 Qiguang Wang1†
Qiguang Wang1† Xuhui Hui
Xuhui Hui Yan Ju
Yan Ju- 1Department of Neurosurgery, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Radiology, Hospital of Chengdu Office of People’s Government of Tibetan Autonomous Region (Hospital C.T), Chengdu, China
- 3Huaxi MR Research Center (HMRRC), Functional and Molecular Imaging Key Laboratory of Sichuan Province, Department of Radiology, West China Hospital, Sichuan University, Chengdu, China
- 4School of Computer Science and Engineering, University of Electronic Science and Technology of China, Chengdu, China
- 5Medical Insurance Office, West China Hospital, Sichuan University, Chengdu, China
Aim: Multifocal desmoplastic infantile ganglioglioma/astrocytoma (DIA/DIG) has rarely been reported. Here, two cases have been presented, reviewing the literature and proposed treatment algorithms for this rare tumor.
Patients and Methods: We report two patients diagnosed with multifocal DIA/DIGs in West China Hospital. In addition, a literature review was performed, in October 2019, on case reports of DIA/DIGs with multifocal lesions. The clinical and radiological features, treatment, and outcome of this rare disease were discussed.
Results: DIA/DIGs with multifocal locations were rare, and only thirteen cases (including ours) had been reported. This series included 8 males and 5 females with a mean age of 31.4 ± 45.7 months (range, 3-144 months). The supratentorial hemisphere, suprasellar region, posterior cranial fossa, and spinal cord were frequently involved. Ten patients (76.9%) received surgical resection for the symptomatic lesions and three patients (23.1%) underwent biopsy. Seven patients received chemotherapy postoperatively. Six individuals had tumor recurrences during the follow-up period, while three patients had tumors that spontaneously regressed. Finally, two patients died of tumor progression and one patient died of respiratory insufficiency and hypothalamic dysfunction.
Conclusions: Multifocal DIA/DIGs have more aggressive clinical behavior and poor outcome despite benign histology. DIA/DIGs should be included in the differential diagnosis of multifocal brain tumors in children. The mainstay of treatment is surgical resection; adjuvant treatment with chemotherapeutic drugs is unknown and requires additional research.
Introduction
Desmoplastic infantile astrocytoma (DIA) and desmoplastic infantile ganglioglioma (DIG) are rare intracranial tumors in infants, and account for only 1.25% of all childhood brain tumors (1). They most commonly manifest as large cystic or solid well-defined supratentorial lesions in infants aged 1 to 24 months (2). The WHO classification (2016) of nervous system tumors categorized DIA and DIG together as one diagnosis, among grade 1 “neuronal and mixed neuronal-glial” entities (3). They frequently present as solitary cortical-surfacing neoplasms, gross total resection can always achieve a favorable outcome (4).
Only 11 cases of DIA/DIGs with multifocal intracranial or intraspinal tumors have been recorded in the English literature (5–11). Its natural history, diagnosis and management remain a dilemma because of the rarity. Here, based on the institutional and literature cases, the largest case series of multifocal DIA/DIGs have been presented, to summarize the clinical features, optimize the treatment and elaborate the nature of this rare disease.
Patients and Methods
Case Presentation
Patient 1
A 12-year-old female was admitted to our hospital with seizures over two months. Cranial magnetic resonance imaging (MRI) depicted five solid tumors in the right and left tentorium, left para sellar, right lateral ventricle, and left frontal lobe respectively, the solid tumors are hypointense in T1 sequences and isodense or hypointense with obvious peritumoral edema, after administration of gadolinium, the tumors enhanced strongly (Figures 1A–D). She didn’t have a family history of genetic disease, but she had a previous surgery history of lung inflammatory myofibroblastic tumor. The tumors in the left frontal lobe and right tentorium were completely resected after admission to obtain a histopathological diagnosis and relieve symptoms (Figures 1E, F). Postoperative histopathological examination supported the diagnosis of DIA/DIG (Figure 1G). The patient was discharged from the hospital with a favorable outcome two weeks later and a close follow-up was achieved.
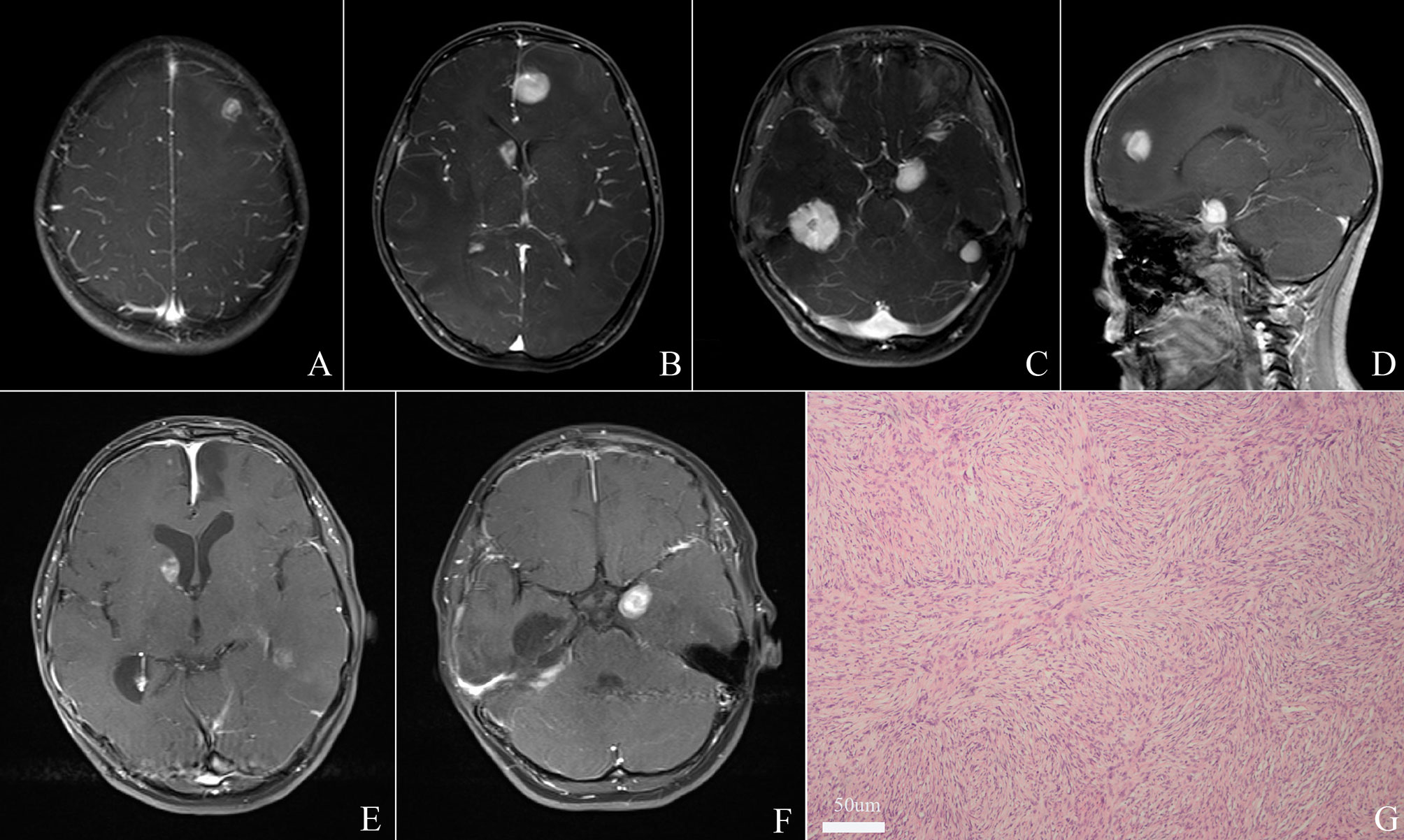
Figure 1 Cranial magnetic resonance imaging (MRI) depicted five lesions located in the right and left tentorium, left para sellar, right lateral ventricle, and left frontal lobe respectively (A–D). Total resection of tumors in the left frontal lobe and right tentorium was achieved (E, F). Histopathological examination revealed a desmoplastic spindle cell tumor with highly eosinophilic collagenous fiber, confirming the diagnosis of DIA/DIGs. (x 200) (G).
Patient 2
A 6-month-year old boy presented to our hospital with a history of bilateral nystagmus, exophthalmos for two months. Multiple lesions were seen on Gd-enhanced cranial MRI in the suprasellar cistern, bilateral cavernous sinus, and hippocampus, intraorbital, and cisterna magna (Figures 2A, E). The suprasellar region tumors contained both solid and cystic components, the solid part showed hypointense in both T1 and T2 sequences, the cystic part showed hypointense on T1 sequences and hyperintense in T2. CT angiography depicted no obvious vessels inside the tumors (Figure 2B), MR Spectroscopy showed the tumor in the suprasellar was featured by an elevated choline peak, a reduced NAA peak, and a lactate peak (Figure 2F). He didn’t get the spinal MRI scan because he didn’t have any related symptoms. He didn’t have a family history of genetic disease. Due to the abundant blood supply and hard tumor texture intraoperatively, a subfrontal craniotomy was performed, and partial resection was obtained (Figures 2C, G). Histologically, the tumor showed abundant collagenous fiber with fibroblastic and astrocytic elements. His parents chose regular follow-up without further adjuvant interventions. The follow-up MRI at 22 months depicted spontaneous regression of the lesions in the medial hippocampus, suprasellar cistern and cisterna magna (Figures 2D, H).
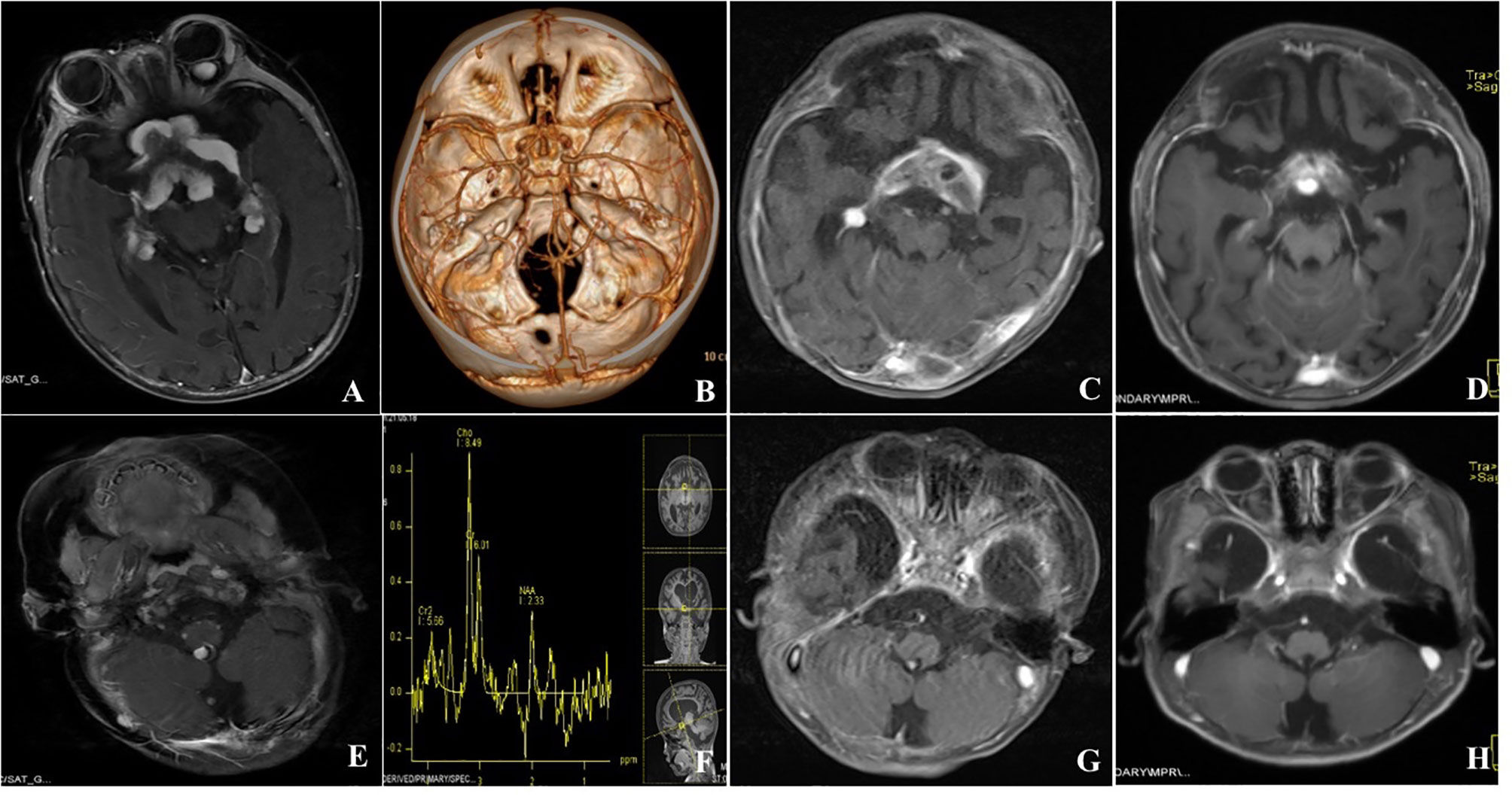
Figure 2 Gd-enhanced cranial MRI depicted multiple enhanced lesions in the suprasellar cistern, bilateral cavernous sinus and hippocampus, intraorbital and cisterna magna (A, E). CT angiography showed that the lesions were not vascular tumors and no obvious vessels were found inside the tumor (B), MR Spectroscopy showed that the lesion in the saddle area was featured by an elevated choline peak, a reduced NAA peak and a lactate peak (F). Postoperative MRI depicted partial resection of suprasellar cistern tumors (C, G). Further MRI was done at 22 months follow-up depicted spontaneous regression of the lesions in the medial hippocampus, suprasellar cistern, and cisterna magna (D, H).
Literature Review
The data of patients with multifocal DIA/DIGs was collected according to the PRISMA (preferred reporting items for systematic reviews and meta-analyses) guidelines (12). The literature review was conducted on PUBMED/MEDLINE database with the following keywords: multiple, multifocal, metastases, desmoplastic infantile astrocytomas, desmoplastic infantile ganglioglioma. The search included only articles or case reports in English until October 2019 without an early date limit.
Case report or article titles and abstracts were evaluated. Only articles are written in English that described cases of multifocal desmoplastic infantile ganglioglioma/astrocytoma were included. The literature review was conducted independently by two authors (QG, W and J, C) using EndNote X7 software (Thomson Reuters, Carlsbad, California, USA). For controversial studies regarding the inclusion, a discussion would be performed until a consensus agreement was reached. The patient characteristics such as demographic data, clinical and radiological features, pathologic diagnosis, treatment and prognosis were extracted from the literature by two authors (QG, W and J, C) independently.
Results
Patient Sample, Clinical and Radiological Features
A total of 13 cases (including ours) Table 1) were included in the present study. The baseline data were summarized in Table 2. This series included eight infants with mean age of 5.7 ± 2.5 months (range, 3-11 months) and five children with average age of 6.2 ± 4.5 years (range, 1.2-12 years). Among them, 8 patients were male and 5 patients were female. The most common symptoms were those related to increased intracranial ICP or macrocephaly (n=5, 38.5%), nystagmus (n=3, 23.1%), seizures (n=4, 30.8%) and sensorimotor deficits (n=2, 15.4%). In this series, each patient had multiple tumors, and the multifocal lesions were found in the supratentorial hemisphere (n=8), suprasellar region (n=8), infratentorial location (n=8) and spinal cord(n=8), other locations include pial and ependymal, periventricular and tentorium. The lesions in first case from our institution were noted in tentorium, left para sellar, right lateral ventricle and left frontal lobe (Figure 1); the second case included tumors in the suprasellar cistern, bilateral cavernous sinus and hippocampus, intraorbital and cisterna magna (Figure 2). The solid lesions frequently showed iso- to hypointense on T1W sequences, predominantly hypointense on T2W sequences, and homogenous enhancement in Gd-enhanced MRI. Sometimes multifocal DIA/DIGs include both cystic and solid lesions, with the cystic component frequently being deep and the solid component being more peripheral; the cyst wall can be enhanced or not (8). In this series, the multifocal tumors were present at diagnosis in 12 cases, in the case presented by Darish et al (9), the multifocal leptomeningeal lesions were found six weeks after the initial diagnosis of DIA/DIGs.
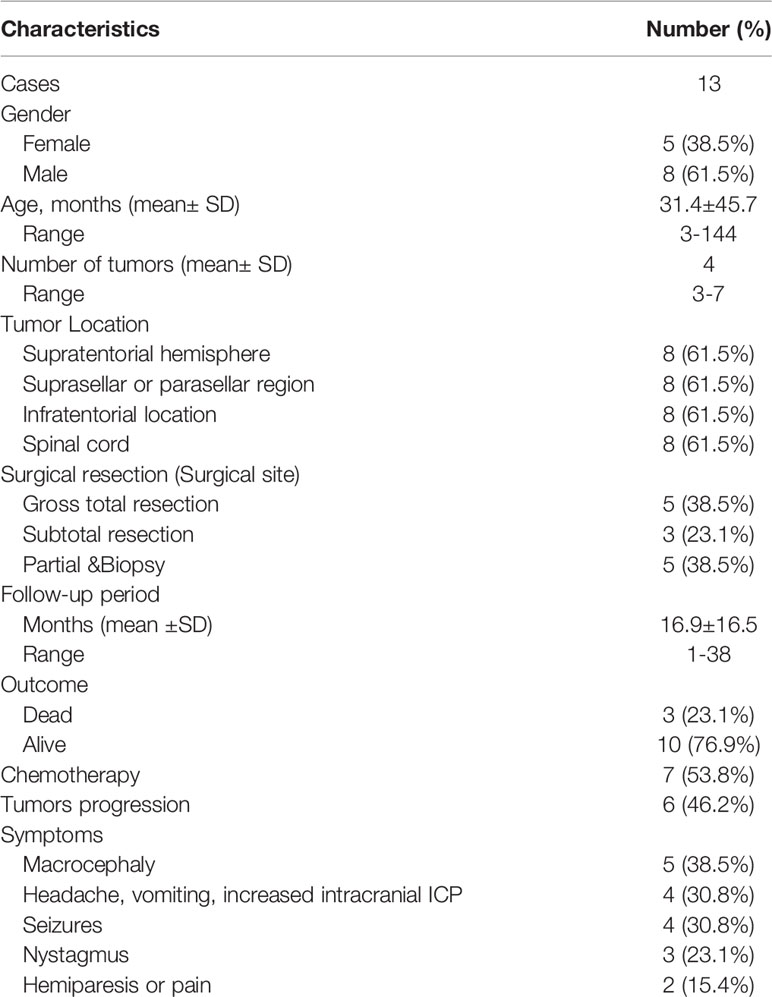
Table 1 Patients' characteristics.
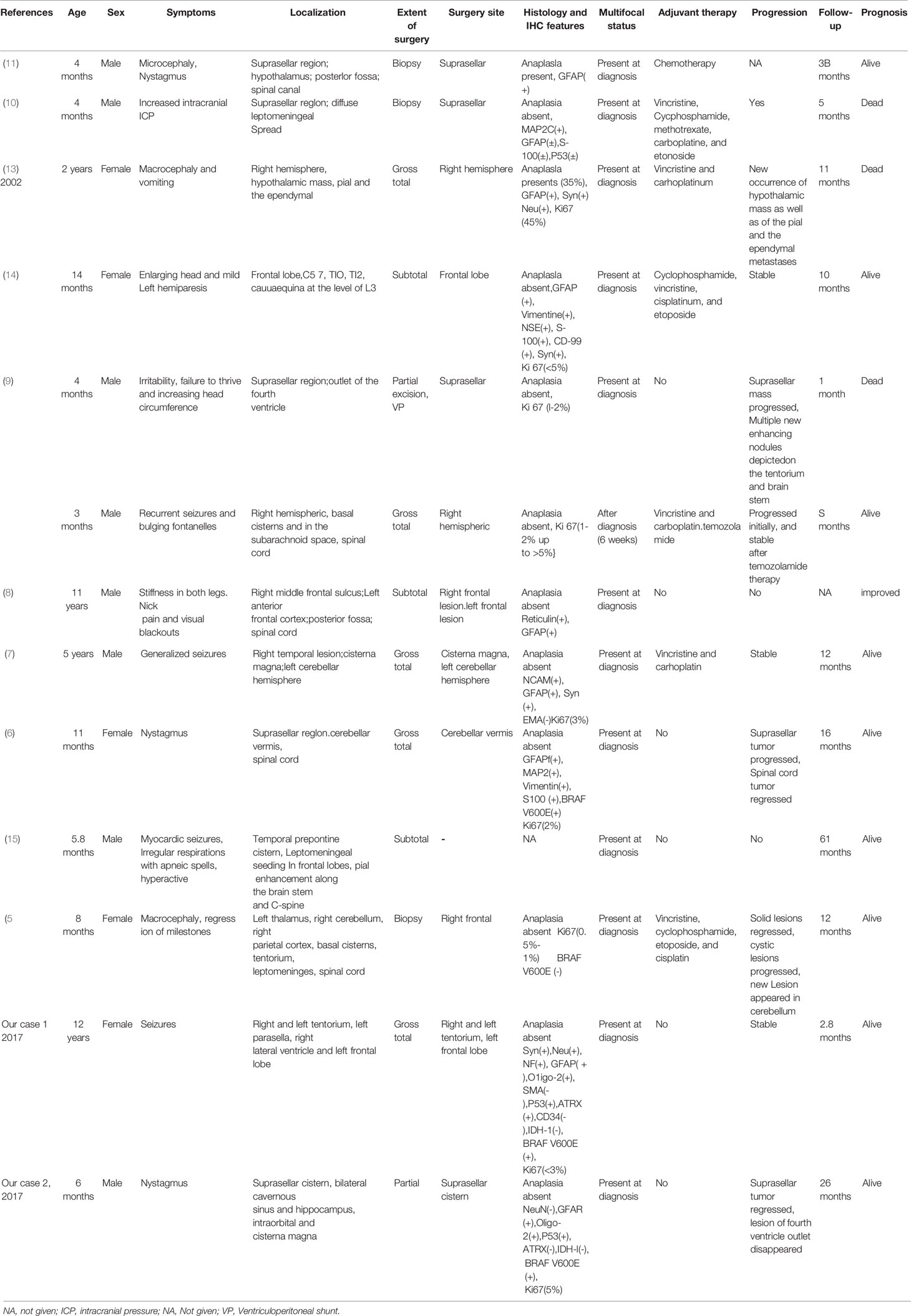
Table 2 Literature review of multifocal DIA/DIGs.
Surgical Management, Outcome and Molecular Histological Analyses
Ten patients (76.9%) in this series received surgical resection for the symptomatic lesions and three patients (23.1%) underwent biopsy. Case 1 has been previously reported by our group member (16), but we updated some opinions and statements. In the case presented by Setty et al (11), the patient received a suprasellar tumor biopsy as well as surveillance for the hypothalamus, posterior fossa, and spinal canal, and the residual tumors remained stable 38 months later. Bock et al (10) reported the patient underwent a biopsy for suprasellar tumor and observation for the diffuse meningeal spread, in spite of chemotherapy, the patient expired at a 5-month follow-up due to progression. In the case reported by De Munnynck et al (13), the patient underwent gross total resection for the right hemisphere tumor, the hypothalamic mass and ependymal lesions received close observation, however, the spectacular growth of the hypothalamic mass as well as of the pial and the ependymal metastases was noted 5 weeks later, the patient died of tumor progression 11 months after initial diagnosis. According to Knapp et al (14), the patient underwent subtotal resection for the frontal lobe tumor and close observation for the spinal lesions, and the leftover tumors remained stable 10 months later following treatment. Darish et al (9) reported a 4-month-old boy who underwent partial resection of the suprasellar tumor and close observation of lesion in the outlet of the fourth ventricle, but the suprasellar tumor progressed 24 days after the surgery and the patient died of respiratory insufficiency and hypothalamic dysfunction 1 week later; the other patient was alive 8 months after gross total resection of the right hemisphere tumors and chemotherapy for the residual basal cisterns and spinal cord lesions. The residual tumors in the case reported by Uro-Coste et al (7) remained stable 12 months after the surgery. Abuharbid et al (6) reported the patient underwent gross total resection for the cerebellar vermis tumor and observation of the suprasellar and spinal cord tumors, during the follow-up of 16 months, the suprasellar tumor progressed but the spinal cord tumors regressed. In the case reported by Jurkiewica et al. (15), the residual tumors were stable 61 months following surgery. In the case presented by Narayan et al (5), the residual solid lesions regressed spontaneously, the cystic tumors progressed and new lesions appeared in the cerebellum.
Based on the postoperative histological investigation, all of the patients were categorized as WHO grade I DIA/DIGs. Histologically, they frequently showed a desmoplastic spindle cell tumor with a densely eosinophilic collagenous substrate (Figure 1G, x 200). In this series, only two patients showed suspected anaplasia histological features, De Munnynck et al (13) reported a case with suspected anaplastic component and high Ki-67 proliferation index (45%). Nine patients reported the Ki-67 proliferation data; eight patients (88.9%) had benign histological phenotype with a very low Ki-67 proliferation rate (0.5-5%). Four patients reported the BRAF V600E status, and three of four patients showed BRAF V600E mutation-positive. Other immunohistochemical markers included NSE (+), GFAP (+), Syn (+), and EMA (-) (Table 2).
Discussion
Taratuto et al. reported the first case of desmoplastic infantile astrocytoma (DIA) in 1984 (17) and Vandenberg et al. reported the first of desmoplastic infantile ganglioglioma (DIG) in 1987 (18). The 2016 WHO classification of Nervous system categorized DIA and DIG together (18, 19) and classified as grade I (3). DIA/DIGs are generally solitary and have a favorable prognosis with long-term disease-free survival after surgical resection (20). DIA/DIGs with multifocal lesions are uncommon, and little is known about their clinical features and management.
DIA/DIGs frequently occur as a supratentorial, contrast-enhanced solid tumor with cystic components (4). And the solid component always manifests as hypointense in T1 and T2 sequences, and the cystic part is hypointense and hyperintense in T2 at brain MRI (15). They frequently involved frontal and parietal lobes; the tumors tend to be large with solid components located at the surface of the cerebral cortex (4, 21). However, we found that multifocal DIA/DIGs are not restricted to the supratentorial region, they can also occur in the suprasellar region, posterior cranial fossa and spinal cord. In this series, we found eight patients (61.5%) had tumors in suprasellar or parasellar region, eight patients (61.5%) in the posterior fossa and eight patients (61.5%) in the spinal cord. Multifocal DIA/DIGs may be confused with fibrous meningioma, inflammatory disease, metachronous or metastatic lesions preoperatively due to its multifocal location and no specific radiological characteristics (5). Hence, neurosurgeons should at least be reminded of the possibility of DIA/DIG when confronting multiple intracranial and spinal lesions in infants and young children.
Our study found no obvious differences regarding histological features between multifocal DIA/DIGs and solitary counterparts. Both of them contained reticulin- and collagen-rich areas with spindle fibroblastic elements, DIGs always show neuronal elements whereas DIAs show astrocytic component (4). Besides, the mitotic activity in both solitary and multifocal DIA/DIGs is low with the Ki67 index ranging between 0.5 and 15% (4).
Some research speculated that multifocal DIA/DIGs were metastases from a single tumor via the CSF pathway, and they identified tumor cells in the CSF of these patients (5, 9, 11, 13). However, the pattern of direct shedding of tumor cells from the initial tumor into CSF, survival and migration with CSF, followed by distal implantation on the pial surface of CNS was only an assumption in the literature, but was poorly supported by empirical evidence (22). Meanwhile, some authors found that the CSF in multiple DIA/DIGs contained inflammatory cells, chiefly macrophages and signet ring-like cells, but no tumor cells (8, 10). The Case 1 in our series showed the multifocal tumors were located in the brain parenchyma, hinting these lesions were not metastases via CSF pathways. Furthermore, twelve patients were found to have multifocal status at the time of diagnosis in this series, excluding the case reported by Darish et al (9), who found multifocal leptomeningeal lesions six weeks after the initial diagnosis of DIA/DIGs. Hence, the mechanism of multifocal DIA/DIGs, metastases, or an inherited genetic disorder, remained to need further study.
Solitary DIA/DIGs were frequently stable and had long relapse-free intervals even after partial resection, hinting at the indolent nature of the tumor (4, 23, 24). But the growth pattern and natural history of multifocal DIA/DIGs were unclear. Despite the WHO I grade and low Ki-67 proliferation rate (0.5%-5%), we found multifocal DIA/DIGs can sometimes be fast-growing after surgical resection and chemotherapy. In this series, 3 patients (27.3%) died of tumor progression and 6 of 13 (46.2%) patients showed residual tumor progression or new lesions occurrence during the follow-up. Hence, long-term careful monitoring is needed. It is worth mentioning that some tumors in three patients spontaneously regressed, and this phenomenon should be taken into consideration when treating multifocal DIA/DIGs.
Gross total resection is always associated with far superior outcomes in solitary DIA/DIG (25, 26). But the treatment of multifocal DIA/DIGs can be more complex due to the multifocal location. It was suggested that surgical treatment can be performed in symptomatic mass to get the histopathological diagnosis and improve quality of life, while close observation for the asymptomatic tumors. As for the adjuvant chemotherapy, despite some studies suggesting that vincristine and carboplatin may be contributing to control DIA/DIGs in the adjuvant setting, but no consensus had reached so far (5, 7, 13). BRAF gene mutation has been found in 43.8% solitary DIA/DIGs recently (27, 28), which hinted DIA/DIGs were another low-grade MAPK pathway-driven neuroepithelial tumor and might allow for the use of targeted molecular therapies, such as vemurafenib (29). In this study, four patients examined the BRAF status, and 3 of 4 patients depicted BRAF V600E mutation, which showed higher occurrence than the solitary counterparts.
Several limitations should be stated in our study. Firstly, due to the limited sample size, it is difficult to conduct a statistical analysis to find the prognostic factors. Secondly, the present study included both institutional series and literature case reports, some intrinsic limitations should attend to. Thirdly, only four patients in this series screened for the BRAF mutation, further multicenter studies were needed to investigate the incidence of BRAF mutation in multifocal DIA/DIGs. Finally, future studies, especially multicenter prospective studies with a large sample size of this rare tumor subset, are needed.
Conclusions
Despite the benign histology of multifocal DIA/DIGs, they had incompatibly aggressive clinical features and poor prognosis. They can occur in the entire neural axis and some of tumors can undergo spontaneous regression. They should also be included in the differential diagnosis of multiple intracranial and spinal cord tumors in infants and young adults. We suggest that symptom-producing tumors be surgically resected with care, and that residual lesions be followed up closely. Due to the high risk of residual tumor progression, regular long-term follow-up is essential.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of West China Hospital, Clinical data for patients were retrospectively sourced after having gained approval. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
QW and JM, conceptualization, methodology, formal analysis, validation, investigation, data curation, writing - original draft, writing - review and editing, and visualization. JC, writing - review and editing, methodology, and funding acquisition. SZ, data curation. XH, QL, and WL, validation and resources. YJ and LS conceptualization, methodology, and validation. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Science and technology project of Sichuan Province (Fund No. 2021YJ0161), the Medical Research project of Sichuan Province (Fund No. Q20042), the Science and technology Project of Tibet Autonomous Region. The central government guides local projects (Fund No. XZ202102YD0032C).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rout P, Santosh V, Mahadevan A, Kolluri VR, Yasha TC, Shankar SK. Desmoplastic Infantile Ganglioglioma–Clinicopathological and Immunohistochemical Study of Four Cases. Childs Nerv Syst (2002) 18(9-10):463–7. doi: 10.1007/s00381-002-0610-3
2. Trehan G, Bruge H, Vinchon M, Khalil C, Ruchoux MM, Dhellemmes P, et al. MR Imaging in the Diagnosis of Desmoplastic Infantile Tumor: Retrospective Study of Six Cases, AJNR. Am J Neuroradiol (2004) 25(6):1028–33.
3. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
4. Bianchi F, Tamburrini G, Massimi L, Caldarelli M. Supratentorial Tumors Typical of the Infantile Age: Desmoplastic Infantile Ganglioglioma (DIG) and Astrocytoma (DIA). Childs Nerv Syst (2016) 32(10):1833–8. doi: 10.1007/s00381-016-3149-4
5. Narayan V, Savardekar AR, Mahadevan A, Arivazhagan A, Appaji L. Unusual Occurrence of Multifocal Desmoplastic Infantile Astrocytoma: A Case Report and Review of the Literature. Pediatr Neurosurg (2017) 52(3):173–80. doi: 10.1159/000455926
6. Abuharbid G, Esmaeilzadeh M, Hartmann C, Hermann EJ, Krauss JK. Desmoplastic Infantile Astrocytoma With Multiple Intracranial and Intraspinal Localizations at Presentation. Childs Nerv Syst (2015) 31(6):959–64. doi: 10.1007/s00381-015-2715-5
7. Uro-Coste E, Ssi-Yan-Kai G, Guilbeau-Frugier C, Boetto S, Bertozzi AI, Sevely A, et al. Desmoplastic Infantile Astrocytoma With Benign Histological Phenotype and Multiple Intracranial Localizations at Presentation. J Neuro-Oncol (2010) 98(1):143–9. doi: 10.1007/s11060-009-0075-2
8. Santhosh K, Kesavadas C, Radhakrishnan VV, Abraham M, Gupta AK. Multifocal Desmoplastic Noninfantile Astrocytoma, Journal of Neuroradiology. J Neuroradiologie (2008) 35(5):286–91. doi: 10.1016/j.neurad.2008.04.003
9. Darwish B, Arbuckle S, Kellie S, Besser M, Chaseling R. Desmoplastic Infantile Ganglioglioma/Astrocytoma With Cerebrospinal Metastasis. J Clin Neurosci (2007) 14(5):498–501. doi: 10.1016/j.jocn.2006.01.024
10. Bock D, Rummele P, Friedrich M, Wolff JE. Multifocal Desmoplastic Astrocytoma, Frontal Lobe Dysplasia, and Simian Crease. J Pediatr (2002) 141(3):445. doi: 10.1067/mpd.2002.126695
11. Setty SN, Miller DC, Camras L, Charbel F, Schmidt ML. Desmoplastic Infantile Astrocytoma With Metastases at Presentation. Mod Pathol (1997) 10(9):945–51.
12. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PloS Med (2009) 6(7):e1000097. doi: 10.1371/journal.pmed.1000097
13. De Munnynck K, Van Gool S, Van Calenbergh F, Demaerel P, Uyttebroeck A, Buyse G, et al. Desmoplastic Infantile Ganglioglioma: A Potentially Malignant Tumor? Am J Surg Pathol (2002) 26(11):1515–22. doi: 10.1097/00000478-200211000-00015
14. Knapp J, Olson L, Tye S, Bethard JR, Welsh CA, Rumbolt Z, et al. Case of Desmoplastic Infantile Ganglioglioma Secreting Ceruloplasmin. J Child Neurol (2005) 20(11):920–4. doi: 10.1177/08830738050200111201
15. Cohen AR. The Great Neurosurgical Masquerader: 3 Cases of Desmoplastic Infantile Ganglioglioma. J Neurosurg Pediatr (2019) 24(3):258–66. doi: 10.3171/2019.5.PEDS19151
16. Wei F, Richard SA, Tan J, Lan Z, Ju Y. Inflammatory Myofibroblastic Lung Tumor Transforming Into Intracranial Desmoplastic Noninfantile Ganglioglioma: A Case Report and Literature Review. Med (Baltimore) (2018) 97(40):e12668. doi: 10.1097/MD.0000000000012668
17. Taratuto AL, Monges J, Lylyk P, Leiguarda R. Superficial Cerebral Astrocytoma Attached to Dura. Report of Six Cases in Infants. Cancer (1984) 54(11):2505–12. doi: 10.1002/1097-0142(19841201)54:11<2505::aid-cncr2820541132>3.0.co;2-g
18. VandenBerg SR, May EE, Rubinstein LJ, Herman MM, Perentes E, Vinores SA, et al. Desmoplastic Supratentorial Neuroepithelial Tumors of Infancy With Divergent Differentiation Potential ("Desmoplastic Infantile Gangliogliomas"). Report on 11 cases of a distinctive embryonal tumor with favorable prognosis. J Neurosurg (1987) 66(1):58–71. doi: 10.3171/jns.1987.66.1.0058
19. Gessi M, Zur Muhlen A, Hammes J, Waha A, Denkhaus D, Pietsch T. Genome-Wide DNA Copy Number Analysis of Desmoplastic Infantile Astrocytomas and Desmoplastic Infantile Gangliogliomas. J Neuropathol Exp Neurol (2013) 72(9):807–15. doi: 10.1097/NEN.0b013e3182a033a0
20. Naylor RM, Wohl A, Raghunathan A, Eckel LJ, Keating GF, Daniels DJ. Novel Suprasellar Location of Desmoplastic Infantile Astrocytoma and Ganglioglioma: A Single Institution's Experience, Journal of Neurosurgery. Pediatrics (2018) 22(4):397–403. doi: 10.3171/2018.4.PEDS17638
21. Samkari A, Alzahrani F, Almehdar A, Algahtani H. Desmoplastic Infantile Astrocytoma and Ganglioglioma: Case Report and Review of the Literature. Clin Neuropathol (2017) 36(2017):31–40. doi: 10.5414/NP300945
22. Garzia L, Kijima N, Morrissy AS, De Antonellis P, Guerreiro-Stucklin A, Holgado BL, et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell (2018) 173(6):1549.
23. VandenBerg SR. Desmoplastic Infantile Ganglioglioma and Desmoplastic Cerebral Astrocytoma of Infancy. Brain Pathol (1993) 3(3):275–81. doi: 10.1111/j.1750-3639.1993.tb00754.x
24. Sperner J, Gottschalk J, Neumann K, Schorner W, Lanksch WR, Scheffner D. Clinical, Radiological and Histological Findings in Desmoplastic Infantile Ganglioglioma. Childs Nerv Syst (1994) 10(7):458–62; discussion 462-3. doi: 10.1007/BF00303613
25. Cohen AR. The Great Neurosurgical Masquerader: 3 Cases of Desmoplastic Infantile Ganglioglioma, Journal of Neurosurgery. Pediatrics (2019) 24(3):258–66. doi: 10.3171/2019.5.PEDS19151
26. Romero FR, Listik S, Fabris PA. Desmoplastic Ganglioglioma: Report of a non-Infantile Case. Arq Neuropsiquiatr (2011) 69(2A):258–9. doi: 10.1590/S0004-282X2011000200022
27. Wang AC, Jones DTW, Abecassis IJ, Cole BL, Leary SES, Lockwood CM, et al. Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities With Frequent BRAFV600 Mutations. Mol Cancer Res MCR (2018) 16(10):1491–8. doi: 10.1158/1541-7786.MCR-17-0507
28. Chatterjee D, Garg C, Singla N, Radotra BD. Desmoplastic non-Infantile Astrocytoma/Ganglioglioma: Rare Low-Grade Tumor With Frequent BRAF V600E Mutation. Hum Pathol (2018) 80:186–91. doi: 10.1016/j.humpath.2018.06.005
Keywords: Multiple DIA/DIGs, treatment, outcome, infant, clinical feature
Citation: Wang Q, Meng J, Cheng J, Zhang S, Hui X, Li Q, Liu W, Ju Y and Sun L (2022) Multifocal Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIA/DIG): An Institutional Series Report and a Clinical Summary of This Rare Tumor. Front. Oncol. 11:608129. doi: 10.3389/fonc.2021.608129
Received: 19 September 2020; Accepted: 22 December 2021;
Published: 09 May 2022.
Edited by:
Theodore Nicolaides, New York University, United StatesReviewed by:
Ibrahim Abdelrahim Qaddoumi, St. Jude Children’s Research Hospital, United StatesAllison Martin, Albert Einstein College of Medicine, United States
Copyright © 2022 Wang, Meng, Cheng, Zhang, Hui, Li, Liu, Ju and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Ju, anV5YW4wODA2QDEyNi5jb20=; Lin Sun, cGlja2Vyc3VuQHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship