 Giada Del Baldo1*
Giada Del Baldo1* Roberto Carta1
Roberto Carta1 Iside Alessi1
Iside Alessi1 Pietro Merli1
Pietro Merli1 Emanuele Agolini2
Emanuele Agolini2 Martina Rinelli2
Martina Rinelli2 Luigi Boccuto3,4
Luigi Boccuto3,4 Giuseppe Maria Milano1Annalisa Serra1
Giuseppe Maria Milano1Annalisa Serra1 Andrea Carai5
Andrea Carai5 Franco Locatelli1,6
Franco Locatelli1,6 Angela Mastronuzzi1
Angela Mastronuzzi1- 1Department of Paediatric Haematology/Oncology, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 2Laboratory of Medical Genetics, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 3JC Self Research Institute, Greenwood Genetic Center, Greenwood, SC, United States
- 4School of Nursing, College of Behavioral, Social and Health Science, Clemson University, Clemson, SC, United States
- 5Department of Neuroscience and Neurorehabilitation, Neurosurgery Unit, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 6Department of Maternal, Infantile, and Urological Sciences, University of Rome La Sapienza, Rome, Italy
Rhabdoid tumors are rare aggressive malignancies in infants and young children with a poor prognosis. The most common anatomic localizations are the central nervous system, the kidneys, and other soft tissues. Rhabdoid tumors share germline and somatic mutations in SMARCB1 or, more rarely, SMARCA4, members of the SWI/SNF chromatin-remodeling complex. Rhabdoid tumor predisposition syndrome (RTPS) is a condition characterized by a high risk of developing rhabdoid tumors, among other features. RTPS1 is characterized by pathogenic variants in the SMARCB1 gene, while RTPS2 has variants in SMARCA4. Interestingly, germline variants of SMARCB1 and SMARCA4 have been identified also in patients with Coffin-Siris syndrome. Children with RTPS typically present with tumors before 1 year of age and in a high percentage of cases develop synchronous or multifocal tumors with aggressive clinical features. The diagnosis of RTPS should be considered in patients with rhabdoid tumors, especially if they have multiple primary tumors and/or in individuals with a family history. Because germline mutations result in an increased risk of carriers developing rhabdoid tumors, genetic counseling, and surveillance for all family members with this condition is recommended.
Introduction
Rhabdoid tumor predisposition syndrome (RTPS) is characterized by an elevated risk of developing malignancies called rhabdoid tumors (RTs). RTs are rare, aggressive tumors, typically diagnosed in infants (1).
Primary rhabdoid tumor sites can include the central nervous system (65%), kidney (9%) and in the remaining 26% of cases: head and neck soft tissues, paravertebral muscles, liver, bladder, mediastinum, retroperitoneum, and pelvis (2).
Immunohistochemical characteristics of these tumors include loss of the BAF47/BRG1 protein (3). Among newly diagnosed cases, 25%–35% will harbor a germline variant of the SMARCB1 gene (OMIM*601607) (4, 5). Recently, pathogenic variants in the SMARCA4 gene (OMIM*603254) have also been associated with RT (6); while the involvement of other genes appears to be exceedingly rare in RTs (7, 8).
The most frequent pediatric tumor associated with RTPS is atypical teratoid/rhabdoid tumor (AT/RT). AT/RTs are rare, accounting for 1%–2% of all brain cancers, 90% of cases being diagnosed in children of less than 3 years of age (9–12), with a slight male predominance (13). At the time of presentation, 65.4% are in the posterior fossa, 31% supratentorial and 3.6% multifocal (14).
Histologically, AT/RT shows areas of rhabdoid phenotype containing rhabdoid cells with eccentric nuclei, prominent nucleoli, abundant eosinophilic cytoplasm, and a mesenchymal component with spindle cells. In the last years, molecular characterization of RT has become increasingly relevant. SMARCB1 and SMARCA4 are tumor suppressor genes playing a critical etiologic role in all rhabdoid tumors including AT/RT, which is linked to somatic and germline mutations of SMARCB1 or, more rarely, SMARCA4.
AT/RTs are biologically heterogeneous. In the last few years, different authors described transcriptional features of AT/RTs that can be summarized in three molecular subgroups (12, 15–17) with different genetic profile, age at onset, prognosis, and brain localization:
1) AT/RT-TYR tumors are characterized by infratentorial location, younger age at diagnosis (<1 year) and overexpression of the melanosomal markers such as DCT, TYR, and MITF and many genes involved in ciliogenesis (DNAH11 and SPEF1). Other pathways described include bone morphogenetic protein (BMP) and orthodenticle homeobox 2 (OTX2). Chromosome 22q loss is the most common cytogenetic anomaly.
2) AT/RT-MYC tumors are generally supratentorial, affected individuals are older (age 4–5 years), and the cluster genes MYC, HOTAIR, and HOX are overexpressed. Focal deletions of SMARCB1 are the most common molecular anomaly. Supratentorial location is the more frequent site. Spinal tumors are included in this subgroup.
3) AT/RT-SHH tumors location may be infratentorial or supratentorial with similar frequency, diagnosis is in the age interval 2 to 5 years. Genes of the sonic hedgehog pathway (GLI2, BOC, PTCHD2) and NOTCH signaling (ASLC1, CBL, HES1) are overexpressed.
Patients outcome for each group is not homogeneous among the different data published to date and prognosis is still unclear (12, 15–17).
The most common extra-cerebral site for the primary onset of an RT is the kidney (48% of cases), followed by head and neck (14%), liver (13%), and other sites such as trunk and arms (25%) (18, 19).
RTs of the kidney account for about 2% of all pediatric renal cancers (20). Renal RT is highly aggressive and has a poor prognosis, with a 12-month survival rate of only 30% (18). Patients presenting with renal RT in the first year of life tend to develop brain tumors in 10%–15% of cases (21). These patients often harbor a germline mutation of SMARCB1 and have a worse prognosis, as compared to those with sporadic RTs (22).
Rhabdoid Tumor Predisposition Syndrome
RTPS is an autosomal dominant cancer predisposition syndrome. When the mutation pathogenic variants occur in the SMARCB1 gene, the syndrome is called RTPS1, and RTPS2 has variants in the SMARCA4 gene.
BAF47/BRG1 proteins encoded by SMARCB1/SMARCA4 genes are key components of the ATP-dependent chromatin-remodeling SWI/SNF complex, which is essential for lineage specification, gene regulation, and maintenance of stem cell pluripotency (23).
RTs are the most frequent malignancies associated with these syndromes, but not the only ones. In most cases these arise de novo but there is a small percentage of familial cases having RTPS. RTs can present in a familial setting, with up to 35% of cases due to germline mutations in SMARCB1 (4) or, in 2%–3% of cases, in SMARCA4 (24, 25).
Children with RTPS typically present with tumors before 12 months of age and in 35% of cases develop synchronous or multifocal tumors with aggressive clinical features (20, 22, 26). RTs can be detected in the prenatal period or during childhood with a median age at onset of 4–7 months (range prenatally – 60 months) (1, 27, 28) versus sporadic RTs that are detected at a median age of 13–30 months (range: age 1 day–228 months). Often RTs in RTPS are synchronous, with advanced stage at diagnosis and clinically aggressive. Progression occurs during chemotherapy in 58% of individuals with RTPS and RTs (24). In the EU-RHAB Registry 28% of cases had synchronous RT: eight individuals AT/RT and extracranial malignant rhabdoid tumors (eMRT), four had AT/RT and rhabdoid tumor of the kidney (RTK), and two AT/RT, multiple eMRT and RTK (28).
Furthermore, other conditions are known to be related to RTPS. Family history of RT or cribriform neuroepithelial tumor (CRINET) and/or combination of RT with one of the following: schwannoma, malignant peripheral nerve sheath tumor, meningioma are highly suggestive for RTPS (29).
The diagnosis of RTPS is established in a proband with a rhabdoid tumor and/or a family history of RT and/or multiple SMARCB1/SMARCA4 deficient tumors (synchronous or metachronous) and identification of a germline pathogenic variant in SMARCB1 or SMARCA4 by genetic testing (30). In Figure 1 are summarized the main clinical and genetics features of RTPS.
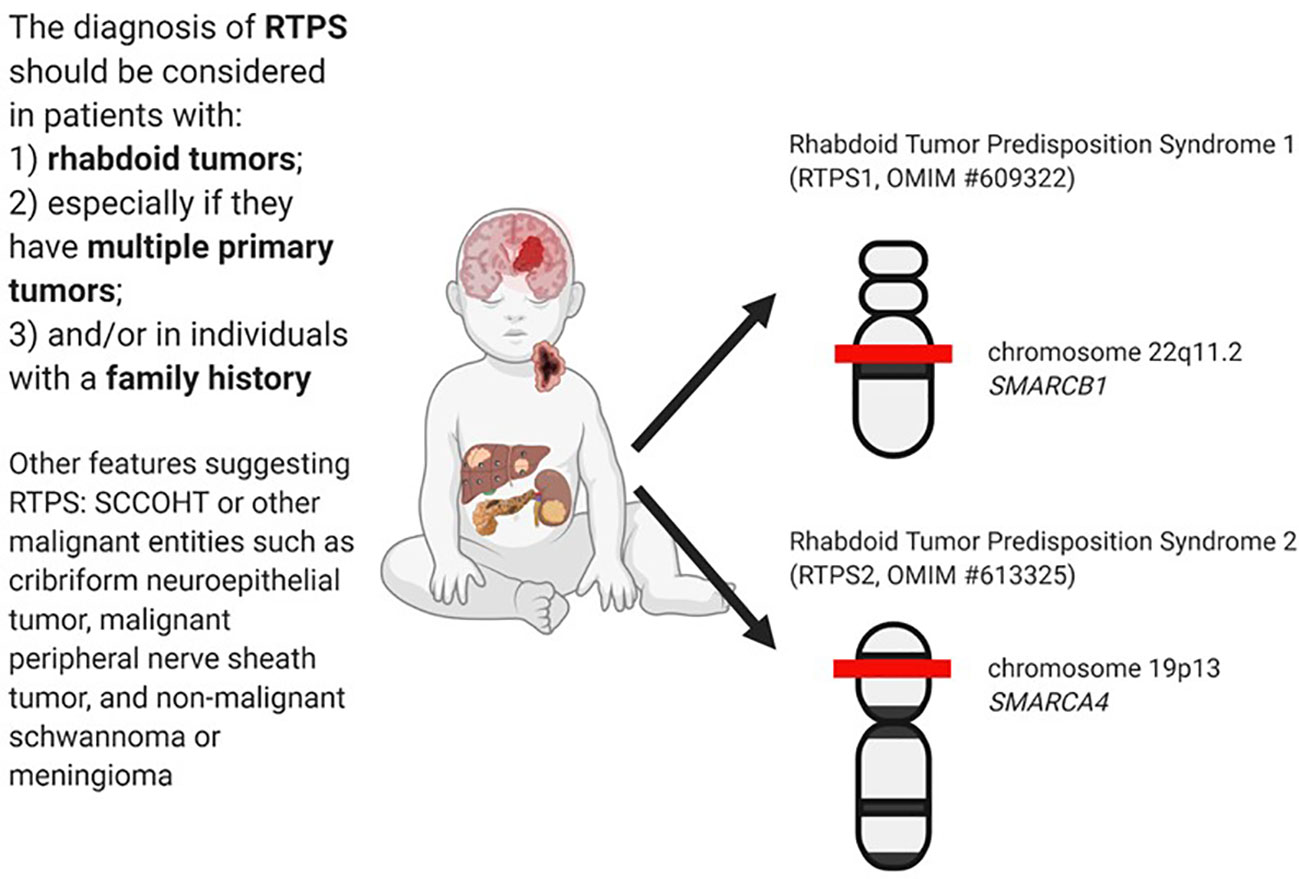
Figure 1 RTPS tumors spectrum and related genes involved.
Rhabdoid Tumor Predisposition Syndrome 1
Rhabdoid Tumor Predisposition Syndrome 1 (RTPS1, OMIM #609322) is caused by heterozygous germline mutations in the SMARCB1 gene, which maps to chromosome 22q11.2 (31). The protein involved is an SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (30).
Clinical Features
As described above, the syndrome predisposes to the development of RTs, including brain tumors, renal and extrarenal cancers. AT/RT is the most frequent brain cancer in patients with SMARCB1 mutations, but other CNS tumors are described (32).
Interestingly, Thomas et al. (33) described a case of RTPS1 in an infant with AT/RT in which supratentorial and infratentorial parts of the tumor demonstrated different DNA methylation profiles suggesting synchronous or metachronous AT/RT with different molecular subgroup and cell of origin.
Recently, the SMARCB1 gene has been found also in familial and sporadic schwannomatosis. Hulsebos et al. (34) described two family members with schwannomatosis and a germline mutation of SMARCB1, suggesting it as a candidate predisposing gene. Swensen et al. reported a family with hereditary schwannomatosis associated with a germline mutation of SMARCB1. Three members of the family developed RTs and died before 2 years of age (35). About 40%–50% of familial schwannomatosis and 8%–10% of sporadic cases harbor a constitutional mutation in SMARCB1 (25). Interestingly, SMARCB1 and NF2 loci map very close to each other on the long arm of chromosome 22 (25).
Furthermore, Schmitz et al. found the same somatic mutation of SMARCB1 in four of 126 meningiomas. The data suggest that SMARCB1 is a tumor suppressor gene that may be important also for the oncogenesis in a subset of meningiomas (36).
Moreover, SMARCB1 mutation carriers may be at risk for developing other tumors such as malignant peripheral nerve sheath tumors and cribriform neuroepithelial tumors (37).
Genetics
SMARCB1 inactivation can be caused by different mechanisms like gross chromosomal aberration or loss of heterozygosity of 22q11.2 or loss-of-function mutations including nonsense, frameshift, splicing and missense mutations (6).
Concerning cytogenetics, the most frequent alteration described in AT/RT is the monosomy of chromosome 22 (14, 38, 39). Biegel et al. described also a rhabdoid tumor with an unbalanced 9;22 translocation (40).
Penetrance. Penetrance may vary according to the mutation type. Incomplete penetrance has been observed in three of nine published families with RTPS due to SMARCB1 mutations (6). Rarely a SMARCB1 pathogenic variant is inherited from an unaffected parent or a parent with late-onset or undiagnosed RTPS (41). Germline mosaicism must be taken into account for at least half of the families with sibs affected by RTPS (30).
Rhabdoid Tumor Predisposition Syndrome 2
Rhabdoid Tumor Predisposition Syndrome 2 (RTPS2, OMIM #613325) is caused by heterozygous germline mutations in the SMARCA4 gene, which maps to chromosome 19p13 (6) and encodes a protein involved in the transcription activator BRG1, a catalytic component of the ATP-dependent SWI/SNF chromatin remodeling complex (30).
Clinical Features
The main tumor resulting from germline pathogenic variants in SMARCA4 is small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) (37, 42). It seems that up to 40% of females with SCCOHT may harbor a germline variant in SMARCA4 (43), therefore the detection of SCCOHT in young women is high evocative for RTPS2 (44–46).
Although more rarely than SMARCB1 mutations, pathogenic germline SMARCA4 variants are found in children with AT/RT and it seems that SMARCA4-mutated AT/RT may be associated with a worse prognosis (24, 47). The risk of other RTs in SMARCA4 germline heterozygotes is unknown, but probably very low.
Other epithelial cancers, such as lung cancer, have been reported in some adults with pathogenic germline variants in SMARCA4, but again, the risks remain unquantified (46).
Recently, a novel entity designated “SMARCA4-deficient thoracic sarcoma” (SDTS) was described by Le Loarer et al. in 19 adult individuals, supporting the carcinogenic effect of SMARCA4 inactivation, with consequences beyond the pediatric age range (48).
Genetics
Among the different SMARCA4 pathogenic variants reported to date, nonsense, and intragenic deletions are the prevalent types, while only a single missense variant has been detected (24).
Penetrance. It appears that SMARCA4 mutations are less penetrant for AT/RT than SMARCB1 ones (37). In contrast to SMARCB1, most reported patients with RTs and a SMARCA4 mutation inherited it from an unaffected parent (30). In SMARCA4-related RTPS, the penetrance for RT in the preceding generation of seven informative families was zero. However, in one family, two sibs with a SMARCA4 pathogenic variant were both affected (6, 24, 30).
Other Rare Manifestations Related to SMARCB1 and SMARCA4 Mutations
Interestingly, germline variants of SMARCB1 and SMARCA4 have been identified also in patients with Coffin-Siris syndrome three (CSS3, OMIM #614608) and four (CSS4, OMIM #614609). CSS is a congenital malformation syndrome characterized by developmental delay, intellectual disability, coarse facial features, feeding difficulties, and hypoplastic or absent fifth fingernails and fifth distal phalanges (49). Individuals with CSS carrying SMARCB1 or SMARCA4 mutations seem to show no predisposition to develop RTs or other forms of tumor. This can be explained by the fact that mutations resulting in CSS3 are non-truncating, implying that they exert gain-of-function or dominant-negative effects (excluding haploinsufficiency as a cause) (50). Very rare exceptions have been described. To date, a single CSS individual with schwannomatosis and a SMARCB1 variant has been reported (51): the SMARCB1 c.1121G>A(p.Arg374Gln) germline transition in exon 9 lead to the inactivation of the second allele in the tumor tissue. More recently, a pediatric patient with mild CSS who concomitantly developed small‐cell carcinoma of the ovary hypercalcaemic type has been found to harbor a germline heterozygous nonsense mutation and a somatic frameshift mutation in SMARCA4 (52).
Genotype-Phenotype Correlation
According to Smith et al. and Holsten et al. a clear genotype-phenotype correlation could be identified (53, 54). Germline SMARCB1 mutations located in the central portion of the gene, involving multiple exon deletions or duplications and truncating mutations, likely responsible for a loss of SMARCB1 protein product, are most frequently associated with rhabdoid tumors. Instead, SMARCB1 mutations located at the ends of the gene, particularly non-truncating alterations, including missense variants, are most frequently associated with non-oncologic diseases and low-grade tumors such as the ones reported in CSS, meningiomas, and schwannomas. Unlike the germline SMARCB1 mutations detected in RT cases, schwannomatosis-associated alterations determine reduced expression levels or a partial loss of function of the SMARCB1 protein (53). Moreover, a correlation was identified between the type of SMARCB1 variant and the time of onset of the disease: truncating variants are associated with early-onset disease, non-truncating variants with late-onset disease.
Surveillance
To date, no universally accepted surveillance recommendations for RTPS carriers have been established. In Table 1 are summarized two surveillance propositions suggested by Foulkes et al. (37) and Teplick et al. (55). Nemes et al. (30) proposed a protocol of surveillance not only in pre-symptomatic RTPS carriers but also in individuals affected by RTs.
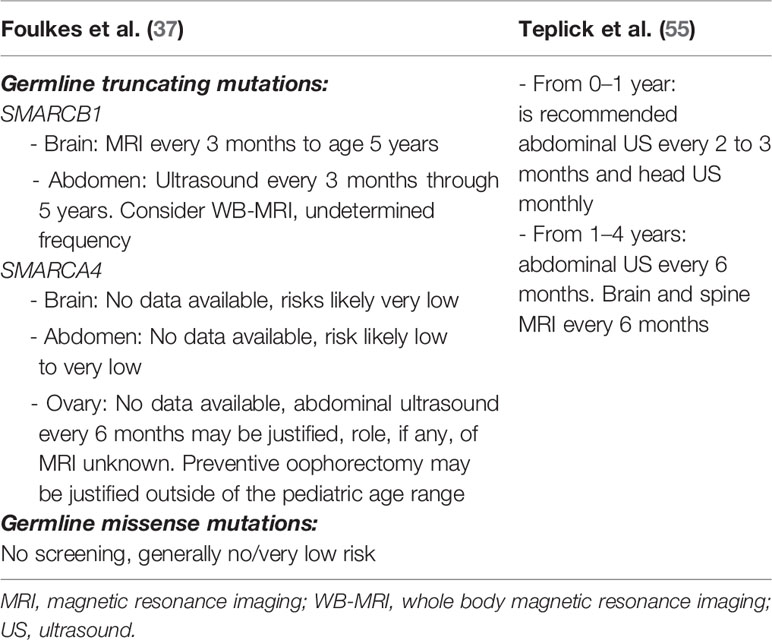
Table 1 Surveillance recommendations for rhabdoid tumor predisposition syndrome (RTPS) carriers.
Foulks et al. (37) give more detailed indications about monitoring of SMARCB1 or SMARCA4 carriers as opposed to Teplick et al. (55), even if they failed to stratify cancer monitoring for age range. They recommended brain MRI in SMARCB1 carriers every 3 months for the first 5 years of life. As known, AT/RTs in RTPS1 arise generally within the first year of life and MRI is an expensive examen, and sedation is needed in young children. After the first year of life, a brain MRI should be performed every 6 months. About abdominal monitoring, they recommended ultrasound every 3 months through 5 years and consider whole-body MRI, with undetermined frequency. Whole-body MRI will guarantee high diagnostic accuracy as opposed to ultrasound, but it is an expensive procedure and requires sedation in little patients.
Regarding SMARCA4 carriers they suggest an abdominal ultrasound every 6 months with no mention of the beginning or end of the follow-up. Considering the rarity of the condition and the very low risk, unfortunately, there is no data available for monitoring of brain and abdominal RTs in SMARCA4 carriers.
Interestingly, Folkes et al. (37) proposed a separated surveillance protocol for germline truncating mutations versus germline missense mutations, underlining that germline missense mutations need no screening for their very low risk of RTs. On the other hand, they proposed MRI surveillance for patients with a germline missense mutation of SMARCB1 to allow the early detection of schwannomas.
Teplick et al. (55) did not take into account the due separated conditions RTPS1 and 2 and different germline kinds of mutations. They suggested the use of ultrasound in the first year of life to monitor the brain and abdomen every 2–3 months. Between 1 and 4 years of age, they suggest extending abdominal ultrasound monitoring every 6 months and using brain and spine MRI to exclude the onset of brain tumors every 6 months. In their proposal, there is no mention of whole-body MRI.
Genetic Test
Molecular genetic testing for RTPS is appropriate in any patients with:
- RTs, familial RTs, multifocal or synchronous tumor, congenital or early-onset disease, other conditions known to be related to RTPS
- SMARCB1- or SMARCA4-deficient tumors with a positive family history.
Point variants of SMARCB1 and SMARCA4 can be identified by Sanger sequencing or next-generation sequencing (NGS). Besides point mutations, other alterations of SMARCB1 and SMARCA4 have also been documented, including deletion of the entire SMARCB1 locus or intragenic deletions involving one or more exons (5). Methods used to detect this kind of alteration may include quantitative PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
Genetic Counseling and Risk to Family Members
Siblings and Parents
When a pathogenic variant of SMARCB1 or SMARCA4 is detected in a proband, molecular genetic evaluation of parents and siblings is required.
As mentioned above, carriers of SMARCA4 mutation inherited a pathogenic variant from an unaffected parent (24), while the vast majority of individuals with RTPS1 have a de novo germline SMARCB1 mutation, and only in extremely rare cases, they inherited a SMARCB1 pathogenic variant from an unaffected parent.
A healthy parent with a pathogenic germline variant has to start surveillance as for siblings, but at longer intervals, as the risk of malignancies is very low.
If the SMARCA4 or SMARCB1 pathogenic variant found in the proband cannot be detected in either parent, it raises the possibility of a de novo pathogenic variant in the proband or germline mosaicism in a parent. Parental germline mosaicism in SMARCB1 has been rarely described (5, 27, 32, 56, 57), while the overall incidence of germline mosaicism in RTPS is unknown.
The cancer risk for the siblings of a proband depends on the genetic status of the proband’s parents:
- 50% risk of inheriting the variant if the proband harbors a SMARCA4 or SMARCB1 pathogenic variant, although penetrance can be incomplete.
- 1% risk of inheriting the variant if the parent is negative for SMARCA4 or SMARCB1 mutations, considering the possibility of parental germline mosaicism (5, 27, 56, 57).
Offspring of a Proband
As mentioned above, patients with RTPS1 die at a young age. Despite it occurs very rarely, it should be considered the cancer risk in offsprings. If children are affected by a de novo germline SMARCB1 mutation and survive to adulthood, they can potentially transmit the mutation to their offspring (25).
The family history of most individuals with RTPS may appear to be negative for many reasons: failure of detection of the disorder in family members, reduced penetrance (more evident in SMARCA4-related RTPS), late onset in the affected parent.
Prevention and Prenatal Diagnosis
There is no possibility of preventing cancer development in patients with RTPS, but in case of detected SMARCB1/SMARCA4 mutations, the advice of surveillance and follow-up must be followed. Prophylactic oophorectomy may be discussed in women with SMARCA4-related RTPS for the high risk to develop SCCOHT (58).
It would also be important to prevent secondary complications related to aggressive treatments.
Once SMARCB1 and SMARCA4 pathogenic variants are detected, prenatal testing for a pregnancy at increased risk and preimplantation genetic diagnosis are possible. The preferred tests used to assess if a product of conception carries a known SMARCB1/SMARCA4 mutation are chorionic villus sampling and amniocentesis.
Conclusion
Germline variants play a role in 8.5%–10% of all pediatric cancer with the prevalence of certain genes such as TP53, APC, NF1, PMS2, RB1, and RUNX1. The increasing implementation and availability of genetic testing lead to the opportunity to identify the risk of cancer and early detection of tumors with the aim of reducing mortality and morbidity (21).
RTPS is characterized by a high risk of developing RTs and other unfrequent conditions. RTs are a rare, aggressive form of malignancies typically diagnosed in young infants that can arise in multiple anatomical sites. About 25%–35% of RTs carry a germline variant of SMARCB1 (4, 5), or more rarely SMARCA4. The diagnosis of RTPS should be taken into account in patients with RTs, especially if early and multiple primary tumors and/or if a positive family history of RTs is present (25).
The ongoing new characterization of AT/RTs and RTs (12) will likely lead to further biological insights that can delineate molecular subtypes and may lead to novel therapeutic options. Despite these promising advancements, surveillance for cancer risk and prevention remains the focus of current management. Further research is needed to increase our understanding of RTs biology and gather further knowledge of the role of SMARCB1/SMARCA4 in RTs development and other rare manifestations.
Author Contributions
GB and RC wrote the manuscript. PM provided the figure. AS, IA, and GM contributed to the finishing of the work. EA and MR contributed to the genetic details of the manuscript. AM, AC, LB, and FL revised it critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Geller JI, Roth JJ, Biegel JA. Biology and Treatment of Rhabdoid Tumor. Rev Crit Rev Oncog (2015) 20(3-4):199–216. doi: 10.1615/CritRevOncog.2015013566
2. Nemes K, Frühwald MC. Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert Opin Ther Targets (2018) 22(4):365–79. doi: 10.1080/14728222.2018.1451839
3. Parham DM, Weeks DA, Beckwith JB. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohis-tochemistry or electron microscopy. Am J Surg (1994) 18(10):1010–29. doi: 10.1097/00000478-199410000-00005
4. Bourdeaut F, Lequin D, Brugières L, Reynaud S, Dufour C, Doz F, et al. Frequent hSNF5/INI1 Germline Mutations in Patients with Rhabdoid Tumor. Hum Cancer Biol (2011) 17(1):31–8. doi: 10.1158/1078-0432.CCR-10-1795
5. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 Mutations in Familial and Sporadic Rhabdoid Tumors. Pediatr Blood Cancer (2011) 56(1):7–15. doi: 10.1002/pbc.22831
6. Schneppenheim R, Frühwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, et al. Germline Nonsense Mutation and Somatic Inactivation of SMARCA4/BRG1 in a Family with Rhabdoid Tumor Predisposition Syndrome. J Home Page Am J Hum Genet (2010) 86(2):279–84. doi: 10.1016/j.ajhg.2010.01.013
7. Lee SR, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest (2012) 122(8):2983–8. doi: 10.1172/JCI64400
8. Kieran MW, Roberts RCMW, SN C, KL L, BE R, MacConaill LE, et al. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer (2012) 59(7):1155–7. doi: 10.1002/pbc.24315
9. Hilden JM, Meerbaum S, Burger P, Finaly J, Janss A, Scheithauer BW, et al. Central Nervous System Atypical teratoid/rhabdoid Tumor: Results of Therapy in Children Enrolled in a Registry. J Clin Oncol (2004) 22(14):2877–84. doi: 10.1200/JCO.2004.07.073
10. Picard D, Miller S, Hawkins CE, Bouffet PE, Rogers HA, Chan TSY, et al. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol (2012) 13(8):838–48. doi: 10.1016/S1470-2045(12)70257-7
11. Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, et al. Atypical Teratoid/Rhabdoid Tumors (ATRT): Improved Survival in Children 3 Years of Age and Older With Radiation Therapy and High-Dose Alkylator-Based Chemotherapy. J Clin Oncol (2005) 23(7):1491–9. doi: 10.1200/JCO.2005.05.187
12. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell (2016) 29(3):379–93. doi: 10.1016/j.ccell.2016.02.001
13. Biswas A, Goyal S, Puri T, Das P, Sarkar C, Julka PK, et al. Atypical teratoid rhabdoid tumor of the brain: case series and review of literature. Childs Nerv Syst (2009) 25(11):1495–500. doi: 10.1007/s00381-009-0903-x
14. Burger P, Yu I-T, Tihan T, Friedman HS, Strother DR, Kepner JL, et al. Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System: A Highly Malignant Tumor of Infancy and Childhood Frequently Mistaken for Medulloblastoma. A Pediatric Oncology Group Study. Am J Surg Pathol (1998) 22(9):1083–92. doi: 10.1097/00000478-199809000-00007
15. Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Frühwald M, et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors—a reinvestigation and current consensus. Neuro Oncol (2020) 22(5):613–24. doi: 10.1093/neuonc/noz235
16. Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell (2016) 30(6):891–908. doi: 10.1016/j.ccell.2016.11.003
17. Kordes U, Gesk S, Frühwald MC, Graf N, Leuschner I, Hasselblatt M, et al. Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer (2010) 49(2):176–81. doi: 10.1002/gcc.20729
18. Brennan B, Stiller C, Bourdeaut F. Extracranial Rhabdoid Tumours: What We Have Learned So Far and Future Directions. Lancet Oncol (2013) 14(8):e329–36. doi: 10.1016/S1470-2045(13)70088-3
19. Wick MR, Ritter JH, Dehner LP. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol (1995) 12(3):233–48.
20. Pastore G, Znaor A, Spreafico F, Graf N, Pritchard-Jones K, Steliarova-Foucher E. Malignant renal tumor incidence and survival in European children (1978–1997): report from the Automated Childhood Cancer Information System project. Eur J Cancer (2006) 42(13):2103–14. doi: 10.1016/j.ejca.2006.05.010
21. Tomlinson GE, Breslow NE, Dome J, Guthrie KA, Norkool P, Li S, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol (2005) 23(30):7641–5. doi: 10.1200/JCO.2004.00.8110
22. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Gogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res (1999) 59(1):74–9.
23. Wilson BG, Roberts CWM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer (2011) 11(7):481–92. doi: 10.1038/nrc3068
24. Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schüller U, et al. SMARCA4−mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol (2014) 128(3):453–6. doi: 10.1007/s00401-014-1323-x
25. Sredni ST, Tomita T. Rhabdoid Tumor Predisposition Syndrome. Pediatr Dev Pathol (2015) 18(1):49–58. doi: 10.2350/14-07-1531-MISC.1
26. Seeringer A, Reinhard H, Hasselblatt M, Schneppenheim R, Siebert R, Bartelheim K, et al. Synchronous Congenital Malignant Rhabdoid Tumor of the Orbit and Atypical teratoid/rhabdoid Tumor–Feasibility and Efficacy of Multimodal Therapy in a Long-Term Survivor. Cancer Genet (2014) 207(9):429–33. doi: 10.1016/j.cancergen.2014.06.028
27. Bruggers CS, Bleyl SB, Pysher T, Barnette P, Afify Z, Walker M, et al. Clinicopathologic Comparison of Familial Versus Sporadic Atypical teratoid/rhabdoid Tumors (AT/RT) of the Central Nervous System. Pediatr Blood Cancer (2011) 56(7):1026–31. doi: 10.1002/pbc.22757
28. Frühwald MC, Biegel JA, Bourdeaut F, Roberts CWM, Chi SN. Atypical teratoid/rhabdoid Tumors-Current Concepts, Advances in Biology, and Potential Future Therapies. Neuro Oncol (2016) 18(6):764–78. doi: 10.1093/neuonc/nov264
29. van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJM. Germline SMARCB1 Mutation Predisposes to Multiple Meningiomas and Schwannomas With Preferential Location of Cranial Meningiomas at the Falx Cerebri. Neurogenetics (2012) 13(1):1–7. doi: 10.1007/s10048-011-0300-y
30. Nemes K, Bens S, Bourdeaut F, Hasselblatt M, Kool M, Johann P, et al. Rhabdoid Tumor Predisposition Syndrome. Seattle, Seattle (WA: Book from University of Washington (2017).
31. Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature (1998) 394(6689):203–6. doi: 10.1038/28212
32. Sévenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional Mutations of the hSNF5/INI1 Gene Predispose to a Variety of Cancers. AJHG (1999) 65(5):1342–8. doi: 10.1086/302639
33. Thomas C, Knerlich-Lukoschus F, Reinhard H, Johann PD, Sturm D, Sahm F, et al. Two molecularly distinct atypical teratoid/rhabdoid tumors (or tumor components) occurring in an infant with rhabdoid tumor predisposition syndrome 1. Acta Neuropathol (2019) 137:847–50. doi: 10.1007/s00401-019-02001-3
34. Hulsebos TLM, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet (2007) 80(4):805–10. doi: 10.1086/513207
35. Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet (2009) 46:68–72. doi: 10.1136/jmg.2008.060152
36. Schmitz U, Mueller W, Weber M, Sévenet N, Delattre O, von Deimling A. INI1 mutations in meningiomas at a potential hotspot in exon 9. BJC (2001) 84(2):199–201. doi: 10.1054/bjoc.2000.1583
37. Foulkes WD, Kamihara J, Evans DGR, Brugières L, Bourdeaut F, Molenaar JJ, et al. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res (2017) 23(12):e62–7. doi: 10.1158/1078-0432.CCR-17-0595
38. Biegel JA, Rorke LB Packer RJ, Emanuel BS. Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg (1990) 73:710–4. doi: 10.3171/jns.1990.73.5.0710
39. Douglass EC, Valentine M, Rowe ST, Parham DM, Wilimas JA, Sanders JM, et al. Malignant rhabdoid tumor: a highly malignant childhood tumor with minimal karyotypic changes. Genes Chromosomes Cancer (1990) 2:210–6. doi: 10.1002/gcc.2870020308
40. Biegel JA, Burk CD, Parmiter AH, Emanuel BS. Molecular analysis of a partial deletion of 22q in a central nervous system rhabdoid tumor. Genes Chromosomes Cancer (1992) 5:104–8. doi: 10.1002/gcc.2870050203
41. Ammerlaan ACJ, Ararou A, Houben MPWA, Baas F, Tijssen CC, Teepen JLJM, et al. Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br J Cancer (2008) 98:474–9. doi: 10.1038/sj.bjc.6604156
42. Witkowski L, Goudie C, Foulkes WD, McCluggage WG. Small-cell carcinoma of the ovary of hypercalcemic type (malignant rhabdoid tumor of the ovary): a review with recent developments on pathogenesis. Surg Pathol Clin (2016) 9:215–26. doi: 10.1016/j.path.2016.01.005
43. Witkowski L, Goudie C, Ramos P, Boshari T, Brunet JS, Karnezis AN, et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol (2016) 141:454–60. doi: 10.1016/j.ygyno.2016.03.013
44. Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small surprise – small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol (2014) 233(3):209–14. doi: 10.1002/path.4362
45. Moes-Sosnowska J, Szafron L, Nowakowska D, Dansonka-Mieszkowska A, Budzilowska A, Konopka B, et al. Germline SMARCA4 Mutations in Patients With Ovarian Small Cell Carcinoma of Hypercalcemic Type. Orphanet J Rare Dis (2015) 10:32. doi: 10.1186/s13023-015-0247-4
46. Witkowski L, Donini N, Byler-Dann R, Knost JA, Albrecht S, Berchuck A, et al. The hereditary nature of small cell carcinoma of the ovary, hypercalcemic type: two new familial cases. Fam Cancer (2017) 16(3):395–9. doi: 10.1007/s10689-016-9957-6
47. Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol (2018) 35(3):193–8. doi: 10.1053/j.semdp.2018.01.002
48. Le Loarer F, Watson S, Pierron, Vincent Thomas de Montpreville VT, Ballet S, Firmin N, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet (2015) 47(10):1200–5. doi: 10.1038/ng.3399
49. Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Matsumoto N, Makita Y, et al. Coffin-Siris Syndrome Is a SWI/SNF Complex Disorder. Clin Genet (2014) 85(6):548–54. doi: 10.1111/cge.12225
50. Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, et al. Mutations Affecting Components of the SWI/SNF Complex Cause Coffin-Siris Syndrome. Nat Genet (2012) 44(4):376–8. doi: 10.1038/ng.2219
51. Gossai N, Biegel JA, Messiaen L, Berry SA, Moertel CL. Report of a Patient With a Constitutional Missense Mutation in SMARCB1, Coffin-Siris Phenotype, and Schwannomatosis. Am J Med Genet A (2015) 167A(12):3186–91. doi: 10.1002/ajmg.a.37356
52. Errichiello E, Mustafa N, Vetro A, Notarangelo LD, de Jonge H, Rinaldi B, et al. SMARCA4 Inactivating Mutations Cause Concomitant Coffin-Siris Syndrome, Microphthalmia and Small-Cell Carcinoma of the Ovary Hypercalcaemic Type. J Pathol (2017) 243(1):9–15. doi: 10.1002/path.4926
53. Smith MJ, Wallace AJ, Bowers NL, Eaton H, Evans DGR. SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genet (2014) 207(9):373–8. doi: 10.1016/j.cancergen.2014.04.001
54. Holsten T, Bens S, Oyen F, Nemes K, Hasselblatt M, Kordes U, et al. Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: a meta-analysis. Eur J Hum Genet (2018) 26(8):1083–93. doi: 10.1038/s41431-018-0143-1
55. Teplick A, Kowalski M, Biegel JA, Nichols KE. Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr (2011) 170(3):285–94. doi: 10.1007/s00431-010-1377-2
56. Biegel JA, Busse TM, Weissman BE. SWI/SNF Chromatin Remodeling Complexes and Cancer. Am J Med Genet C Semin Med Genet (2014) 166C(3):350–66. doi: 10.1002/ajmg.c.31410
57. Gigante L, Paganini I, Frontali M, Ciabattoni S, Sangiuolo FC, Papi L. Rhabdoid tumor predisposition syndrome caused by SMARCB1 constitutional deletion: prenatal detection of new case of recurrence in siblings due to gonadal mosaicism. Fam Cancer (2016) 15(1):123–6. doi: 10.1007/s10689-015-9836-6
Keywords: rhabdoid tumors, atypical teratoid/rhabdoid tumors, cancer surveillance, genetic test, cancer risk, cancer predisposition syndromes
Citation: Del Baldo G, Carta R, Alessi I, Merli P, Agolini E, Rinelli M, Boccuto L, Milano GM, Serra A, Carai A, Locatelli F and Mastronuzzi A (2021) Rhabdoid Tumor Predisposition Syndrome: From Clinical Suspicion to General Management. Front. Oncol. 11:586288. doi: 10.3389/fonc.2021.586288
Received: 22 July 2020; Accepted: 05 January 2021;
Published: 22 February 2021.
Edited by:
Michael C. Fruehwald, Augsburg University Hospital, GermanyReviewed by:
Andrea Di Cataldo, University of Catania, ItalyJhon A. Guerra, HIMA San Pablo Oncologic, United States
Franck Bourdeaut, Institut Curie, France
Copyright © 2021 Del Baldo, Carta, Alessi, Merli, Agolini, Rinelli, Boccuto, Milano, Serra, Carai, Locatelli and Mastronuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giada Del Baldo, Z2lhZGEuZGVsYmFsZG9Ab3BiZy5uZXQ=