergy of T cells (112). Adoptive Cell Therapies (ACTs) that form the other arm of direct immunotherapy target HCC via ex vivo genetic modifications of autologous immune cells (113).
It is known that HCC-associated inflammation contributes to genomic instability, epigenetic modification, induction of cancer cell proliferation, enhancement of anti-apoptotic pathways, stimulation of angiogenesis, and eventually, cancer dissemination (30). Since immune cells are an essential player of HCC-related inflammation, efforts have focused on understanding how these cells impact tumor fate in different stages of disease: early neoplastic transformation, clinically detected tumors, metastatic dissemination, and therapeutic intervention (28). The aforementioned approaches on modulating the immune environment to treat HCC demonstrate limited feasibility of available therapies and offer opportunities for mechanistic explorations and development of effective HCC treatment measures. Monotherapy with ICIs, ACT, etc. have largely failed to meet the primary clinical endpoints of antitumor responses and decreased tumor size (114). Development of resistance, heterogeneity of tumors, circumvention of inhibitory mechanisms that prevent anti-tumor responses, altered TME, hypervascularity, hypoxia, severe adverse events, potential transplant rejection, etc. further complicate the use of ICIs in effectively managing HCC (115). Furthermore, these strategies mostly temper a singular population of cells and oversimplify the complex and multifaceted immune responses in the TME. As the current immunotherapies rely mostly on modulating adaptive immune responses, deciphering novel mechanisms involving innate immunity can improve therapeutic efficacy and reduce HCC burden. Newer treatment protocols may therefore take advantage of combining novel therapeutic agents with existing first and second-line therapies. One such area of mechanistic investigation and approach garnering significant attention is modulation of the multiple components of the Complement cascade (“CC” or “C”). CC is a critical and integral arm of the innate immune response involving the Complement system (C) that not only enhances the effects of antibodies and eliminates cellular debris, foreign intruders, and dead cells but also tightly regulates liver injury, inflammation, and regenerative responses (116).
Complement System: Regulation of Immune Responses
The complement system is an integral part of the innate immune response with abilities to discriminate self from non-self, and rapidly eliminate invasive pathogens while causing minimal injury to the host (117). It is an intricate system with broader functions in immune surveillance and homeostasis, controlled through a balance of activating and regulatory proteins (118). Complement activation occurs via three major pathways: classical pathway (CP), lectin pathway (LP), and alternative pathway (AP), which merge into a common terminal pathway to activate C3. CP and LP are activated by antibodies and other pattern recognition molecules whereas AP is continually activated in plasma through a process called tick-over via continuous formation of C3b (119). Activation of C3 leads to formation of C5 convertase which cleaves component 5 (C5) into C5a and C5b. C5b then binds to C6 and C7 to form the C5b–C6–C7 complex. This complex interacts with C8 and C9 to form the membrane attack complex (MAC), resulting in antibody-mediated complement-dependent cytotoxicity (CDC) when inserted into a membrane. These activated proteins can then be deposited on cell surfaces or released into body fluids to interact with specific receptors, leading to lysis of foreign cells via cytoplasmic swelling and rupture of cell membranes, which are classical characteristics of necrosis (120). However, recent studies provide new perspectives on the immunosuppressive functions of complement components. Over the last decade, studies have demonstrated that these complement components could contribute to regulating the functions and tumor-suppressing immune responses (121). Recent findings suggest an insidious relationship between complement and cancer, tumorigenic competency of the complement system, cellular proliferation, and regeneration. Complement principally plays a protective role against tumor formation in humans (122) while also contributing to a large variety of divergent inflammatory and immune processes (123). Since HCC has underlying origins of chronic and ectopic inflammatory states, premature complement activation can be envisaged as a potential driver of onco-inflammatory processes. Indeed, altered or enhanced complement activation underlies a wide spectrum of inflammatory diseases including asthma (124), kidney and cardiac diseases (125, 126), multiple sclerosis (127), and rheumatoid arthritis (128). In addition, complement regulates several key biological processes including liver injury and regeneration (116), cellular proliferation (129), angiogenesis (130), epithelial mesenchymal transition (131), and metastasis (132). An overview of the etiopathogenic events in HCC triggering complement activation is shown in Figure 1.
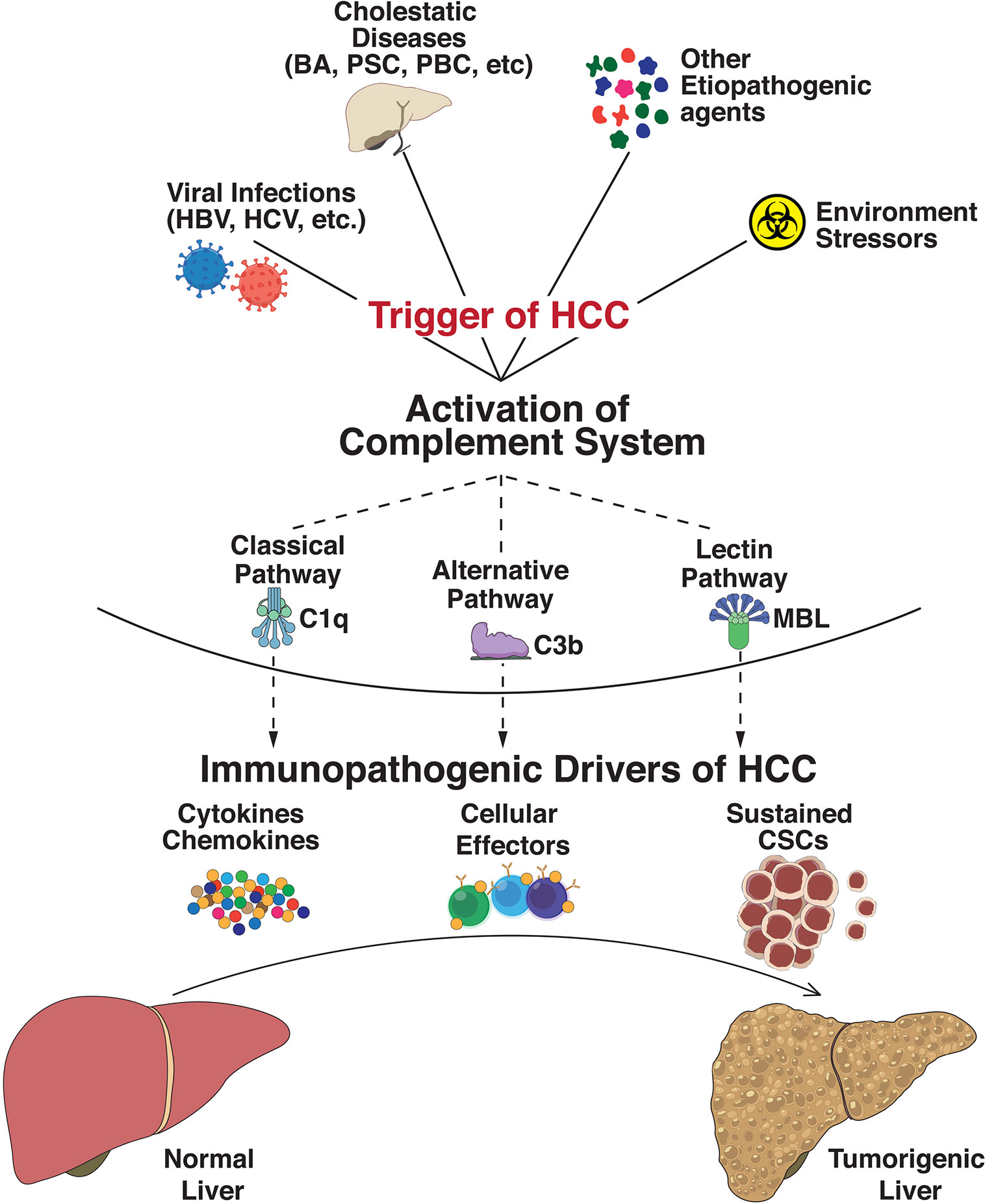
Figure 1 Immunopathogenic complement activation regulates progression to hepatocellular carcinoma. Exposure of the hepatic milieu to several triggers linked either to viral infections (Hepatitis B virus, Hepatitis C virus, etc.), severe obstructive and cholestatic diseases (Biliary atresia, Primary Sclerosing Cholangitis, Primary Biliary Cirrhosis, etc.), environmental stressors/toxin exposures (polychlorinated biphenyls, arsenic, androgenic steroids, etc.), and other etiopathogenic agents (aflatoxins, oral contraceptives, vinyl chloride, etc.) dictate the evolution of hepatocellular carcinoma (HCC). These triggers activate the innate immune complement cascade via classical (involving C1q complex), alternative (C3b-dependent activation), or lectin (triggered by carbohydrates) pathways. Abnormal activation of these complement pathways modulates functional effects of intrahepatic immune and epithelial cell compartments and disseminates significant perturbation of effector innate and adaptive cells, cytokine and chemokine expressions, and sustained cancer stem cell (CSC) activities. The collective net result of these processes defines the progression of HCC tumorigenesis.
Complement Proteins in HCC
Despite substantial research on the role of inflammatory cells and their immunosurveillance within the TME, little attention has been given to the tumor propagating properties of the complement cascade. Although increased levels of complement proteins in malignant tumors promote proliferative tumorigenesis, the exact role of complement in HCC remains unclear. The relevance of the complement system is further underscored by its ability to principally regulate the cellular and molecular events in HCC including TAMs, TANs, Tregs, MDSCs, DCs, NK cells, and cytokine (IL-1, IL-2, TNF-α, IL-4, IL-10, etc.) and chemokine axes (CXCL12–CXCR4, CCL2–CCR2, etc). Complement activation therefore can promote HCC via enhanced angiogenesis, protection of tumor cells from immunosurveillance, increased mitogenic signaling, activation of anti-apoptotic mechanisms and aberrant cell proliferation, invasion, and migration (133). It is only recently that the complement proteins have garnered interests in cancer through immunosuppression and their roles in promoting HCC are being discovered. Complement activation has also been linked with the development and spread of several cancers, raising the possibility that impaired complement regulation could be a risk factor for oncogenesis (119). In this context, recent work by Mittal et al., discussed the ability of the immune system to act against tumor progression in an “immune-editing process” composed of three distinct phases: elimination, equilibrium, and escape (134). The authors showed that the immunological responses were able to prevent tumor progression in elimination & equilibrium phase whereas the acquired adaptations of malignant cells and the host immune system allowed for expansion of the tumor cell population during the escape phase. The complement system, an integral component of the antitumor immune response, acts as an intrinsic effector mechanism to form a functional bridge between the innate and the adaptive immune system thereby promoting or suppressing tumor development. Complement activation within the liver may therefore contribute to the development of HCC by several mechanisms, for example, via activation of NF-κB in Kupffer cells and STAT3 in hepatocytes. While these events facilitate recovery of liver after acute injury, the sustained chronic activation promotes hepatocyte proliferation and development of HCC (135–137). Figure 2 depicts the loss of homeostasis resulting in dysregulated complement activation, immune responses and biological processes promoting amplified hepatic oncogenic responses in HCC. In the following section, the roles of several components of the complement system in the etiology, pathogenesis, and therapeutic modulation of HCC are discussed.
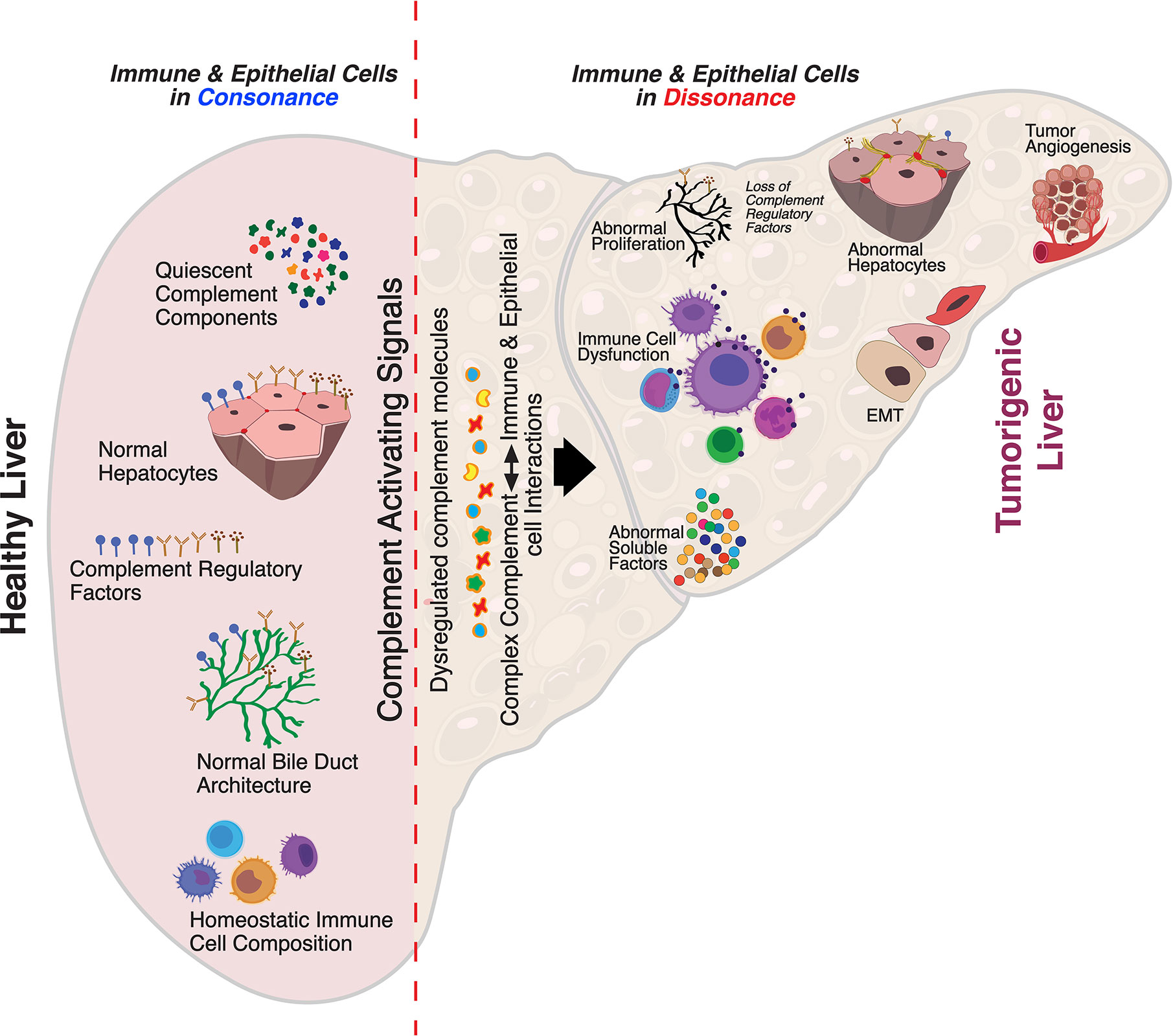
Figure 2 Aberrant complement activation: a driver for disease progression in hepatocellular carcinoma. A schematic of the liver microenvironment depicting the transition of a healthy immunologically quiescent intrahepatic microenvironment to dysregulated immune status following activation cues to the complement system. In a healthy liver, immune and epithelial cells function in synergy to preserve normal architecture of bile ducts, quiescence of complement molecules, and homeostasis of immune cells. Complement activating disease-triggering signals orchestrate the evolution of dysregulated complement molecules (increased C3, C5, etc) and altered complement regulatory factors (CFH, etc). These acute and/or chronic sustenance of dysregulated complement molecules and their complex interaction with the immune and epithelial cell compartments drive the progression of hepatocellular carcinoma (HCC). Loss of complement regulatory factors and divergent activation of the complement system leads to abnormal hepatocyte architecture, deranged cellular and effector functions, and reactive bile duct profiles. Cumulatively, these events lead to epithelial to mesenchymal transition (EMT) and tumor angiogenesis which worsen the disease, resulting in poor clinical outcomes and death.
Complement Factor H
CFH is a soluble complement protein expressed constitutively in the liver (138) by epithelial (139) and endothelial (140) cells, platelets (141), etc. CFH regulates the activation of AP by accelerating the decay of AP C3 convertase and inactivating C3b (142). A recent study using CFH-deficient mice demonstrated the importance of CFH in controlling hepatobiliary complement activation, absence of which resulted in chronic inflammation and development of HCC (119). CFH-deficient mice showed extensive complement activation and hepatocellular inflammation as early as 3 months of age and developed liver steatosis and chronic hepatic injury followed by HCC in >50% of mice by 15 months of age, confirming the role of activated AP in HCC (119). The tumor-suppressive effects of CFH in liver carcinogenesis were further confirmed by analyzing gene expression and methylation profiles in patients with HCC (143). Bioinformatic analysis by Laskowski et al., revealed that patients with mutations in the CFH gene were reported to have a median disease/progression-free time of less than a year compared to almost 2 years for those without mutations (119). In addition to rendering the hepatic environment susceptible to carcinogenesis, Seol et al. reported the co-operativity of CFH and complement component C7 in maintaining the stemness and replication competency of tumor-initiating hepatocarcinoma cells (144). Using tumor-sphere cultures, the authors showed that absence of C7 and CFH abrogated tumor-sphere formation with restored stem cell proliferation in liver cells that overexpress these components. The ability of CFH and C7 to maintain cancer cell stemness was mediated through the induction of Late SV-40 factor (LSF-1) which plays a prominent oncogenic role in HCC and is overexpressed in >90% of patients with HCC (145). Inhibition of LSF significantly attenuated metastasis of HCC in nude mice while overexpression promoted aggressiveness and angiogenic and metastatic potentials of HCC tumors. It is important to note that LSF regulates a string of proteins involved in hepatocarcinogenesis, including osteopontin (OPN) (146). OPN sequesters CFH on the surface of the tumor cells and hinders the formation of membrane attack complex (MAC), effectively preventing complement-mediated lysis and enabling tumor cells to escape immune surveillance (146). Several recent studies have also investigated the role of Complement factor H-related 3 (CFHR3) in HCC, which until recently, remained unexplored. CFHR3, a member of the human factor H family, exhibited significantly lower mRNA and protein levels in HCC tumor tissue. Studies by Liu et al. showed overexpression of CFHR3 blocked cellular proliferation and viability, and enhanced apoptosis (147). In patients with HCC, differential expression levels of CFHR3 correlated with better prognosis (148). Gene enrichment analysis showed decreased CFHR3 expressions with pathways upregulated in tumorigenesis including regulation of cell activation cycle and WNT and NOTCH signaling pathways. Searching for novel prognostic biomarkers of HCC, Pan et al. identified a panel of 10 differentially expressed genes between cohorts of patients with high and low infiltrations of immune and stromal cells (149). The 10-gene signature predicted a favorable outcome of overall survival in HCC patients. The presence of CFHR3 in conjunction with other markers in the TME may therefore serve as a prognostic predictor for targeted therapeutics in HCC. Similarly, RNASeq data analysis of HCC patients identified 17 genes with significant effect on HCC prognosis (150). Of these, a set of seven genes that contained CFHR1 defined a clinical prognostic signature that predicts the survival of HCC patients. Collectively, these data point towards treatment options that enhance CFH/CFHR levels either by gene therapies or by CFH reconstitution to lower tumor burden in HCC.
CD59
CD59 is another mCRP that is involved in restricting initiation and progression of complement activation on cell surfaces. In general, downregulation of CD59 promotes the activation of complement‐mediated cell lysis while increased expression can confer resistance to cancer cells (151). Low levels of CD59 are also linked to several autoimmune diseases including rheumatic diseases (152), autoimmune thrombocytopenia (153), diabetes (154), and multiple sclerosis (155). In some instances, increased expression of CD59 correlates with overall decreased survival rates in patients with colorectal cancer (156), prostate cancer (157), and B cell lymphoma (158) while low CD59 expression in breast tumors correlates with increased invasiveness and poor survival (159). Regardless of the spectrum of expression, extreme variations in the levels of CD59 result in pathologic outcomes of oncogenesis or cancer progression (160). Recent studies have linked CD59 to pathogenesis of HCC by prevention of complement mediated apoptosis. Abdel-Latif et al. showed increased mRNA and protein expressions of CD59 in a DEN-induced rat model of HCC that promoted enhanced tumor growth (161). In rats with HCC, increased levels of serum CD59 were not linked to phospholipase D (PLD)-dependent cleavage of CD59 (162) but rather to inflammation driven shedding from membrane lipid rafts (163, 164). Treatment with CoenzymeQ10 resulted in decreased CD59 and proinflammatory responses, providing protection against HCC. Using ChIP assays to study the role of the Hepatitis B virus (HBV) X protein (HBx) in HCC development, Shan et al. reported the upregulation of CD59 levels and protection from complement-dependent cytotoxicity (CDC) (165). siRNA mediated downregulation of CD59 sensitized the HBx-positive tumor cells and rendered them susceptible to CDC, suggesting new therapeutic avenues in HBV–HCC patients (165). Recent work by Lan et al. has shown that CD59 can function as a potential oncogenic driver in HCC and metastasis. Liver specimens from HCC patients showed high expressions of CD59 that correlated with poor overall and disease-free survival. Mechanistically, the authors showed that loss of CD59 impaired in vitro and in vivo tumorigenic and metastatic capacities via excessive Smad7 formation and abolishment of Smad2/3 phosphorylation. Therefore, CD59 facilitates HCC pathogenesis via suppression of CDC and modulation of TGF-β signaling; it may serve as an effective prognostic biomarker and potential therapeutic target in HCC (166).
C1q
C1q forms the recognition element of complement component C1 as a complex with the proteases C1r and C1s involved in activation of the classical pathway (167). The C1q complex is not only involved in recognition of complement activating elements, but also in regulation of autoimmune diseases (168) and in prostate cancer via the activation of tumor suppressor molecule WOX1 (WW-domain containing oxidoreductase) (169). Unlike other complement proteins, C1q is synthesized by several cell types relevant to the pathophysiology of oncogenesis, including epithelial and mesenchymal cells (170), monocytes/macrophages (171), dendritic cells (172), fibroblasts (173), and endothelial cells (174). In addition, the human hepatoma-derived cell line HepG2 secretes functional complement proteins C1r, C1s, C2, C3, C4, C5, etc. (175). Emerging data shows the involvement of C1q in progression and survival of cancer cells. Similar to requirements of increased expressions in preventing autoimmune diseases (176), C1q sustains WOX1 in blocking cell proliferation and hyperplasia in prostate cancer (169). C1q interacts with cell surface binding proteins cC1q-R and gC1q-R (177) that show divergent roles in cancer, with cC1q-R showing tumor suppressive activity (178) and gC1q-R promoting tumor cell progression and metastasis (179). Earlier studies have shown measurement of C1q-binding serum factors as a useful method in monitoring tumor growth in experimental animals (180) and enhanced C1q inhibition activity in sera of patients with HCC (181). In a similar approach, C1q solid phase assays were used to detect hepatitis B surface antigen (HBsAg) in HBsAg+ and HBsAg– patients with primary HCC (182) and to detect increased levels of CICs in HCC patients (183). Similar to C1q, Yao et al. showed the ability of HCV core/gC1qR interactions to suppress T cell immune responses, resulting in persistent infection (184). Takeuchi et al. further showed that C1qTNF6 is overexpressed in HCC tissue specimens and contributes to tumor angiogenesis by activating Akt pathway (185). A direct functional role for C1q in the tumor microenvironment was demonstrated in wild-type mice that showed early C1q deposition, high vascular density, and increased lung metastasis compared to C1qa-deficient mice. Results showed that C1q directly regulates complement activation, cancer cell adhesion, migration, and proliferation (186). Recent seminal work by Lee et al. showed that the collagen-like portion of C1q induces activation and upregulation of discoidin domain receptor 1 (DDR1), a collagen receptor, resulting in enhanced migration and invasion of HepG2 cells. C1q induced activation of MAPKs and PI3K/Akt signaling, and increased MMP2 and MMP9 expressions, strongly suggesting C1q–DDR1 interactions in the progression of HCC (187). In this context, MMP2 and MMP9 have been shown to regulate the migrative and invasive capacities of HepG2 cells (188). Independent of these primary functions of C1q, pioneering work by Ho et al. showed that C1q released from macrophages provided an unconventional signal that activated the β–catenin pathway and induced expansion and de-differentiation of periportal hepatic progenitor cells (HPCs). Treatment with C1q inhibitors blocked the β–catenin pathway and expansion of liver tumors, identifying a hitherto unknown pathway of hepatocarcinogenesis (189). Recognition of these novel regulatory pathways for C1q is expected to further expedite mechanistic understandings and design of new approaches for HCC treatment.
Complement Component C2 (C2)
Complement C2 is an important component of the complement cascade necessary for the formation of C3 convertase, a serine protease significantly associated with HCC. Analogous to CFH, Ning et al. reported that higher expressions of C2 were associated significantly with better prognosis in HCC patients, implicating a protective role for C2. Investigations showed that C2 influenced HCC prognosis via several mechanisms, including higher levels of tumor infiltrating CD4+ T and M0 macrophage cells in HCC patients with higher and lower levels of C2, respectively (190). These findings are important considering the association of high mortality rates and reduced survival time in HCC patients with loss of CD4+ cytotoxic T cells (191) and M2 polarization of TAMs that promote tumorigenesis, angiogenesis, and metastasis (40). The ability of C2 to suppress HCC and regulate multiple biological processes was supported by the identification of pathways linked to cell cycle, complement and coagulation cascades, AMPK, and PPAR signaling pathways in patients with elevated C2 expressions. The importance of C2 is further exemplified by associations of single nucleotide polymorphisms (SNP) with disease severity of HCC. While C2 SNP rs9267665 is associated with increased risk of developing HCC, the SNP rs10947223 affords protection from HCC (192) (193). Higher expressions of C2 are therefore beneficial for HCC prognosis and modulating complement C2 levels can afford novel therapeutic avenues.
Complement Component C3 (C3)
C3, the central component of the complement system is also activated in the milieu of oncogenic development. Under normal physiologic and homeostatic conditions, C3 is primarily produced by hepatocytes and restricted mostly to the extracellular space. Several lines of evidence have now confirmed that C3 is generated locally as well as intracellularly by almost all cell types including myeloid, lymphocytic, fibroblastic, and epithelial cells (194). Within the TME, C3 is produced either systematically by tumors (195), or by tumor infiltrating CD8+ T cells (196). This tumor cell-derived C3 imparts an immunosuppressive TME by regulating the activity of TAMs via C3a–C3AR–PI3Kγ signaling and suppressing antitumor responses (197). Increased activation of intracellular C3 significantly suppressed anti-tumor activity of CD8+ T cells, enhanced T-cell exhaustion, promoted an environment rich in immunosuppressive M2 macrophages, and provided resistance to cell lines against anti-PD-L1 treatment. However, blocking tumor cell derived complement C3 enhanced antitumor functions by enhancing the efficacy of anti–PD-L1 treatment, suggesting C3 in combination with ICIs as a potential target for HCC therapy. In HCC, hepatic stellate cells (HSCs) promote complement C3 mediated immunosuppression by restricting proliferation and enhancing T-cell apoptosis, decreasing DC maturation, and amplifying expansion of MDSCs (198). Blocking or modulating C3 functions may not only augment existing treatments, but also dampen cellular responses promoting fibrosis. Equally important is HSC-driven maturation of DCs into MDSCs, a function critically dependent on the presence of C3; complement C3-deficient HSCs, however, fail to induce MDSCs. This immunosuppressive function was linked to HSC derived factor B and factor D, resulting in C3 cleavage to iC3b and C3d; addition of iC3b also promoted differentiation of immunosuppressive MDSCs (199). In HCC, MDSCs promote angiogenesis and immunosuppression. Several clinical studies show the translational importance of MDSC activities (200), providing rationale for future studies that simultaneously target C3 and MDSCs. Furthermore, levels of immunosuppressive iNOS, Arg-1, and IL-4Ra were augmented in HSC-induced MDSCs via activation of the COX2–PGE2–EP4 signaling pathway. Inhibition of PGE2 blocked HCC growth by decreasing HSC-induced MDSC accumulation. Complement C3 and PGE2 may also participate in M2 polarization of macrophages in the TME to enhance anti-inflammatory effects (201). The biological roles of C3, however, precede its identification in serum of patients with HCC of HCV origin by MALDI-TOF and complement component C4 as potential biomarkers (202). In addition, other studies identified the diagnostic roles of serum complement C3a in HCC. Using proteomics analysis, Leung et al. identified lower levels of C3a C-terminal truncated fragment in HCC serum via SELDI technology, suggesting its value as a serum biomarker for HCC (203). Using a related technology of SELDI-TOF MS analysis, Lee et al. identified complement C3a to be specifically and differentially elevated in patients with chronic hepatitis C and HCV-related HCC (204). These findings were further corroborated by a recent study by Kanmura et al. who aimed to identify novel diagnostic markers for HCC using ProteinChip SELDI system (205). Results showed that a combination of complement C3a fragment, AFP, and des-gamma-carboxy prothrombin (DCP) resulted in 98% positive identification rate. These recent advances in complement-based diagnostic markers are of clinical significance since AFP is the only diagnostic marker indicative of HCC, albeit in about 60% of cases. Complement C3, therefore, plays a central role in biological functions and as a potential biomarker and therapeutic modality.
Complement Component C5 (C5)
Complement component C5 forms the terminal and an integral component of the complement cascade (206) and is expressed by and interacts with C5AR1 on several cells including lymphocytes, macrophages/monocytes, myeloid cells, hematopoietic stem cells, epithelial cells, and more importantly cells undergoing oncogenic transformations (207). In the context of cancer, the C5-C5AR1 signaling modulates proliferative, anti-apoptotic and prosurvival pathways (208). Upon activation, complement component C5 generates C5a, an anaphylatoxin and a leukocyte chemoattractant, and plays a crucial role in TME by promoting metastasis of cancer cells. In patients with chronic HBV infection, serum complement component C5a is upregulated, predisposing the patients to develop HCC. Tumor cells from HCC patients as well as HCC cell lines show significant upregulation of the complement C5a receptor, C5AR1 (209). Activation of C5aR by C5a enhances the dissemination of circulating tumor cells (CTCs) in HCC via upregulation of INHBA/Activin and induction of EMT/MMP by phosphorylation of Smad2/3 (210). Hu et al. demonstrated that C5a ligation of C5aR resulted in activation of the ERK1/2 pathway and induced EMT by increasing Snail expression and downregulating E-cadherin and Claudin-1 expressions (209). While not much of the pathobiological role of complement C5/C5a–C5AR1 axis is known in HCC, its ability to critically influence and control signaling processes relevant to HCC is largely evident from several studies. Activation of the C5a-C5AR1 axis mediates tumorigenic polarization of TAMs via NF-κB pathway in metastatic liver lesions (211), while suppressing IL-12 production (212) and promoting immunosuppressive TME via C5aR1+ macrophages (213). Increased C5ar1 expression also facilitates recruitment of other myeloid cells like neutrophils via IL-1 production (214) and leukotriene B4 (LTB4) (215), while C5a stimulates neutrophil derived tissue factor (TF) synthesis, enhancing tumor growth and metastasis formation (216). As a potent chemoattractant of MDSCs to primary tumors (217), C5a can augment disease severity in HCC by suppressing CD8+ T cell function via immunosuppressive MDSCs (218). In Lewis lung cancer model, blockade of C5aR reduced MDSCs and inhibited tumor growth (219). Additionally, signaling via C5a–C5aR promotes Treg expansion and suppresses T cell responses in breast cancer metastasis (220), and increases expression of MCP-1, IL-10, Arg-1, and TGF-β1 in colon cancer tumor metastasis (221). Progressive HCC is typified by EMT with matrix metalloproteinases (MMPs) expressed in the TME predominating an important role in this process. C5a expressed by tumor cells triggers expression of MMPs, enhancing tumor invasiveness, release of pro-angiogenic factors, and cell migration (222). Collectively, complement C5a–C5AR1 axis plays a central role as a regulator of innate and adaptive immunity in the TME and a plausible target for development of novel therapeutics for HCC.
Complement Receptor 1 (CR1)
Complement receptor 1 (CR1, CD35) is a glycoprotein expressed either on the membrane or in soluble form on erythrocytes, DCs, monocytes, neutrophils, and B and T cells (223). CR1 inhibits both classical and alternative pathways of complement activation by binding C1q, cleaved C3b and C4b, MBL-2, collectins, and ficolins (224–226) on altered cell surfaces to prevent the formation of terminal membrane attack complex (MAC). Erythrocyte CR1 (E-CR1) is important for processing and removal of circulating immune complexes (CICs) to prevent tissue deposition (227). In HCC, serum CIC levels are abnormally high, with pathological implications (183). The ability of CR1 to bind CICs is particularly important in HCC, with underlying viral etiologies where free and IC-associated HCV binds to E-CR1, differentially driving HCV-IC related features of the disease (228). Kanto et al. showed an inverse correlation between low E-CR1 levels and higher C3d immune complexes. Incidentally, low E-CR1 correlated with severe liver inflammation, cirrhosis, and HCC than those with mild inflammation, demonstrating the relationship between IC and HCV disease severity (229). Similarly, low E-CR1 and high levels of IC were observed in patients with chronic hepatitis and liver cirrhosis (230), emphasizing the importance of defective CIC clearance by altered CR1 functions. A recent study by Luo et al. analyzed genetic polymorphisms and found that two SNPs in CR1 gene (rs3811381 and rs2274567) can potentially predispose subgroups of males, alcohol drinkers, and nonsmokers to HBV-HCC and HBV-chronic hepatitis B risks, while decreasing the risk to HBV-liver cirrhosis in females (231). In contrast, soluble sCR1 levels are increased in liver cirrhosis, end-stage renal failure, and hematologic malignancies (232). In addition, increased levels of sCR1 have been found in patients with increasing grades of cirrhosis and decreased liver functions (233). Since sCR1 levels are elevated in these inflammatory conditions, it is envisaged to play important regulatory and anti-inflammatory roles and act as a potential therapeutic target. Preclinical efficacies of a recombinant form of sCR1 with binding sites for C3b and C4b have been assessed in autoimmune and inflammatory disorders with a potential clinical use in HCC (234).
Mannose Binding Lectin (MBL)
Mannose-binding lectin is an important component involved in the lectin pathway of complement activation (235). MBL functions as a pattern recognition molecule in senescent fibroblast sensing (236), autoimmunity (237), and apoptotic/necrotic cell clearing (238). Further, MBL regulates anti-cancer immunity, plays a diverse role in TME, and contributes to either development or inhibition of tumor growth, depending on the type of cancer (239). MBL2 is primarily produced and secreted by liver cells with significantly elevated levels found in HCC and in HepG2 cell lines (240). Proteomic analysis of serum in patients with pancreatic cancer showed increased levels of MBL2 as a marker of potential diagnostic value (241). Using seven publicly available protein and gene databases, Awan et al. performed enrichment analysis and identified 6 proteins, including MBL2, that defined the biomarkers of HCC (242). This study also identified MBL2 to be a target of 11 circulating and 48 deregulated miRNAs, suggesting MBL2 as a strong candidate for biomarker discovery in HCC (242). Exploring the little-known roles of MBL in TME, Li et al. showed that the genetic loss of MBL in a murine model of HCC triggered enhanced tumorigenesis compared to wild-type mice (243). MBL-deficient mice showed increased accumulation of MDSCs, Treg induction, impaired CD8+ T cell function, COX-2 expression, and PGE2 production in tumor tissues. Mechanistically, MBL inhibited hepatic stellate cell activation via downregulation of ERK/COX-2/PGE2 signaling pathway. Restoring MBL in these mice significantly reduced HCC progression by inhibiting HSC activation, suggesting MBL to be a potential therapeutic option for HCC. Jalal et al. explored circulating liver-derived lectins and found elevated serum binding activities of ficolin-2 and MBL as potential biomarkers of HCC development in chronic HCV infection (244). Interestingly, dysregulation of miRNAs has been associated with progressive HCC. miR-942-3p was found to be increased in HCC tissue and cell lines and was associated with tumor metastasis and poor patient prognosis. In cell lines, ectopic expression of miR-942-3p resulted in enhanced proliferation and invasiveness while restoration of MBL2 blocked progression of HCC and tumorigenic responses (245). Several studies have also investigated associations of genetic polymorphisms in MBL and altered functionality with HCC. MBL rs7096206 polymorphism was associated with polymorphisms in VDR/VEGF and IL-18 which collectively conferred susceptibility to HCC in Asian populations (246), while MBL2 polymorphisms tended to influence the outcomes of HCC susceptibility, progressive tumor development, and clinical outcomes in patients infected with HBV (247). Mutations in MBL2 are also proposed to predispose patients to elevated HCC risk with significantly reduced serum MBL2 and increased IL-6 and IL-1β levels in HCC (248). Similar analysis in HBV-related cirrhotic patients with HCC suggested that MBL2 SNP rs11003123 was a potential risk factor for HCC development in the Chinese population (249). The importance of MBL gene polymorphisms in progressive forms of severe hepatitis B and liver cirrhosis was further supported by a larger meta-analysis study (250). While some patients with chronic hepatitis B or C infection showed lowered levels of MBL (251), studies by Segat et al. showed no significant associations of MBL2 and MASP2 polymorphisms with either HBV/HCV infection dependent HCC or for HCC alone (252).
Mannose-Binding Lectin (MBL)-Associated Serine Protease-2 (MASP-2)
The MBL-associated protease MASP-2 predominantly promotes activation of the lectin complement pathway. While MASP-2 and lectin pathway components are highly conserved in immune defenses, loss of MASP-2 regulates infectious or autoimmune diseases, immunodeficiency of which are significantly associated with pyogenic bacterial infections, inflammatory lung disease, and autoimmunity (253). In oncogenic environments, increased levels of serum MASP and related lectin pathway molecules have been found to be associated with poor overall survival, disease progression, recurrence, and worse disease prognosis in patients with colorectal (254–256), ovarian (257), and cervical (258) cancers. Similar increases in MASP-2 protein were also associated with advanced clinical stages and nodal metastasis in esophageal squamous cell carcinoma (259). In agreement with these findings, serum MASP-2 levels were higher in pediatric patients with acute lymphoblastic leukemia, non-Hodgkin lymphoma, and CNS tumors (260). However, significant variations between pediatric and adult patients have been documented. In contrast to the severe disease pathogenesis defined by elevated levels of MASP-2, MASP-2 deficiency in leukemic children on chemotherapy was associated with increased risk of febrile neutropenia (FN), antimicrobial therapy, and prolonged duration of hospitalization (261). Similarly, higher serum levels of MASP-2 were associated with impaired event-free survival in pediatric patients with lymphoma (262). Schlapbach et al. further showed that MASP-2 deficient children had significantly increased risk of developing FN in children on chemotherapy (263). These studies show MASP-2 deficiency as a potential risk factor for infections. In the context of HCC, analysis of patient secretomes derived from cancer and adjacent normal tissues using integrative transcriptomics and proteomics identified chitinase‐3‐like protein 1 (CHI3L1) and MASP2 as biomarkers in HCC diagnosis. However, when diagnosed in combination, the detection for HCC was further enhanced (264). Analyzing patients with moderate and severe chronic hepatitis C, Tulio et al. identified five SNPs in regions critical for formation of MBL/MASP-2 complexes and C4 cleavage of MASP2 gene that resulted in high plasma levels of MASP-2 in hepatitis C patients (265). Mechanistic investigations into the determinants regulating MASP-2 expression via in silico analysis of the MASP2 promoter regions revealed conservation of two putative Stat binding sites, StatA and StatB. In vitro analysis of hepatoma cell line HepG2 revealed double stranded StatB oligonucleotides, suggesting interaction of lectin and STAT signaling in liver diseases including fatty liver, fibrosis, and HCC (266). The diverse roles of MASP-2 documented in malignancies other than HCC warrant detailed further analysis into the roles of MASP-2 in adult and pediatric HCC.
C4b-Binding Protein (C4BP)
C4BP is a fluid-phase regulatory component with potent inhibitory properties of the classical and lectin pathways of complement system (267) by providing cofactor activity for factor I-dependent degradation of C4b and C3b (268) (269) and accelerating decay of C3-convertases (269, 270). C4BP is synthesized primarily by hepatocytes (271) and activated monocytes (272). Synthesis of C4BP is enhanced in the presence of inflammatory cytokines such as IFN-γ, IL-1, IL-6, and TNF-α (273), with increased levels of C4BP in inflammatory diseases (274–277). Searching for biomarkers of colorectal cancer (CRC), especially the asymptomatic nascent tumors, Kopylov et al. (278) identified increased levels of C4BP as a potential biomarker in patients with CRC. In patients with non-metastatic CRC, C4BP levels correlated with several coagulation factors, suggesting risk factors for intravascular coagulation activation (279). Elevated levels of fully sialylated C4BP are also found in patients with epithelial ovarian cancer and can distinguish early cases of ovarian clear cell carcinoma from endometriomas (280). Profiling the pre-therapy serum proteome of patients with non-small cell lung cancer (NSCLC) to discover biomarkers and for patient-tailored therapeutics, Liu et al. applied shotgun and targeted proteomic analysis to identify relapse-related gene signatures. Results from the analysis identified a combination of C4BP, LRG1, and SAA or C4BP alone as determinants of disease prognosis, treatment optimization, and overtreatment prevention in patients with NSCLC (281). Indeed, NSCLC cells produce C4BP and provide significant protection from complement mediated tumor cell death (282). Another study also found increased serum C4BP levels in patients with NSCLC and showed strong associations with clinical staging (283). Thus, the ability of C4BP to regulate tumorigenesis in multiple organs and the liver as a primary source strongly suggests a role for C4BP in HCC. The proinflammatory cytokines IL-1, IL-6, and Oncostatin M all significantly upregulated C4BP expressions in the HepG-2 hepatoma cell line (284), suggesting an interplay between inflammation-driven regulation of complement components shielding the tumor from cytotoxic effectors. Tomes et al. further showed that C4BP not only binds strongly to necrotic cells but also limits DNA release from necrotic cells, inhibiting complement activation in both events. Persistence of necrotic core due to C4BP binding may have serious implications in cancer patients, manifesting with poor prognosis, enhanced tumorigenesis, progressive metastases, and emergence of chemoresistance (285). In keeping with the protective and tumor-augmenting roles of C4BP, Williams et al. showed that C4BP binds to CD154 and prevents CD40 mediated cholangiocyte apoptosis. Livers of patients with HCC showed enhanced expression and co-localization of C4BP and CD40, suggesting modulation of cholangiocyte survival in conditions of chronic inflammation and malignancy (286). Similarly, the hepatitis B virus X protein (HBx) affords protection of hepatoma cells from complement attack by upregulating C4BPα via activation of the transcription factor Sp1 (287). In tissues from patients with HCC, C4BPα expressions positively correlated with HBx, suggesting tumor-enhancing properties. Using protein-protein interaction networks and gene expression data from patient populations, Ardakani, et al. (288) identified C4BP as an important component of a common molecular relationship between HCC and liver cirrhosis. Identification of such networks and associated molecular connections are expected to serve as novel biomarkers and/or aid in the development of novel treatment strategies. The integral roles played by C4BP in regulating processes critical to tumor growth and progression make it an attractive target for developing interventional therapeutics.
Complement Component C4 (C4)
C4 is the fourth component of the complement cascade, vital to several key roles in defense mechanisms, innate immune function, clearance of CICs, regulation of apoptotic bodies, and autoimmune processes (289, 290). Differentially altered levels of C4 are linked to inflammation in chronic liver diseases (291), metabolic syndrome (292), chronic urticaria (293), and autoimmune processes (294). Serum C4 levels can be used in early detection of HCC, particularly in HCV-infected patients with liver cirrhosis. Serum levels of complement C4 were detected at notably higher levels in the HCC group than in controls. Further analysis showed that a combination of AFP and C4 significantly improved the detection of HCC in HCV-related liver cirrhosis patients (295). More importantly, HCV proteins transcriptionally repress complement C4 expression in liver biopsy specimens from patients with HCV infection. mRNA levels of the two C4 isoforms C4a and C4b are also decreased in hepatocytes transfected with HCV RNA and in HCV core transgenic mice. Thus, the suppression of complement mediated immune responses promotes chronic HCV infection, fibrosis, and HCC (296). Investigating the impact of HBV infection on expression of serum C4 levels, Zhu et al. found that HBV similarly inhibits the expression of complement C3 and C4 in vitro and in vivo (297). Since AFP alone is used in clinical practice as a biomarker of HCC, Kim et al. performed global data mining using HCC proteomic databases to identify novel biomarkers. Alongside AFP, the data analysis revealed a set of other biomarkers including C4a (with ANLN and FLNB) that were proposed to further improve the screening of patients with HCC (298). Serum C4a/C4b also constitute clinically relevant candidate biomarkers in association with KNG1 and HPX, distinguishing patients with HCC and liver cirrhosis (299). Complement C4 also represents a component that can distinguish HCC and liver cirrhosis with the highest accuracy (300, 301). Increased levels of serum C4a were also found in HCV-infected alcoholic patients with progressive cirrhosis and HCC (302). As a precursor to development of HCC, patients with HBV or HCV infection are at greater risk, necessitating a specific biomarker with increased sensitivity. Dalal et al. identified increased C4a/C4b levels as a reliable marker in patients with HCV related end-stage liver disease (303). Thus, the direct participations in biological regulation of immune responses in HCC and the ability of differential expressions to distinguish patient populations as biomarkers signify complement C4/C4a/C4b as important targets for disease modulation and therapeutic targeting.
Complement Factor H-Related Protein 1 (CFHL1)
Similar to CFH, CFHL1 is an immunoregulatory complement component produced primarily in the liver (304). CFHL1 is derived via alternative splicing of the N-terminal domain and shares negative regulatory functions of the alternative complement pathway similar to N terminus of CFH (305). Along with CFHL1, the complement factor H-related protein 1 (CFHR1) functions as a complement regulator by blocking C5 convertase activity and C5b surface deposition (306). CFHR1 also competes with CFHL1 for binding to C3b during CFH-regulation of immune processes (307). The role of CFHR in bladder cancer has been documented, showing the importance of the CFH family of proteins in oncogenesis (308, 309). In surgically resected tissues from HCC patients, decreased CFH mRNA expressions correlated with increased CpG site methylations (143). Furthermore, reduced CFHR3 expression was associated with tumorigenesis, cell proliferation, and activation of WNT and NOTCH signaling pathways (148). In this context, Feng et al. recently demonstrated that CFHL1 can be used as a potential prognostic biomarker in HCC. Analysis of tumor and peritumor specimens from patients with HCC showed downregulation of CFHL1 that was associated with worse time-to-recurrence of the cancer and reduced patient survival rates. This signifies the high prognostic value and potential biomarker capacity of CFHL1 in postoperative patients with HCC (310). The importance of CFHL1 in tumor biology, particularly in HCC, has recently been explored. Future studies will expectedly investigate the clinical efficacies of restoring CFHL1 levels to counter progressive oncogenesis.
Complement Component 8A (C8A)
Complement component 8 alpha (C8A) is a late-phase component of the complement cascade and, along with C5, is involved in the formation of membrane attack complex (MAC). C8A is a liver-specific protein whose expression is regulated by hepatocyte nuclear factor 1α (HNF1α) (311). With relevance to HCC, C8A has been identified in the secretome of an HCC cell line, HEP3B. C8A was also identified as a putative biomarker in a study that investigated HCC–specific proteins enriched for cancer secretome followed by interactome analysis (242). Using genome-wide transcriptional profiling of patient specimens, 439 differentially expressed mRNAs (DEGs) and 214 long non-coding RNAs (lncRNAs; DELs) were identified in HCC. Multiple DELs correlated with tumor cell differentiation, thrombosis, AFP levels, and co-expressions of DEGs of complement cascade, including complement C8A (312). Similarly, Zhe L. et al. (313) utilized publicly available gene expression profiling datasets from the gene expression omnibus (GEO) to identify differentially expressed genes between tumor and adjacent healthy tissue, and found significant enrichment of genes involved in complement activation and coagulation cascade including C8a, C8b, and C6, in HCC specimens. Performing Ingenuity Pathway Analysis (IPA) of HCC gene expression data sets, Yin et al. identified uniquely decreased expression of C8A. Corresponding decreases in expression levels of other complement components including C1S, C2, C5, C6, C7, C8B, C8G, C9 were identified, strongly suggesting downregulation of key complement molecules during early stages of HCC (314). Mu Di et al. used ONCOMINE and TIMER to identify C6 as a candidate gene in diagnosis and prognosis that was associated with significantly decreased overall survival in patients with HCC (315). The regulatory roles of C8A participating in key functions of MAC formation and governing the fate of the tumor cell death can potentially be harnessed in understanding terminal complement processes and/or design of targeted therapeutics.
CD46
CD46 is a membrane-bound complement regulatory protein (mCRP) expressed on the cell surfaces that restrains over-activation of the complement system and protects tissues from injury. CD46 primarily controls the alternative over classical pathway of complement activation. Besides its role as an mCRP, CD46 uniquely functions as a regulator of T cell mediated immune responses that may be relevant in the pathophysiology of HCC invasion and progression. Binding of CD46 on CD4+ T cells promotes differentiation to T regulatory phenotype (316) and dysregulated IL-10 production (317). While expression of CD46 on unconventional γδ T cells suppresses the production of IFN-γ and TNFα (318), CD4+ T cell ligation of CD46 results in production of IFN-γ (319). Thus, the duality of CD46 signaling in anti- and pro-tumoral functions necessitates a careful evaluation of its function in oncogenesis. In patients with ovarian and breast cancer, expression of CD46 is linked to shorter relapse periods and worse prognosis (320) (321); similar outcomes are observed in patients with CRC (322) and multiple myeloma (320). In patients with HBV-HCC, the HBx protein upregulates CD46 in hepatoma and human immortalized liver cells and affords protection from complement mediated cell lysis mechanisms (323). Investigations into CD46 distribution and expression patterns in HCC specimens showed a non-polarized membrane localization of CD46 in contrast to the basolateral expression in non-cancerous livers. This divergent expression pattern may allow HCC cells to escape complement-dependent cytotoxicity (324). In this regard, intratumoral and IV therapies that utilize the nonpathogenic oncolytic measles virus Emonston strain (MV-Edm) showed significant inhibition of tumor growth, survival benefits, and tumor regression in susceptible mice via CD46 targeting. This approach, therefore, represents a novel HCC gene therapy system (325). A similar approach using a fiber chimeric oncolytic adenovirus that targets CD46, SG635-p53, showed antitumor activity in Hep3B subcutaneous xenograft tumor models. Intratumoral injections of the adenovirus resulted in significant inhibition of tumor growth and survival of animals, suggesting a safe approach for HCC treatment (326). CD46 was also targeted using another oncolytic adenovirus, SG511, which was fused to the human RANTES/CCL5 gene and regulated by oxygen-dependent degradation domain (ODD). The chimeric SG511-CCL5-ODD showed significantly enhanced antitumor efficacy in HCC xenograft models in nude mice (327). The importance of the CD46 signaling pathway association with miRNA signatures in HCC was demonstrated via bioinformatic analysis. The authors performed complement-related gene expression profiling in tissue samples and found a total of 37 differentially regulated miRNA. Unsupervised hierarchical clustering analysis identified high CD46 expressions in HCC tissues, which negatively correlated with let-7b and miR-17 expression in HepG2 cells, suggesting important regulatory roles of CD46 in HCC via modulation of miRNA activities (328). Of note, upregulation of let-7 (329) and miR17 (330) has been associated with progressive carcinogenesis and poor prognosis of HCC. More importantly, the CD46 SNP rs2796267 was recently found to contribute to susceptibility and disease outcomes in HCC by modifying promoter activity. The rs2796267 AG/GG genotype was found to be associated with worse prognosis of resected patients with HCC (331). Table 2 summarizes the various SNPs found in complement proteins relevant to the pathogenesis of HCC. To overcome the limitations associated with using monoclonal antibodies in cancer immunotherapies due to increased expressions of mCRPs, Geis et al. designed siRNAs for posttranscriptional gene knock down of CD46, CD55, and CD59 in tumor cell lines. The approach successfully reduced CD46 protein expression by 80% with a corresponding increase in CDC by 20%–30%, demonstrating sensitization of malignant cells to complement attack via siRNA mediated inhibition of mCRP as a means of cancer therapy (332). A concise summary of the complement proteins together with their biological functions and clinical implications is provided in Table 3.
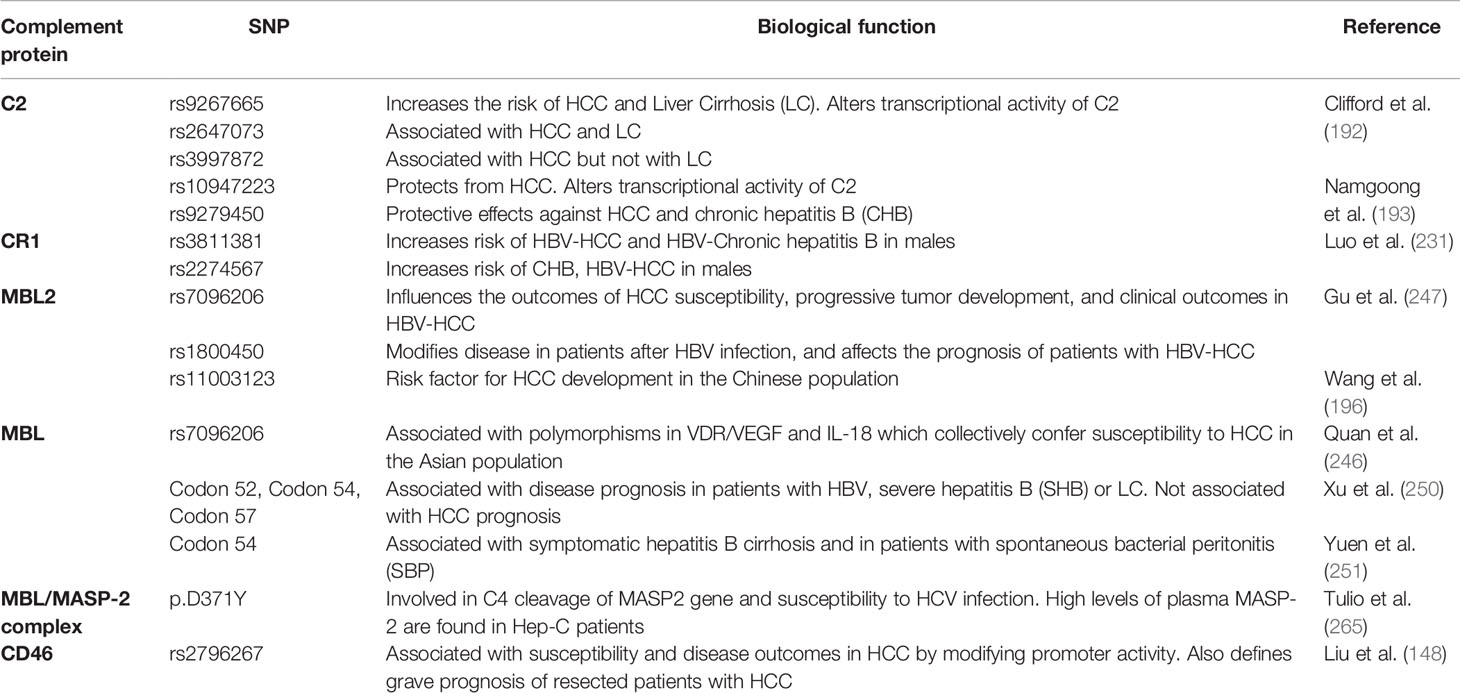
Table 2 Summary of complement protein single nucleotide polymorphisms (SNPs) in hepatocellular carcinoma (HCC).
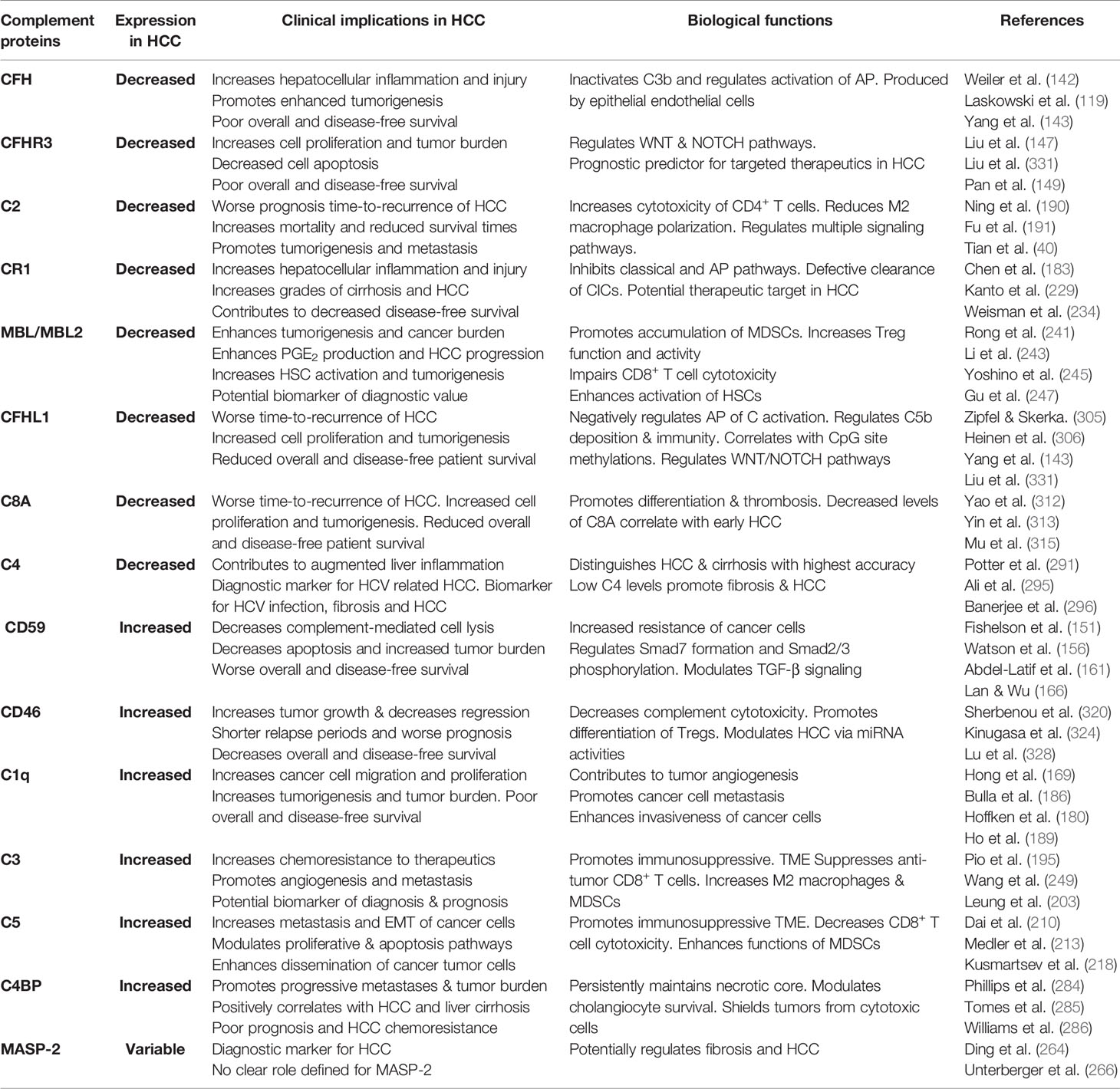
Table 3 Summary of clinical and biological roles of complement proteins in hepatocellular carcinoma (HCC).
Complement Proteins as Regulators of Liver Metastases
The aforementioned components and receptors of the complement cascade not only regulate hepatic neoplasia but promote early events of metastases involving increased tumor cell motility, invasiveness, and intravasation. The extra- and intra-hepatic metastatic spread remains one of the major hurdles in improving health related quality of life and long-term survival in patients with metastatic HCC and therefore is one of the most prevalent form of cancers with poor prognosis. HCC cells that survive immune-mediated clearance continue to proliferate and reserve the capacity to generate secondary tumors. Within this framework, perturbation of the complement cascade facilitates dissemination of the tumor cells via triggering intracellular EMT pathways and transition to a highly motile cellular phenotype. Recent studies have correlated the C5a/C5AR1 axis with increased angiogenesis and metastasis promoting factors that induce EMT (333) and liver metastasis (334). In HCC, C5AR1 increases cell invasiveness by enhancing Snail and decreasing E-cadherin and Claudin-1 expressions (209). The ability of C5AR1 to facilitate metastasis was also linked to suppression of CD8+ and CD4+ T-cell responses via recruitment of immature myeloid cells and generation of Tregs. This study also showed that pharmacologic blockade or genetic ablation of C5AR1 prevented metastatic potential of cancer cells (220). Expression of C5AR1 on TAMs conferred M2 polarization in colon cancer and enhanced liver metastatic lesions affirming a central role for C5AR1 in metastatic spread; importantly, genetic loss of C5ar1 severely impaired the metastatic ability of colon cancer cells (211). Genetic ablation of other complement proteins such as C3 was also shown to have profound inhibitory effects on primary tumor growth and metastasis correlating to increased numbers of IFNγ+/TNFα+/IL10+ CD4+ and CD8+ T-cells (335). The ability of complement C3 to function in conjunction with EMT contributing towards metastasis is shown by the ability of TWIST1 to regulate C3 expression in tumor cells (209). Cumulatively, the complement components work in synchrony as a “dark knight–a watchful protector” offering immune surveillance and regulating tumorigenesis and metastatic potential of the transformed oncogenic cells.
Juxtaposing these components are the membrane-bound and soluble complement regulatory factors that protect tumor cells from immune mediated cytotoxicity. Incidentally, high expressions of CD46, CD55, and CD59 are homogenously expressed and positively correlate with increased metastatic tumor cells in the liver of patients with colorectal (336) and other cancers with poor prognosis (321, 337). Inhibitory factors such as CFH were also shown to be highly expressed in exosomes of the metastatic cells (EV-CFH) resulting in increased migratory and invasive capacity of liver cancer cells. Blocking EV-CFH with a tumor specific anti-CFH antibody showed reduction in liver tumor promoting potentials and a potential therapeutic target (338). Directly “complementing” these pro-oncogenic functions, the complement cascade also interacts with the coagulation system resulting in a hyper-coagulable state and survival of tumor cells. In this context, C5a stimulates neutrophils to release tissue factor (339) while C3a induces platelet aggregation and activation (340), both processes culminating in a prothrombotic environment. Furthermore, the ability of neutrophil derived C3AR1 to form neutrophil extracellular trap (NET) drives tumorigenesis (341) and potentially enhances metastatic capacities. It is therefore important to consider these interactive mutually synergistic pathways in the design of novel therapeutics targeting HCC.
Therapeutic Targeting of Complement System: A Reality?
The myriad effects of complement molecules in regulating the TME and molecular and cellular effectors of immunopathogenic mechanisms driving HCC may offer new avenues to develop complement-based therapeutics. Figure 3 depicts the influence of differential complement protein expressions in regulating key pathobiological functions promoting oncogenesis in HCC and provides a platform for therapeutic interventions. In particular, the immune-based therapies have raised concerns and skepticism over failures to produce clinically meaningful disease-modulating effects in cancers. Some anticancer immunotherapies that inhibit PD-1 and/or PD-L1, such as Nivolumab and Pembrolizumab, are currently used to treat unresectable HCC. They can also induce complement activation due to their affinities for C1q and Fc receptors (342, 343); increased C1q levels have been shown to augment liver damage. In this context, C1-INH, approved by the FDA for treatment of hereditary angioedema, has been shown to block the classical activation pathway via C1q inhibition (344). Blocking C1q activity may thus represent a beneficial approach in regulating tumorigenesis in HCC while preserving the functions of other complement pathways. In addition to C1q, the roles of other downstream complement molecules such as C5a have been extensively studied in HCC. Inhibition of C5a within the TME without deleterious effects on additional complement dependent defenses has previously been proposed (344). In HCC, C5a activation was shown to induce EMT via inhibition of claudin-1 and activation of ERK1/2 pathway. Therefore, targeting C5a generation via anti-C5 antibodies (Eculizumab) or blocking C5a-C5aR interaction using a receptor antagonist (PMX-53) that are currently in clinical trials for acute myocardial infarction or rheumatoid arthritis respectively, may serve as promising therapeutic candidates for HCC. Other components of the complement cascade such as C3, C3a/C3b could also be targeted using inhibitors such as the Compostatin/POT-4, currently in clinical trials to treat age related macular degeneration (123). These studies highlight the importance of therapeutic targeting of complement as a novel therapeutic strategy for HCC. In parallel, complement dependent cytotoxicity (CDC) and antibody dependent cellular cytotoxicity (ADCC) are the leading cause of cell death when treating tumors/cancer cells with monoclonal antibodies. Several factors and/or etiopathogenic agents have been associated with tumor progression in HCC. Circulating apoptosis inhibitor of macrophage (AIM) is one such element that was recently described by Maehara et al. to play a role in activation of the complement cascade on the cell surface of tumorigenic, not normal, hepatocytes due to defective endocytosis (345). The authors showed that membrane bound AIM accumulation resulted in C3 activation in vivo and was detrimental for viability of cancer cells in HCC via CDC cascade. The fundamental dogma in complement biology is a skewing towards enhanced inflammation, with therapeutic approaches designed primarily towards inactivating the complement cascade; however, in situations of tumorigenesis, local stimulation of complement may be advantageous. In this regard the recently described anti-CD20 mAb currently in development: HuMax CD20, HuMax CD38, and HuMaX ZP3, have been demonstrated to increase CDC potency. Anti-CD20 mAb such as Rituximab may therefore prove beneficial in patients with HCC (346–348).
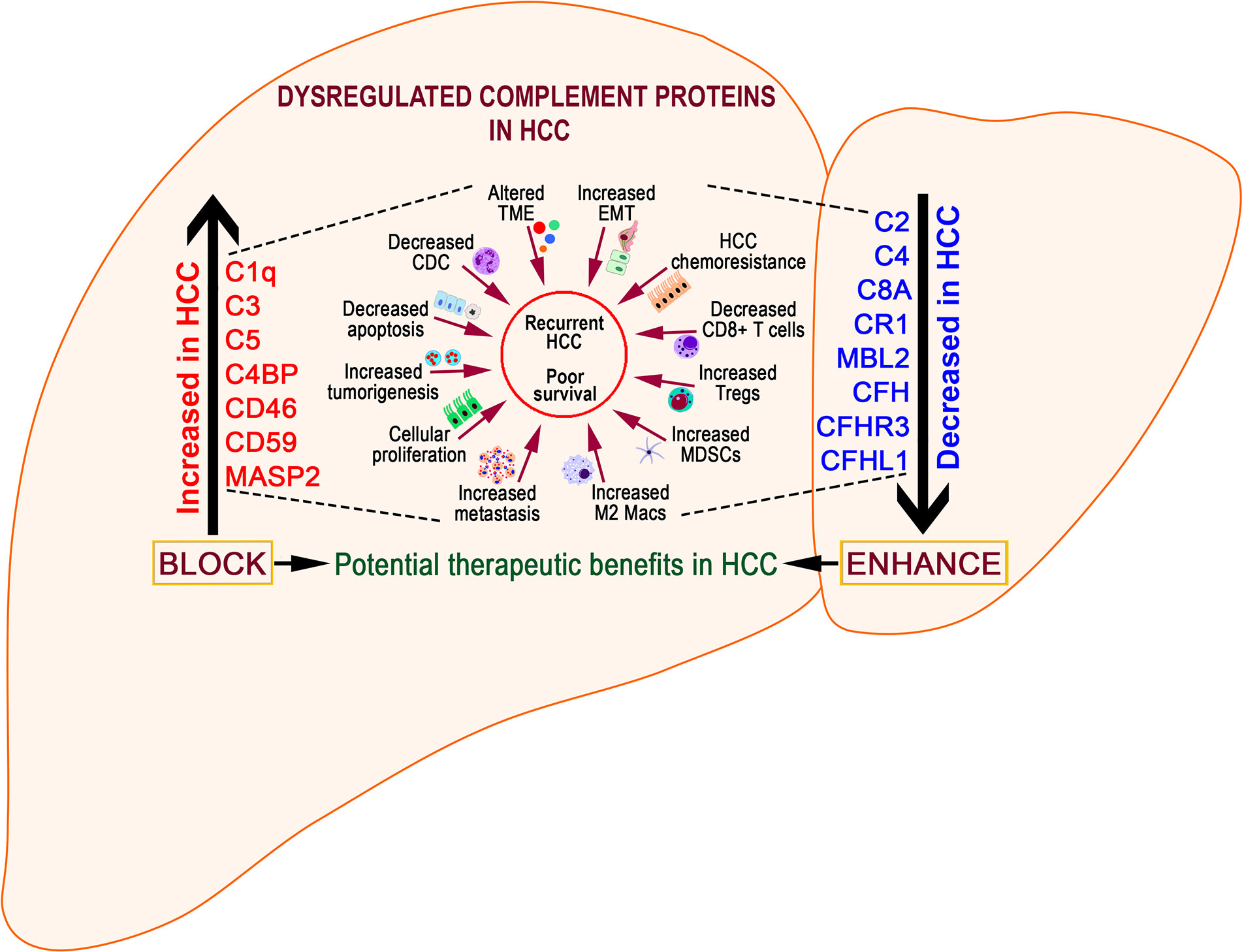
Figure 3 Dysregulated expressions of complement components orchestrate the pathobiology of hepatocellular carcinoma. Breakdown of the tightly controlled activation and regulatory component signals of the complement system results in dysregulation of the normal homeostatic cellular processes within the liver microenvironment. Increased levels of C1q, C3, C5, C4BP, CD46, CD59, and MASP2 and decreased levels of C2, C4, C8A, CR1, MBL2, CFH, CFHR3, and CFHL1 with potential immunoregulatory functions contribute to increased cell proliferation, metastasis, EMT, altered immune cell functions, etc. resulting in recurrent episodes and/or poor overall or disease-free survival in patients with HCC. Images from Motifolio drawing toolkit software (http://motifolio.com) were used for rendering the figure.
As discussed in the previous section, several factors contribute to the etiopathogenesis of HCC. One such factor, the oncolytic viruses, have been associated with tumorigenesis in HCC and are known to activate complement cascade. Recently, Kim et al., utilized Pexastimogene devacirepvec (Pexa-vec), an oncolytic virus, and showed its ability to induce complement-mediated cancer cell cytotoxicity in rabbits, resulting in improved survival in tumor bearing animals (349). Survival benefits were also achieved in patients with advanced hepatocellular carcinoma that were treated with Pexa-Vec (350). Due to significant success and promising clinical activity of the oncolytic virus, Pexa-vec has recently been tested along with Nivolumab to target tumor cells in HCC (348, 351). In conjunction with our review on the factors of complement cascade and the role of activated components in promoting HCC oncogenesis, we provide a platform identifying various molecules of the complement pathway as potential therapeutic targets in treating patients with HCC and fostering improved survival.
Concluding Remarks and the Impact of COVID-19
HCC continues to be a grave prognostic feature for patients with advanced liver disease of varying etiologies. While early diagnosis remains the mainstay of appropriate medical and surgical approaches, the mostly uncharacteristic features of HCC circumvent early disease diagnosis. The worldwide prevalence, lack of available therapeutic modalities, and rapid progression to severely compromised liver functions urgently necessitate identification and interrogation of newer mechanisms towards better treatment approaches. This urgent need is also dictated by the only available treatment of HCC that relies on liver transplantation, which inherently suffers from shortage of donor livers, higher costs, risk of tumor recurrence, etc.
The demand and need for non-surgical systemic therapies to effectively manage and treat HCC are greater than ever due to the evolving COVID-19 pandemic that has greatly overwhelmed the healthcare system. HCC patients are especially vulnerable due to the decreased allocation of healthcare resources including limited access to operating rooms, deferrals and delays in curative surgery and ablation therapies. The ever-changing scenario of the pandemic, disparity amongst nations in infection rates and limited data of COVID-19 infected HCC patients dictates ongoing efforts in liver oncology. HCC patients require repeated hospital visits, experience social and nosocomial contacts, risks posed by the prevalence of asymptomatic COVID-19 carriers in the community, treatment-related immunosuppression and more importantly treatment delays (352). A recent study reported 21.5% of patients with HCC experienced a significant treatment delay: longer than 1 month in 2020 compared to 2019 (353) as well as a significant drop in number of follow up patients visits (354). Moreover, the significant burden on healthcare providers and resource-intensive protocols have offered little guidance in addressing treatment strategies (352). Therefore, care providers must ensure appropriate surveillance, treatment, and monitoring of patients with HCC and continue to provide therapeutic avenues as in non-COVID-19 pandemic. A system to triage HCC patients where resources are limited should be adapted along with efforts to eliminate the virus in patients with confirmed COVID-19 infection (355). Amongst the many etiopathogenic factors known to cause or promote HCC, the real impact of COVID-19 pandemic or the SARS-CoV-2 virus itself in HCC patients remains unknown. Recent reports have indicated about 15%–54% of patients infected with the virus have hepatic injury and elevated levels of transaminases (356). It is therefore plausible that HCC patients infected with COVID-19 may experience exacerbated disease symptoms and predisposed to increased risk of secondary infections leading to significant morbidities or early mortality. Indeed, the risk factors that predict higher overall mortality in patients with chronic liver disease and COVID-19 are alcohol-related liver disease, decompensated cirrhosis and HCC (357). Using retrospective cohorts, many studies have associated increased biomarkers of liver injury (ALT, AST, GGT) to SARS-CoV-2 infection (358–360) with worsened disease responses in HCC and other cancers. Although the fundamental and intrinsic regulators remain unknown, increased injury responses have been ascribed to direct cytopathic effects of the SARS-CoV-2 virus on hepatocytes and/or cholangiocytes, hypoxia, immune-mediated hepatitis, etc. (356, 361). Standard care treatments such as antivirals and antibiotics prescribed to treat COVID-19 infection have also been linked with increased risk for hepatotoxicity and elevated liver enzymes. To overcome these new challenges and design effective treatment strategies, combination therapies that utilize existing or newly designed immunomodulators targeting complement cascade proteins described herein with immune checkpoint inhibitors may hold significant promise and provide novel therapeutic strategies to treat HCC patients with superimposed COVID-19 infection.
HCC is an immunogenic cancer characterized by chronic inflammation, fibrosis, and cirrhosis. Dysregulated immune responses constitute a major risk factor for HCC. The chronic inflammation, secondary to persistent liver damage, promotes immune cell activation and increased apoptosis. These events enhance tumorigenesis via cell stress, epigenetic modifications, altered mitochondrial metabolism, and activation of cellular senescence pathways. Combined, these biological phenomena directly regulate the high density of liver resident macrophages, NK cells, innate lymphocytes, etc. constituting the immune system as a potential target for managing and treating HCC. Central to these processes, recent studies have assigned multifaceted roles for complement molecules in the immunoregulation of HCC-TME. In this review, we have aimed to decipher the mechanistic roles of complement system in immune dysregulation and oncogenesis in HCC. Our review also describes several components of the complement cascade as potential targets for development of therapeutics. This is highly relevant in the context of limited chemotherapeutic measures that include Sorafenib, Lenvatinib and Regorafenib which currently form the standard care for advanced non-resectable HCC.
We have made an effort to elucidate how complement system regulates the activation of cellular and molecular responses including NK cells, DCs, MDSCs, TAMs, TANs, cytokines, and chemokines that form the first line of defense. Impaired complement activation alters the anti-tumorigenic immunological responses involved in halting the progression and expansion of the tumor, leading to HCC. Efforts to treat HCC via modulation of the pro-tumorigenic immune response in TME have been explored, but failure of these therapies in producing clinically meaningful effects limits their use. Elevated expressions of various complement components including C1q, C8a, and anaphylatoxins have been demonstrated in HCC tumors. Targeting these components either by specific inhibitors or antagonists in synergism with existing therapies has a great potential to treat HCC. In contrast to promoting tumor progression, components such as C2 and CFH exhibit tumor-suppressive effects and better prognosis in HCC patients. Furthermore, complement C3, C3a, C4, C4a, and C7 have been identified as biomarkers in HCC diagnosis while C7 and CFH are recognized for their critical roles in mediating stemness of tumor-initiating cells. Similarly, complement C5 regulates EMT, cell migration, and invasion, MBL activates stellate cells, CR1 clears complement CICs, C4BP acts as a complement inhibitor, and CD59 suppresses C3 activation and MAC formation while loss of CFHL1 corresponds to poor time-to-recurrence and overall survival rates. Given these divergent roles, future efforts should be directed toward developing strategies that selectively target or inhibit the tumorigenic effects of complement components while promoting or retaining their anti-tumorigenic effects. Development of novel and targeted therapeutics also benefits from an array of integrated bioinformatics analysis using either a biologically validated known set of genes or a discovery module that employs either large-scale transcriptomic or proteomic techniques. As an initial approach to identify molecular partners and biological processes linked to Complement, we subjected complement components found in HCC (C1q, C2, C3, C4BP, C5, C7, C8A, CD46, etc.) to bioinformatics analysis. Analyzing the genes using ToppGene database (http://toppgene.cchmc.org) with a threshold of P<0.05, we identified several biological processes that were further clustered using CIMminer (https://discover.nci.nih.gov/cimminer/home.do). A significant number of complement molecules defined several dominant pathways linked to activation and regulation of innate and adaptive immune systems emphasizing the importance of complement-driven immunopathogenesis (Figure 4). Protein-protein interaction network (Figure 5) generated using ToppGenet (https://toppgene.cchmc.org) and complement genes showed close associations with several molecules that either positively (ADM, APOA1, ATP12A, BIRC5, CFB, CHD1, DLG4, GRB2, ITGA2, ITGB1, KRAS, LCK) or negatively (APCS, BRCA2, CASK, CD27, CD81, CD82, DMP1, FCN2, GRK2, LLGL1) regulate the pathogenesis of HCC. Regulation of these diverse processes by complement molecules may further propel identification of novel interventional targets for drug development and therapeutic interventions. Thus, the complement cascade serves as a link between the innate and adaptive immune system, activating immune cells critical to drive HCC pathogenesis. With the current understandings of complement molecules as oncogenic drivers, targeted therapies could be developed independently or in combination with existing first and second line of HCC therapies. In summary, a deeper understanding of the mechanistic role of tumor complement components in these pro- and anti-tumorigenic pathways supplemented by advanced bioinformatics approaches are expected to foster the design and development of effective clinical treatments for HCC.
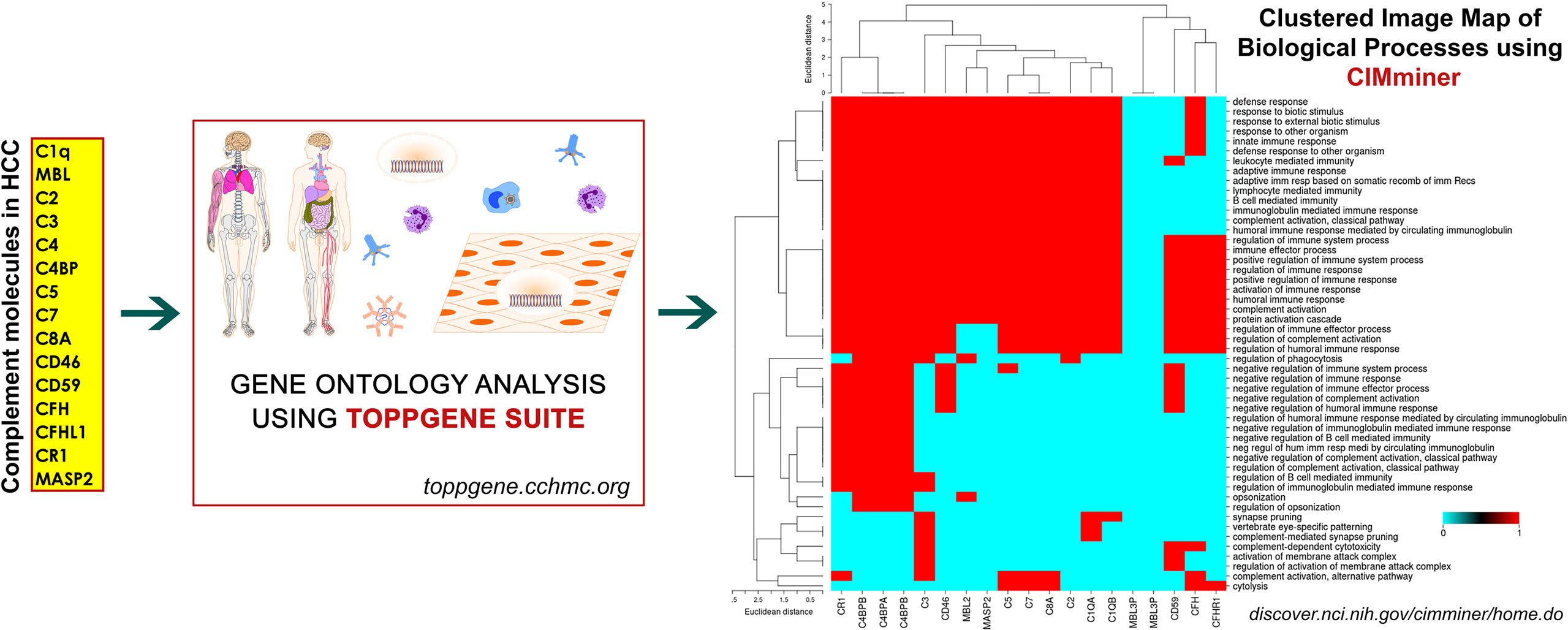
Figure 4 Bioinformatics analysis identifies dominant signatures of immune–mediated biological processes. Supervised gene ontology (GO) annotation analysis of complement components in hepatocellular carcinoma (HCC) was performed via ToppGene Suite portal (http://toppgene.cchmc.org) for in-silico enrichment of biological processes with a threshold False Discovery Rate (FDR) corrected P value of <0.05. ToppGene Suite is a freely available online tool used for functional enrichment, prioritization of candidate genes using transcriptome, ontology, phenotype, proteome, and functional annotations. GO: biological processes identified by ToppGene were further subjected to functional enrichment using CIMminer (https://discover.nci.nih.gov/cimminer/home.do). Red areas depicted in the heatmap show closely related biological processes linked to immunity that are shared by a major group of complement molecules shown on the horizontal axis.
Figure 5 Protein-protein interaction network analysis identifies biological relatedness of complement components to regulators of hepatocellular carcinoma (HCC) pathogenesis. Complement molecules of relevance to HCC were subjected to protein-protein interaction (PPI) network analysis using the network-based gene prioritization algorithm, ToppGenet of the ToppGene Suite (http://toppgene.cchmc.org). ToppGenet identifies and prioritizes candidate genes based on functional annotations, similar expressions, and network and topographical features. A Step Size of 6 and the Prioritization method of k-Step Markov were used as default analytical parameters. The Cytoscape-compatible ToppGenet output file was used to generate the graphical network. The first shell of 41 interacting proteins (grey color) associated directly with the input complement proteins (blue) in the PPI were generated by Cytoscape.
Author Contributions
AM and UT equally contributed towards design and manuscript drafting. SA, RM, SN, and AB compiled the literature and assisted in drafting the manuscript. RM analyzed complement genes using bioinformatics tools. PS conceived the original idea and completed the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by NIH P30 DK078392 of the Digestive Diseases Research Core Center in Cincinnati.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the assistance of Mr. Chris Woods, Pathology Research Core at the Cincinnati Children’s Hospital Medical Center for his help with the graphical design of Figures.
References
1. Buendia MA, Neuveut C. Hepatocellular carcinoma. Cold Spring Harb Perspect Med (2015) 5(2):a021444. doi: 10.1101/cshperspect.a021444
2. Collaboration GBoDC. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol (2018) 4(11):1553–68. doi: 10.1001/jamaoncol.2018.2706
3. Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology (2015) 61(1):191–9.
4. Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol (2015) 9(6):765–79. doi: 10.1586/17474124.2015.1028363
5. Cazzagon N, Trevisani F, Maddalo G, Giacomin A, Vanin V, Pozzan C, et al. Rise and fall of HCV-related hepatocellular carcinoma in Italy: a long-term survey from the ITA.LI.CA centres. Liver Int (2013) 33(9):1420–7. doi: 10.1111/liv.12208
6. Goh GB-B, Li JW, Chang P-E, Chow K-Y, Tan C-K. Deciphering the epidemiology of hepatocellular carcinoma through the passage of time: A study of 1,401 patients across 3 decades. Hepatol Commun (2017) 1(6):564–71. doi: 10.1002/hep4.1059
7. EASL. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol (2018) 69(1):182–236. doi: 10.1016/j.jhep.2018.03.019
8. El–Serag HB, Rudolph KL. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology (2007) 132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061
9. Welzel TM, Graubard BI, Quraishi S, Zeuzem S, Davila JA, El-Serag HB, et al. Population-Attributable Fractions of Risk Factors for Hepatocellular Carcinoma in the United States. Off J Am Coll Gastroenterol ACG (2013) 108(8):1314–21. doi: 10.1038/ajg.2013.160
10. Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA: A Cancer J Clin (2010) 60(5):277–300. doi: 10.3322/caac.20073
11. CLIP. A new prognostic system for hepatocellular carcinoma: a retrospective study of 435 patients: the Cancer of the Liver Italian Program (CLIP) investigators. Hepatology (1998) 28(3):751–5. doi: 10.1002/hep.510280322
12. ICGHN. Pathologic diagnosis of early hepatocellular carcinoma: a report of the international consensus group for hepatocellular neoplasia. Hepatology (2009) 49(2):658–64. doi: 10.1002/hep.22709
13. Czigany Z, Schöning W, Ulmer TF, Bednarsch J, Amygdalos I, Cramer T, et al. Hypothermic oxygenated machine perfusion (HOPE) for orthotopic liver transplantation of human liver allografts from extended criteria donors (ECD) in donation after brain death (DBD): a prospective multicentre randomised controlled trial (HOPE ECD-DBD). BMJ Open (2017) 7(10):e017558. doi: 10.1136/bmjopen-2017-017558
14. Lo CM, Ngan H, Tso WK, Liu CL, Lam CM, Poon RTP, et al. Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology (2002) 35(5):1164–71.
15. Salem R, Gabr A, Riaz A, Mora R, Ali R, Abecassis M, et al. Institutional decision to adopt Y90 as primary treatment for hepatocellular carcinoma informed by a 1,000-patient 15-year experience. Hepatology (2018) 68(4):1429–40. doi: 10.1002/hep.29691
16. Lasley FD, Mannina EM, Johnson CS, Perkins SM, Althouse S, Maluccio M, et al. Treatment variables related to liver toxicity in patients with hepatocellular carcinoma, Child-Pugh class A and B enrolled in a phase 1-2 trial of stereotactic body radiation therapy. Pract Radiat Oncol (2015) 5(5):e443–e9. doi: 10.1016/j.prro.2015.02.007
17. Ahmed M, Brace CL, Lee FT Jr., Goldberg SN. Principles of and advances in percutaneous ablation. Radiology (2011) 258(2):351–69. doi: 10.1148/radiol.10081634
18. Chok KSH, Cheung TT, Lo RCL, Chu FSK, Tsang SHY, Chan ACY, et al. Pilot study of high-intensity focused ultrasound ablation as a bridging therapy for hepatocellular carcinoma patients wait-listed for liver transplantation. Liver Transplant (2014) 20(8):912–21. doi: 10.1002/lt.23892
19. Lencioni R, Crocetti L. Local-regional treatment of hepatocellular carcinoma. Radiology (2012) 262(1):43–58. doi: 10.1148/radiol.11110144
20. Lurje I, Czigany Z, Bednarsch J, Roderburg C, Isfort P, Neumann UP, et al. Treatment Strategies for Hepatocellular Carcinoma - a Multidisciplinary Approach. Int J Mol Sci (2019) 20(6):1465. doi: 10.3390/ijms20061465
21. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in Advanced Hepatocellular Carcinoma. N Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
22. Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet (2018) 391(10126):1163–73. doi: 10.1016/S0140-6736(18)30207-1
23. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
24. Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017) 389(10064):56–66. doi: 10.1016/S0140-6736(16)32453-9
25. Abou-Alfa GK, Meyer T, Cheng A-L, El-Khoueiry AB, Rimassa L, Ryoo B-Y, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med (2018) 379(1):54–63. doi: 10.1056/NEJMoa1717002
26. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res (2004) 64(19):7099–109. doi: 10.1158/0008-5472.Can-04-1443
27. Sieber OM, Heinimann K, Tomlinson IP. Genomic instability—the engine of tumorigenesis? Nat Rev Cancer (2003) 3(9):701–8.
28. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev (2018) 32(19-20):1267–84. doi: 10.1101/gad.314617.118
29. Huang H, Lu Y, Zhou T, Gu G, Xia Q. Innate Immune Cells in Immune Tolerance After Liver Transplantation. Front Immunol (2018) 9:2401. doi: 10.3389/fimmu.2018.02401
30. Yang YM, Kim SY, Seki E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Semin Liver Dis (2019) 39(1):26–42. doi: 10.1055/s-0038-1676806
31. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med (2010) 49(11):1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006
32. Hymel L, Nielsen M, Gietzen K. Target sizes of human erythrocyte membrane Ca2+-ATPase and Mg2+-ATPase activities in the presence and absence of calmodulin. Biochim Biophys Acta (1985) 815(3):461–7. doi: 10.1016/0005-2736(85)90374-8
33. Yu Y, Dai Y, Fang M, Chen X. ADPRT-mediated decrease of cellular NAD content and the detection of chemically induced DNA damage–development of a new short-term screening test for mutagens. Proc Chin Acad Med Sci Peking Union Med Coll (1990) 5(1):19–24.
34. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
35. Tian Z, Chen Y, Gao B. Natural killer cells in liver disease. Hepatology (2013) 57(4):1654–62. doi: 10.1002/hep.26115
36. Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol (2008) 129(3):428–37. doi: 10.1016/j.clim.2008.08.012
37. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front Immunol (2019) 10:771. doi: 10.3389/fimmu.2019.00771
38. Sachdeva M, Chawla YK, Arora SK. Immunology of hepatocellular carcinoma. World J Hepatol (2015) 7(17):2080–90. doi: 10.4254/wjh.v7.i17.2080
39. Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) Macrophages in Inflammatory and Malignant Diseases. Int J Mol Sci (2020) 21(15):5497–529. doi: 10.3390/ijms21155497
40. Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and hepatocellular carcinoma. Cell Biosci (2019) 9:79. doi: 10.1186/s13578-019-0342-7
41. Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J, et al. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol (2000) 164(2):762–7. doi: 10.4049/jimmunol.164.2.762
42. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci (2011) 7(5):651–8. doi: 10.7150/ijbs.7.651
43. Lu C, Rong D, Zhang B, Zheng W, Wang X, Chen Z, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer (2019) 18(1):130. doi: 10.1186/s12943-019-1047-6
44. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology (2016) 150(7):1646–58.e17. doi: 10.1053/j.gastro.2016.02.040
45. Zhou SL, Yin D, Hu ZQ, Luo CB, Zhou ZJ, Xin HY, et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology (2019) 70(4):1214–30. doi: 10.1002/hep.30630
46. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer (2019) 120(1):16–25. doi: 10.1038/s41416-018-0333-1
47. Gao XH, Tian L, Wu J, Ma XL, Zhang CY, Zhou Y, et al. Circulating CD14(+) HLA-DR(-/low) myeloid-derived suppressor cells predicted early recurrence of hepatocellular carcinoma after surgery. Hepatol Res (2017) 47(10):1061–71. doi: 10.1111/hepr.12831
48. Nguyen HN, Sevin BU, Averette HE, Perras J, Donato D, Penalver M. Comparative evaluation of single and combination chemotherapy in uterine cancer cell lines. Gynecol Oncol (1991) 42(3):227–32. doi: 10.1016/0090-8258(91)90350-e
49. Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology (2015) 62(4):1304–12. doi: 10.1002/hep.27867
50. Li W, Han J, Wu H. Regulatory T-cells promote hepatitis B virus infection and hepatocellular carcinoma progression. Chronic Dis Transl Med (2016) 2(2):67–80. doi: 10.1016/j.cdtm.2016.09.001
51. Yuan CH, Sun XM, Zhu CL, Liu SP, Wu L, Chen H, et al. Amphiregulin activates regulatory T lymphocytes and suppresses CD8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget (2015) 6(31):32138–53. doi: 10.18632/oncotarget.5171
52. Huang Y, Wang FM, Wang T, Wang YJ, Zhu ZY, Gao YT, et al. Tumor-infiltrating FoxP3+ Tregs and CD8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion (2012) 86(4):329–37. doi: 10.1159/000342801
53. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi: 10.1038/nature06306
54. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol (2010) 11(12):1093–101. doi: 10.1038/ni.1952
55. Fu YP, Yi Y, Cai XY, Sun J, Ni XC, He HW, et al. Overexpression of interleukin-35 associates with hepatocellular carcinoma aggressiveness and recurrence after curative resection. Br J Cancer (2016) 114(7):767–76. doi: 10.1038/bjc.2016.47
56. Zhao HQ, Li WM, Lu ZQ, Yao YM. Roles of Tregs in development of hepatocellular carcinoma: a meta-analysis. World J Gastroenterol (2014) 20(24):7971–8. doi: 10.3748/wjg.v20.i24.7971
57. Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. J Leukoc Biol (2006) 80(6):1197–213. doi: 10.1189/jlb.0506297
58. Saxena R, Kaur J. Th1/Th2 cytokines and their genotypes as predictors of hepatitis B virus related hepatocellular carcinoma. World J Hepatol (2015) 7(11):1572–80. doi: 10.4254/wjh.v7.i11.1572
59. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med (2008) 359(19):1995–2004. doi: 10.1056/NEJMoa0804525
60. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
61. Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell (2006) 10(2):99–111. doi: 10.1016/j.ccr.2006.06.016
62. Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology (2011) 54(3):900–9. doi: 10.1002/hep.24486
63. Beckebaum S, Zhang X, Chen X, Yu Z, Frilling A, Dworacki G, et al. Increased levels of interleukin-10 in serum from patients with hepatocellular carcinoma correlate with profound numerical deficiencies and immature phenotype of circulating dendritic cell subsets. Clin Cancer Res (2004) 10(21):7260–9. doi: 10.1158/1078-0432.CCR-04-0872
64. Pan QZ, Pan K, Zhao JJ, Chen JG, Li JJ, Lv L, et al. Decreased expression of interleukin-36alpha correlates with poor prognosis in hepatocellular carcinoma. Cancer Immunol Immunother (2013) 62(11):1675–85. doi: 10.1007/s00262-013-1471-1
65. Zhao JJ, Pan QZ, Pan K, Weng DS, Wang QJ, Li JJ, et al. Interleukin-37 mediates the antitumor activity in hepatocellular carcinoma: role for CD57+ NK cells. Sci Rep (2014) 4:5177. doi: 10.1038/srep05177
66. Liu R, Tang C, Shen A, Luo H, Wei X, Zheng D, et al. IL-37 suppresses hepatocellular carcinoma growth by converting pSmad3 signaling from JNK/pSmad3L/c-Myc oncogenic signaling to pSmad3C/P21 tumor-suppressive signaling. Oncotarget (2016) 7(51):85079–96. doi: 10.18632/oncotarget.13196
67. Kohga K, Tatsumi T, Tsunematsu H, Aono S, Shimizu S, Kodama T, et al. Interleukin-1beta enhances the production of soluble MICA in human hepatocellular carcinoma. Cancer Immunol Immunother (2012) 61(9):1425–32. doi: 10.1007/s00262-012-1208-6
68. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206(6):1327–37. doi: 10.1084/jem.20082173
69. Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int J Mol Sci (2018) 19(8):2155–89. doi: 10.3390/ijms19082155
70. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology (2018) 67(5):1872–89. doi: 10.1002/hep.29681
71. Ge NL, Ye SL, Zheng N, Sun RX, Liu YK, Tang ZY. Prevention of hepatocellular carcinoma in mice by IL-2 and B7-1 genes co-transfected liver cancer cell vaccines. World J Gastroenterol (2003) 9(10):2182–5. doi: 10.3748/wjg.v9.i10.2182
73. Xu QG, Yu J, Guo XG, Hou GJ, Yuan SX, Yang Y, et al. IL-17A promotes the invasion-metastasis cascade via the AKT pathway in hepatocellular carcinoma. Mol Oncol (2018) 12(6):936–52. doi: 10.1002/1878-0261.12306
74. Li J, Lau GK, Chen L, Dong SS, Lan HY, Huang XR, et al. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PLoS One (2011) 6(7):e21816. doi: 10.1371/journal.pone.0021816
75. Gu FM, Li QL, Gao Q, Jiang JH, Zhu K, Huang XY, et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer (2011) 10:150. doi: 10.1186/1476-4598-10-150
76. Brunner SM, Rubner C, Kesselring R, Martin M, Griesshammer E, Ruemmele P, et al. Tumor-infiltrating, interleukin-33-producing effector-memory CD8(+) T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology (2015) 61(6):1957–67. doi: 10.1002/hep.27728
77. Ikeguchi M, Hirooka Y. Interleukin-2 gene expression is a new biological prognostic marker in hepatocellular carcinomas. Onkologie (2005) 28(5):255–9. doi: 10.1159/000084695
78. Easom NJW, Stegmann KA, Swadling L, Pallett LJ, Burton AR, Odera D, et al. IL-15 Overcomes Hepatocellular Carcinoma-Induced NK Cell Dysfunction. Front Immunol (2018) 9:1009. doi: 10.3389/fimmu.2018.01009
79. Chang CM, Lo CH, Shih YM, Chen Y, Wu PY, Tsuneyama K, et al. Treatment of hepatocellular carcinoma with adeno-associated virus encoding interleukin-15 superagonist. Hum Gene Ther (2010) 21(5):611–21. doi: 10.1089/hum.2009.187
80. Cheng L, Du X, Wang Z, Ju J, Jia M, Huang Q, et al. Hyper-IL-15 suppresses metastatic and autochthonous liver cancer by promoting tumour-specific CD8+ T cell responses. J Hepatol (2014) 61(6):1297–303. doi: 10.1016/j.jhep.2014.07.004
81. Li W, Gomez E, Zhang Z. Immunohistochemical expression of stromal cell-derived factor-1 (SDF-1) and CXCR4 ligand receptor system in hepatocellular carcinoma. J Exp Clin Cancer Res (2007) 26(4):527–33.
82. Sutton A, Friand V, Brule-Donneger S, Chaigneau T, Ziol M, Sainte-Catherine O, et al. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol Cancer Res (2007) 5(1):21–33. doi: 10.1158/1541-7786.MCR-06-0103
83. Liu H, Pan Z, Li A, Fu S, Lei Y, Sun H, et al. Roles of chemokine receptor 4 (CXCR4) and chemokine ligand 12 (CXCL12) in metastasis of hepatocellular carcinoma cells. Cell Mol Immunol (2008) 5(5):373–8. doi: 10.1038/cmi.2008.46
84. Chu H, Zhou H, Liu Y, Liu X, Hu Y, Zhang J. Functional expression of CXC chemokine recepter-4 mediates the secretion of matrix metalloproteinases from mouse hepatocarcinoma cell lines with different lymphatic metastasis ability. Int J Biochem Cell Biol (2007) 39(1):197–205. doi: 10.1016/j.biocel.2006.07.008
85. Schimanski CC, Bahre R, Gockel I, Muller A, Frerichs K, Horner V, et al. Dissemination of hepatocellular carcinoma is mediated via chemokine receptor CXCR4. Br J Cancer (2006) 95(2):210–7. doi: 10.1038/sj.bjc.6603251
86. Garcia-Irigoyen O, Latasa MU, Carotti S, Uriarte I, Elizalde M, Urtasun R, et al. Matrix metalloproteinase 10 contributes to hepatocarcinogenesis in a novel crosstalk with the stromal derived factor 1/C-X-C chemokine receptor 4 axis. Hepatology (2015) 62(1):166–78. doi: 10.1002/hep.27798
87. Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology (2012) 56(6):2242–54. doi: 10.1002/hep.25907
88. Bertran E, Crosas-Molist E, Sancho P, Caja L, Lopez-Luque J, Navarro E, et al. Overactivation of the TGF-beta pathway confers a mesenchymal-like phenotype and CXCR4-dependent migratory properties to liver tumor cells. Hepatology (2013) 58(6):2032–44. doi: 10.1002/hep.26597
89. Li X, Li P, Chang Y, Xu Q, Wu Z, Ma Q, et al. The SDF-1/CXCR4 axis induces epithelial-mesenchymal transition in hepatocellular carcinoma. Mol Cell Biochem (2014) 392(1-2):77–84. doi: 10.1007/s11010-014-2020-8
90. Yang W, Wang C, Lin Y, Liu Q, Yu LX, Tang L, et al. OV6(+) tumor-initiating cells contribute to tumor progression and invasion in human hepatocellular carcinoma. J Hepatol (2012) 57(3):613–20. doi: 10.1016/j.jhep.2012.04.024
91. Huang P, Xu X, Wang L, Zhu B, Wang X, Xia J. The role of EGF-EGFR signalling pathway in hepatocellular carcinoma inflammatory microenvironment. J Cell Mol Med (2014) 18(2):218–30. doi: 10.1111/jcmm.12153
92. Zhou SL, Zhou ZJ, Hu ZQ, Li X, Huang XW, Wang Z, et al. CXCR2/CXCL5 axis contributes to epithelial-mesenchymal transition of HCC cells through activating PI3K/Akt/GSK-3beta/Snail signaling. Cancer Lett (2015) 358(2):124–35. doi: 10.1016/j.canlet.2014.11.044
93. Ma S, Cheng Q, Cai Y, Gong H, Wu Y, Yu X, et al. IL-17A produced by gammadelta T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res (2014) 74(7):1969–82. doi: 10.1158/0008-5472.CAN-13-2534
94. Ren Y, Poon RT, Tsui HT, Chen WH, Li Z, Lau C, et al. Interleukin-8 serum levels in patients with hepatocellular carcinoma: correlations with clinicopathological features and prognosis. Clin Cancer Res (2003) 9(16 Pt 1):5996–6001.
95. Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol (2011) 54(5):948–55. doi: 10.1016/j.jhep.2010.08.041
96. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem (2009) 284(10):6038–42. doi: 10.1074/jbc.C800207200
97. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61(3):416–26. doi: 10.1136/gutjnl-2011-300304
98. Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Möckel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut (2014) 63(12):1960–71.
99. Shih YT, Wang MC, Zhou J, Peng HH, Lee DY, Chiu JJ. Endothelial progenitors promote hepatocarcinoma intrahepatic metastasis through monocyte chemotactic protein-1 induction of microRNA-21. Gut (2015) 64(7):1132–47. doi: 10.1136/gutjnl-2013-306302
100. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest (2009) 119(7):1858–70. doi: 10.1172/jci37444
101. Rubie C, Frick VO, Wagner M, Rau B, Weber C, Kruse B, et al. Enhanced expression and clinical significance of CC-chemokine MIP-3 alpha in hepatocellular carcinoma. Scand J Immunol (2006) 63(6):468–77. doi: 10.1111/j.1365-3083.2006.001766.x
102. Uchida H, Iwashita Y, Sasaki A, Shibata K, Matsumoto T, Ohta M, et al. Chemokine receptor CCR6 as a prognostic factor after hepatic resection for hepatocellular carcinoma. J Gastroenterol Hepatol (2006) 21(1 Pt 1):161–8. doi: 10.1111/j.1440-1746.2005.04157.x
103. Tang L, Hu HD, Hu P, Lan YH, Peng ML, Chen M, et al. Gene therapy with CX3CL1/Fractalkine induces antitumor immunity to regress effectively mouse hepatocellular carcinoma. Gene Ther (2007) 14(16):1226–34. doi: 10.1038/sj.gt.3302959
104. Harsha C, Banik K, Ang HL, Girisa S, Vikkurthi R, Parama D, et al. Targeting AKT/mTOR in Oral Cancer: Mechanisms and Advances in Clinical Trials. Int J Mol Sci (2020) 21(9):3285–311. doi: 10.3390/ijms21093285
105. Fondevila F, Mendez-Blanco C, Fernandez-Palanca P, Gonzalez-Gallego J, Mauriz JL. Anti-tumoral activity of single and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers. Exp Mol Med (2019) 51(9):1–15. doi: 10.1038/s12276-019-0308-1
106. Li D, Sedano S, Allen R, Gong J, Cho M, Sharma S. Current Treatment Landscape for Advanced Hepatocellular Carcinoma: Patient Outcomes and the Impact on Quality of Life. Cancers (Basel) (2019) 11(6):841-59. doi: 10.3390/cancers11060841
107. Personeni N, Pressiani T, Rimassa L. Lenvatinib for the treatment of unresectable hepatocellular carcinoma: evidence to date. J Hepatocell Carcinoma (2019) 6:31–9. doi: 10.2147/JHC.S168953
108. Marino D, Zichi C, Audisio M, Sperti E, Di Maio M. Second-line treatment options in hepatocellular carcinoma. Drugs Context (2019) 8:212577. doi: 10.7573/dic.212577
109. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
110. Pan C, Liu H, Robins E, Song W, Liu D, Li Z, et al. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol (2020) 13(1):29. doi: 10.1186/s13045-020-00862-w
111. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
112. Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JH, et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A (2019) 116(45):22699–709. doi: 10.1073/pnas.1821218116
113. Tagliamonte M, Mauriello A, Cavalluzzo B, Ragone C, Manolio C, Petrizzo A, et al. Tackling hepatocellular carcinoma with individual or combinatorial immunotherapy approaches. Cancer Lett (2020) 473:25–32. doi: 10.1016/j.canlet.2019.12.029
114. Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res (2020) 30(6):507–19. doi: 10.1038/s41422-020-0337-2
115. Onuma AE, Zhang H, Huang H, Williams TM, Noonan A, Tsung A. Immune Checkpoint Inhibitors in Hepatocellular Cancer: Current Understanding on Mechanisms of Resistance and Biomarkers of Response to Treatment. Gene Expr (2020) 20(1):53–65. doi: 10.3727/105221620X15880179864121
116. Thorgersen EB, Barratt-Due A, Haugaa H, Harboe M, Pischke SE, Nilsson PH, et al. The Role of Complement in Liver Injury, Regeneration, and Transplantation. Hepatology (2019) 70(2):725–36. doi: 10.1002/hep.30508
117. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest (2015) 125(9):3347–55. doi: 10.1172/JCI80007
118. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9(10):729–40. doi: 10.1038/nri2620
119. Laskowski J, Renner B, Pickering MC, Serkova NJ, Smith-Jones PM, Clambey ET, et al. Complement factor H–deficient mice develop spontaneous hepatic tumors. J Clin Invest (2020) 130(8):4039–54. doi: 10.1172/JCI135105
120. Hess C, Kemper C. Complement-Mediated Regulation of Metabolism and Basic Cellular Processes. Immunity (2016) 45(2):240–54. doi: 10.1016/j.immuni.2016.08.003
121. Zhang R, Liu Q, Li T, Liao Q, Zhao Y. Role of the complement system in the tumor microenvironment. Cancer Cell Int (2019) 19:300. doi: 10.1186/s12935-019-1027-3
122. Mamidi S, Hone S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology (2017) 222(1):45–54. doi: 10.1016/j.imbio.2015.11.008
123. Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol (2007) 25(11):1265–75. doi: 10.1038/nbt1342
124. Humbles AA, Lu B, Nilsson CA, Lilly C, Israel E, Fujiwara Y, et al. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature (2000) 406(6799):998–1001. doi: 10.1038/35023175
126. Niculescu F, Rus H. The role of complement activation in atherosclerosis. Immunol Res (2004) 30(1):73–80. doi: 10.1385/IR:30:1:073
127. Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol (1998) 43(4):465–71. doi: 10.1002/ana.410430409
128. Linton SM, Morgan BP. Complement activation and inhibition in experimental models of arthritis. Mol Immunol (1999) 36(13-14):905–14. doi: 10.1016/s0161-5890(99)00113-3
129. Rutkowski MJ, Sughrue ME, Kane AJ, Ahn BJ, Fang S, Parsa AT. The complement cascade as a mediator of tissue growth and regeneration. Inflamm Res (2010) 59(11):897–905. doi: 10.1007/s00011-010-0220-6
130. Khan MA, Assiri AM, Broering DC. Complement and macrophage crosstalk during process of angiogenesis in tumor progression. J BioMed Sci (2015) 22:58. doi: 10.1186/s12929-015-0151-1
131. Goswami MT, Reka AK, Kurapati H, Kaza V, Chen J, Standiford TJ, et al. Regulation of complement-dependent cytotoxicity by TGF-beta-induced epithelial-mesenchymal transition. Oncogene (2016) 35(15):1888–98. doi: 10.1038/onc.2015.258
132. Kochanek DM, Ghouse SM, Karbowniczek MM, Markiewski MM. Complementing Cancer Metastasis. Front Immunol (2018) 9:1629. doi: 10.3389/fimmu.2018.01629
133. Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res (2010) 8(11):1453–65. doi: 10.1158/1541-7786.MCR-10-0225
134. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
135. Cressman DE, Diamond RH, Taub R. Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology (1995) 21(5):1443–9.
136. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2010) 140(2):197–208. doi: 10.1016/j.cell.2009.12.052
137. He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell (2010) 17(3):286–97. doi: 10.1016/j.ccr.2009.12.048
138. Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol (1987) 17(10):1485–9. doi: 10.1002/eji.1830171015
139. Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res (2007) 84(4):635–45. doi: 10.1016/j.exer.2006.11.015
140. Brooimans RA, van der Ark AA, Buurman WA, van Es LA, Daha MR. Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J Immunol (1990) 144(10):3835–40.
141. Licht C, Pluthero FG, Li L, Christensen H, Habbig S, Hoppe B, et al. Platelet-associated complement factor H in healthy persons and patients with atypical HUS. Blood (2009) 114(20):4538–45. doi: 10.1182/blood-2009-03-205096
142. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U S A (1976) 73(9):3268–72. doi: 10.1073/pnas.73.9.3268
143. Yang JD, Seol SY, Leem SH, Kim YH, Sun Z, Lee JS, et al. Genes associated with recurrence of hepatocellular carcinoma: integrated analysis by gene expression and methylation profiling. J Korean Med Sci (2011) 26(11):1428–38. doi: 10.3346/jkms.2011.26.11.1428
144. Seol HS, Lee SE, Song JS, Rhee JK, Singh SR, Chang S, et al. Complement proteins C7 and CFH control the stemness of liver cancer cells via LSF-1. Cancer Lett (2016) 372(1):24–35. doi: 10.1016/j.canlet.2015.12.005
145. Yoo BK, Emdad L, Gredler R, Fuller C, Dumur CI, Jones KH, et al. Transcription factor Late SV40 Factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proc Natl Acad Sci U S A (2010) 107(18):8357–62. doi: 10.1073/pnas.1000374107
146. Jain A, Karadag A, Fohr B, Fisher LW, Fedarko NS. Three SIBLINGs (small integrin-binding ligand, N-linked glycoproteins) enhance factor H’s cofactor activity enabling MCP-like cellular evasion of complement-mediated attack. J Biol Chem (2002) 277(16):13700–8. doi: 10.1074/jbc.M110757200
147. Liu H, Zhang L, Wang P. Complement factor Hrelated 3 overexpression affects hepatocellular carcinoma proliferation and apoptosis. Mol Med Rep (2019) 20(3):2694–702. doi: 10.3892/mmr.2019.10514
148. Liu J, Li W, Zhao H. CFHR3 is a potential novel biomarker for hepatocellular carcinoma. J Cell Biochem (2020) 121(4):2970–80. doi: 10.1002/jcb.29551
149. Pan L, Fang J, Chen MY, Zhai ST, Zhang B, Jiang ZY, et al. Promising key genes associated with tumor microenvironments and prognosis of hepatocellular carcinoma. World J Gastroenterol (2020) 26(8):789–803. doi: 10.3748/wjg.v26.i8.789
150. Xue F, Yang L, Dai B, Xue H, Zhang L, Ge R, et al. Bioinformatics profiling identifies seven immune-related risk signatures for hepatocellular carcinoma. PeerJ (2020) 8:e8301. doi: 10.7717/peerj.8301
151. Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol (2003) 40(2-4):109–23. doi: 10.1016/s0161-5890(03)00112-3
152. Asimakopoulos JV, Terpos E, Papageorgiou L, Kampouropoulou O, Christoulas D, Giakoumis A, et al. The presence of CD55- and/or CD59-deficient erythrocytic populations in patients with rheumatic diseases reflects an immune-mediated bone-marrow derived phenomenon. Med Sci Monit (2014) 20:123–39. doi: 10.12659/MSM.889727
153. Ruiz-Delgado GJ, Vazquez-Garza E, Mendez-Ramirez N, Gomez-Almaguer D. Abnormalities in the expression of CD55 and CD59 surface molecules on peripheral blood cells are not specific to paroxysmal nocturnal hemoglobinuria. Hematology (2009) 14(1):33–7. doi: 10.1179/102453309X385089
154. Krus U, King BC, Nagaraj V, Gandasi NR, Sjolander J, Buda P, et al. The complement inhibitor CD59 regulates insulin secretion by modulating exocytotic events. Cell Metab (2014) 19(5):883–90. doi: 10.1016/j.cmet.2014.03.001
155. Ingram G, Hakobyan S, Robertson NP, Morgan BP. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin Exp Immunol (2009) 155(2):128–39. doi: 10.1111/j.1365-2249.2008.03830.x
156. Watson NF, Durrant LG, Madjd Z, Ellis IO, Scholefield JH, Spendlove I. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol Immunother (2006) 55(8):973–80. doi: 10.1007/s00262-005-0055-0
157. Xu C, Jung M, Burkhardt M, Stephan C, Schnorr D, Loening S, et al. Increased CD59 protein expression predicts a PSA relapse in patients after radical prostatectomy. Prostate (2005) 62(3):224–32. doi: 10.1002/pros.20134
158. Song G, Cho WC, Gu L, He B, Pan Y, Wang S. Increased CD59 protein expression is associated with the outcome of patients with diffuse large B-cell lymphoma treated with R-CHOP. Med Oncol (2014) 31(7):56. doi: 10.1007/s12032-014-0056-y
159. Madjd Z, Pinder SE, Paish C, Ellis IO, Carmichael J, Durrant LG. Loss of CD59 expression in breast tumours correlates with poor survival. J Pathol (2003) 200(5):633–9. doi: 10.1002/path.1357
160. Maio M, Brasoveanu LI, Coral S, Sigalotti L, Lamaj E, Gasparollo A, et al. Structure, distribution, and functional role of protectin (CD59) in complement-susceptibility and in immunotherapy of human malignancies (Review). Int J Oncol (1998) 13(2):305–18. doi: 10.3892/ijo.13.2.305
161. Abdel-Latif M, Saidan S, Morsy BM. Coenzyme Q10 attenuates rat hepatocarcinogenesis via the reduction of CD59 expression and phospholipase D activity. Cell Biochem Funct (2020) 38(4):490–9. doi: 10.1002/cbf.3487
162. Jian-Hua T, Xiao-Jie Z, Yi-Dan W, Wen-Kai L, Wang-Jiao H, Cheng-Hong W, et al. Effect of glycosylphosphatidylinositol specific phospholipase D gene expression levels on complement mediated killing of leukemic cells in patients with chronic myeloid leukemia. Clin Chim Acta (2005) 359(1-2):115–24. doi: 10.1016/j.cccn.2005.03.045
163. Sangiorgio V, Pitto M, Palestini P, Masserini M. GPI-anchored proteins and lipid rafts. Ital J Biochem (2004) 53(2):98–111.
164. Lehto T, Honkanen E, Teppo AM, Meri S. Urinary excretion of protectin (CD59), complement SC5b-9 and cytokines in membranous glomerulonephritis. Kidney Int (1995) 47(5):1403–11. doi: 10.1038/ki.1995.197
165. Shan C, Zhang S, Cui W, You X, Kong G, Du Y, et al. Hepatitis B virus X protein activates CD59 involving DNA binding and let-7i in protection of hepatoma and hepatic cells from complement attack. Carcinogenesis (2011) 32(8):1190–7. doi: 10.1093/carcin/bgr106
166. Lan T, Wu H. CD59 facilitates tumor progression through activating TGF-β/Smad signaling pathway in hepatocellular carcinoma. Cancer Res (2020) 80(16):2932.
167. Arlaud GJ, Gaboriaud C, Thielens NM, Rossi V, Bersch B, Hernandez JF, et al. Structural biology of C1: dissection of a complex molecular machinery. Immunol Rev (2001) 180:136–45. doi: 10.1034/j.1600-065x.2001.1800112.x
168. Hoekzema R, Hannema AJ, Swaak TJ, Paardekooper J, Hack CE. Low molecular weight C1q in systemic lupus erythematosus. J Immunol (1985) 135(1):265–71.
169. Hong Q, Sze CI, Lin SR, Lee MH, He RY, Schultz L, et al. Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PLoS One (2009) 4(6):e5755. doi: 10.1371/journal.pone.0005755
170. Morris KM, Colten HR, Bing DH. The first component of complement. A quantitative comparison of its biosynthesis in culture by human epithelial and mesenchymal cells. J Exp Med (1978) 148(4):1007–19. doi: 10.1084/jem.148.4.1007
171. Hoekzema R, Brouwer MC, de Graeff-Meeder ER, van Helden HP, Hack CE. Biosynthesis of normal and low-molecular-mass complement component C1q by cultured human monocytes and macrophages. Biochem J (1989) 257(2):477–86. doi: 10.1042/bj2570477
172. Reis ES, Barbuto JA, Isaac L. Complement components, regulators and receptors are produced by human monocyte-derived dendritic cells. Immunobiology (2007) 212(3):151–7. doi: 10.1016/j.imbio.2006.11.010
173. Reid KB, Solomon E. Biosynthesis of the first component of complement by human fibroblasts. Biochem J (1977) 167(3):647–60. doi: 10.1042/bj1670647
174. Ripoche J, Mitchell JA, Erdei A, Madin C, Moffatt B, Mokoena T, et al. Interferon gamma induces synthesis of complement alternative pathway proteins by human endothelial cells in culture. J Exp Med (1988) 168(5):1917–22. doi: 10.1084/jem.168.5.1917
175. Morris KM, Aden DP, Knowles BB, Colten HR. Complement biosynthesis by the human hepatoma-derived cell line HepG2. J Clin Invest (1982) 70(4):906–13. doi: 10.1172/jci110687
176. Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet (1998) 19(1):56–9. doi: 10.1038/ng0598-56
177. Ghebrehiwet B, Lu PD, Zhang W, Keilbaugh SA, Leigh LE, Eggleton P, et al. Evidence that the two C1q binding membrane proteins, gC1q-R and cC1q-R, associate to form a complex. J Immunol (1997) 159(3):1429–36.
178. Pike SE, Yao L, Setsuda J, Jones KD, Cherney B, Appella E, et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood (1999) 94(7):2461–8.
179. Kim KB, Yi JS, Nguyen N, Lee JH, Kwon YC, Ahn BY, et al. Cell-surface receptor for complement component C1q (gC1qR) is a key regulator for lamellipodia formation and cancer metastasis. J Biol Chem (2011) 286(26):23093–101. doi: 10.1074/jbc.M111.233304
180. Hoffken K, Price MR, McLaughlin PJ, Moore VE, Baldwin RW. Circulating immune complexes in rats bearing chemically induced tumors. I. Sequential determination during the growth of tumours at various body sites. Int J Cancer (1978) 21(4):496–504. doi: 10.1002/ijc.2910210416
181. Narumoto J. Studies on circulating soluble immune complexes of the liver disease. 6. Comparative studies of 125I-pRF inhibition assay, 125I-Clq inhibition assay and 125I-Clq binding assay. Gastroenterol Jpn (1981) 16(2):174–85. doi: 10.1007/BF02774392
182. Brown SE, Howard CR, Steward MW, Ajdukiewicz AB, Whittle HC. Hepatitis B surface antigen containing immune complexes occur in seronegative hepatocellular carcinoma patients. Clin Exp Immunol (1984) 55(2):355–9.
183. Chen YM, Hu CP, Wong FH, Chen PH, Tsai YT, Lee SD, et al. Circulating immune complexes in the sera and ascites of hepatocellular carcinoma or chronic hepatitis patients. Zhonghua Min Guo Wei Sheng Wu Ji Mian Yi Xue Za Zhi (1986) 19(1):34–45.
184. Yao ZQ, Ray S, Eisen-Vandervelde A, Waggoner S, Hahn YS. Hepatitis C virus: immunosuppression by complement regulatory pathway. Viral Immunol (2001) 14(4):277–95. doi: 10.1089/08828240152716547
185. Takeuchi T, Adachi Y, Nagayama T. Expression of a secretory protein C1qTNF6, a C1qTNF family member, in hepatocellular carcinoma. Anal Cell Pathol (Amst) (2011) 34(3):113–21. doi: 10.3233/ACP-2011-009
186. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun (2016) 7:10346. doi: 10.1038/ncomms10346
187. Lee JH, Poudel B, Ki HH, Nepali S, Lee YM, Shin JS, et al. Complement C1q stimulates the progression of hepatocellular tumor through the activation of discoidin domain receptor 1. Sci Rep (2018) 8(1):4908. doi: 10.1038/s41598-018-23240-6
188. Zhao X, Yang W, Pei F, Ma W, Wang Y. Downregulation of matrix metalloproteinases contributes to the inhibition of cell migration and invasion in HepG2 cells by sodium valproate. Oncol Lett (2015) 10(1):531–5. doi: 10.3892/ol.2015.3203
189. Ho TC, Wang EY, Yeh KH, Jeng YM, Horng JH, Wu LL, et al. Complement C1q mediates the expansion of periportal hepatic progenitor cells in senescence-associated inflammatory liver. Proc Natl Acad Sci U S A (2020) 117(12):6717–25. doi: 10.1073/pnas.1918028117
190. Ning G, Huang YL, Zhen LM, Xu WX, Li XJ, Wu LN, et al. Prognostic Value of Complement Component 2 and Its Correlation with Immune Infiltrates in Hepatocellular Carcinoma. BioMed Res Int (2020) 2020:3765937. doi: 10.1155/2020/3765937
191. Fu J, Zhang Z, Zhou L, Qi Z, Xing S, Lv J, et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology (2013) 58(1):139–49. doi: 10.1002/hep.26054
192. Clifford RJ, Zhang J, Meerzaman DM, Lyu MS, Hu Y, Cultraro CM, et al. Genetic variations at loci involved in the immune response are risk factors for hepatocellular carcinoma. Hepatology (2010) 52(6):2034–43. doi: 10.1002/hep.23943
193. Namgoong S, Shin JG, Cheong HS, Kim LH, Kim JO, Seo JY, et al. Genetic association of complement component 2 variants with chronic hepatitis B in a Korean population. Liver Int (2018) 38(9):1576–82. doi: 10.1111/liv.13675
194. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity (2013) 39(6):1143–57. doi: 10.1016/j.immuni.2013.10.018
195. Pio R, Corrales L, Lambris JD. The role of complement in tumor growth. Adv Exp Med Biol (2014) 772:229–62. doi: 10.1007/978-1-4614-5915-6_11
196. Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, et al. Autocrine Complement Inhibits IL10-Dependent T-cell-Mediated Antitumor Immunity to Promote Tumor Progression. Cancer Discov (2016) 6(9):1022–35. doi: 10.1158/2159-8290.CD-15-1412
197. Zha H, Wang X, Zhu Y, Chen D, Han X, Yang F, et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol Res (2019) 7(2):193–207. doi: 10.1158/2326-6066.CIR-18-0272
198. Xu Y, Huang Y, Xu W, Zheng X, Yi X, Huang L, et al. Activated Hepatic Stellate Cells (HSCs) Exert Immunosuppressive Effects in Hepatocellular Carcinoma by Producing Complement C3. Onco Targets Ther (2020) 13:1497–505. doi: 10.2147/OTT.S234920
199. Hsieh CC, Chou HS, Yang HR, Lin F, Bhatt S, Qin J, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood (2013) 121(10):1760–8. doi: 10.1182/blood-2012-06-440214
200. Lu LC, Chang CJ, Hsu CH. Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: current state and future perspectives. J Hepatocell Carcinoma (2019) 6:71–84. doi: 10.2147/JHC.S159693
201. Xu Y, Zhao W, Xu J, Li J, Hong Z, Yin Z, et al. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget (2016) 7(8):8866–78. doi: 10.18632/oncotarget.6839
202. Goldman R, Ressom HW, Abdel-Hamid M, Goldman L, Wang A, Varghese RS, et al. Candidate markers for the detection of hepatocellular carcinoma in low-molecular weight fraction of serum. Carcinogenesis (2007) 28(10):2149–53. doi: 10.1093/carcin/bgm177
203. Leung H-CE, Leow C-K, Tsai H-H, George J, Lai PB, Chang AY, et al. Serum complement C3a C-terminal truncated protein fragment - a potential marker for hepatocellular carcinoma. Cancer Res (2005) 65(9 Supplement):1130–1.
204. Lee IN, Chen CH, Sheu JC, Lee HS, Huang GT, Chen DS, et al. Identification of complement C3a as a candidate biomarker in human chronic hepatitis C and HCV-related hepatocellular carcinoma using a proteomics approach. Proteomics (2006) 6(9):2865–73. doi: 10.1002/pmic.200500488
205. Kanmura S, Uto H, Sato Y, Kumagai K, Sasaki F, Moriuchi A, et al. The complement component C3a fragment is a potential biomarker for hepatitis C virus-related hepatocellular carcinoma. J Gastroenterol (2010) 45(4):459–67. doi: 10.1007/s00535-009-0160-5
206. Hugli TE. The structural basis for anaphylatoxin and chemotactic functions of C3a, C4a, and C5a. Crit Rev Immunol (1981) 1(4):321–66.
207. Ward PA. Functions of C5a receptors. J Mol Med (Berl) (2009) 87(4):375–8. doi: 10.1007/s00109-009-0442-7
208. Ajona D, Ortiz-Espinosa S, Pio R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin Cell Dev Biol (2019) 85:153–63. doi: 10.1016/j.semcdb.2017.11.023
209. Hu WH, Hu Z, Shen X, Dong LY, Zhou WZ, Yu XX. C5a receptor enhances hepatocellular carcinoma cell invasiveness via activating ERK1/2-mediated epithelial-mesenchymal transition. Exp Mol Pathol (2016) 100(1):101–8. doi: 10.1016/j.yexmp.2015.10.001
210. Dai Q, Zhu J, Shen M, Jiang H, Pan B, Zhou J, et al. C5aR correlated with the dissemination capacity of circulating tumor cells in hepatocellular carcinoma by targeting INHBA-p-smad2/3-EMT/MMPs axis. Am Soc Clin Oncol (2020) 38:e16649. doi: 10.1200/JCO.2020.38.15
211. Piao C, Zhang WM, Li TT, Zhang CC, Qiu S, Liu Y, et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp Cell Res (2018) 366(2):127–38. doi: 10.1016/j.yexcr.2018.03.009
212. Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Kohl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity (2005) 22(4):415–26. doi: 10.1016/j.immuni.2005.02.006
213. Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell (2018) 34(4):561–78.e6. doi: 10.1016/j.ccell.2018.09.003
214. Khameneh HJ, Ho AW, Laudisi F, Derks H, Kandasamy M, Sivasankar B, et al. C5a Regulates IL-1beta Production and Leukocyte Recruitment in a Murine Model of Monosodium Urate Crystal-Induced Peritonitis. Front Pharmacol (2017) 8:10. doi: 10.3389/fphar.2017.00010
215. Allendorf DJ, Yan J, Ross GD, Hansen RD, Baran JT, Subbarao K, et al. C5a-mediated leukotriene B4-amplified neutrophil chemotaxis is essential in tumor immunotherapy facilitated by anti-tumor monoclonal antibody and beta-glucan. J Immunol (2005) 174(11):7050–6. doi: 10.4049/jimmunol.174.11.7050
216. Kourtzelis I, Markiewski MM, Doumas M, Rafail S, Kambas K, Mitroulis I, et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood (2010) 116(4):631–9. doi: 10.1182/blood-2010-01-264051
217. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol (2008) 9(11):1225–35. doi: 10.1038/ni.1655
218. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol (2004) 172(2):989–99. doi: 10.4049/jimmunol.172.2.989
219. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol (2012) 189(9):4674–83. doi: 10.4049/jimmunol.1201654
220. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res (2014) 74(13):3454–65. doi: 10.1158/0008-5472.CAN-14-0157
221. Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a Enhances Hepatic Metastases of Colon Cancer via Monocyte Chemoattractant Protein-1-mediated Inflammatory Cell Infiltration. J Biol Chem (2015) 290(17):10667–76. doi: 10.1074/jbc.M114.612622
222. Ajona D, Ortiz-Espinosa S, Pio R, Lecanda F. Complement in Metastasis: A Comp in the Camp. Front Immunol (2019) 10:669. doi: 10.3389/fimmu.2019.00669
223. Oliveira LC, Kretzschmar GC, Dos Santos ACM, Camargo CM, Nisihara RM, Farias TDJ, et al. Complement Receptor 1 (CR1, CD35) Polymorphisms and Soluble CR1: A Proposed Anti-inflammatory Role to Quench the Fire of “Fogo Selvagem” Pemphigus Foliaceus. Front Immunol (2019) 10:2585. doi: 10.3389/fimmu.2019.02585
224. Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity (1997) 7(3):345–55. doi: 10.1016/s1074-7613(00)80356-8
225. Ghiran I, Barbashov SF, Klickstein LB, Tas SW, Jensenius JC, Nicholson-Weller A. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J Exp Med (2000) 192(12):1797–808. doi: 10.1084/jem.192.12.1797
226. Jacquet M, Lacroix M, Ancelet S, Gout E, Gaboriaud C, Thielens NM, et al. Deciphering complement receptor type 1 interactions with recognition proteins of the lectin complement pathway. J Immunol (2013) 190(7):3721–31. doi: 10.4049/jimmunol.1202451
227. Khera R, Das N. Complement Receptor 1: disease associations and therapeutic implications. Mol Immunol (2009) 46(5):761–72. doi: 10.1016/j.molimm.2008.09.026
228. Salam KA, Wang RY, Grandinetti T, De Giorgi V, Alter HJ, Allison RD. Binding of Free and Immune Complex-Associated Hepatitis C Virus to Erythrocytes Is Mediated by the Complement System. Hepatology (2018) 68(6):2118–29. doi: 10.1002/hep.30087
229. Kanto T, Hayashi N, Takehara T, Katayama K, Kato M, Akiyama M, et al. Low expression of erythrocyte complement receptor type 1 in chronic hepatitis C patients. J Med Virol (1996) 50(2):126–34. doi: 10.1002/(SICI)1096-9071(199610)50:2<126::AID-JMV5>3.0.CO;2-C
230. Miyaike J, Iwasaki Y, Takahashi A, Shimomura H, Taniguchi H, Koide N, et al. Regulation of circulating immune complexes by complement receptor type 1 on erythrocytes in chronic viral liver diseases. Gut (2002) 51(4):591–6. doi: 10.1136/gut.51.4.591
231. Luo J, Chen S, Wang J, Ou S, Zhang W, Liu Y, et al. Genetic polymorphisms in complement receptor 1 gene and its association with HBV-related liver disease: A case-control study. Gene (2019) 688:107–18. doi: 10.1016/j.gene.2018.11.082
232. Pascual M, Duchosal MA, Steiger G, Giostra E, Pechere A, Paccaud JP, et al. Circulating soluble CR1 (CD35). Serum levels in diseases and evidence for its release by human leukocytes. J Immunol (1993) 151(3):1702–11.
233. Di Bona D, Montalto G, Clemenza L, Bascone F, Accardo P, Bellavia D, et al. Soluble complement receptor type 1 (sCR1) in chronic liver diseases: serum levels at different stages of liver diseases. Clin Exp Immunol (1998) 114(1):102–5. doi: 10.1046/j.1365-2249.1998.00707.x
234. Weisman HF, Bartow T, Leppo MK, Boyle MP, Marsh HC Jr., Carson GR, et al. Recombinant soluble CR1 suppressed complement activation, inflammation, and necrosis associated with reperfusion of ischemic myocardium. Trans Assoc Am Phys (1990) 103:64–72.
235. Worthley DL, Bardy PG, Gordon DL, Mullighan CG. Mannose-binding lectin and maladies of the bowel and liver. World J Gastroenterol (2006) 12(40):6420–8. doi: 10.3748/wjg.v12.i40.6420
236. Tomaiuolo R, Ruocco A, Salapete C, Carru C, Baggio G, Franceschi C, et al. Activity of mannose-binding lectin in centenarians. Aging Cell (2012) 11(3):394–400. doi: 10.1111/j.1474-9726.2012.00793.x
237. Pradhan V, Surve P, Ghosh K. Mannose binding lectin (MBL) in autoimmunity and its role in systemic lupus erythematosus (SLE). J Assoc Phys India (2010) 58:688–90.
238. Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol (2003) 33(10):2853–63. doi: 10.1002/eji.200323888
239. Swierzko AS, Kilpatrick DC, Cedzynski M. Mannan-binding lectin in malignancy. Mol Immunol (2013) 55(1):16–21. doi: 10.1016/j.molimm.2012.09.005
240. Slany A, Haudek VJ, Zwickl H, Gundacker NC, Grusch M, Weiss TS, et al. Cell characterization by proteome profiling applied to primary hepatocytes and hepatocyte cell lines Hep-G2 and Hep-3B. J Proteome Res (2010) 9(1):6–21. doi: 10.1021/pr900057t
241. Rong Y, Jin D, Hou C, Hu J, Wu W, Ni X, et al. Proteomics analysis of serum protein profiling in pancreatic cancer patients by DIGE: up-regulation of mannose-binding lectin 2 and myosin light chain kinase 2. BMC Gastroenterol (2010) 10:68. doi: 10.1186/1471-230X-10-68
242. Awan FM, Naz A, Obaid A, Ali A, Ahmad J, Anjum S, et al. Identification of Circulating Biomarker Candidates for Hepatocellular Carcinoma (HCC): An Integrated Prioritization Approach. PLoS One (2015) 10(9):e0138913. doi: 10.1371/journal.pone.0138913
243. Li J, Li H, Yu Y, Liu Y, Liu Y, Ma Q, et al. Mannan-binding lectin suppresses growth of hepatocellular carcinoma by regulating hepatic stellate cell activation via the ERK/COX-2/PGE2 pathway. Oncoimmunology (2019) 8(2):e1527650. doi: 10.1080/2162402X.2018.1527650
244. Jalal PJ, King BJ, Saeed A, Adedeji Y, Mason CP, Ball JK, et al. Elevated serum activity of MBL and ficolin-2 as biomarkers for progression to hepatocellular carcinoma in chronic HCV infection. Virology (2019) 530:99–106. doi: 10.1016/j.virol.2019.02.002
245. Yoshino T, Kimoto F, Shiotani K, Hashimoto H, Ito Y. [Congenital broncho-esophageal fistula with a narrow trachea and double esophageal diverticula]. Kyobu Geka (1987) 40(2):149–51.
246. Quan Y, Yang J, Qin T, Hu Y. Associations between twelve common gene polymorphisms and susceptibility to hepatocellular carcinoma: evidence from a meta-analysis. World J Surg Oncol (2019) 17(1):216. doi: 10.1186/s12957-019-1748-8
247. Gu X, Ji Q, Wang H, Jiang M, Yang J, Fang M, et al. Genetic variants of mannose-binding lectin 2 gene influence progression and prognosis of patients with hepatitis B virus infection in China. Clin Res Hepatol Gastroenterol (2016) 40(5):614–21. doi: 10.1016/j.clinre.2015.12.015
248. Su C, Lin Y, Cai L, Mao Q, Niu J. Association between mannose-binding lectin variants, haplotypes and risk of hepatocellular carcinoma: A case-control study. Sci Rep (2016) 6:32147. doi: 10.1038/srep32147
249. Wang PS, Kuai J, Li H, Wang CG, Shi BJ, Zhong L. Mannose-binding lectin 2 rs11003123 polymorphism is associated with the development of hepatocellular carcinoma in patients with hepatitis B-related cirrhosis in the Chinese population. Hepatobiliary Pancreat Dis Int (2016) 15(3):282–8. doi: 10.1016/s1499-3872(16)60050-4
250. Xu HD, Zhao MF, Wan TH, Song GZ, He JL, Chen Z. Association between Mannose-binding lectin gene polymorphisms and hepatitis B virus infection: a meta-analysis. PLoS One (2013) 8(10):e75371. doi: 10.1371/journal.pone.0075371
251. Yuen MF, Lau CS, Lau YL, Wong WM, Cheng CC, Lai CL. Mannose binding lectin gene mutations are associated with progression of liver disease in chronic hepatitis B infection. Hepatology (1999) 29(4):1248–51. doi: 10.1002/hep.510290417
252. Segat L, Fabris A, Padovan L, Milanese M, Pirulli D, Lupo F, et al. MBL2 and MASP2 gene polymorphisms in patients with hepatocellular carcinoma. J Viral Hepat (2008) 15(5):387–91. doi: 10.1111/j.1365-2893.2007.00965.x
253. Garcia-Laorden MI, Hernandez-Brito E, Munoz-Almagro C, Pavlovic-Nesic S, Rua-Figueroa I, Briones ML, et al. Should MASP-2 Deficiency Be Considered a Primary Immunodeficiency? Relevance of the Lectin Pathway. J Clin Immunol (2020) 40(1):203–10. doi: 10.1007/s10875-019-00714-4
254. Ytting H, Christensen IJ, Steffensen R, Alsner J, Thiel S, Jensenius JC, et al. Mannan-binding lectin (MBL) and MBL-associated serine protease 2 (MASP-2) genotypes in colorectal cancer. Scand J Immunol (2011) 73(2):122–7. doi: 10.1111/j.1365-3083.2010.02480.x
255. Ytting H, Christensen IJ, Thiel S, Jensenius JC, Nielsen HJ. Pre- and postoperative levels in serum of mannan-binding lectin associated serine protease-2 -a prognostic marker in colorectal cancer. Hum Immunol (2008) 69(7):414–20. doi: 10.1016/j.humimm.2008.05.005
256. Ytting H, Christensen IJ, Thiel S, Jensenius JC, Nielsen HJ. Serum mannan-binding lectin-associated serine protease 2 levels in colorectal cancer: relation to recurrence and mortality. Clin Cancer Res (2005) 11(4):1441–6. doi: 10.1158/1078-0432.CCR-04-1272
257. Swierzko AS, Szala A, Sawicki S, Szemraj J, Sniadecki M, Sokolowska A, et al. Mannose-Binding Lectin (MBL) and MBL-associated serine protease-2 (MASP-2) in women with malignant and benign ovarian tumours. Cancer Immunol Immunother (2014) 63(11):1129–40. doi: 10.1007/s00262-014-1579-y
258. Maestri CA, Nisihara R, Mendes HW, Jensenius J, Thiel S, Messias-Reason I, et al. MASP-1 and MASP-2 Serum Levels Are Associated With Worse Prognostic in Cervical Cancer Progression. Front Immunol (2018) 9:2742. doi: 10.3389/fimmu.2018.02742
259. Verma A, Matta A, Shukla NK, Deo SV, Gupta SD, Ralhan R. Clinical significance of mannose-binding lectin-associated serine protease-2 expression in esophageal squamous cell carcinoma. Int J Cancer (2006) 118(12):2930–5. doi: 10.1002/ijc.21721
260. Fisch U, Zehnder A, Hirt A, Niggli F, Simon A, Ozsahin H, et al. Mannan-binding lectin (MBL) and MBL-associated serine protease-2 in children with cancer. Swiss Med Wkly (2011) 141:w13191. doi: 10.4414/smw.2011.13191
261. Nazari S, Ebrahimi M, Abdollah Gorji F, Abadi A, Fahimzad A. Association between serum levels of MASP-2 and neutropenic febrile attacks in children with leukemia. Arch Iran Med (2012) 15(10):625–8.
262. Zehnder A, Fisch U, Hirt A, Niggli FK, Simon A, Ozsahin H, et al. Prognosis in pediatric hematologic malignancies is associated with serum concentration of mannose-binding lectin-associated serine protease-2 (MASP-2). Pediatr Blood Cancer (2009) 53(1):53–7. doi: 10.1002/pbc.22028
263. Schlapbach LJ, Aebi C, Otth M, Leibundgut K, Hirt A, Ammann RA. Deficiency of mannose-binding lectin-associated serine protease-2 associated with increased risk of fever and neutropenia in pediatric cancer patients. Pediatr Infect Dis J (2007) 26(11):989–94. doi: 10.1097/INF.0b013e31811ffe6a
264. Ding W, Qiu Q, Liu G, Liu J, Mao R, Lin B. Metadata checklist: identification of CHI3L1 and MASP2 as a biomarker pair for liver cancer through integrative secretome and transcriptome analysis. OMICS (2014) 18(10):658–61. doi: 10.1089/omi.2014.0090
265. Tulio S, Faucz FR, Werneck RI, Olandoski M, Alexandre RB, Boldt AB, et al. MASP2 gene polymorphism is associated with susceptibility to hepatitis C virus infection. Hum Immunol (2011) 72(10):912–5. doi: 10.1016/j.humimm.2011.06.016
266. Unterberger C, Hanson S, Klingenhoff A, Oesterle D, Frankenberger M, Endo Y, et al. Stat3 is involved in control of MASP2 gene expression. Biochem Biophys Res Commun (2007) 364(4):1022–5. doi: 10.1016/j.bbrc.2007.10.114
267. Blom AM, Villoutreix BO, Dahlback B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol Immunol (2004) 40(18):1333–46. doi: 10.1016/j.molimm.2003.12.002
268. Blom AM, Kask L, Dahlback B. CCP1-4 of the C4b-binding protein alpha-chain are required for factor I mediated cleavage of complement factor C3b. Mol Immunol (2003) 39(10):547–56. doi: 10.1016/s0161-5890(02)00213-4
269. Dahlback B, Hildebrand B. Degradation of human complement component C4b in the presence of the C4b-binding protein-protein S complex. Biochem J (1983) 209(3):857–63. doi: 10.1042/bj2090857
270. Gigli I, Fujita T, Nussenzweig V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc Natl Acad Sci U S A (1979) 76(12):6596–600. doi: 10.1073/pnas.76.12.6596
271. Kusada-Funakoshi M, Sasaki J, Takada Y, Soji T, Arakawa K. Evidence that C4b-binding protein (proline-rich protein) is synthesized by hepatocytes. Biochem Med Metab Biol (1991) 45(3):350–4. doi: 10.1016/0885-4505(91)90040-r
272. Lappin DF, Whaley K. Interferon-induced transcriptional and post-transcriptional modulation of factor H and C4 binding-protein synthesis in human monocytes. Biochem J (1990) 271(3):767–72. doi: 10.1042/bj2710767
273. Moffat GJ, Tack BF. Regulation of C4b-binding protein gene expression by the acute-phase mediators tumor necrosis factor-alpha, interleukin-6, and interleukin-1. Biochemistry (1992) 31(49):12376–84. doi: 10.1021/bi00164a012
274. Marcovina SM, Zoppo A, Vigano-D’Angelo S, Di Cola G, D’Angelo A. Determination of serum levels of complement component C4b-binding protein: influence of age and inflammation. Int J Clin Lab Res (1991) 21(2):171–5. doi: 10.1007/BF02591638
275. Saeki T, Hirose S, Nukatsuka M, Kusunoki Y, Nagasawa S. Evidence that C4b-binding protein is an acute phase protein. Biochem Biophys Res Commun (1989) 164(3):1446–51. doi: 10.1016/0006-291x(89)91832-9
276. Barnum SR, Dahlback B. C4b-binding protein, a regulatory component of the classical pathway of complement, is an acute-phase protein and is elevated in systemic lupus erythematosus. Complement Inflamm (1990) 7(2):71–7. doi: 10.1159/000463131
277. Garcia de Frutos P, Alim RI, Hardig Y, Zoller B, Dahlback B. Differential regulation of alpha and beta chains of C4b-binding protein during acute-phase response resulting in stable plasma levels of free anticoagulant protein S. Blood (1994) 84(3):815–22.
278. Kopylov AT, Stepanov AA, Malsagova KA, Soni D, Kushlinsky NE, Enikeev DV, et al. Revelation of Proteomic Indicators for Colorectal Cancer in Initial Stages of Development. Molecules (2020) 25(3):619–39. doi: 10.3390/molecules25030619
279. Battistelli S, Stefanoni M, Lorenzi B, Dell’Avanzato R, Varrone F, Pascucci A, et al. Coagulation factor levels in non-metastatic colorectal cancer patients. Int J Biol Markers (2008) 23(1):36–41. doi: 10.5301/JBM.2008.4255
280. Mikami M, Tanabe K, Matsuo K, Miyazaki Y, Miyazawa M, Hayashi M, et al. Fully-sialylated alpha-chain of complement 4-binding protein: Diagnostic utility for ovarian clear cell carcinoma. Gynecol Oncol (2015) 139(3):520–8. doi: 10.1016/j.ygyno.2015.10.012
281. Liu YS, Luo XY, Li QR, Li H, Li C, Ni H, et al. Shotgun and targeted proteomics reveal that pre-surgery serum levels of LRG1, SAA, and C4BP may refine prognosis of resected squamous cell lung cancer. J Mol Cell Biol (2012) 4(5):344–7. doi: 10.1093/jmcb/mjs050
282. Okroj M, Hsu YF, Ajona D, Pio R, Blom AM. Non-small cell lung cancer cells produce a functional set of complement factor I and its soluble cofactors. Mol Immunol (2008) 45(1):169–79. doi: 10.1016/j.molimm.2007.04.025
283. Luo X, Liu Y, Wang R, Hu H, Zeng R, Chen H. A high-quality secretome of A549 cells aided the discovery of C4b-binding protein as a novel serum biomarker for non-small cell lung cancer. J Proteomics (2011) 74(4):528–38. doi: 10.1016/j.jprot.2011.01.011
284. Phillips DJ, Novinger MS, Evatt BL, Hooper WC. TNF-alpha suppresses IL-1 alpha and IL-6 upregulation of C4b-binding protein in HepG-2 hepatoma cells. Thromb Res (1996) 81(3):307–14. doi: 10.1016/0049-3848(96)00002-3
285. Tomes L, Emberley E, Niu Y, Troup S, Pastorek J, Strange K, et al. Necrosis and hypoxia in invasive breast carcinoma. Breast Cancer Res Treat (2003) 81(1):61–9. doi: 10.1023/A:1025476722493
286. Williams KT, Young SP, Negus A, Young LS, Adams DH, Afford SC. C4b binding protein binds to CD154 preventing CD40 mediated cholangiocyte apoptosis: a novel link between complement and epithelial cell survival. PLoS One (2007) 2(1):e159. doi: 10.1371/journal.pone.0000159
287. Feng G, Li J, Zheng M, Yang Z, Liu Y, Zhang S, et al. Hepatitis B virus X protein up-regulates C4b-binding protein alpha through activating transcription factor Sp1 in protection of hepatoma cells from complement attack. Oncotarget (2016) 7(19):28013–26. doi: 10.18632/oncotarget.8472
288. Ehsani Ardakani MJ, Safaei A, Arefi Oskouie A, Haghparast H, Haghazali M, Mohaghegh Shalmani H, et al. Evaluation of liver cirrhosis and hepatocellular carcinoma using Protein-Protein Interaction Networks. Gastroenterol Hepatol Bed Bench (2016) 9(Suppl1):S14–22.
289. Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol (1998) 16:545–68. doi: 10.1146/annurev.immunol.16.1.545
290. Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Med (Baltimore) (1984) 63(5):243–73.
291. Potter BJ, Trueman AM, Jones EA. Serum complement in chronic liver disease. Gut (1973) 14(6):451–6. doi: 10.1136/gut.14.6.451
292. Liu Z, Tang Q, Wen J, Tang Y, Huang D, Huang Y, et al. Elevated serum complement factors 3 and 4 are strong inflammatory markers of the metabolic syndrome development: a longitudinal cohort study. Sci Rep (2016) 6:18713. doi: 10.1038/srep18713
293. Kasperska-Zajac A, Grzanka A, Machura E, Misiolek M, Mazur B, Jochem J. Increased serum complement C3 and C4 concentrations and their relation to severity of chronic spontaneous urticaria and CRP concentration. J Inflammation (Lond) (2013) 10(1):22. doi: 10.1186/1476-9255-10-22
294. Yu CY, Whitacre CC. Sex, MHC and complement C4 in autoimmune diseases. Trends Immunol (2004) 25(12):694–9. doi: 10.1016/j.it.2004.10.006
295. Ali OS, Abo-Shadi MA, Hammad LN. The biological significance of serum complements C3 and C4 in HCV-related chronic liver diseases and hepatocellular carcinoma. Egypt J Immunol (2005) 12(2):91–9.
296. Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Transcriptional repression of C4 complement by hepatitis C virus proteins. J Virol (2011) 85(9):4157–66. doi: 10.1128/JVI.02449-10
297. Zhu C, Song H, Xu F, Yi W, Liu F, Liu X. Hepatitis B virus inhibits the expression of complement C3 and C4, in vitro and in vivo. Oncol Lett (2018) 15(5):7459–63. doi: 10.3892/ol.2018.8223
298. Kim H, Kim K, Yu SJ, Jang ES, Yu J, Cho G, et al. Development of biomarkers for screening hepatocellular carcinoma using global data mining and multiple reaction monitoring. PLoS One (2013) 8(5):e63468. doi: 10.1371/journal.pone.0063468
299. Barefoot ME, Varghese RS, Zhou Y, Poto CD, Ferrarini A, Ressom HW. Multi-omic Pathway and Network Analysis to Identify Biomarkers for Hepatocellular Carcinoma. Annu Int Conf IEEE Eng Med Biol Soc (2019) 2019:1350–4. doi: 10.1109/EMBC.2019.8856576
300. Chang WY, Chuang WL. Complements as new diagnostic tools of hepatocellular carcinoma in cirrhotic patients. Cancer (1988) 62(2):227–32. doi: 10.1002/1097-0142(19880715)62:2<227::aid-cncr2820620202>3.0.co;2-d
301. Takezaki E, Murakami S, Nishibayashi H, Kagawa K, Ohmori H. [A clinical study of complements as a marker of a hepatocellular carcinoma]. Gan No Rinsho (1990) 36(12):2119–22.
302. Ferrin G, Rodriguez-Peralvarez M, Aguilar-Melero P, Ranchal I, Llamoza C, Linares CI, et al. Plasma protein biomarkers of hepatocellular carcinoma in HCV-infected alcoholic patients with cirrhosis. PLoS One (2015) 10(3):e0118527. doi: 10.1371/journal.pone.0118527
303. Dalal K, Khorate P, Dalal B, Chavan R, Bhatia S, Kale A, et al. Differentially expressed serum host proteins in hepatitis B and C viral infections. Virusdisease (2018) 29(4):468–77. doi: 10.1007/s13337-018-0484-y
304. Zipfel PF, Skerka C, Hellwage J, Jokiranta ST, Meri S, Brade V, et al. Factor H family proteins: on complement, microbes and human diseases. Biochem Soc Trans (2002) 30(Pt 6):971–8. doi: 10.1042/bst0300971
305. Zipfel PF, Skerka C. FHL-1/reconectin: a human complement and immune regulator with cell-adhesive function. Immunol Today (1999) 20(3):135–40. doi: 10.1016/s0167-5699(98)01432-7
306. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood (2009) 114(12):2439–47. doi: 10.1182/blood-2009-02-205641
307. Hannan JP, Laskowski J, Thurman JM, Hageman GS, Holers VM. Mapping the Complement Factor H-Related Protein 1 (CFHR1):C3b/C3d Interactions. PLoS One (2016) 11(11):e0166200. doi: 10.1371/journal.pone.0166200
308. Heicappell R, Wettig IC, Schostak M, Muller M, Steiner U, Sauter T, et al. Quantitative detection of human complement factor H-related protein in transitional cell carcinoma of the urinary bladder. Eur Urol (1999) 35(1):81–7. doi: 10.1159/000019822
309. Malkowicz SB. The application of human complement factor H-related protein (BTA TRAK) in monitoring patients with bladder cancer. Urol Clin North Am (2000) 27(1):63–73, ix. doi: 10.1016/s0094-0143(05)70235-4
310. Feng H, Fang F, Yuan L, Xiao M, Yang XY, Huang Y. Downregulated expression of CFHL1 is associated with unfavorable prognosis in postoperative patients with hepatocellular carcinoma. Exp Ther Med (2019) 17(5):4073–9. doi: 10.3892/etm.2019.7455
311. Pontoglio M, Pausa M, Doyen A, Viollet B, Yaniv M, Tedesco F. Hepatocyte nuclear factor 1alpha controls the expression of terminal complement genes. J Exp Med (2001) 194(11):1683–9. doi: 10.1084/jem.194.11.1683
312. Yao J, Wu L, Meng X, Yang H, Ni S, Wang Q, et al. Profiling, clinicopathological correlation and functional validation of specific long non-coding RNAs for hepatocellular carcinoma. Mol Cancer (2017) 16(1):164. doi: 10.1186/s12943-017-0733-5
313. Li Z, Xu J, Cui H, Song J, Chen J, Wei J. Bioinformatics analysis of key biomarkers and potential molecular mechanisms in hepatocellular carcinoma induced by hepatitis B virus. Medicine (Baltimore) (2020) 99(20):e20302. doi: 10.1097/MD.0000000000020302
314. Yin L, Chang C, Xu C. G2/M checkpoint plays a vital role at the early stage of HCC by analysis of key pathways and genes. Oncotarget (2017) 8(44):76305–17. doi: 10.18632/oncotarget.19351
315. Mu D, Qin F, Li B, Zhou Q. Identification of the Sixth Complement Component as Potential Key Genes in Hepatocellular Carcinoma via Bioinformatics Analysis. Biomed Res Int (2020) 2020:7042124. doi: 10.1155/2020/7042124
316. Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest (2006) 116(12):3252–7. doi: 10.1172/JCI29251
317. Xu YQ, Gao YD, Yang J, Guo W. A defect of CD4+CD25+ regulatory T cells in inducing interleukin-10 production from CD4+ T cells under CD46 costimulation in asthma patients. J Asthma (2010) 47(4):367–73. doi: 10.3109/02770903.2010.481340
318. Cardone J, Le Friec G, Vantourout P, Roberts A, Fuchs A, Jackson I, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol (2010) 11(9):862–71. doi: 10.1038/ni.1917
319. Sanchez A, Feito MJ, Rojo JM. CD46-mediated costimulation induces a Th1-biased response and enhances early TCR/CD3 signaling in human CD4+ T lymphocytes. Eur J Immunol (2004) 34(9):2439–48. doi: 10.1002/eji.200324259
320. Sherbenou DW, Aftab BT, Su Y, Behrens CR, Wiita A, Logan AC, et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J Clin Invest (2016) 126(12):4640–53. doi: 10.1172/JCI85856
321. Maciejczyk A, Szelachowska J, Szynglarewicz B, Szulc R, Szulc A, Wysocka T, et al. CD46 Expression is an unfavorable prognostic factor in breast cancer cases. Appl Immunohistochem Mol Morphol (2011) 19(6):540–6. doi: 10.1097/PAI.0b013e31821a0be9
322. Liu Y, Aickelin U, Feyereisl J, Durrant LG. Wavelet feature extraction and genetic algorithm for biomarker detection in colorectal cancer data. Knowledge-Based Syst (2013) 37:502–14. doi: 10.1016/j.knosys.2012.09.011
323. Zhang S, Shan C, Cui W, You X, Du Y, Kong G, et al. Hepatitis B virus X protein protects hepatoma and hepatic cells from complement-dependent cytotoxicity by up-regulation of CD46. FEBS Lett (2013) 587(6):645–51. doi: 10.1016/j.febslet.2013.01.019
324. Kinugasa N, Higashi T, Nouso K, Nakatsukasa H, Kobayashi Y, Ishizaki M, et al. Expression of membrane cofactor protein (MCP, CD46) in human liver diseases. Br J Cancer (1999) 80(11):1820–5. doi: 10.1038/sj.bjc.6690604
325. Blechacz B, Splinter PL, Greiner S, Myers R, Peng KW, Federspiel MJ, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology (2006) 44(6):1465–77. doi: 10.1002/hep.21437
326. Chen W, Wu Y, Liu W, Wang G, Wang X, Yang Y, et al. Enhanced antitumor efficacy of a novel fiber chimeric oncolytic adenovirus expressing p53 on hepatocellular carcinoma. Cancer Lett (2011) 307(1):93–103. doi: 10.1016/j.canlet.2011.03.021
327. Li J, Liu H, Li L, Wu H, Wang C, Yan Z, et al. The combination of an oxygen-dependent degradation domain-regulated adenovirus expressing the chemokine RANTES/CCL5 and NK-92 cells exerts enhanced antitumor activity in hepatocellular carcinoma. Oncol Rep (2013) 29(3):895–902. doi: 10.3892/or.2012.2217
328. Lu Z, Zhang C, Cui J, Song Q, Wang L, Kang J, et al. Bioinformatic analysis of the membrane cofactor protein CD46 and microRNA expression in hepatocellular carcinoma. Oncol Rep (2014) 31(2):557–64. doi: 10.3892/or.2013.2877
329. Shi W, Zhang Z, Yang B, Guo H, Jing L, Liu T, et al. Overexpression of microRNA let-7 correlates with disease progression and poor prognosis in hepatocellular carcinoma. Med (Baltimore) (2017) 96(32):e7764. doi: 10.1097/MD.0000000000007764
330. Lu Z, Li X, Xu Y, Chen M, Chen W, Chen T, et al. microRNA-17 functions as an oncogene by downregulating Smad3 expression in hepatocellular carcinoma. Cell Death Dis (2019) 59:1423–55. doi: 10.1038/s41419-019-1960-z
331. Liu F, Luo L, Liu Z, Wu S, Zhang W, Li Q, et al. A genetic variant in the promoter of CD46 is associated with the risk and prognosis of hepatocellular carcinoma. Mol Carcinog (2020) 59:1243–55. doi: 10.1002/mc.23252
332. Geis N, Zell S, Rutz R, Li W, Giese T, Mamidi S, et al. Inhibition of membrane complement inhibitor expression (CD46, CD55, CD59) by siRNA sensitizes tumor cells to complement attack in vitro. Curr Cancer Drug Targets (2010) 10(8):922–31. doi: 10.2174/156800910793357952
333. Ajona D, Zandueta C, Corrales L, Moreno H, Pajares MJ, Ortiz-Espinosa S, et al. Blockade of the Complement C5a/C5aR1 Axis Impairs Lung Cancer Bone Metastasis by CXCL16-mediated Effects. Am J Respir Crit Care Med (2018) 197(9):1164–76. doi: 10.1164/rccm.201703-0660OC
334. Nitta H, Shimose T, Emi Y, Imamura T, Ohnishi K, Kusumoto T, et al. Expression of the anaphylatoxin C5a receptor in gastric cancer: implications for vascular invasion and patient outcomes. Med Oncol (2016) 33(11):118. doi: 10.1007/s12032-016-0834-9
335. Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, et al. Complement Activation via a C3a Receptor Pathway Alters CD4(+) T Lymphocytes and Mediates Lung Cancer Progression. Cancer Res (2018) 78(1):143–56. doi: 10.1158/0008-5472.CAN-17-0240
336. Hosch SB, Scheunemann P, Luth M, Inndorf S, Stoecklein NH, Erbersdobler A, et al. Expression of 17-1A antigen and complement resistance factors CD55 and CD59 on liver metastasis in colorectal cancer. J Gastrointest Surg (2001) 5(6):673–9. doi: 10.1016/s1091-255x(01)80111-6
337. Meng ZW, Liu MC, Hong HJ, Du Q, Chen YL. Expression and prognostic value of soluble CD97 and its ligand CD55 in intrahepatic cholangiocarcinoma. Tumour Biol (2017) 39(3):1010428317694319. doi: 10.1177/1010428317694319
338. Mao X, Zhou L, Tey SK, Ma APY, Yeung CLS, Ng TH, et al. Tumour extracellular vesicle-derived Complement Factor H promotes tumorigenesis and metastasis by inhibiting complement-dependent cytotoxicity of tumour cells. J Extracellular Vesicles (2020) 10(1):e12031. doi: 10.1002/jev2.12031
339. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol (2006) 177(7):4794–802. doi: 10.4049/jimmunol.177.7.4794
340. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med (1983) 158(2):603–15. doi: 10.1084/jem.158.2.603
341. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun (2016) 7:11037. doi: 10.1038/ncomms11037
342. Fessas P, Lee H, Ikemizu S, Janowitz T. A molecular and preclinical comparison of the PD-1-targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin Oncol (2017) 44(2):136–40. doi: 10.1053/j.seminoncol.2017.06.002
343. Weber JS, Sznol M, Sullivan RJ, Blackmon S, Boland G, Kluger HM, et al. A Serum Protein Signature Associated with Outcome after Anti-PD-1 Therapy in Metastatic Melanoma. Cancer Immunol Res (2018) 6(1):79–86. doi: 10.1158/2326-6066.CIR-17-0412
344. Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol (2013) 25(1):54–64. doi: 10.1016/j.smim.2013.04.001
345. Maehara N, Arai S, Mori M, Iwamura Y, Kurokawa J, Kai T, et al. Circulating AIM prevents hepatocellular carcinoma through complement activation. Cell Rep (2014) 9(1):61–74. doi: 10.1016/j.celrep.2014.08.058
346. Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol (2006) 177(1):362–71. doi: 10.4049/jimmunol.177.1.362
347. Lim SH, Beers SA, French RR, Johnson PW, Glennie MJ, Cragg MS. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica (2010) 95(1):135–43. doi: 10.3324/haematol.2008.001628
348. Pinato DJ, Guerra N, Fessas P, Murphy R, Mineo T, Mauri FA, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene (2020) 39(18):3620–37. doi: 10.1038/s41388-020-1249-9
349. Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med (2013) 5(185):185ra63. doi: 10.1126/scitranslmed.3005361
350. Evgin L, Acuna SA, Tanese de Souza C, Marguerie M, Lemay CG, Ilkow CS, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther (2015) 23(6):1066–76. doi: 10.1038/mt.2015.49
351. Cheng H, Sun G, Chen H, Li Y, Han Z, Li Y, et al. Trends in the treatment of advanced hepatocellular carcinoma: immune checkpoint blockade immunotherapy and related combination therapies. Am J Cancer Res (2019) 9(8):1536–45.
352. Bennett S, Soreide K, Gholami S, Pessaux P, Teh C, Segelov E, et al. Strategies for the delay of surgery in the management of resectable hepatobiliary malignancies during the COVID-19 pandemic. Curr Oncol (2020) 27(5):e501–e11. doi: 10.3747/co.27.6785
353. Thomas N, Bennett R, Jones CN. A comparison of immunocytochemical staining enhancement methods using a rapid microtitre immunocytochemistry assay (MIA). J Immunol Methods (1987) 104(1-2):201–7. doi: 10.1016/0022-1759(87)90505-9
354. Toyoda H, Huang DQ, Le MH, Nguyen MH. Liver Care and Surveillance: The Global Impact of the COVID-19 Pandemic. Hepatol Commun (2020) 4:1751–7. doi: 10.1002/hep4.1579
355. Shiina S, Gani RA, Yokosuka O, Maruyama H, Nagamatsu H, Payawal DA, et al. APASL practical recommendations for the management of hepatocellular carcinoma in the era of COVID-19. Hepatol Int (2020) 14:92–9. doi: 10.1007/s12072-020-10103-4
356. Chan SL, Kudo M. Impacts of COVID-19 on Liver Cancers: During and after the Pandemic. Liver Cancer (2020) 9(5):491–502. doi: 10.1159/000510765
357. Kim D, Adeniji N, Latt N, Kumar S, Bloom PP, Aby ES, et al. Predictors of Outcomes of COVID-19 in Patients with Chronic Liver Disease: US Multi-center Study. Clin Gastroenterol Hepatol (2020) In press:1–32. doi: 10.1016/j.cgh.2020.09.027
358. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
359. Schmid J, Bauer E, Daneck K, Franke H, Koss FW. Lack of detectable N-nitroso-N-methyl-N-cyclohexylamine in humans after administration of bromhexine. Arzneimittelforschung (1988) 38(9):1359–64.
360. Fan Z, Chen L, Li J, Cheng X, Yang J, Tian C, et al. Clinical Features of COVID-19-Related Liver Functional Abnormality. Clin Gastroenterol Hepatol (2020) 18(7):1561–6. doi: 10.1016/j.cgh.2020.04.002
Keywords: hepatocellular carcinoma, HCC and COVID-19, complement activation, complement proteins, prognostic markers, inflammatory factors, complement-based therapeutics, immunotherapy
Citation: Malik A, Thanekar U, Amarachintha S, Mourya R, Nalluri S, Bondoc A and Shivakumar P (2021) “Complimenting the Complement”: Mechanistic Insights and Opportunities for Therapeutics in Hepatocellular Carcinoma. Front. Oncol. 10:627701. doi: 10.3389/fonc.2020.627701
Received: 10 November 2020; Accepted: 22 December 2020;
Published: 24 February 2021.
Edited by:
Prasanna K. Santhekadur, JSS Academy of Higher Education and Research, IndiaReviewed by:
Kiran Kumar Mudnakudu N., JSS Academy of Higher Education and Research, IndiaGopi Sundaram, Central Food Technological Research Institute (CSIR), India
Samudyata C. Prabhuswamimath, All India Institute of Speech and Hearing (AIISH), India
Copyright © 2021 Malik, Thanekar, Amarachintha, Mourya, Nalluri, Bondoc and Shivakumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pranavkumar Shivakumar, cHJhbmF2LnNoaXZha3VtYXJAY2NobWMub3Jn
†These authors have contributed equally to this work