 Sandra Ortiz-Cuaran
Sandra Ortiz-Cuaran Jebrane Bouaoud
Jebrane Bouaoud Andy Karabajakian
Andy Karabajakian Jérôme Fayette
Jérôme Fayette Pierre Saintigny
Pierre Saintigny- 1Univ Lyon, Université Claude Bernard Lyon 1, INSERM 1052, CNRS 5286, Centre Léon Bérard, Centre de Recherche en Cancérologie de Lyon, Lyon, France
- 2Department of Medical Oncology, Centre Léon Bérard, Lyon, France
- 3Department of Maxillofacial Surgery and Stomatology, Pitié-Salpêtrière University Hospital, Pierre et Marie Curie University, Sorbonne University, Paris, France
Head and neck squamous cell carcinoma (HNSCC) is the sixth most incident cancer worldwide. More than half of HNSCC patients experience locoregional or distant relapse to treatment despite aggressive multimodal therapeutic approaches that include surgical resection, radiation therapy, and adjuvant chemotherapy. Before the arrival of immunotherapy, systemic chemotherapy was previously employed as the standard first-line protocol with an association of cisplatin or carboplatin plus 5-fluorouracil plus cetuximab (anti-EFGR antibody). Unfortunately, acquisition of therapy resistance is common in patients with HNSCC and often results in local and distant failure. Despite our better understanding of HNSCC biology, no other molecular-targeted agent has been approved for HNSCC. In this review, we outline the mechanisms of resistance to the therapeutic strategies currently used in HNSCC, discuss combination treatment strategies to overcome them, and summarize the therapeutic regimens that are presently being evaluated in early- and late-phase clinical trials.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most incident cancer worldwide, responsible for more than 700,000 cases worldwide per year and around 350,000 deaths, making it a particularly fatal disease (1).
Squamous cell cancers of the oral cavity, the pharynx, and the larynx (the most frequent) are linked to smoking and alcohol consumption, and squamous cell carcinomas of the oropharynx are most commonly associated with human papilloma virus (HPV) infection, especially for young or nonsmoker patients. The incidence of the latter is rising, mostly among men (2). Cigarette- or alcohol-related and HPV-induced cancers are described by the 2017 World Health Organization (WHO) as two different clinical entities with different oncogenic pathways and prognostics (3). Other anatomical localizations of head and neck cancers include the sinus cavities and nasal fossae, which are rare and rather linked to professional and environmental exposures.
More than half of HNSCC patients experience locoregional or distant relapse despite aggressive multimodal therapeutic approaches that include surgical resection (often with neck dissection), radiation therapy (exclusive or postoperative), and adjuvant chemotherapy given as a radiosensitizer (4). After relapse, treatment options are often limited due to a high risk of complications (e.g., fistulas, dysphagia, spinal cord myelopathy) if surgery or reirradiation are attempted. If a salvage surgery (with R0 resection) or reirradiation is indeed deemed unfeasible, then systemic treatment options (detailed in this review) are proposed. Before the arrival of immunotherapy, systemic chemotherapy was employed as the standard first-line protocol with an association of cisplatin or carboplatin and 5-fluorouracil plus cetuximab (anti-EFGR antibody), known as the EXTREME protocol, which confers a dismal median overall survival (OS) of around 10 months (5).
In this review, we outline the mechanisms of resistance to the therapeutic strategies currently used in HNSCC, discuss combination treatment strategies to overcome them, and summarize the therapeutic regimens that are presently being evaluated in early- and late-phase clinical trials.
Mechanisms of Resistance to Chemotherapy
Chemotherapy is currently used as the therapeutic option for advanced HNSCC tumors (T3 or T4), concurrent to radiation, if surgical resection is deferred in the primary setting. For recurrent or metastatic disease and for cases in which first-line treatment with immunotherapy is not feasible, first-line systemic chemotherapy is advised with a protocol that includes cisplatin or carboplatin plus 5-fluorouracil and cetuximab (5, 6). Unfortunately, acquisition of chemotherapy resistance is common in patients with HNSCC and often results in local and distant failure.
Cancer Stem Cells and EMT
Epithelial-to-mesenchymal transition (EMT) is a reversible embryonic transdifferentiation program that allows partial or complete transition from an epithelial to a mesenchymal state (7). Although EMT was initially considered to be involved in invasion and metastatic spread, its key role in the initiation and development of primary tumors as well as in resistance to therapy is also demonstrated (8).
Nasopharyngeal carcinoma (NPC) is a highly invasive head–neck cancer derived from the nasopharyngeal epithelium. Preclinical studies in NPC cells demonstrate that resistance to radiotherapy and adjuvant cisplatin (DDP) chemotherapy is associated with morphological and molecular marker changes consistent with EMT. Mechanistically, depletion of NEDD4 in resistant cells leads to a partial reversion of the EMT phenotype, suggesting that NEDD4 promotes EMT features and chemoresistance of NPC in vitro (9). In a subsequent study, analysis of parental HNE1 and cisplatin-resistant HNE1/DDP NPC cells reveals that the upregulation of miR-139-5p expression inhibits proliferation, invasion, migration, and EMT. In these cells, miR-139-5p expression levels positively correlate with DDP-induced apoptosis, suggesting that miR-139-5p is associated with DDP resistance in human NPC by modulating the EMT (10).
More recently, it was demonstrated that epithelial mesenchymal crosstalk (EMC), which constitutes the interaction of the tumor stroma and associated fibroblasts with epithelial cancer cells, induces a hybrid epithelial–mesenchymal phenotype in HNSCC cells that is associated with chemotherapy resistance, via IL-6/STAT3 pathway activation (11). Interestingly, transcriptome analyses of HNSCC cell lines reveals that STAT1 and STAT3 activation enable aldo-keto reductase family 1 member C1 (AKR1C1)-induced resistance to cisplatin, which can be overcome by ruxolitinib treatment (12).
Cisplatin-resistant oral squamous cell carcinoma (OSCC) cells exhibit an enriched putative cancer stem–like signature with increased expression of CD44 and Oct-4 and enhanced sphere-forming ability, coupled with the acquisition of an EMT phenotype. This study also reveals that, irrespective of drug treatment, cell migration is significantly increased in cisplatin-resistant cell lines compared with drug-sensitive cells. In line with these observations, bioinformatic analysis of miRNA–mRNA networks in cisplatin-resistant OSCC cells reveals the upregulation of ATP-binding cassette (ABC) transporter genes, genes associated with inhibition of apoptosis (e.g., BIRC family) and cancer stem cell (CSC) marker CD44 (13).
A subpopulation of CSCs characterized by high levels of CD44v3 and aldehyde dehydrogenase-1 (ALDH1) expression has been identified in HNSCC-derived HSC-3 cells and HNSCC patient samples. In HSC-3 cells, it is shown that hyaluronan (HA) stimulates the interaction of CD44v3 with Oct-4-Sox2-Nanog, which results in the nuclear translocation of these three CSC transcription factors. Notably, it is demonstrated that Oct-4-Sox2-Nanog– dependent activation of miR-302 promotes the upregulation of the survival proteins cIAP-1, cIAP-2, and XIAP, leading to self-renewal and cisplatin resistance. In this context, transfection with an anti-miR-302 inhibitor is shown to downregulate the expression of these survival proteins and to abrogate the HA-CD44v3–mediated sphere formation and chemoresistance (14). It is noteworthy that the histone methyltransferase DOT1L is also upregulated by HA in CSCs isolated from HSC-3 cells and results in the overexpression of RhoGTPases and survival proteins involved in cell invasion and cisplatin resistance (15).
Inhibition of the aldehyde dehydrogenase 1 family member A1 (ALDH1A1) in cisplatin-resistant HNSCC cells results in downregulation of CSC markers that are diminished in migratory, self-renewal, and tumorigenic potential and resensitizes HNSCC cells to cisplatin. These observations are further validated in four ex vivo explants from HNSCC patients in which combined treatment of cisplatin and NCT-501, a theophylline-based inhibitor of ALDH1A1, results in a significant decrease in proliferating cells as compared with monotherapy (16). In a subsequent study, gene set enrichment analysis identified enhanced FGF2 expression in cisplatin-resistant ALDHhigh/CD44high HNSCC cells. Pharmacological inhibition of FGF signaling using BGJ398 preferentially targeted the ALDHhigh/CD44high subpopulation, suggesting that FGFR signaling plays a key role in in vitro stemness and in cisplatin resistance in HNSCC cells (17).
Of note, preclinical studies show that long noncoding RNA FOXD2-AS1 regulates therapeutic resistance in laryngeal squamous cell carcinoma (LSCC) by acting as an upstream activator of STAT3, which is essential to maintain cancer stemness. In LSCC patients, FOXD2-AS1 expression was predictive of poor prognosis in chemotherapy-resistant patients (18).
Overall, these studies show that the acquisition of CSC properties and the transition to a mesenchymal phenotype mediate chemotherapy resistance of HNSCC.
DNA Damage
Cisplatin triggers the formation of phosphorylated histone H2AX (γ H2AX)-positive foci at the site of DNA damage (19), dependent on ATR and via the activation of downstream CHEK1/2 (20). In HNSCC, as well as in other cancer types, altered DNA damage response signaling has been associated with resistance to chemotherapies (21).
Indeed, functional depletion of DDR effectors WDHD1, DSCC1, CSNK2B, POLR2I, and RAD54L in HNSCC cells treated with cisplatin results in decreased ATR serine/threonine kinase (ATR) phosphorylation and reduces cisplatin-induction of γ H2AX foci, suggesting that impaired DDR signaling is a driving mechanism of cisplatin resistance in HNSCC in vitro (21). Moreover, gene expression analysis of pretreatment biopsy specimens from 64 HNSCC patients treated with 5−FU/cisplatin identified that ERCC1 expression is a significant predictor of response to chemotherapy, further indicating that DNA repair is a pivotal mechanism implicated in response to chemotherapy in HNSCC (22).
At present, clinical trials are evaluating the efficacy of targeting DNA damage response in HNSCC. ATR acts as a DNA damage sensor, activating cell cycle checkpoint signaling upon DNA stress. Pharmacological inhibition of ATR using M6620 is currently being tested in combination with cisplatin and radiation therapy in the setting of locally advanced HNSCC (NCT02567422). Similarly, a modular, phase-I/Ib, open-label trial is ongoing to evaluate the efficacy of ceralasertib (AZD6738, ATR inhibitor) in combination with carboplatin (NCT02264678).
Along the same lines, because PARP is involved in DNA repair, inhibition of PARP may enhance the damaging effects of chemotherapy on tumor DNA. A phase-I/II study recently reported the safety and efficacy of veliparib, a PARP inhibitor, in combination with carboplatin-paclitaxel chemotherapy in patients with locoregionally advanced HNSCC (23). The WEE1 tyrosine kinase maintains genomic stability and regulates G2–M transition, particularly in p53-deficient tumors, protecting cells against replication stress and subsequent cell death. A phase-I clinical trial evaluating the triplet combination of AZD1775 (WEE1 inhibitor), cisplatin and docetaxel reported satisfactory results in terms of safety and tolerability as well as promising antitumor efficacy in patients with stage-III/IVB HNSCC (i.e., partial response in 5 patients and stable disease in 4 patients) (24).
Epigenetic Modifications
Resistance to cisplatin-based chemotherapy can be also modulated by epigenetic alterations. Indeed, hypermethylation of the promoter CpG islands of the neurofilament light polypeptide (NEFL) gene is associated with resistance to cisplatin-based chemotherapy in HNSCC cell lines. Functional analyses showed that NEFL interacts with tuberous sclerosis complex 1 (TSC1) at the protein level. Because TSC1 is a negative regulator of the mTOR pathway, it is suggested that NEFL downregulation results in functional activation of the mTOR pathway and, thus, cisplatin resistance. Interestingly, in this study, analysis of more than 50 HNSCC patient samples evidenced that NEFL promoter hypermethylation predicted diminished OS and disease-free survival in patients treated with cisplatin-based chemotherapy (25). A recent clinical trial evaluated the impact of mTOR pathway inhibition in HNSCC patients in the neoadjuvant setting. This study reports that rapamycin treatment was well tolerated, reduced mTOR signaling (i.e., phosphorylation of S6, AKT, and 4EBP) and tumor growth, and resulted in significant clinical responses in 4/16 of patients (1 complete response, 3 partial responses, and 12 stable disease) (26).
Histone modifiers are essential for chromatin dynamics and gene expression, and their dysregulated function may alter gene regulation in favor of oncogenic growth. Elevated expression of p21-activated kinase 2 (PAK2), a binding partner of the Rho GTPases that are implicated in chromatin remodeling, cell proliferation, and apoptosis, is correlated with chemoresistance and is associated with the poor clinical outcome of HNSCC patients. Mechanistically, PAK2 upregulates c-Myc expression, which, in turn, transcriptionally activates and induces pyruvate kinase M2 (PKM2) expression, resulting in reduced aerobic glycolysis, proliferation, and chemotherapeutic resistance of HNSCC cells (27).
Small noncoding RNAs are also key effectors of transcriptional gene silencing in HNSCC. Analysis of global miRNA expression in CD44-expressing HNSCC cells reveals that miR-629-3p expression promotes cell migration and inhibits apoptotic cell death upon cisplatin treatment. Of note, miR-629-3p-transfected cells display a significant enrichment of gene sets associated with drug metabolism and EMT. Interestingly, the role of miR-629-3p in conferring resistance to cisplatin was also observed in a xenograft model, and the expression of miR-629-3p was associated with decreased survival in HNSCC patients, potentially suggesting a physio-pathological role of miR-629-3p in resistance to cisplatin in HNSCC (28).
Enhanced expression of miR-96-5p is shown to promote cell migration but not cell proliferation, in p53-mutant HNSCC cell lines and to drive resistance to radiotherapy and cisplatin-based chemotherapy in vitro (29). Of note, this study identified PTEN, a negative regulator of the intracellular levels of phosphatidylinositol-3,4,5-trisphosphate as a direct target of miR-96-5p through the binding to its cognate site on the 3’UTR of PTEN. Interestingly, functional experiments performed in vitro shows that PTEN depletion recapitulates the biological effects of miR-96-5p overexpression in HNSCC cells as they were less prone to cisplatin-induced cell killing (29).
At present, a noninterventional clinical trial (NCT03953443) is evaluating the impact of expression and epigenetic silencing of microRNAs for predicting therapeutic response and prognosis of HPV-negative HNSCC.
Further knowledge on the epigenetic alterations that promote HNSCC chemoresistance can open the possibility for the development of therapeutic strategies that can be used as an adjuvant therapy associated with conventional chemotherapeutic drugs to enhance treatment effectiveness.
Evasion of apoptosis
The adaptive response to chemotherapy in HNSCC is modulated by changes in the expression of pro- or anti-apoptotic proteins and include defects in cellular responses caused by mutations of tumor suppressor gene TP53 (30, 31).
Survivin (BIRC5), a member of the inhibitor of apoptosis (IAP) gene family, is shown to be significantly upregulated in HNSCC primary tumors and cell lines and to be particularly highly expressed in HPV-negative patients who generally respond poorly to cisplatin treatment (32). Immunohistochemical and mutational analyses on HNSCC biopsies from patients displaying high levels of nuclear survivin (BIRC5) identified the presence of the somatic mutation c.278T>C (p.Phe93Ser). Functional characterization of this mutant by ectopic expression and microinjection experiments revealed that it attenuates the cytoprotective activity of survivin against chemoradiation-induced apoptosis. Therefore, genetic inactivation of survivin may promote an increased therapy response in cancer patients (33).
Interestingly, pharmacological inhibition of survivin using the small molecule YM155, either as a single agent or in combination with cisplatin, evidenced a significant dose-dependent decrease in cell proliferation and the reversion of cisplatin resistance in in vitro and in vivo models of HNSCC. Mechanistically, YM155 induced a rapid reduction of survivin in the cytoplasm, which is key for its antiapoptotic function (32). Thus, survivin inhibition might potentially be a novel strategy to enhance the effectiveness of chemotherapy in HNSCC.
The x-linked inhibitor of apoptosis (XIAP) is an E3 ubiquitin protein ligase that functions through binding to tumor necrosis factor receptor–associated factors TRAF1 and TRAF2 to inhibit apoptosis. Analysis of XIAP expression in tumor samples from 60 patients with advanced HNSCC, before and after chemotherapy, evidenced that XIAP is a predictor of cisplatin response and prognosis for patients with advanced HNSCC. Interestingly, preclinical experiments show that inhibiting XIAP expression with siRNA in XIAP-high HNSCC cells markedly increased their sensitivity to cisplatin treatment (30). Of note, the dual cIAP/XIAP antagonist ASTX660 significantly delays growth of both HPV- and HPV+ human tumor xenografts in combination with radiotherapy.
Resistance to the activity of TNF-related apoptosis inducing ligand (TRAIL), an effector of tumor cell–specific apoptosis, is associated with HPV positivity in HNSCC in vitro. HPV-positive HNSCC cell lines were sensitized to TRAIL-induced cell death by bortezomib-mediated proteasome inhibition via the activation of caspases 8, 9, and 3; increased membrane expression of TRAIL-R2; and G2/M arrest. Of note, XIAP depletion also augmented HPV-positive HNSCC cell death in response to TRAIL alone and in combination with bortezomib (34).
Tumor Microenvironment (TME)
HNSCC tumors are commonly associated with hypoxia, which is characterized by an acute or chronic decline in oxygen tension.
Activin receptor–like kinase (ALK)-1 represents a promising target for antiangiogenic therapy in solid tumors. activin receptor–like kinase-1 ligand trap (ALK1-Fc) is a chimeric protein consisting of the ALK1 extracellular domain fused to the Fc-part of an antibody. ALK1-Fc prevents the binding of BMP9 and BMP10 to the endothelial ALK1 receptor, which results in decreasing angiogenic responses (35). Therapeutic combination of ALK1-Fc with cisplatin is shown to inhibit tumor growth in HNSCC in vivo models more efficiently than chemotherapy alone. Treatment of mice with ALK1-Fc strongly decreased the microvascular density of tumors, increased the pericyte coverage of the remaining tumor vessels, and decreased the hypoxia within the tumor (36). Interestingly, results of an early-phase clinical trial show that the ALK1-Fc displayed promising antitumor activity in HNSCC patients with advanced refractory cancer (35).
Signaling via the SDF-1/CXCR4 axis, a chemokine-receptor pathway, is involved in cancer progression due to its roles in modulation of dendritic cells, enhanced matrix metalloproteinase activity, and the induction of TNF-alpha production and angiogenesis. Analysis of the expression of SDF-1 and CXCR4 in a cohort of 221 patients with locally advanced HNSCC evidenced that SDF-1 is associated with resistance to adjuvant radiotherapy concurrent with cisplatin-based chemotherapy (37). In this study, neither SDF-1 nor CXCR4 expression were associated with distant metastasis or with OS. The functional basis of these observations as well as the potential role of SDF-1/CXCR4 as a therapeutic target to overcome treatment resistance in HNSCC remains to be determined.
In preclinical models of OSCC, combination therapy of cisplatin and inhibitors of VEGFR (i.e., pazopanib and nintedanib) was more potent than treatment with chemotherapy alone (38). The efficacy and toxicity of docetaxel with or without vandetanib, an inhibitor of VEGFR, RET, and EGFR, was investigated in patients with advanced recurrent or metastatic HNSCC. This trial shows only a minor trend toward improved PFS for the combination arm (39). Of note, a current clinical trial is ongoing to evaluate the combination of atezolizumab (humanized IgG1 antibody against PD-L1) and bevacizumab (monoclonal antibody developed against VEGF) in patients with recurrent or metastatic HNSCC (ATHENA, NCT03818061).
The TME also constitutes a reservoir of cancer-associated fibroblasts (CAFs) which, in a close crosstalk with tumor cells, enhance the production of growth factors, cytokines, chemokines, and inflammatory mediators to promote tumor growth (40). CAFs are observed in both primary and metastatic HNSCC, and oral CAFs are reported to acquire rapid growth and increased proliferation and viability compared with normal oral fibroblasts (40). CAF-secreted paracrine factors increase HNSCC migration, invasion, and proliferation in vitro and promote tumor growth and metastases in vivo (i.e., orthotopic floor-of-the-mouth tumor model) (41).
CAFs are also known to mediate resistance to anticancer drugs in HNSCC. In HNSCC cell lines, culture with conditioned medium from a tumor cell/CAF coculture induced cisplatin resistance and increased their colony-formation capacity (11). Interestingly, exosomal miR-196a released by CAFs targets CDKN1B and ING5 and, thus, confers cisplatin resistance in vitro (42). Interestingly, in this context, high levels of plasma exosomal miR-196a are clinically correlated with poor OS and chemoresistance in HNSCC patients. In line with these observations, it is demonstrated that, in OSCC patients, CAFs secrete increased levels of midkine (a heparin-binding growth factor that promotes carcinogenesis and chemoresistance) and abrogated cisplatin-induced cell death (43). Finally, analysis of tumor specimens obtained from 60 OSCC patients who underwent surgery following 5-fluorouracil-based chemoradiotherapy revealed that higher numbers of CAFs and tumor-associated macrophages (TAMs) were significantly correlated with a poor prognosis, suggesting their potential as biomarkers for predicting the clinical response to 5-FU-based chemoradiotherapy (44).
Understanding how CAFs contribute to drug resistance, proliferation, invasion, and metastasis might open up new strategies for the diagnosis, prognosis, and therapy of HNSCC.
Mechanisms of Resistance to Cetuximab
Initially described in 1962 by Cohen (45, 46), the epidermal growth factor receptor (EGFR) is a transmembrane receptor with tyrosine kinase activity (47). Several ligands bind specifically EGFR (e.g., epidermal growth factor [EGF], tumor growth factor-alpha [TGF-alpha], and amphiregulin), and others (betacellulin, heparin-binding growth factor [HB-EGF], and epiregulin) bind to both EGFR and ErbB4 (48–50). Ligand binding induces the homo- or hetero-dimerization of EGFR, which is followed by the activation of downstream signaling, mainly via the RAS–RAF–MEK–ERK, the PI3K–AKT–mTOR, and the JAK–STAT cascades (51). These pathways are involved in the carcinogenesis and invasiveness of many cancer types (52).
Because EGFR is overexpressed in 80%–90% of HNSCC cases, tumors are often addicted to EGFR signaling for sustained survival and proliferation, and this overexpression is correlated with poor prognosis and treatment outcomes (53–55), therapies targeting EGFR have been widely evaluated for HNSCC (56–58): first, intravenous anti-EGFR antibodies that bind to the extracellular domain of the receptor causing its internalization to prevent its activation by other ligand–receptor interactions (59) while favoring antibody-dependent cell-mediated cytotoxicity (i.e., ADCC, which refers to the linking to innate and adaptive antitumor immune responses via NK cells and antigen-presenting cells that lead to EGFR-specific T cells) (60–63) and ii-oral EGFR tyrosine kinase inhibitors (TKI) binding to the intracellular domain of EGFR inhibiting its autophosphorylation (blocking of the ATP binding to the intracellular tyrosine kinase domain of EGFR) and downstream signaling (56, 64, 65).
Cetuximab (CTX), a monoclonal antibody targeting the EGFR extracellular domain, is to date the only targeted therapy that has demonstrated benefits in OS in combination with both radiotherapy for patients with locally advanced HNSCC (66) and chemotherapy (platinum, 5-FU, and CTX) as the first-line treatment of patients with recurrent and/or metastatic HNSCC (5, 67). Of note, CTX has never proven to be effective postoperatively (56, 58, 68).
Despite our better understanding of HNSCC biology (51, 69–71), no other molecular-targeted agent has been approved for HNSCC (12). Furthermore, CTX has shown limited efficacy in HNSCC with an overall response rate of 10%–20%, contrasting with the high rates of EGFR overexpression (51, 72). This underlines the existence of resistance mechanisms, remaining unresolved, but for which several hypotheses have been proposed (48, 56, 64, 67, 73–81) (4, 12, 21, 24, 29–37). The different type of resistance mechanisms to CTX could be defined as follows: alterations of the EGFR-ligand binding, alterations of the EGFR downstream signaling effectors, parallel/bypass pathway activation, alterations of proteins involved in classic cancer pathways, EMT, epigenetic alterations and establishment of an immunosuppressive TME (Figure 1). In this review and for each CTX resistance mechanism, we report preclinical (based on HNSCC cell lines/xenograft) and clinical evidence of CTX resistance as well as ongoing clinical trials of CTX-based combined therapies to overcome CTX resistance (Table 1).
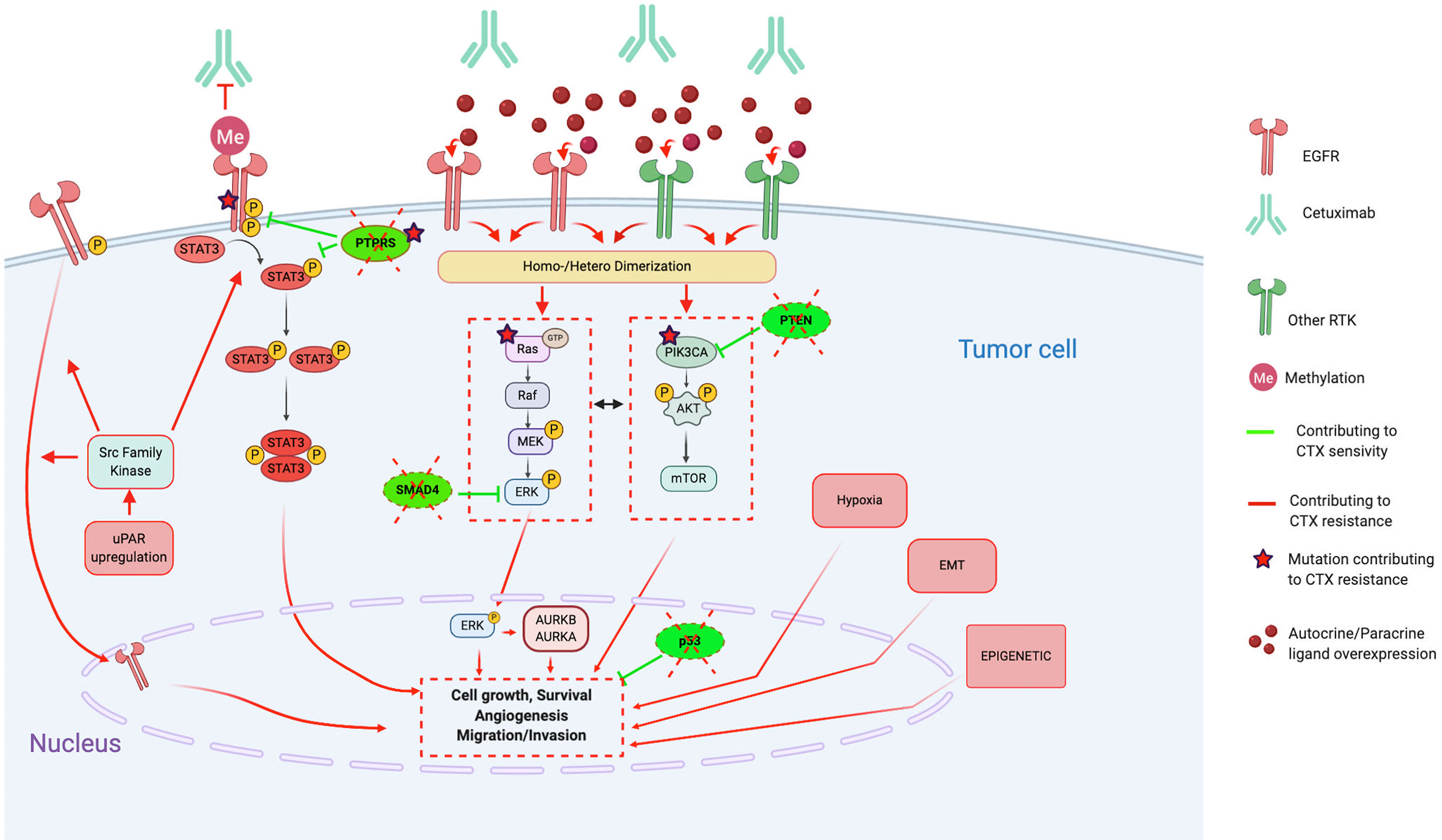
Figure 1 Molecular mechanisms contributing to Cetuximab resistance, in particular through alterations of the EGFR pathways, activation of bypass pathways and alterations of downstream signaling effectors. Red lines and arrows show mechanisms contributing to Cetuximab resistance, and green lines and arrows show mechanisms contributing to Cetuximab sensitivity. (CTX, Cetuximab; EGFR, Epidermal Growth Factor Receptor; RTK, Tyrosine Kinase Receptor; EMT, epithelial-mesenchymal-transition; uPAR, urokinase-type plasminogen activator receptor; STAT3, signal transducer and activator of transcription 3; PTPRS, Transmembrane Protein Tyrosine Phosphatase RPTPsigma; PTEN, phosphatase and tensin homolog; AURKA, Aurora Kinase A; AURKB, Aurora Kinase B).
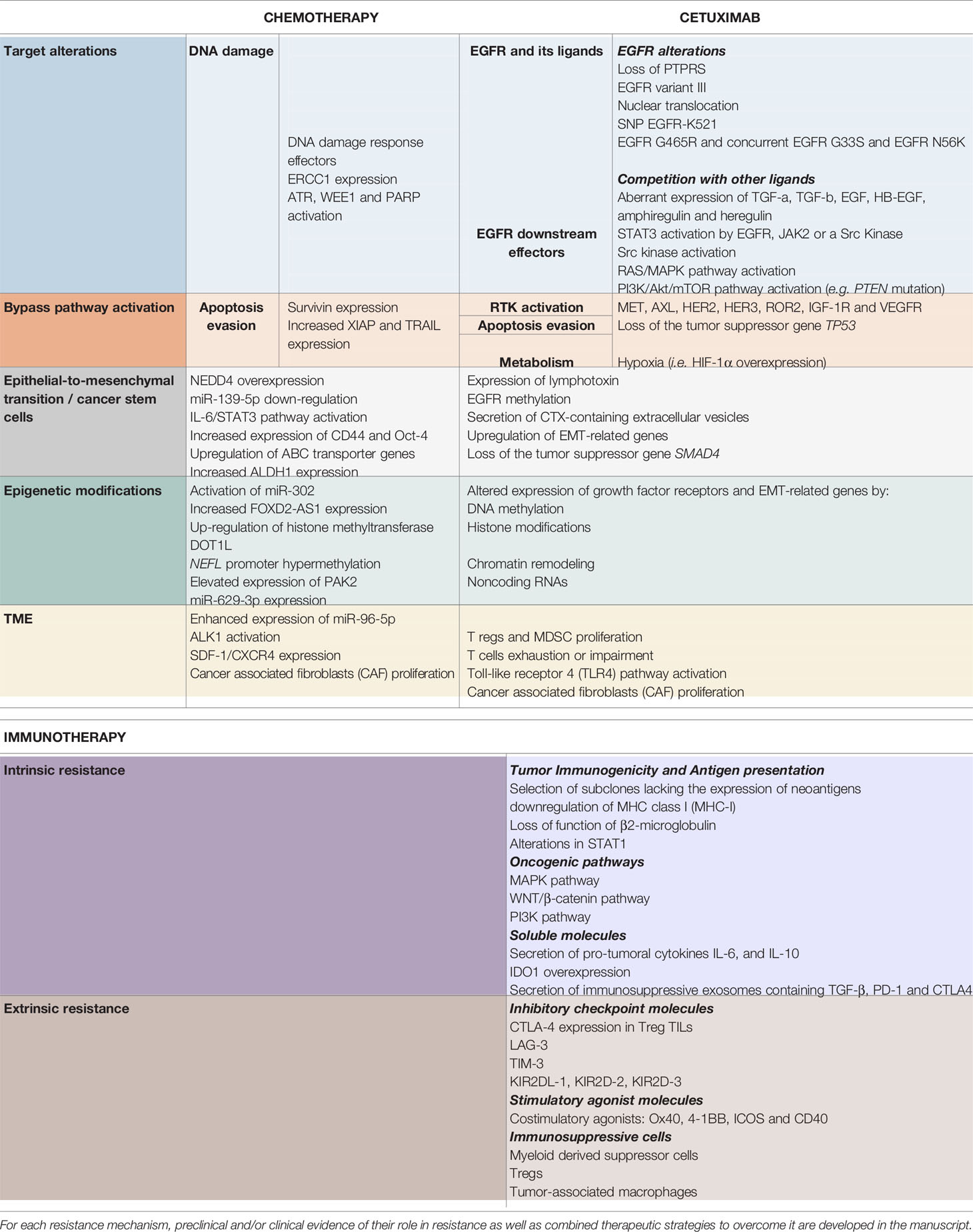
Table 1 Resistance mechanisms to chemotherapy (left), cetuximab (middle) and immunotherapy (right), described in head and neck squamous cell carcinoma.
Alterations in EGFR and Its ligands
Alterations of the antibody–receptor interactions can be induced by either alteration of the EGFR (82–92) or via competition with other EGFR ligands (87, 93–98).
EGFR Alterations
Several EGFR alterations have been reported in preclinical studies. First, loss of the EGFR phosphatase transmembrane protein tyrosine phosphatase RPTP sigma (PTPRS), which is known to directly interact, dephosphorylate, and inactivate EGFR (99), enhances EGFR-induced transformation and promotes EGFR/PI3K pathway activation, resulting in resistance to EGFR inhibition (88). Furthermore, constitutive activation of the EGFR, such as the EGFR variant III (EGFRvIII, activating mutation), results in activation of several downstream modulators (preferentially the PI3K pathway) and participates in increasing tumorigenicity and CTX resistance (92).
On the other hand, several CTX resistance mechanisms are based on the perturbation of CTX binding on EGFR. Indeed, contrary to the classic EGFR located on the plasma membrane, the nuclear EGFR (translocation mediated by the Src family kinases) cannot be targeted by CTX and, thus, functions as a transcription factor for several factors, inducing proliferation (cyclin D1, iNOS, B-myb, and aurora kinase A) (91). Moreover, the single nucleotide polymorphism encoding EGFRK521 (K-allele), which is expressed in >40% of HNSCC cases, has been shown to reduce stability of the EGFR and, thus, the affinity for CTX binding (85). Finally, the two concurrent, nonsynonymous missense G33S and N56K mutations in the extracellular domain of EGFR restrict adoption of a fully closed (tethered) and inactive EGFR conformation, thus, not permitting binding of CTX to the EGFR (82, 86).
Besides the preclinical evidence, clinical studies support some of the previously cited CTX-resistance mechanisms. Based on analysis of n=31 HNSCC (oral cavity) cases, Morris et al. find significant PTPRS loss or deletion in 32% of cases (88). They observed pathway activation (elevated levels of phospho-EGFR and phospho-AKT) in tumors with PTPRS deletion but not in tumors lacking PTPRS deletion. Smilek et al. show that a somatic EGFR mutation located in exon 19 may contribute to the limited clinical response to therapy with CTX plus radiotherapy (n=2/29 patients with advanced HNSCC) (86). Moreover, the high EGFRvIII expression, detected in 17%–42% of HNSCC tumors, was significantly and independently associated with shorter progression-free survival in patients with recurrent or metastatic HNSCC treated by CTX + Docetaxel (87, 92). For some authors, the role of this EGFR polymorphism in CTX resistance remains limited (100). The EGFR extracellular domain mutation G465R is reported to confer resistance to CTX by altering its binding to EGFR in a patient with a regional neck recurrence of an oral cavity HNSCC (83).
CTX-based combined therapy has been tested in the preclinical as well as clinical setting to overcome the previously cited resistance mechanisms. Dasatinib (BMS-354825), a tyrosine kinase inhibitor (TKI), limits the nuclear EGFR translocation (by blocking the Src family kinases), which leads to increased EGFR on the plasma membrane and restores sensitivity to CTX (90). Thus, Dasatinib is currently evaluated in combination with CTX in patients with recurrent HNSCC (NCT01488318, phase 2) (84) as well as in combination with CTX/cisplatin/RT (NCT00882583, phase 1). Interestingly, in the CTX + Dasatinib combination setting, patients with low serum IL6 have shown clinical benefit and improved OS (NCT01488318) (84).
Aberrant Expression of EGFR Ligands
The CTX-EGFR interactions are reduced in the context of competitive interaction with the overexpression of some ligands, such as TGF-α, TGF-β, EGF, HB-EGF, amphiregulin, and the aberrant HER3 ligand heregulin-expression (87, 93–98). Thus, this autocrine/paracrine growth factor production reduces CTX effectiveness in several HNSCC cell lines.
Indeed, based on the analysis of tumor biopsies from n=47 recurrent/metastatic HNSCC, the amphiregulin overexpression (representing 47% of cases) was a statistically significant prognostic factor of worse OS and progression-free survival (87). Yonesaka et al. report that n=2/28 HNSCC tumor samples that presented aberrant heregulin expression comparable to that of HNSCC CTX-resistant cell lines (FaDuCR cells) were resistant to CTX (94).
Interestingly, FaDuCR recovered the sensitivity to CTX in combination with Pertuzumab (anti-HER2 antibody) (94). Indeed, Pertuzumab prevents the binding of HER2 with its ligand (heregulin), avoiding the coupling of HER2/HER3, thus, resulting in the absence of the HER3-AKT pathway activation, which is responsible for inducing CTX resistance.
Alterations of EGFR Downstream Signaling Effectors
Activation of downstream signaling effectors, such as STAT3 (signal transducer and activator of transcription 3) (77, 84, 101–106), Src Kinases (64, 107, 108), RAS/MAPK pathways (86, 96, 109–115), and PI3K/Akt/mTOR pathway (102, 105, 116–127) could induce CTX resistance independently of the EGFR-ligand activation.
STAT3 Activation
STAT3, a member of the STAT family of transcription factors, is considered as an oncogene activated in several cancers, including HNSCC (128). Its activation could be driven by EGFR as well as in an independent EGFR way by another growth factor receptor, the Janus kinase 2 (JAK2) or by the Src kinase family. Furthermore, loss of the PTPRS tumor suppressor gene that dephosphorylates STAT3 may lead to permanent activation of STAT3. Several studies report that hyperactivation of STAT3 is implicated in CTX treatment resistance.
Indeed, several HNSCC cells lines that develop acquired resistance to CTX are characterized by increased total STAT3 expression (77). The role of STAT3 in HNSCC cell CTX that acquires resistance is supported by recovering increased sensitivity to CTX (greater antiproliferative effects and cytotoxicity) when STAT3 is knocked down (104) or by blocking JAK2–STAT3 signaling (using miR-204) (101). Moreover, analysis of n=22 samples from patients with HNSCC tumors that recurred following CTX treatment finds increased phosphorylated STAT3 (103).
Regarding CTX-based combinations, guggulsterone, a natural compound contained in the Commiphora mukul plant resin used in Indian ayurvedic medicine and considered as an anti-STAT3 agent, enhances the efficacy of CTX when combined with CTX (106). Moreover, the combination of CTX + JAK2 inhibitor (miR-204) inhibits STAT3 activation, resulting in inhibition of angiogenesis and promotion of in vivo CTX sensitivity (101). Given that STAT3 may be activated by the Src kinase family, Dasatinib (SRC inhibitor), which is tested in combination with CTX in phase-2 (84) and phase-1 clinical trials (NCT01488318 and NCT00882583, respectively), could provide some insight about the utility of the STAT3 inhibition in overcoming CTX resistance.
Activation of Src Kinases
Src family kinases are frequently overexpressed and/or activated in several cancers, including those arising in the head and neck (85). These nonreceptor protein tyrosine kinases play critical roles in signaling pathways, regulating cell division, motility, adhesion, angiogenesis, and survival (129). Thus, activation of Src kinases could be involved in proliferation/migration/invasion of cancer cells as well as in treatment resistance.
Based on gene expression profiles of CTX-resistant OSCC cells as well as of publicly available data sets, Uzawa et al. identify a 12-gene expression signature of CTX resistance, including the urokinase-type plasminogen activator receptor (uPAR) (107). They show that CTX resistance could be mediated by uPAR upregulation. Indeed, through the uPAR/integrin β1/Src/FAK signal circuit, the uPAR upregulation activates ERK1/2 phosphorylation to maintain cell proliferation/invasion resulting in CTX resistance in vitro and in vivo even in the absence of EGFR overexpression or acquired activating mutations. Src kinases could also induce CTX resistance by EGFR-ligand independent transactivation (cell-substratum adhesion), which phosphorylates ErbB3 to form a heterodimer complex, inducing proliferation via AKT (108).
Based on previous evidence, the CTX-based combination with the Src inhibitor-1 or resveratrol (uPAR inhibitor) are shown to overcome CTX resistance in vitro and in vivo (tumor growth suppression and uPAR downstream protein downregulation), respectively.
Activation of the RAS/MAPK Pathway
The family of mitogen-activated protein kinases (MAPK) are a family of serin-threonin kinases implicated in the regulation of the majority of physiological cellular processes, including proliferation, differentiation, and apoptosis in response to changes in the cellular environment (130). In particular, the Ras/Raf/MEK/ERK1/2 (extracellular signal-regulated protein kinases) cascade is the MAPK signaling cascade most frequently associated with carcinogenesis of several cancer types (131). Regarding HNSCC, this signaling cascade can be activated by several tyrosine kinase receptors, such as EGFR, as well as independently of them by alterations of the Ras/Raf oncogenes. This highlights the variety of CTX-resistance mechanisms involving this signaling cascade.
First, the MAPK signaling pathway activation related to the HNSCC CTX resistance could involve a RAS-activating mutation (G12V HRAS) (113). The restored sensitivity to CTX by silencing H-Ras in H-Ras mutant HNSCC cell lines reinforces this observation (132). However, activation of the RAS/MAPK pathway even in the absence of constitutive gene mutations could lead to CTX resistance (111). Indeed, overexpression the K-Ras, H-Ras, and N-Ras proteins (96) leads to CTX resistance. Furthermore, dysregulation of the regulating proteins of the RAS/MAPK pathway could also contribute to CTX resistance as supported by the low expression of DUSP5 and DUSP6 (negative regulators of ERK1/2) and upregulation of AURKB (100) and AURKA (114), which are key regulators in mitosis.
In the clinical setting, Braig et al. show that acquisition of RAS mutant clones (KRAS G12S, G13C; NRAS Q61K, NRAS A146P; HRAS G13R) correlates significantly with clinical resistance to CTX in a cohort of n=20 patients treated by CTX/platinum/5-fluorouracil treatment with monitoring of the circulating tumoral DNA (ctDNA) (110). The role of the KRAS p.Gly12Val mutation in CTX resistance previously found in vitro, is also demonstrated in only one patient carrying this mutation (among the n=29 studied) associated with an absence of response to treatment (86). Rampias et al. confirm that the HRAS mutation (n=7/50 patients with HNSCC) is associated with de novo resistance to CTX-based therapy (113). Overall, Bossi et al.’s observations (cohort of n=40 recurrent/metastatic HNSCC) are in accordance with others and show that overactivation of the RAS pathway leads to CTX/platinum resistance (111).
On the basis of their results, especially the interesting observation of the crosstalk between the RAS/RAF/MAPK and PI3K/AKT pathways, Rampias et al. tested the combination of CTX + a PI3K inhibitor (LY294002) in an H-Ras mutated cell line and found a marked reduction of their viability (113). Apigenin, an ERK 1/2 inhibitor, in combination with CTX resulted in a significant decrease of HNSCC CTX-resistant cell survival (112). Interestingly, the combination of CTX + tipifarnib (farnesyltransferase inhibitor) showed enhancement of the tipifarnib antitumor effect through concomitant ERK inhibition in vitro and in vivo (109). Finally, a combination of CTX with inhibition of the ERK upregulators, i.e., aurora kinase knockdown (siRNA) and inhibitor (the pan aurora kinase inhibitor R763), showed inhibition of proliferation and increased apoptosis in HNSCC cells lines (112, 114, 115).
PI3K/Akt/mTOR Pathway
The phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathways are involved in several physiological as well as pathological cellular processes, including proliferation, differentiation, survival, and motility (133). In HNSCC, PI3K/AKT/mTOR signaling is active in more than 90% of HNSCC as a result of EGFR activation, PI3K overexpression, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PI3KCA) mutations/amplifications, and PTEN mutation (116, 134). Activated PI3K/AKT/mTOR signaling is related to radiotherapy and cytostatic drug resistance, likely through enhanced DNA-repair mechanisms.
Several genetic alterations causing PI3K/AKT/mTOR activation, such as activating mutations in the oncogene PI3KCA or inactivating mutations in the tumor suppressor protein PTEN, are driving CTX resistance in different HNSCC cell lines (125, 127). Indeed, Izumi et al. show that loss of PTEN conferred independence from EGFR activity and resistance to EGFR inhibition by CTX in terms of downstream signaling, proliferation, and tumor growth both in vitro and in in vivo xenograft models (119).
Moreover, Eze et al. recently reported the analysis of PTEN and PIK3CA expression in samples from patients with recurrent or metastatic HNSCC enrolled in two trials of cetuximab-based therapy (n=48 patients in the E5397 trial and n=37 in the NCI-8070 trial) (117). Patients with low PTEN expression had significantly worse survival.
Thus, CTX-based combined therapy has been realized using ATP-competitive PI3K inhibitors as well as mTOR inhibitors (Rapamycin and analogues). Regarding ATP-competitive PI3K inhibitors, the CTX combination with Buparlisib or BKM120 demonstrates the highest antiproliferative effect and inhibition of PI3K/protein kinase B, AKT/mTOR signaling pathways in vitro (122) and in vivo (121). The BYL719 (PI3Kα specific inhibitor), namely Alpelisib (123) and the Copanlisib (highly selective, pan-class I PI3K inhibitors) (120), are shown to improve CTX-induced tumor inhibition in HNSCC CTX-resistant cell lines and PDX. Interestingly, the combination of CTX plus the PKI-587 (PI3K/mTOR inhibitor), namely Gedatolisib, which restored sensitivity to CTX in resistant HNSCC cell lines and xenografts (124), is found to have a greater synergistic enhancement of the CTX effectiveness, especially in basal-like HNSCC cells with mutated CDKN2A (118). Regarding mTOR inhibitors, Rapamycin (Rad001) (126) and Temsirolimus (105) show improving CTX antiproliferative effects in xenografts.
Interestingly, combinations of CTX with PI3K/Akt/mTOR inhibitors are widely investigated in clinical trials. Regarding ATP-competitive PI3K inhibitors, there are several phase-1 and -2 trials enrolling patients with HNSCC to be treated by CTX + Buparlisib (BKM 120) (NCT01816984, phases 1 and 2), Alpelisib (BYL719) (NCT01602315, phases 1b and 2) (126), and Copanlisib (NCT02822482, phases 1b and 2, COPAN-ORL06, specifically for patients harboring a PI3KCA mutation/amplification and/or a PTEN loss). PX-866, a noncompetitive PI3K inhibitor, was also tested in combination with CTX (NCT01252628, phases 1 and 2). Analogues of the rapamycin, temsirolimus (NCT01256385, phase 2, MAESTRO HN) and everolimus (NCT01637194, phase 1; NCT01283334, phases 1 and 2) (57) have already brought some interesting results to overcome CTX resistance.
Bypass-Pathway Activation
Another resistance mechanism involves the abnormal activation of parallel signaling pathways to counteract the blockade of the EGFR signaling by CTX. Thus, cancer cell survival is ensured by increased expression/activation of alternative receptor tyrosine kinases (RTK) (50, 77, 79, 123, 135–150), ensuring the activation of several parallel pathways, including the VEGF pathway (72, 151–153).
Receptor Tyrosine Kinases
Among the growth-factor receptor family, RTK are transmembrane receptors implicated in several physiological as well as pathological (oncogenesis) processes (154). The binding ligand-extracellular domain induces receptor dimerization, activation of the intrinsic tyrosine-kinase activity of the RTK, and activation of downstream signaling cascades implicated in cell proliferation, differentiation, motility, survival, and cell–cell communication (155). Thus, activation of these RTKs is a mechanism of resistance to CTX during HNSCC treatment (50, 77, 79, 123, 135–150).
Indeed, the increased expression and activation of RTK, such as MET, AXL, HER2, HER3, and ROR2, are reported in several CTX-resistant cell lines (148) as well as in vivo (PDX). For example, MET/HGF (146) as well as AXL (123, 139, 148) overexpression and activation stimulate cell proliferation despite CTX treatment in vitro and in vivo, especially through MAPK downstream signaling. Recently, McDaniel et al. investigated the AXL-mediated CTX-resistance mechanisms in HNSCC and report that the tyrosine 821 of AXL mediates resistance to CTX by activation of c-ABL (oncoprotein) (156).
Other ErbB family members, ErbB2 (HER2) (102) and ErbB3 (HER3), could also be implicated in CTX resistance. Indeed, Yonesaka et al. report the persistence of ERK 1/2 signaling caused by the permanent activation of ErbB2 signaling (amplification of the receptor ErbB2 or upregulation of the ligand heregulin) induces CTX resistance in HNSCC cell lines (141). The restoration of CTX sensitivity through inhibition of ErbB2 or disruption of ErbB2/ErbB3 heterodimerization reinforce their observations. On the other hand, this heterodimerization also highlights the role of HER3 activation in resistance to CTX treatment of some HNSCC cell lines (135). The permanent activation of ErbB3/Akt signaling could be caused by an autocrine neuregulin expression (autocrine loop) as well as by aberrant HER3 ligand heregulin expression (94). Furthermore, increased activity of the IGF1R signaling pathways has been reported in several CTX-resistant HNSCC cell lines (136, 137, 143). IGF1R and HER3 activations with partial EGFR persistent activity are intertwined during CTX resistance as supported by the ability of a multitarget mAb mixture against EGFR, HER3, and IGF1R to overcome CTX resistance.
These preclinical observations are supported by clinical evidence. Indeed, in a retrospective cohort of n=57 patients with recurrent/metastatic HNSCC, patients who presented HGF/MET pathway overexpression and activation had worse prognoses (138). Moreover, Chung et al. report the case of a patient with recurrent HNSCC who presented an interesting response to AMG-479 (a monoclonal antibody against IGF-1R) after CTX resistance. Tumor sample analysis suggests the potential benefit of a combined therapy using AMG-479 plus CTX (142).
To overcome CTX-resistance, several combined therapies using RTK inhibitors have been tested. The BET inhibitor JQ1, which binds preferentially to the bromodomains of BRD4, abrogates the expression of the alternative RTK (HER3 and AXL), resulting in significantly delayed acquired resistance in two PDX models of HNSCC (148). Combined with CTX, MET inhibitor PHA-665752 is also shown to restore CTX sensitivity in vitro and in vivo, especially by decreasing akt and ERK1/2 phosphorylation (146, 147). Inhibition of the AXL receptor is explored by using imatinib (which targets c-Abl) in CTX-resistant HNSCC PDX (156). This led to complete tumor regression and a prolonged effect (no recurrence up to 3 months after cessation of treatment). Moreover, the Lida et al. experiment brings general support to the implication of several ErbB family members in CTX resistance. They find that the pan-HER mixture of six antibodies targeting EGFR, HER2, and HER3 decreases HER family receptors in acquired CTX-resistant HNSCC cells lines and overcomes CTX resistance in PDX (98). More precisely, a dual EGFR/HER2 inhibitor with CTX plus Afatinib shows significantly improved tumor volume reduction in CTX-resistant xenografts compared with either agent alone in monotherapy (140). ErbB3 inhibition has also been realized in vitro and in vivo using MM-121 (97, 113) as well as CDX-3379 (ErbB3-specific blocking antibody) (93). These combinations inhibit proliferation through inhibition of PI3K/Akt and ERK signaling pathways. When combined with CTX, the anti-IGF-IR antibody (IMC-A12) A12 provides important inhibitions of cell proliferation and migration in vitro and in vivo (regression of tongue cancer cell xenografts) (144). Although the rationale of dual VEGF and EGFR inhibition is proposed in several other cancers (157), Argiris et al. show that combined targeting of EGFR with CTX and VEGF with bevacizumab enhances growth inhibition both in vitro and in vivo (153).
Given that ErbB3 activation induced by heregulin is previously described as a CTX-resistance mechanism, the combination of Patritumab (U3-1287), an anti-HER3 monoclonal antibody, and CTX with platinum-based therapy was evaluated in a randomized, double-blind, phase-II study of first-line treatment of patients with recurrent or metastatic HNSCC (NCT02633800) (158). Although tolerable, the combination Patritumab + CTX + platinum was not superior to CTX + platinum. Based on the previous rationale as well as on a phase-I study (149), Deeken et al. evaluate the combination of lapatinib (which blocks both EGFR and ErbB2) plus CTX (NCT01184482) in patients with advanced solid malignant tumors, including HNSCC. Results were interesting with an overall response rate of 17% and a clinical benefit rate of 67%.
CDX-3379, an anti-ErbB3 monoclonal antibody, has been recently reported to inhibit tumor ErbB3 phosphorylation in HNSCC and induce measurable tumor regression and was well tolerated (93). Thus, a phase-2 clinical trial (NCT03254927) is ongoing and aims to determine the clinical benefit, safety, and tolerability of combining CDX-3379 and CTX in patients with advanced HNSCC who have previously received CTX and progressed.
IGF-1R inhibitors are also widely explored in combination with CTX for recurrent/metastatic HNSCC. Glisson et al. as well as Ferrarotto et al. report no improvement of progression-free survival and OS using the Cixutumumab + CTX compared with CTX alone (159, 160). The OSI-906, a dual kinase inhibitor of both IGF-1R and insulin receptor was evaluated in combination with CTX among patients with HNSCC (NCT01427205, phase 2), but results are not available. More recently, the combination of CTX plus the anti-IGF-1R antibody A12 (IMC-A12) was evaluated in the neoadjuvant setting for patients with HNSCC NCT00957853 (Phase 2).
Finally, VEGF inhibitors combined with CTX have been also investigated. Although some results are not available (NCT00906360, phase 1, CTX + Sunitinib), others are contradictory. Indeed, although some trials report that bevacizumab + cisplatin + CTX + intensity-modulated radiation therapy (IMRT) in locally advanced HNSCC is associated with favorable efficacy outcomes (NCT00968435, phase 2), Argiris et al. find that adding bevacizumab increases toxicity without apparent improvement in efficacy (NCT00703976, phase 2) (151). Thus, the potential clinical benefit of combined EGFR–VEGF targeting is not clearly established.
Other Signal Transducers
Several proteins involved in classic cancer pathways, such as proliferation, apoptosis, invasion, and metastasis, could be altered and implicated in CTX resistance during HNSCC treatment (64, 115, 161–165).
Among all somatic genomic alterations in HNSCC, the tumor suppressor gene TP53 is the most frequent (166), highlighting its importance in carcinogenesis and progression. Indeed, although the tumor suppressor protein p53 has a critical role in cell cycle arrest, apoptosis, and senescence, loss of its function is linked to disease progression and treatment response (64). Regarding CTX, comparative analysis of sensitive vs. CTX-resistant HNSCC cells reveals the central role of the loss of p53 in the development of acquired resistance to CTX (163).
The precise role of hypoxia in acquired resistance to cetuximab is not clearly established, and further studies are needed. Indeed, Boeckx et al. find that the sensitivity to CTX is not altered but increased in HNSCC cells exposed to prolonged hypoxia (164). On the other hand, Lu et al. report that HNSCC cells with acquired CTX resistance express a high level of the alpha subunit of the hypoxia-inducible factor-1 (HIF-1α) and are highly glycolytic (aerobic glycolysis, i.e., the Warburg effect). Furthermore, the experimental overexpression of HIF-1α confers resistance to CTX as well as abolishes CTX-mediated radiosensitization in HNSCC cells (161).
These preclinical observations have caused Lu et al. to explore the inhibition of hypoxia and its relationship with CTX efficacy. Downregulation of HIF-1α by siRNA or a small molecule inhibitor (1-methyl 1, 9 PA) enhances response of CTX-resistant HNSCC cells to CTX plus radiotherapy (161). Finally, Lu et al. confirm that CTX inhibits HNSCC cell proliferation through inhibition of glycolysis and that the combination of CTX + oxamate (inhibition of LDH-A, an enzyme catalyzing the conversion of pyruvate to lactate in anaerobic conditions) improves the therapeutic effect of CTX in cancer cells (162). As a continuation of their work on the role of hypoxia and glycolysis during HNSCC treatment, Lu et al. explore the role in CTX resistance of the mitochondrial enzyme pyruvate dehydrogenase kinase-1 (PDK1), known to allow the switching glucose metabolism toward aerobic glycolysis in cancer cells (165). They found that the combination of CTX plus PDK1 knockdown (siRNA) or with pharmacological inhibition of PDK1 with dichloroacetic acid (DCA) overcomes CTX-resistance in vitro and in vivo (xenografts) thanks to the overproduction of reactive oxygen species (ROS) and the subsequent apoptosis.
Epithelial-to-Mesenchymal Transition
The importance of EMT in human disease, especially in carcinogenesis, has been reviewed elsewhere (167). The EMT can be considered as a continuum of multiple and dynamic transitional states whereby cells exhibit epithelial, intermediate, and mesenchymal phenotypes. Regarding HNSCC, acquisition of an EMT phenotype (modulation of cell polarity and adhesion) by cancer cells is involved in disease progression as well as in CTX resistance (168–179).
Indeed, several authors report that HNSCC cells exhibiting a mesenchymal-like phenotype are resistant to CTX treatment in vitro and in vivo (xenografts) (174, 176, 177). Several potential mechanisms implicated in this EMT-induced CTX resistance are observed, such as (i) expression of lymphotoxin-b; (ii) methylation of EGFR that promotes the EGFR ligand-binding ability and dimerization (EGFR persistent activity) (169); (iii) secretion of CTX-containing extracellular vesicles, which lead to cancer cell protection (179); (iv) upregulation of EMT-related genes (133), especially by epigenetic regulation (170, 180); and (v) loss of the tumor suppressor gene SMAD4, which induces JNK and MAPK pathway activation (172, 173). Indeed, Ozawa et al. find that SMAD4 loss is associated with CTX resistance and poor survival in HPV-negative patients (cohort of n=130 newly diagnosed and n=43 patients with recurrent HNSCC) (172). Thus, Ozawa et al. tested the combination of CTX + JNK inhibitor (SP600125) or MAPK/MEK inhibitor (U0126) and show that it contributes to overcome CTX resistance in vitro.
Moreover, the development of CTX resistance could also be accompanied by increasing hedgehog pathway transcription factor expression in vitro (175). Thus, Keysar et al. tested the combination of CTX and IPI-926 (hedgehog pathway inhibitor) in four different PDX models. This combination forced tumor cells into an EGFR-dependent state and blocked tumor recurrences.
Epigenetic Modifications
Epigenetic alterations, including DNA methylation, histone modifications, chromatin remodeling, and noncoding RNAs, are frequently involved in head and neck carcinogenesis, tumor progression, and resistance to therapy (137), especially to CTX (75).
As previously described, Kagohara et al. report that genes associated with CTX resistance in HNSCC cell lines, including TFAP2A, which regulates growth factor receptors and EMT, are epigenetically regulated (170). Stein-O’Brien et al. show that FGFR1 demethylation is associated with CTX resistance and this type of epigenetic alteration might stabilize the resistant phenotype (171). Interestingly, Shimizu et al. recently reported that nicotine (one of the main tobacco components) contributes to CTX resistance in vitro as well as in vivo (xenografts) (181). Indeed, they show that nicotine induces, through the nicotinic acetylcholine receptor, both EGFR phosphorylation (and the subsequent Akt and mTOR downstream cascade activation) and nuclear translocation of the phosphorylated EGFR.
Establishment of an Immunosuppressive TME
In addition to all the previously cited intrinsic resistance mechanisms, there are also extrinsic resistance mechanisms, i.e., involving the TME (182). These mechanisms encompass, in fact, the cancer cell–TME crosstalk, contributing to CTX resistance (40, 75). In HNSCC, establishment of an immunosuppressive microenvironment is an important resistance mechanism to treatment, especially to CTX (6, 70, 183, 184) (Figure 2).
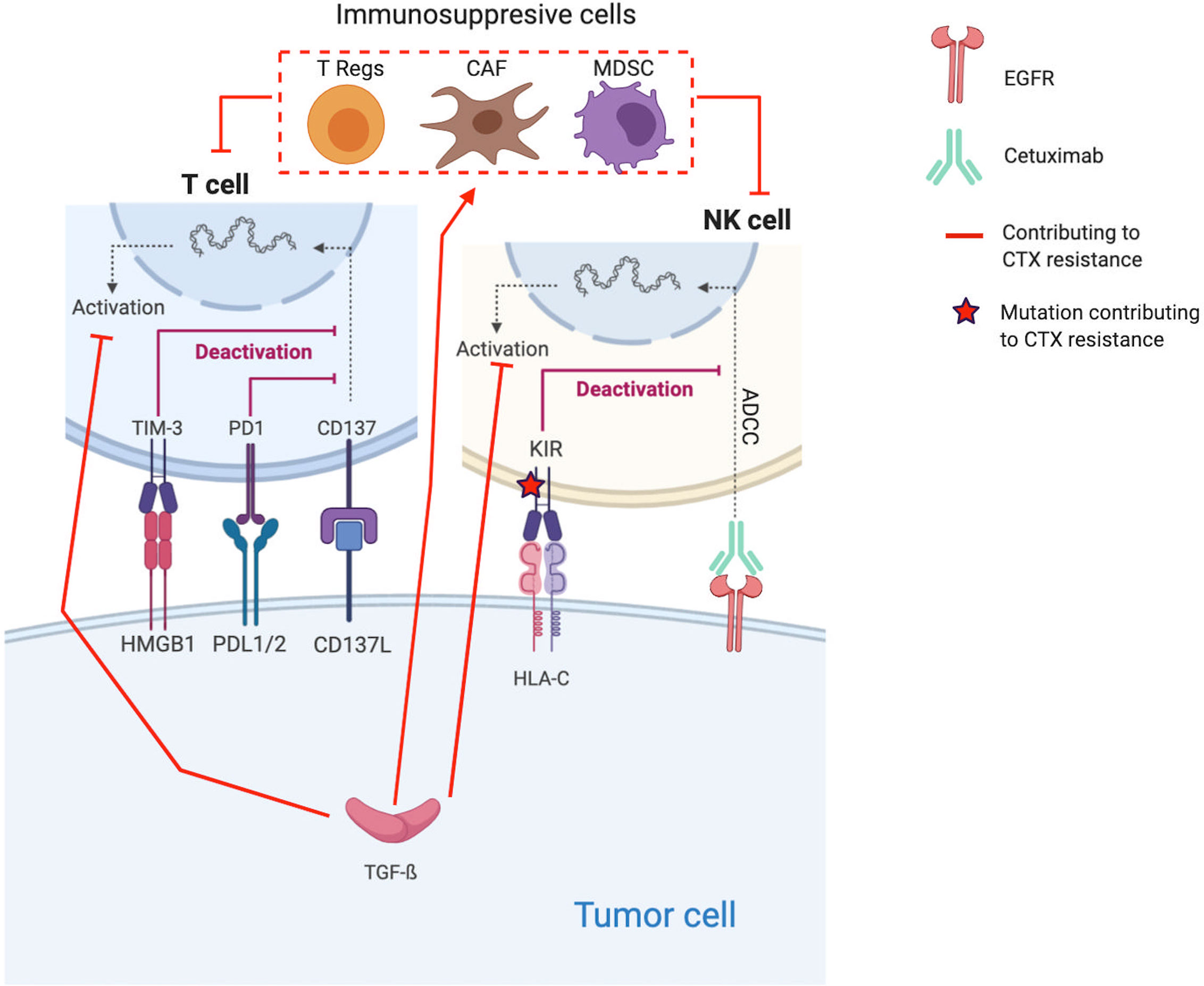
Figure 2 Molecular mechanisms contributing to Cetuximab resistance through the establishment of an immunosuppressive TME. Red lines and arrows show mechanisms contributing to Cetuximab resistance, and green lines and arrows show mechanisms contributing to Cetuximab sensitivity. (EGFR, Epidermal Growth Factor Receptor; NK, Natural Killer; PD1, Programmed death 1; PDL1, Programmed Death Ligand 1; KIR, Killer Immunoglobulin-like Receptor; ADCC, Antibody-dependent cellular cytotoxicity; HLA-C, Human leukocyte antigen-C; TGF, transforming growth factor; Treg, regulatory T-cells; MDSC, Myeloid-derived suppressor cells).
Regulatory T Cells (Tregs) and Myeloid-Derived Suppressor Cells (MDSC) Proliferation
Proliferation of immunosuppressive cells, such T regs and MDSC, in response to CTX treatment is one of the key resistance mechanisms (185–188). Indeed, several coculture experiments show that CTX expands CTLA-4+FOXP3+ Tregs in part by inducing dendritic cell maturation. These Tregs lead to CTX resistance by suppressing the CTX-mediated ADCC (cytolytic functions of NK cells) (188). Based on the analysis of blood samples from CTX-treated patients with locally advanced/metastatic (stage III/IV) HNSCC (n=22 patients, NCT 00226239 and n=18, NCT 01218048), Jie et al. confirm their in vitro observations (188). Indeed, they find that CTX increased the frequency of intratumoral Tregs expressing the inhibitory checkpoint cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which is known to inhibit T cell activation (6). Thus, Tregs suppress the CTX-mediated ADCC. Their presence is correlated with poor clinical outcomes in these cohorts. Based on these results, Jie et al. tested CTX in combination with ipilimumab, a monoclonal antibody that also induces NK cell–mediated ADCC. Ipilimumab treatment enhanced the CTX-mediated ADCC by eliminating Tregs (targeting CTLA-4), allowing effector T cell activation and restoration of the cytolytic functions of NK cells (n=6 HNSCC tumors) (188).
Furthermore, Shayan et al. hypothesized that the combination of CTX plus motolimod, a small-molecule TLR8 agonist that can activate monocytes, DCs, and NK cells (189), might enhance T cell stimulation and CTX effects (187). They find that the TLR8 stimulation through motolimod skewed monocytes toward an antitumor M1 phenotype and reversed MDSC suppression of T cell proliferation. These in vitro observations are confirmed in patients. Indeed, in a phase-Ib trial (NCT02124850) enrolling n=14 patients with previously untreated stage-III/IV HNSCC, Shayan et al. tested the combination of CTX plus motolimod (formerly VTX-2337) (187). The combination reversed MDSC-induced immunosuppression and improved antitumor immunity with increased circulating tumor antigen-specific T cells (EGFR specific) and increased the number and function of tumor-infiltrating CD8 T cells. These encouraging results are confirmed in another phase-Ib study (n=13 patients with recurrent/metastatic HNSCC, NCT01334177) demonstrating the significant increasing antitumor activity of this combination (increased plasma cytokines and activated circulating NK cells). To finish, the potential benefit of adding motolimod to the standard EXTREME regimen (CTX + platinum + fluorouracil) was evaluated in a phase-2 trial enrolling n=195 patients with recurrent/metastatic HNSCC (NCT01836029) (185). Ferris et al. find that the combination fails to prove a benefit for survival when considering the intent-to-treat population, but significant benefits are observed when considering only the selected subgroup of patients with HPV-positive tumors and injection site reactions.
T Cell Exhaustion/Impairment
HNSCCs are among the most immune-infiltrated cancers, and several mechanisms are implemented by tumor cells to escape to the host immune defense system (190, 191). The immune-modulatory effect of CTX treatment, in particular ADCC, might be inhibited by cancer cells through several mechanisms inducing T cell exhaustion/impairment and all CTX resistance (67, 97, 192–204). Indeed, to counteract the antitumor activity of CTX, tumor cells express TGF-β, which inhibits the expression of cytotoxic effector molecules in immune cells (Apo2L/TRAIL, CD95L/FasL, granzyme B, and IFN-γ) and suppresses their ability to induce cetuximab-mediated ADCC (97).
Moreover, in a cohort of n=18 patients with stage-III/IV HNSCC treated by CTX alone (NCT 01218048), Jie et al. find that the increased frequency of PD-1+ and TIM-3+ tumor-infiltrating lymphocytes (TILs) during CTX treatment inversely correlates with objective response (200). Besides PD-1 and TIM immune checkpoint (ICP) receptors, KIR, the ICP on NK cells that modulate their activation, is also indirectly implicated in CTX resistance. Indeed, Faden et al. report a statistically significant increase of missense mutations and loss of heterozygosity in HLA-C (the ligand for KIR) in patients not responding to CTX compared with responders (196).
In accordance with the previously cited in vitro observations, several CTX combinations have been tested in the preclinic as well as clinic setting.
For example, Bedi et al. explored the combined effect of CTX and TGF-β blocking in vivo (PDX). Although CTX alone forced the selection of resistant clones, i.e., TGF-β–overexpressing tumor cells, the combined treatment prevented it and induced complete tumor regression (97). In the same way, Faden et al. observed increased HNSCC cell killing when combining CTX and lirilumab, a monoclonal antibody that blocks NK inhibitory KIR signaling (196). Moreover, the upregulation of CD137 or 4-1BB (a member of the TNF-receptor superfamily, which is broadly induced and activated on several effective immune cells) was corelated to clinical response to neoadjuvant CTX (NCT01218048). Srivastava et al. tested the effect on several HNSCC cell lines of the combination CTX plus urelumab (BMS-663513, CD137-agonist monoclonal antibody) (201). This combination enhanced the CTX-mediated ADCC as supported by the increased NK-cell survival, DC maturation, and tumor antigen cross-presentation. Regarding other ICP inhibitory signals, Concha-Benavente et al. explored several HNSCC cell lines and found that the programmed death ligand-1 (PD-L1), which limits the function of activated T lymphocytes when they interact with the ICP receptor programmed death-1 (PD-1), is expressed by tumor cells in an EGFR- and JAK2/STAT1-dependent manner (159). Thus, they tested the combination of CTX and JAK2 inhibition. They found that JAK2 inhibition prevented tumor PD-L1 expression and that the combination enhanced the CTX NK-mediated killing via ADCC against PD-L1+ HNSCC cells.
The better comprehension of immune evasion mechanisms as well as of the immune-modulatory effect of CTX, i.e., CTX-mediated ADCC, brings evidence to support the evaluation of combined approaches with ICP inhibitors in both locally advanced and recurrent/metastatic HNSCC (6, 67, 192). Among the tested agents, inhibition of the PD-1/PD-L1 synapse is widely explored. The anti PD-1 Nivolumab, for which efficacy and safety prior to CTX in HNSCC has been recently reported (CheckMate 141) (205), is actually tested in combination with CTX in phase-1 and -2 trials for patients with recurrent/metastatic HNSCC (NCT03370276). Sacco et al. recently reported some preliminary results of the first trial evaluating the antitumor activity of anti-PD-1 Pembrolizumab combined with CTX in n=33 patients with platinum-refractory/ineligible, recurrent/metastatic HNSCC (NCT03082534, phase 2) (197). Results are promising with an observed 41% response rate. Regarding anti PD-L1, Durvalumab combined with CTX and radiotherapy is currently tested in a phase-I/II clinical trial (NCT03051906, DUCRO) (193). Based on previous safety studies (154, 163, 167), several clinical trials are testing the anti PD-L1 Avelumab in combination with CTX (NCT03494322, phase 2 EACH) (206) +/- radiotherapy (NCT02999087 phase 3 REACH) or Palbociclib (a selective CDK4/6 inhibitor) (NCT03498378 phase 1 and NCT02101034 phase 2) (195). Other interesting combinations involve the 4-1BB agonist Urelumab (NCT02110082) (201), the anti-CTLA-4 monoclonal antibody ipilimumab (NCT01935921, phase 1) (202) and Monalizumab (anti NKG2A receptors expressed on TIL-CD8+ and NK cells) (NCT02643550) (199).
Toll-Like Receptor 4 (TLR4) Pathway Activation
TLR4 is an innate immune receptor involved in defense against microbial agents by recognizing inflammation-associated microbial ligands (such as lipopolysaccharide) and promoting the activity of innate immune cells (207). TLR4 are also expressed by tumor cells, and the role of the TLR4 signaling pathway in the TME has been reviewed elsewhere (208). Unfortunately, by inducing immunosuppressive cytokines, apoptosis resistance, and EMT, the TLR4 signaling pathway can promote cancer cells’ immune escape in several cancer types (including lung, pancreas, and ovarian cancers, HNSCC) as well as resistance to therapy (paclitaxel in ovarian cancer) (209–212).
Indeed, Ju et al. recently reported that the crosstalk between the EGFR and TLR4 pathways could participate in CTX resistance in vitro and in vivo (xenograft) (213). They found that EGFR inhibition led to decreased MyD88 degradation, and thus, MyD88 could activate TLR4 (receptor homodimerization). TLR4 activation induced activation of NF-κB and MAPK signaling pathways, resulting in the release of proinflammatory cytokines (TNF-α, iNOS, COX2, PGE2, NO) favoring EGFR permanent activation as well as the release of anti-apoptosis proteins (Bcl-2, Bcl-xl) allowing tumor cell survival. Overall, the TLR4 signaling pathway leads to CTX-resistance. Thus, Ju et al. tested the combination of CTX and a TLR4 inhibitor (TAK242). They find that this combination overcomes acquired CTX resistance in vivo, in particular by decreasing the secretion of pro-inflammatory cytokines (TNF-α, PGE2, and NO) (175).
CAF Proliferation
Among the different components of the HNSCC TME, CAFs are among the most critical elements contributing to proliferation, invasion, and metastasis (214), in particular by altering the antitumor immune response (215, 216). Furthermore, CAFs have been shown to contribute to drug resistance in HNSCC (e.g., platinum and CTX) (42, 217–219).
Indeed, during CTX treatment, CAFs, especially those activated by TGF-β (218), participate in resistance by secreting immunosuppressive factors, such as IL-6, HGF, and metalloproteinases (219). Thus, co-inhibition of TGF-β and HNSCC cells by the combination of CTX + SIS3 (an inhibitor of the TGF-β pathway), delayed tumor progression and lowered tumor volume/weight (HNSCC PDX) (218). Johansson et al. also tested the combined effect of CTX + MMP inhibitor III (inhibiting MMP-1, -2, -3, -7, and -13), which significantly reduced the protective effect of CAFs (219).
Immunotherapy in HNSCC
HNSCC is among the most inflamed, immune-infiltrated cancers, especially with CD8+ TILs and NK cells (183). A high genetic instability and somatic mutation rate is often observed (220, 221) (about 180 somatic mutations per mega base), in either HPV positive or negative tumors (222, 223). Immunogenicity of HNSCC can result from the overexpressed but nonmutated native proteins that have escaped central tolerance, neo-antigens derived from mutated proteins (224), or HPV-induced viral antigens (e.g., oncogenic drivers E6 and E7) (225). The microenvironment can also vary in terms of intensity or constitution depending on carcinogens or localization (226).
Genetic and epigenetic alterations in cancer cells create a vast array of neoepitopes potentially recognizable by the immune system. However, a key feature of malignant cells is their ability to escape recognition by the immune system, and the dysregulation of immune checkpoints, such as PD-L1, in tumors appears to be a major immune-resistance mechanism affecting T cell response. It was, hence, shown that the reversal of the anergic state of T lymphocytes is possible via the blockade of coinhibitory signals (227), and research initially focused mainly on immune checkpoint inhibitors (ICI) of the PD-L1/PD-1 axis. These antibodies (Abs) have since completely transformed the treatment of R/M HNSCC.
The expression of PDL1 is positive in most cases, estimated at almost 60%–70% with a higher expression in HPV+ compared to HPV- tumors (228, 229). It results either from an immune-adaptive phenomenon induced by IFNγ or from intrinsic oncogenic events, such as the mutation/deletion of the PTEN suppressor gene or the deregulation of the AKT/mTOR, NF-kB and mitogen-activated protein kinase (MAPK) pathways (230). PD-1 binding with PD-L1/PD-L2 causes immunosuppression via reduced t cell receptor (TCR) signaling, reduced cytokine production, reduced target cell lysis, altered lymphocyte motility, and metabolic reprogramming (231).
After showing antitumor activity in multiple other tumor types, nivolumab was the first anti-PD-1 agent to improve OS in recurrent/metastatic (R/M) HNSCC progressing after a first-line platinum-based therapy in the Checkmate 141 trial with a 32% reduction in the risk of death (205, 232). OS was 7.7 months compared with 5.1 months with chemotherapy. Benefit was greater in ≥1% PD-L1 positive (PD-L1+) TPS (tumor cell membrane positivity for PD-L1 or tumor proportion score) patients with an OS of 8.2 versus 4.7 months. Pembrolizumab is the other agent to show efficacy in the second line in the Keynote 0-40 trial (233). Median OS was 8.4 vs 6.9 months with a hazard ratio (HR) of 0.80. Contrary to the Checkmate 141 essay, crossover was allowed. Subgroup analysis shows that, for PD-L1 TPS ≥ 50% patients, survival was significantly increased from 7.9 to 11.6 months with immunotherapy, whereas there was no difference for the PD-L1<50% population.
This same agent is the new standard of front-line therapy in R/M HNSCC following the results of the Keynote-048 trial comparing pembrolizumab alone or in combination with platinum-based and 5FU chemotherapy to the EXTREME standard of care protocol (cisplatin or carboplatin, 5-fluorouracile (5FU) and cetuximab) (234). Survival was significantly increased with pembrolizumab compared with the EXTREME regimen for PD-L1 ≥20 CPS (expression on both tumor cells and immune cells in the microenvironment or combined positive score) patients (14.7 vs. 11 months) and PD-L1 ≥1 CPS patients (12.3 vs. 10.4 months) but not in the total population (11.5 vs. 10.7 months). It is important to note that the experimental treatment was deleterious for some patients in the beginning with more deaths occurring in the first 6 months. Pembrolizumab added to chemotherapy significantly improved OS in the total population with safety comparable to the EXTREME arm (13 vs. 10.7 months). The median duration of response was impressive with pembrolizumab at 22.6 months (vs. 4.5 for EXTREME). Tolerance was also far better with immunotherapy. The FDA approved pembrolizumab for use in combination with platinum and fluorouracil for all patients and as a single agent for patients with a CPS ≥1. In Europe, the EMA approved the use of pembrolizumab alone or in combination for patients with a CPS ≥1. Unlike PD-L1, blockade of cytotoxic T-lymphocyte antigen 4 (CTLA4) as monotherapy has not proven beneficial in HNSCC.
Evidently, monotherapy with ICI seems to be a losing battle despite providing substantial clinical improvements over the previous standards of care. The majority of patients do not respond to treatment, and durable responses are observed only in a minority (generally less than one third) of patients.
Resistance to Immune Checkpoint Inhibitors
Resistance to ICI can be primary (never-responder patients) or secondary (acquired after a certain amount of time of response). It can also be classified as intrinsic to tumor cells (cancer cells directly induce immune resistance via various mechanisms) or extrinsic (other cells or factors mediate immune resistance).
HNSCC hijacks numerous cellular and molecular immunomodulatory pathways to evade recognition and eradication by the immune system. Mechanisms of immune evasion include direct T cell suppression with surface or soluble inhibitory factors, decreased immune stimulation, and the recruitment of immuno-suppressive cell populations (231). In this section, we review the different types of resistance reported in HNSCC and present some of the currently studied strategies to overcome them.
Intrinsic Resistance
Tumor Immunogenicity and Antigen Presentation
HNSCC is one of the cancers with the highest levels of tumor mutational burden (TMB), accompanying elevated neoantigen expression (220). These tumoral neoantigens that derive from nonsynonymous mutations drive (T lymphocytes) TL cytotoxic response against tumor cells. In that sense, a positive correlation between response and TMB was found in a recent meta-analysis (235). Constant interactions between immune and cancer cells can result in a selection of subclones lacking the expression of neoantigens, subsequently resulting in poor immunogenicity and decreasing efficacy of ICI (236). This could explain how some HNSCC tumors with high TMB are unresponsive to ICI.
Furthermore, deficiencies in antigen presentation can result in primary or acquired resistance to ICI as shown in multiple studies (237, 238). This includes downregulation of MHC class I (MHC-I) and loss of function (e.g., truncating mutations) of β2-microglobulin (238). HNSCC has been shown to alter neoantigen presentation and processing by altering key genes, such as signal transducer and activator of signal (STAT) 1 and other antigen processing machinery components (239, 240).
Combining ICI with radiation therapy is a promising strategy as radiotherapy leads to an increased rate of neoantigens and antigen presentation induced by DC activation, increased cytokine production, and tumor cell death, promoting a TIL phenotype (241, 242). Chemotherapy increases antigen release upon cell death and, thus, the priming of cytotoxic TL (243), and this was the rational in combining platinum-based chemotherapy with ICI in the Keynote-048 trial that resulted in added benefit (234).
Emerging novel therapies include oncolytic virus therapy and cancer vaccines with tumor peptides or DCs (244, 245). Their aim is to enhance antigen presentation and TL priming. Oncolytic viruses can also directly infect and induce lysis of tumor cells. Talimogene laherparepvec (TVEC), which is derived from herpes simplex virus type 1, is currently under evaluation in combination with pembrolizumab in the Keynote-137 trial in R/M HNSCC patients. Other novel therapeutics, such as toll-like receptor (TLR) agonists (NCT02521870) and adoptive cell therapy (NCT03247309), are also being evaluated in this same context.
Oncogenic Pathways
Aberrations in canonical oncogenic pathways can change the TME by altering cytokine production and immune cell composition. These include the MAPK (246), WNT/β-catenin (247), and PI3K pathways (248). The activation of the latter creates an immunosuppressive TME. Combined inhibition of PD-1 and PI3K in a preclinical model of HNSCC demonstrates a synergistic growth inhibitory effect and increased survival of mice by activating an immunostimulatory transcriptional program, enhancing T cell cytotoxicity and expression of proinflammatory cytokines (249).
Soluble Molecules
HNSCC cells can also avoid T cell rejection by secreting immunosuppressive exosomes containing transforming growth factor (TGF) β, PD-1, and CTLA4, which impair T and NK cell functions and upregulate Tregs (250). They can also produce and secrete protumoral cytokines, including TGF-β, interleukin (IL)-6, and IL-10 (251). Tumor cells can overexpress Indoleamine 2,3-dioxygenase 1 (IDO1), a rate-limiting enzyme that converts tryptophan to kynurenine, leading to an immune suppression through T cell apoptosis and loss of function. In a study of the immune microenvironment of HPV-negative OSCC from never-smoker and never-drinker (NSND) patients, it was suggested that blockade of IDO1 and PD-1/PD-L1 could insure a higher clinical benefit. However, a phase-III clinical trial evaluating Epacadostat, an IDO inhibitor, in combination with Pembrolizumab was halted after a similar trial in melanoma revealed no improvement compared with the control arm (252).
Extrinsic Resistance
Inhibitory Checkpoint Molecules
Overexpression of alternative immune checkpoints can be a source of adaptive resistance to ICI. These receptors serve to limit effector functions of the immune system to prevent autoimmunity in a normal state. Multiple inhibitory immune checkpoint receptors with different cell distributions and expression patterns have been described, including PD-L1, CTLA4, lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin, mucin domain-3 protein (TIM-3), B and T lymphocyte attenuator (BTLA), V-domain immunoglobulin-containing suppressor of T cell activation (VISTA), and T cell immunoreceptor tyrosine-based inhibition motif domain (TIGIT) (253, 254).
PD-L1 status has been shown to be partially correlated with response to ICI in HNSCC, but complete responses have been observed in PD-L1-negative patients (232–234). CTLA4 is upregulated in HNSCC tumor cells and enriched on Treg TILs (255). These cells are a subset of CD4+ T cells with immunosuppressive effects through various humoral and cellular mechanisms, such as CTLA4-mediated suppression of antigen-presenting cells (256). LAG-3 is expressed on activated CD4+ and CD8+ T cells, NK cells, B cells, and DCs (257). It binds with major histocompatibility complex class II (MHCII) and is highly expressed on Tregs. It was shown that blockade of LAG-3 decreases the inhibitory function of these cells (257). TIM-3 is expressed on both T and NK cells and binds with galectin-9 (258). When specifically coexpressed with PD-1, TIM-3 is the signature of an exhausted T cell phenotype (259).
Because these alternate coinhibitory receptors induce T-cell exhaustion (231), they have been identified as a putative strategy to overcome resistance to PD-1 in previous and many ongoing studies. Based on these observations, two essays of ICI anti-PD1 and anti-CTLA4 combination in HNSCC have been reported to date (260). The CONDOR trial compared outcomes of patients who had low/negative PD-L1 (TPS<25%) tumors and had progressed after first-line platinum-containing therapy. Patients were treated with either durvalumab, an anti-PDL1 Ab, or tremelimumab, an anti-CTLA4 Ab, or the combination (260). Results were deceiving as there was no significant increase in response rate (RR) (7.8% vs. 9.2%), PFS (2 vs. 1.9 months) or OS (7.6 vs. 6 months) compared with durvalumab alone. One toxic death from acute respiratory failure was attributed to the combination regimen. The same combination failed to improve survival regardless of PD-L1 status in the EAGLE trial (261). Monotherapy with the anti-CTLA4 agent in CONDOR appeared clearly inefficient with a 1.6% RR, a 1.9 and 5.5 median PFS and OS, respectively (260). The authors hypothesize that the lack of efficacy of tremelimumab may be in part related to its mechanism of action, which, as an IgG2 Ab, does not cause lysis of regulatory T cells through ADCC, contrary to what is observed with ipilimumab, another anti-CTLA4 agent (262).
Inhibitors of other checkpoint molecules, such as TIM-3 and LAG-3, are still in earlier phases of development. For example, blockade of TIM-3, whose expression is synonymous to T cell exhaustion, is efficient in producing an antitumoral T cell response in a mouse model of HNSCC (263).
Inhibitory checkpoints can also be expressed on the surface of innate immune cells, such as NK cells. The inhibitory killer immunoglobulin-like receptor (KIR) 2DL-1, -2, -3 receptors, which partially control NK cell activation upon binding with their ligands, primarily human leukocyte antigen-C (HLA-C) molecules, can be targeted by Lirilumab, a fully human IgG4 monoclonal Ab. PD-1 blocking on T cells can induce the release of cytokines, such as IL-2, that enhance NK cell function, whereas blockade of KIR can result in the secretion of IFN-γ that may boost T cell–mediated antitumor responses (264). This rationale of NK–T cell crosstalk led to the testing of the combination in phase-1/2 trials (265).
Stimulatory Agonist Molecules
The balance between coinhibitory and costimulatory signals is what determines the state of the immune response. Costimulatory agonists include Ox40, 4-1BB, inducible T cell co-stimulator (ICOS), and CD40. Ox40 is expressed on the surface of T cells and promotes proliferation and IFN-y production. It is shown to be present in HNSCC, but expression of its ligand (Ox40L) is reduced, rendering this pathway ineffective (266). ICOS is expressed on the same cells, promoting a Th2 response [62], and 4-1BB, present on the surface of activated T cells, NK cells, and DCs, is shown to be downregulated in HNSCC patients (267).
Many promising ongoing trials are evaluating receptor agonists in order to reverse resistance to ICI and augment durable responses. For example, agonists of ICOS are being evaluated in combination with anti-PD-1, anti-CTLA-4, and chemotherapy in various tumors, including HNSCC (NCT03693612, NCT02904226). In addition, urelumab (an agonistic 4-1BB monoclonal antibody), is evaluated in combination with cetuximab (NCT02110082) in R/M HNSCC patients.
Immunosuppressive Cells
In addition to all the complex interactions between coinhibitory and costimulatory pathways, immunosuppressive cells can modulate the immune response and create a protumoral environment via multiple diverse mechanisms (268). Their recruitment to the TME is regulated by HNSCC cells. MDSCs, Tregs, and TAMs all modulate NK and T cell responses to various degrees.
MDSCs are an immature myeloid cell population that promotes HNSCC invasiveness, angiogenesis, and metastasis (269, 270) by secreting immunosuppressive enzymes, such as enzymes arginase 1 (Arg-1) and nitric oxide synthase (iNOS) (251). Their presence correlates with poor outcomes with ICI as shown in melanoma patients (271). Monoclonal Ab and small molecule inhibitors that inhibit MSDC functions are currently investigated in R/M HNSCC patients in various clinical trials.
Tregs facilitate self-tolerance through direct contact and inhibitory cytokines, such as IL-10 and TGF-β (272), they also play a key role in immune evasion in HNSCC. These cells preferentially express CCR4 (believed to have a major role in the recruitment of Tregs to the TME), which is being targeted with an anti-CCR4 monoclonal antibody, mogamulizumab, in different tumor types, including HNSCC.
TAMs, particularly M2 macrophages, enhance tumor angiogenesis, motility, growth, and immune evasion by secreting protumoral cytokines in the TME [69]. Their presence in HNSCC is associated with poor prognosis (273). Antibodies and small molecules that inhibit colony-stimulatory factor 1 receptor (CSF1R) binding with CSF1, which serves recruitment to the tumor of M2 TAMs, are currently underway in various advanced solid tumors, including HNSCC (NCT02526017).
Conclusions and Perspectives
Treatment of the majority of patients with HNSCC requires multimodality approaches. Currently, cetuximab is used in the clinical routine as a radiation sensitizer alone or in combination with chemotherapy for the treatment of patients with recurrent or metastatic disease. More recently, pembrolizumab was approved as a first-line therapy in patients who present with metastatic disease, and treatment with either pembrolizumab or nivolumab is used in the setting of cisplatin-refractory recurrent or metastatic HNSCC. Despite the encouraging results observed in some patients, tumor responses observed in most patients are only partial and are systematically followed by acquired resistance due the reactivation of oncogenic signaling, leading to tumor regrowth, as discussed in this review.
Most of the developments toward understanding HNSCC have fallen short of clinically meaningful discoveries, highlighting an urgent need for more effective therapies to improve treatment outcomes. The increasing knowledge on the genomic driver alterations in HNSCC enables their use as predictive markers of targeted therapy regimens, currently evaluated in clinical trials, which are shown to improve survival and tumor response in subgroups of patients (274, 275). For instance, late-phase clinical trials show that HRAS-mutant HNSCC patients (8% of HNSCC) treated with tipifarnib, a selective farnesyltransferase inhibitor, shows promising outcomes with an overall response rate (ORR) of 42.9% with a median duration of response of 14.7 months (275). The Akt/mTOR axis is activated in most HNSCC, particularly in surrounding normal mucosa, and is associated with recurrences. In this context, a phase-II trial (NCT01111058) shows significant improvement in 1-year PFS in patients with locally advanced HNSCC treated with everolimus (276). More recently, Xevinapant, an investigational inhibitor of apoptosis protein (IAP) blocker, showed prolonged OS when added to standard chemoradiotherapy for locally advanced head and neck squamous cell carcinoma. Based on these results, Xevinapant received breakthrough therapy status from the FDA for the first-line treatment of HNSCC in September 2020.
The molecular heterogeneity of HNSCC has hampered the identification of specific targets and, thus, the development of targeted therapies for this group of tumors (51). Indeed, much of the difficulty in studying and treating HNSCC lies in the fact that they are a heterogeneous group of cancers arising from distinct anatomic subsites that display distinct molecular features and are associated with diverse risk factors. However, these diseases are treated uniformly and with limited success.
Genome-wide expression profiling led to the identification of four robust molecular classes of HNSCC (277–279). In this classification, the “classical,” “basal,” and “mesenchymal” subtypes exhibit canonical genomic alterations, such as nuclear factor erythroid 2−related factor 2 (NFE2L2) mutations and high expression of genes in oxidative stress response pathways, high frequency of HRAS mutations, and upregulation of EMT-related genes, respectively (278, 279). Of note, multiple findings have led to increased interest in the mechanisms by which cancer cells undergoing EMT or oscillating within the EMT spectrum might contribute to immune escape through various routes. The “atypical” subtype contains a high proportion of HPV+ tumors, who themselves are very heterogenous and can be subclassified into HPV-KRT (HPV-keratinocyte differentiation and oxidative reduction process) or HPV-IMU (HPV-immune response and mesenchymal cell differentiation) tumors (69). These different subtypes of HNSCC may harbor different patterns of sensitivity to oncogenic-driven targeted therapy and radiotherapy (280, 281); however, the clinical implication of these subtypes is currently unknown.
More recently, based on the gene expression profiles of 1368 patients with SCC in the Cancer Genome Atlas (TCGA), Li B et al. (282) proposed six immune subtypes, including an immune-cold subtype, an immune-hot subtype, a subtype dominated by M2-polarized macrophages, and three other immune subtypes with more diverse immunologic features. Their association with response or resistance to immunotherapy is unclear.
Complementary strategies to assess the molecular programs that are specific to each histological subtype or anatomical location of HNSCC may benefit from comprehensive analyses of patient samples (283) to identify molecular vulnerabilities and, thus, enable rapid clinical deployment to guide therapeutic decisions. Furthermore, single-cell transcriptomics may help revealing intratumoral heterogeneity (ITH) (284, 285) with subtypes as well as identifying cell populations that drive drug resistance. Spatial transcriptomics might also be an informative approach to enable simultaneous capture of the distribution and localization of the different components of the TME and, thus, better understand its interaction in response to treatment. Finally, the establishment of relevant preclinical models of HNSCC (ref) that reflect the disease at the genetic, histological, and functional level may provide a tool to study the molecular modifiers of response to therapies currently used in the clinical routine or tested in clinical trials.
Overall, understanding the molecular vulnerabilities of HNSCC may contribute to identify and therapeutically target residual disease and prevent or delay the evolution of acquired resistance. Of note, acknowledging that drug resistance depends not only upon cancer cells but also upon the TME might enable the identification of potential drug targets to limit cancer cell adaptation to therapy.
Author Contributions
SO-C, JB, AK, JF, and PS: contributed to this paper with the design. SO-C, JB, AK: literature search. SO-C, JB, AK, JF, and PS: drafting. SO-C, JB, AK, JF, and PS: revision and editing. SO-C, JB, AK, JF, and PS: final approval. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the Integrated Cancer Research Site LYriCAN (INCa-DGOS-Inserm_12563). SO-C received funding from the Fondation ARC pour la Recherche sur le Cancer (ARCPJA22020060002212). JB received funding from Nuovo Soldati Foundation (2019) and ITMO Cancer 2020, Formation à la Recherche Fondamentale et Translationnelle en Cancérologie.
Conflict of Interest
PS receives research grants from HTG Molecular Diagnostics, Inivata, Bristol-Myers Squibb, Roche Molecular Diagnostics, Roche, AstraZeneca, Novartis, Bristol-Myers Squibb Foundation, and Illumina.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
EGFR, Epidermal Growth Factor Receptor; PTPRS, The protein tyrosine phosphatase receptor S (interacts with EGFR and inactivates EGFR); CTX, cetuximab; TGF-α, transforming growth factor alpha; TGF-ß, transforming growth factor beta; EGF, Epidermal Growth Factor; HB-EGF, heparin-binding growth factor; JAK2, Janus kinase 2; MAPK, Mitogen-activated protein kinases; MET, Mesenchymal Epithelial Transition; HER2 or ERbB2, human epidermal growth factor receptor 2; HER3 or ERbB3, human epidermal growth factor receptor 3; ROR2, receptor tyrosine kinase-like orphan receptor 2; IGF-1R, Insulin growth factor type 1 receptor; VEGFR, vascular endothelial growth factor receptor; PI3K, Phosphoinositide 3-kinases; mTOR, Mechanistic Target Of Rapamycin Kinase; SMAD4, Mothers against decapentaplegic homolog 4; DNA, deoxyribonucleic acid; RNA, ribonucleic acid; EMT, Epithelial-to-mesenchymal transition; T cells, T lymphocyte; T regs, regulatory T lymphocyte.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J Clin Oncol Off J Am Soc Clin Oncol [Internet] (2015) 33:3235–42. doi: 10.1200/JCO.2015.61.6995
3. Young D, Xiao CC, Murphy B, Moore M, Fakhry C, Day TA. Increase in head and neck cancer in younger patients due to human papillomavirus ({HPV}). Oral Oncol (2015) 51:727–30. doi: 10.1016/j.oraloncology.2015.03.015
4. Karabajakian A, Toussaint P, Neidhardt E-M, Paulus V, Saintigny P, Fayette J. Chemotherapy for recurrent/metastatic head and neck cancers. Anticancer Drugs (2017) 28:357–61. doi: 10.1097/CAD.0000000000000473
5. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer. N Engl J Med (2008) 359:1116–27. doi: 10.1056/NEJMoa0802656
6. Horton JD, Knochelmann HM, Day TA, Paulos CM, Neskey DM. Immune Evasion by Head and Neck Cancer: Foundations for Combination Therapy. Trends Cancer [Internet] (2019) 5:208–32. doi: 10.1016/j.trecan.2019.02.007
7. Morel A-P, Hinkal GW, Thomas C, Fauvet F, Courtois-Cox S, Wierinckx A, et al. EMT Inducers Catalyze Malignant Transformation of Mammary Epithelial Cells and Drive Tumorigenesis towards Claudin-Low Tumors in Transgenic Mice. Horwitz MS, editor. PloS Genet (2012) 8:e1002723. doi: 10.1371/journal.pgen.1002723
8. Caramel J, Ligier M, Puisieux A. Pleiotropic roles for ZEB1 in cancer. Cancer Res (2018) 78:30–5. doi: 10.1158/0008-5472.CAN-17-2476
9. Feng S, Yang G, Yang H, Liang Z, Zhang R, Fan Y, et al. NEDD4 is involved in acquisition of epithelial-mesenchymal transition in cisplatin-resistant nasopharyngeal carcinoma cells. Cell Cycle (2017) 16:869–78. doi: 10.1080/15384101.2017.1308617
10. Shao Q, Zhang P, Ma Y, Lu Z, Meng J, Li H, et al. MicroRNA-139-5p affects cisplatin sensitivity in human nasopharyngeal carcinoma cells by regulating the epithelial-to-mesenchymal transition. Gene (2018) 652:48–58. doi: 10.1016/j.gene.2018.02.003
11. Steinbichler TB, Metzler V, Pritz C, Riechelmann H, Dudas J. Tumor-associated fibroblast-conditioned medium induces CDDP resistance in HNSCC cells. Oncotarget (2015) 7:2508–18. doi: 10.18632/oncotarget.6210
12. Chang WM, Chang YC, Yang YC, Lin SK, Chang PMH, Hsiao M. AKR1C1 controls cisplatin-resistance in head and neck squamous cell carcinoma through cross-talk with the STAT1/3 signaling pathway. J Exp Clin Cancer Res (2019) 38:245. doi: 10.1186/s13046-019-1256-2
13. Ghosh RD, Ghuwalewala S, Das P, Mandloi S, Alam SK, Chakraborty J, et al. MicroRNA profiling of cisplatin-resistant oral squamous cell carcinoma cell lines enriched with cancer-stem-cell-like and epithelial-mesenchymal transition-type features. Sci Rep (2016) 6:23932. doi: 10.1038/srep23932
14. Bourguignon LYW, Wong G, Earle C, Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem J Biol Chem (2012) 287:32800–24. doi: 10.1074/jbc.M111.308528
15. Bourguignon LYW, Wong G, Shiina M. Up-regulation of histone methyltransferase, DOT1L, by matrix hyaluronan promotes MicroRNA-10 expression leading to tumor cell invasion and chemoresistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem (2016) 291:10571–85. doi: 10.1074/jbc.M115.700021
16. Kulsum S, Sudheendra HV, Pandian R, Ravindra DR, Siddappa G, Nisheena R, et al. Cancer stem cell mediated acquired chemoresistance in head and neck cancer can be abrogated by aldehyde dehydrogenase 1 A1 inhibition. Mol Carcinog (2017) 56:694–711. doi: 10.1002/mc.22526
17. McDermott SC, Rodriguez-Ramirez C, McDermott SP, Wicha MS, Nör JE. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget (2018) 9:25148–65. doi: 10.18632/oncotarget.25358
18. Li R, Chen S, Zhan J, Li X, Liu W, Sheng X, et al. Long noncoding RNA FOXD2-AS1 enhances chemotherapeutic resistance of laryngeal squamous cell carcinoma via STAT3 activation. Cell Death Dis (2020) 11:41. doi: 10.1038/s41419-020-2232-7
19. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol (2000) 10:886–95. doi: 10.1016/S0960-9822(00)00610-2
20. Pabla N, Huang S, Mi QS, Daniel R, Dong Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem (2008) 283:6572–83. doi: 10.1074/jbc.M707568200
21. Gaponova AV, Deneka AY, Beck TN, Liu H, Andrianov G, Nikonova AS, et al. Identification of evolutionarily conserved DNA damage response genes that alter sensitivity to cisplatin. Oncotarget (2017) 8:19156–71. doi: 10.18632/oncotarget.13353
22. Hasegawa Y, Goto M, Hanai N, Ozawa T, Hirakawa H. Predictive biomarkers for combined chemotherapy with 5−fluorouracil and cisplatin in oro− and hypopharyngeal cancers. Mol Clin Oncol Spandidos Publ (2017) 8:378–86. doi: 10.3892/mco.2017.1521
23. Jelinek MJ, Foster NR, Zoroufy AJ, De Souza JA, Schwartz GK, Munster PN, et al. A phase I/II trial adding poly(ADP-ribose) polymerase (PARP) inhibitor veliparib to induction carboplatin-paclitaxel (Carbo-Tax) in patients with head and neck squamous cell carcinoma (HNSCC) Alliance A091101. J Clin Oncol (2018) 36:6031–1. doi: 10.1200/JCO.2018.36.15_suppl.6031
24. Mendez E, Rodriguez CP, Kao MC, Raju S, Diab A, Harbison RA, et al. A phase I clinical trial of AZD1775 in combination with neoadjuvant weekly docetaxel and cisplatin before definitive therapy in head and neck squamous cell carcinoma. Clin Cancer Res (2018) 24:2740–8. doi: 10.1158/1078-0432.CCR-17-3796
25. Chen B, Chen J, House MG, Cullen KJ, Nephew KP, Guo Z. Role of neurofilament light polypeptide in head and neck cancer chemoresistance. Mol Cancer Res (2012) 10:305–15. doi: 10.1158/1541-7786.MCR-11-0300
26. Day TA, Shirai K, O’Brien PE, Matheus MG, Godwin K, Sood AJ, et al. Inhibition of mTOR signaling and clinical activity of rapamycin in head and neck cancer in a window of opportunity trial. Clin Cancer Res (2019) 25:1156–64. doi: 10.1158/1078-0432.CCR-18-2024
27. Gupta A, Ajith A, Singh S, Panday RK, Samaiya A, Shukla S. PAK2–c-Myc–PKM2 axis plays an essential role in head and neck oncogenesis via regulating Warburg effect. Cell Death Dis (2018) 9:1–15. doi: 10.1038/s41419-018-0887-0
28. Chikuda J, Otsuka K, Shimomura I, Ito K, Miyazaki H, Takahashi RU, et al. CD44S induces MiR-629-3p expression in association with cisplatin resistance in head and neck cancer cells. Cancers (Basel) (2020) 12:856. doi: 10.3390/cancers12040856.
29. Vahabi M, Pulito C, Sacconi A, Donzelli S, D’Andrea M, Manciocco V, et al. MiR-96-5p targets PTEN expression affecting radio-chemosensitivity of HNSCC cells. J Exp Clin Cancer Res (2019) 38:141. doi: 10.1186/s13046-019-1119-x
30. Yang XH, Feng ZE, Yan M, Hanada S, Zuo H, Yang CZ, et al. Xiap is a predictor of Cisplatin-based chemotherapy response and prognosis for patients with advanced head and neck cancer. PloS One (2012) 7:e31601. doi: 10.1371/journal.pone.0031601
31. Michaud WA, Nichols AC, Mroz EA, Faquin WC, Clark JR, Begum S, et al. Bcl-2 blocks cisplatin-induced apoptosis and predicts poor outcome following chemoradiation treatment in advanced oropharyngeal squamous cell carcinoma. Clin Cancer Res (2009) 15:1645–54. doi: 10.1158/1078-0432.CCR-08-2581
32. Kumar B, Yadav A, Lang JC, Cipolla MJ, Schmitt AC, Arradaza N, et al. YM155 reverses cisplatin resistance in head and neck cancer by decreasing cytoplasmic survivin levels. Mol Cancer Ther (2012) 11:1988–98. doi: 10.1158/1535-7163.MCT-12-0167
33. Knauer SK, Unruhe B, Karczewski S, Hecht R, Fetz V, Bier C, et al. Functional Characterization of Novel Mutations Affecting Survivin (BIRC5)-Mediated Therapy Resistance in Head and Neck Cancer Patients. Hum Mutat (2013) 34:395–404. doi: 10.1002/humu.22249
34. Bullenkamp J, Raulf N, Ayaz B, Walczak H, Kulms D, Odell E, et al. Bortezomib sensitises TRAIL-resistant HPV-positive head and neck cancer cells to TRAIL through a caspase-dependent, E6-independent mechanism. Cell Death Dis (2014) 5:e1489. doi: 10.1038/cddis.2014.455
35. Bendell JC, Gordon MS, Hurwitz HI, Jones SF, Mendelson DS, Blobe GC, et al. Safety, pharmacokinetics, pharmacodynamics, and antitumor activity of dalantercept, an activin receptor-like kinase-1 ligand trap, in patients with advanced cancer. Clin Cancer Res (2014) 20:480–9. doi: 10.1158/1078-0432.CCR-13-1840
36. Hawinkels LJAC, De Vinuesa AG, Paauwe M, Kruithof-De Julio M, Wiercinska E, Pardali E, et al. Activin receptor-like kinase 1 ligand trap reduces microvascular density and improves chemotherapy efficiency to various solid tumors. Clin Cancer Res (2016) 22:96–106. doi: 10.1158/1078-0432.CCR-15-0743
37. De-Colle C, Mönnich D, Welz S, Boeke S, Sipos B, Fend F, et al. SDF-1/CXCR4 expression in head and neck cancer and outcome after postoperative radiochemotherapy. Clin Transl Radiat Oncol (2017) 5:28–36. doi: 10.1016/j.ctro.2017.06.004
38. Brands RC, De Donno F, Knierim ML, Steinacker V, Hartmann S, Seher A, et al. Multi-kinase inhibitors and cisplatin for head and neck cancer treatment in vitro. Oncol Lett (2019) 18:2220–31. doi: 10.3892/ol.2019.10541
39. Limaye S, Riley S, Zhao S, O’Neill A, Posner M, Adkins D, et al. A randomized phase II study of docetaxel with or without vandetanib in recurrent or metastatic squamous cell carcinoma of head and neck (SCCHN). Oral Oncol (2013) 49:835–41. doi: 10.1016/j.oraloncology.2013.04.010
40. Koontongkaew S. The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J Cancer (2013) 4:66–83. doi: 10.7150/jca.5112
41. Wheeler SE, Shi H, Lin F, Dasari S, Bednash J, Thorne S, et al. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck (2014) 36:385–92. doi: 10.1002/hed.23312
42. Qin X, Guo H, Wang X, Zhu X, Yan M, Wang X, et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol (2019) 20:12. doi: 10.1186/s13059-018-1604-0
43. Zhang D, Ding L, Li Y, Ren J, Shi G, Wang Y, et al. Midkine derived from cancer-associated fibroblasts promotes cisplatin-resistance via up-regulation of the expression of lncRNA ANRIL in tumour cells. Sci Rep (2017) 7:16231. doi: 10.1038/s41598-017-13431-y
44. Matsuoka Y, Yoshida R, Nakayama H, Nagata M, Hirosue A, Tanaka T, et al. The tumour stromal features are associated with resistance to 5-FU-based chemoradiotherapy and a poor prognosis in patients with oral squamous cell carcinoma. APMIS (2015) 123:205–14. doi: 10.1111/apm.12344
45. Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem (1962) 237:1555–62. doi: 10.1016/S0021-9258(19)83739-0
46. Cohen S. The stimulation of epidermal proliferation by a specific protein (EGF). Dev Biol (1965) 12:394–407. doi: 10.1016/0012-1606(65)90005-9
47. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys (2004) 59:21–6. doi: 10.1016/j.ijrobp.2003.11.041
48. Bertotti A, Sassi F. Molecular Pathways: Sensitivity and Resistance to Anti-EGFR Antibodies. Clin Cancer Res (2015) 21:3377–83. doi: 10.1158/1078-0432.CCR-14-0848
49. Wee P, Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) (2017) 9:52. doi: 10.3390/cancers9050052
50. Baro M, Lopez Sambrooks C, Burtness BA, Lemmon MA, Contessa JN. Neuregulin Signaling Is a Mechanism of Therapeutic Resistance in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther (2019) 18:2124–34. doi: 10.1158/1535-7163.MCT-19-0163
51. Alsahafi E, Begg K, Amelio I, Raulf N, Lucarelli P, Sauter T, et al. Clinical update on head and neck cancer: molecular biology and ongoing challenges. Cell Death Dis (2019) 10:1–17. doi: 10.1038/s41419-019-1769-9
52. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol (2018) 12:3–20. doi: 10.1002/1878-0261.12155@10.1002/(ISSN)1878-0261.reviews
53. Solomon LW, Frustino JL, Loree TR, Brecher ML, Alberico RA, Sullivan M. Ewing sarcoma of the mandibular condyle: Multidisciplinary management optimizes outcome. Head Neck (2008) 30:405–10. doi: 10.1002/hed.20692/abstract
54. Ang KK, Berkey BA, Tu X, Zhang H-Z, Katz R, Hammond EH, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res (2002) 62:7350–6.
55. Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst (1998) 90:824–32. doi: 10.1093/jnci/90.11.824
56. Gougis P, Moreau Bachelard C, Kamal M, Gan HK, Borcoman E, Torossian N, et al. Clinical Development of Molecular Targeted Therapy in Head and Neck Squamous Cell Carcinoma. JNCI Cancer Spectr (2019) 3:pkz055. doi: 10.1093/jncics/pkz055
57. Sturgeon K, Digiovanni L, Good J, Salvatore D, Fenderson D, Domchek S, et al. Exercise-Induced Dose-Response Alterations in Adiponectin and Leptin Levels Are Dependent on Body Fat Changes in Women at Risk for Breast Cancer. Cancer Epidemiol Biomarkers Prev (2016) 25:1195–200. doi: 10.1158/1055-9965.EPI-15-1087
58. Sacco AG, Worden FP. Molecularly targeted therapy for the treatment of head and neck cancer: A review of the ErbB family inhibitors. Onco Targets Ther (2016) 9:1927–43. doi: 10.2147/OTT.S93720
59. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med (2008) 358:1160–74. doi: 10.1056/NEJMra0707704
60. Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci (2007) 98:1275–80. doi: 10.1111/j.1349-7006.2007.00510.x
61. Patel D, Guo X, Ng S, Melchior M, Balderes P, Burtrum D, et al. IgG isotype, glycosylation, and EGFR expression determine the induction of antibody-dependent cellular cytotoxicity in vitro by cetuximab. Hum Antibodies (2010) 19:89–99. doi: 10.3233/HAB-2010-0232
62. Lee SC, Srivastava RM, López-Albaitero A, Ferrone S, Ferris RL. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res (2011) 50:248–54. doi: 10.1007/s12026-011-8231-0
63. Lattanzio L, Denaro N, Vivenza D, Varamo C, Strola G, Fortunato M, et al. Elevated basal antibody-dependent cell-mediated cytotoxicity (ADCC) and high epidermal growth factor receptor (EGFR) expression predict favourable outcome in patients with locally advanced head and neck cancer treated with cetuximab and radiotherapy. Cancer Immunol Immunother CII (2017) 66:573–9. doi: 10.1007/s00262-017-1960-8
64. Boeckx C, Baay M, Wouters A, Specenier P, Vermorken JB, Peeters M, et al. Anti-Epidermal Growth Factor Receptor Therapy in Head and Neck Squamous Cell Carcinoma: Focus on Potential Molecular Mechanisms of Drug Resistance. Oncologist (2013) 18:850–64. doi: 10.1634/theoncologist.2013-0013
65. Schmitz S, Ang KK, Vermorken J, Haddad R, Suarez C, Wolf GT, et al. Targeted therapies for squamous cell carcinoma of the head and neck: Current knowledge and future directions. Cancer Treat Rev (2014) 40:390–404. doi: 10.1016/j.ctrv.2013.09.007
66. Bonner JA, Azarnia N, Jones CU, Jassem J, Baselga J, Rowinsky EK. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2006) 12:567–78. doi: 10.1056/NEJMoa053422
67. Taberna M, Oliva M, Mesía R. Cetuximab-containing combinations in locally advanced and recurrent or metastatic head and neck squamous cell carcinoma. Front Oncol (2019) 9:383. doi: 10.3389/fonc.2019.00383
68. Azoury SC, Gilmore RC, Shukla V. Molecularly targeted agents and immunotherapy for the treatment of head and neck squamous cell cancer (HNSCC). Discovery Med (2016) 21:507–16.
69. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer (2018) 18:269–82. doi: 10.1038/nrc.2018.11
70. Canning M, Guo G, Yu M, Myint C, Groves MW, Byrd JK, et al. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front Cell Dev Biol (2019) 7. doi: 10.3389/fcell.2019.00052
71. Lawrence MS, Sougnez C, Lichtenstein L, Cibulskis K, Lander E, Gabriel SB, et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
72. Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based the. J Clin Oncol Off J Am Soc Clin Oncol (2007) 25:2171–7. doi: 10.1200/JCO.2006.06.7447
73. Chen LF, Cohen EEW, Grandis JR. New Strategies in Head and Neck Cancer: epidermal growth factor receptor inhibition in head and neck cancer. Clin Cancer Res (2010) 16:2489–95. doi: 10.1158/1078-0432.CCR-09-2318
74. Cohen RB. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat Rev (2014) 40:567–77. doi: 10.1016/j.ctrv.2013.10.002
75. Jagadeeshan S, Prasad M, Ortiz-Cuaran S, Gregoire V, Saintigny P, Elkabets M. Adaptive Responses to Monotherapy in Head and Neck Cancer: Interventions for Rationale-Based Therapeutic Combinations. Trends Cancer (2019) 5:365–90. doi: 10.1016/j.trecan.2019.04.004
76. López-Verdín S, Lavalle-Carrasco J, Carreón-Burciaga RG, Serafín-Higuera N, Molina-Frechero N, González-González R, et al. Molecular Markers of Anticancer Drug Resistance in Head and Neck Squamous Cell Carcinoma: A Literature Review. Cancers (Basel) (2018) 10:376. doi: 10.3390/cancers10100376
77. Willey CD, Anderson JC, Trummell HQ, Naji F, de Wijn R, Yang ES, et al. Differential escape mechanisms in cetuximab-resistant head and neck cancer cells. Biochem Biophys Res Commun (2019) 517:36–42. doi: 10.1016/j.bbrc.2019.06.159
78. Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med (2013) 19:1389–400. doi: 10.1038/nm.3388
79. Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene (2008) 27:3944–56. doi: 10.1038/onc.2008.19
80. Le Tourneau C, Faivre S, Siu LL. Molecular targeted therapy of head and neck cancer: Review and clinical development challenges. Eur J Cancer (2007) 43:2457–66. doi: 10.1016/j.ejca.2007.08.016
81. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer (2005) 5:341–54. doi: 10.1038/nrc1609
82. Nair S, Trummell HQ, Rajbhandari R, Thudi NK, Nozell SE, Warram JM, et al. Novel EGFR ectodomain mutations associated with ligand-independent activation and cetuximab resistance in head and neck cancer. PloS One (2020) 15:e0229077. doi: 10.1371/journal.pone.0229077
83. Khattri A, Sheikh N, Acharya R, Tan Y-HC, Kochanny S, Lingen MW, et al. Mechanism of acquired resistance to cetuximab in head and neck cancer. J Clin Oncol (2018) 36:e18061–1. doi: 10.1200/JCO.2018.36.15_suppl.e18061
84. Stabile LP, Egloff AM, Gibson MK, Gooding WE, Ohr J, Zhou P, et al. IL6 is associated with response to dasatinib and cetuximab: Phase II clinical trial with mechanistic correlatives in cetuximab-resistant head and neck cancer. Oral Oncol (2017) 69:38–45. doi: 10.1016/j.oraloncology.2017.03.011
85. Braig F, Kriegs M, Voigtlaender M, Habel B, Grob T, Biskup K, et al. Cetuximab Resistance in Head and Neck Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res (2017) 77:1188–99. doi: 10.1158/0008-5472.CAN-16-0754
86. Smilek P, Neuwirthova J, Jarkovsky J, Dusek L, Rottenberg J, Kostrica R, et al. Epidermal growth factor receptor (EGFR) expression and mutations in the EGFR signaling pathway in correlation with anti-EGFR therapy in head and neck squamous cell carcinomas. Neoplasma (2012) 59:508–15. doi: 10.4149/neo_2012_065
87. Tinhofer I, Klinghammer K, Weichert W, Knödler M, Stenzinger A, Gauler T, et al. Expression of Amphiregulin and EGFRvIII Affect Outcome of Patients with Squamous Cell Carcinoma of the Head and Neck Receiving Cetuximab–Docetaxel Treatment. Clin Cancer Res (2011) 17:5197–204. doi: 10.1158/1078-0432.CCR-10-3338
88. Morris LGT, Taylor BS, Bivona TG, Gong Y, Eng S, Brennan CW, et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc Natl Acad Sci U.S.A. (2011) 108:19024–9. doi: 10.1073/pnas.1111963108
89. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nat Rev Clin Oncol (2010) 7:493–507. doi: 10.1038/nrclinonc.2010.97
90. Li C, Iida M, Dunn EF, Wheeler DL. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol (2010) 97:330–7. doi: 10.1016/j.radonc.2010.06.010
91. Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR Contributes to Acquired Resistance to Cetuximab. Oncogene (2009) 28:3801–13. doi: 10.1038/onc.2009.234
92. Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res Off J Am Assoc Cancer Res (2006) 12:5064–73. doi: 10.1158/1078-0432.CCR-06-0913
93. Duvvuri U, George J, Kim S, Alvarado D, Neumeister VM, Chenna A, et al. Molecular and Clinical Activity of CDX-3379, an Anti-ErbB3 Monoclonal Antibody, in Head and Neck Squamous Cell Carcinoma Patients. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:5752–8. doi: 10.1158/1078-0432.CCR-18-3453
94. Yonesaka K, Tanaka K, Kitano M, Kawakami H, Hayashi H, Takeda M, et al. Aberrant HER3 ligand heregulin-expressing head and neck squamous cell carcinoma is resistant to anti-EGFR antibody cetuximab, but not second-generation EGFR-TKI. Oncogenesis (2019) 8:54. doi: 10.1038/s41389-019-0164-9
95. Ansell A, Jedlinski A, Johansson A-C, Roberg K. Epidermal growth factor is a potential biomarker for poor cetuximab response in tongue cancer cells. J Oral Pathol Med (2016) 45:9–16. doi: 10.1111/jop.12310
96. Saki M, Toulany M, Rodemann HP. Acquired resistance to cetuximab is associated with the overexpression of Ras family members and the loss of radiosensitization in head and neck cancer cells. Radiother Oncol (2013) 108:473–8. doi: 10.1016/j.radonc.2013.06.023
97. Bedi A, Chang X, Noonan K, Pham V, Bedi R, Fertig EJ, et al. Inhibition of TGF-β Enhances the In Vivo Antitumor Efficacy of EGF Receptor–Targeted Therapy. Mol Cancer Ther (2012) 11:2429–39. doi: 10.1158/1535-7163.MCT-12-0101-T
98. Hatakeyama H, Cheng H, Wirth P, Counsell A, Marcrom SR, Wood CB, et al. Regulation of Heparin-Binding EGF-Like Growth Factor by miR-212 and Acquired Cetuximab-Resistance in Head and Neck Squamous Cell Carcinoma. PloS One (2010) 5:e12702. doi: 10.1371/journal.pone.0012702
99. Suárez Pestana E, Tenev T, Gross S, Stoyanov B, Ogata M, Böhmer FD. The transmembrane protein tyrosine phosphatase RPTPsigma modulates signaling of the epidermal growth factor receptor in A431 cells. Oncogene (1999) 18:4069–79. doi: 10.1038/sj.onc.1202794
100. Boeckx C, Weyn C, Vanden Bempt I, Deschoolmeester V, Wouters A, Specenier P, et al. Mutation analysis of genes in the EGFR pathway in Head and Neck cancer patients: implications for anti-EGFR treatment response. BMC Res Notes (2014) 7:337. doi: 10.1186/1756-0500-7-337
101. Wu Q, Zhao Y, Wang P. miR-204 inhibits angiogenesis and promotes sensitivity to cetuximab in head and neck squamous cell carcinoma cells by blocking JAK2-STAT3 signaling. BioMed Pharmacother (2018) 99:278–85. doi: 10.1016/j.biopha.2018.01.055
102. Saba NF, Hurwitz SJ, Magliocca K, Kim S, Owonikoko TK, Harvey D, et al. Phase 1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer (2014) 120:3940–51. doi: 10.1002/cncr.28965
103. Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, et al. Targeting Stat3 Abrogates EGFR Inhibitor Resistance in Cancer. Clin Cancer Res (2012) 18:4986–96. doi: 10.1158/1078-0432.CCR-12-0792
104. Bonner JA, Yang ES, Trummell HQ, Nowsheen S, Willey CD, Raisch KP. Inhibition of STAT-3 results in greater cetuximab sensitivity in head and neck squamous cell carcinoma. Radiother Oncol J Eur Soc Ther Radiol Oncol (2011) 99:339–43. doi: 10.1016/j.radonc.2011.05.070
105. Bozec A, Etienne-Grimaldi M-C, Fischel J-L, Sudaka A, Toussan N, Formento P, et al. The mTOR-targeting drug temsirolimus enhances the growth-inhibiting effects of the cetuximab–bevacizumab–irradiation combination on head and neck cancer xenografts. Oral Oncol (2011) 47:340–4. doi: 10.1016/j.oraloncology.2011.02.020
106. Leeman-Neill RJ, Seethala RR, Singh SV, Freilino ML, Bednash JS, Thomas SM, et al. Inhibition of EGFR-STAT3 signaling with erlotinib prevents carcinogenesis in a chemically-induced mouse model of oral squamous cell carcinoma. Cancer Prev Res (Phila) (2011) 4:230–7. doi: 10.1158/1940-6207.CAPR-10-0249
107. Uzawa K, Amelio AL, Kasamatsu A, Saito T, Kita A, Fukamachi M, et al. Resveratrol Targets Urokinase-Type Plasminogen Activator Receptor Expression to Overcome Cetuximab-Resistance in Oral Squamous Cell Carcinoma. Sci Rep (2019) 9:1–13. doi: 10.1038/s41598-019-48717-w
108. Nozaki M, Yasui H, Ohnishi Y. Ligand-Independent EGFR Activation by Anchorage-Stimulated Src Promotes Cancer Cell Proliferation and Cetuximab Resistance via ErbB3 Phosphorylation. Cancers (Basel) (2019) 11:1552. doi: 10.20944/preprints201909.0037.v1
109. Shu L, Wang D, Nannapaneni S, Shin DM, Saba NF, Chen GZ. Abstract 4834: Effective treatment of squamous cell carcinoma of the head and neck (HNSCC) using a combined regimen of tipifarnib and cetuximab. Cancer Res (2019) 79:4834–4. doi: 10.1158/1538-7445.AM2019-4834
110. Braig F, Voigtlaender M, Schieferdecker A, Busch C-J, Laban S, Grob T, et al. Liquid biopsy monitoring uncovers acquired RAS-mediated resistance to cetuximab in a substantial proportion of patients with head and neck squamous cell carcinoma. Oncotarget (2016) 7:42988–95. doi: 10.18632/oncotarget.8943
111. Bossi P, Bergamini C, Siano M, Cossu Rocca M, Sponghini AP, Favales F, et al. Functional genomics uncover the biology behind the responsiveness of head and neck squamous cell cancer patients to cetuximab. Clin Cancer Res (2016) 22:3961–70. doi: 10.1158/1078-0432.CCR-15-2547
112. Boeckx C, Op de Beeck K, Wouters A, Deschoolmeester V, Limame R, Zwaenepoel K, et al. Overcoming cetuximab resistance in HNSCC: The role of AURKB and DUSP proteins. Cancer Lett (2014) 354:365–77. doi: 10.1016/j.canlet.2014.08.039
113. Rampias T, Giagini A, Siolos S, Matsuzaki H, Sasaki C, Scorilas A, et al. RAS/PI3K crosstalk and cetuximab resistance in head and neck squamous cell carcinoma. Clin Cancer Res (2014) 20:2933–46. doi: 10.1158/1078-0432.CCR-13-2721
114. Pickhard A, Siegl M, Baumann A, Huhn M, Wirth M, Reiter R, et al. The response of head and neck squamous cell carcinoma to cetuximab treatment depends on Aurora kinase A polymorphism. Oncotarget (2014) 5:5428–38. doi: 10.18632/oncotarget.2117
115. Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora Kinase Inhibition Overcomes Cetuximab Resistance in Squamous Cell Cancer of the Head and Neck. Oncotarget (2011) 2:599–609. doi: 10.18632/oncotarget.311
116. Marquard FE, Jücker M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem Pharmacol (2020) 172:113729. doi: 10.1016/j.bcp.2019.113729
117. Eze N, Lee J-W, Yang D-H, Zhu F, Neumeister V, Sandoval-Schaefer T, et al. PTEN loss is associated with resistance to cetuximab in patients with head and neck squamous cell carcinoma. Oral Oncol (2019) 91:69–78. doi: 10.1016/j.oraloncology.2019.02.026
118. Job S, de Reyniès A, Heller B, Weiss A, Guérin E, Macabre C, et al. Preferential Response of Basal-Like Head and Neck Squamous Cell Carcinoma Cell Lines to EGFR-Targeted Therapy Depending on EREG-Driven Oncogenic Addiction. Cancers (Basel) (2019) 11:795. doi: 10.3390/cancers11060795
119. Izumi H, Wang Z, Goto Y, Ando T, Wu X, Zhang X, et al. Pathway-specific genome editing of PI3K/mTOR tumor suppressor genes reveals that PTEN loss contributes to cetuximab resistance in head and neck cancer. Mol Cancer Ther (2020) 19:1562–71. doi: 10.1158/1535-7163.MCT-19-1036
120. Klinghammer KF, Politz O, Eder T, Otto R, Raguse JD, Hoffmann J, et al. Preclinical efficacy of copanlisib in cetuximab sensitive and resistant tumors of HNSCC. J Clin Oncol (2019) 37:6031. doi: 10.1200/JCO.2019.37.15_suppl.6031
121. Bozec A, Ebran N, Radosevic-Robin N, Chamorey E, Ben Yahia H, Marcie S, et al. Combination of phosphotidylinositol-3-kinase targeting with cetuximab and irradiation: A preclinical study on an orthotopic xenograft model of head and neck cancer. Head Neck (2017) 39:151–9. doi: 10.1002/hed.24560
122. Lattanzio L, Tonissi F, Monteverde M, Vivenza D, Russi E, Milano G, et al. Treatment effect of buparlisib, cetuximab and irradiation in wild-type or PI3KCA-mutated head and neck cancer cell lines. Invest New Drugs (2015) 33:310–20. doi: 10.1007/s10637-015-0210-1
123. Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell (2015) 27:533–46. doi: 10.1016/j.ccell.2015.03.010
124. D’Amato V, Rosa R, D’Amato C, Formisano L, Marciano R, Nappi L, et al. The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. Br J Cancer (2014) 110:2887–95. doi: 10.1038/bjc.2014.241
125. Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Sol Degese M, et al. mTOR Co-Targeting in Cetuximab Resistance in Head and Neck Cancers Harboring PIK3CA and RAS Mutations. JNCI J Natl Cancer Inst (2014) 106. doi: 10.1093/jnci/dju215
126. Razak ARA, Ahn M-J, Yen C-J, Solomon BJ, Lee S-H, Wang H-M, et al. Phase lb/ll study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN). J Clin Oncol (2014) 32:6044. doi: 10.1200/jco.2014.32.15_suppl.6044
127. Rebucci M, Peixoto P, Dewitte A, Wattez N, De Nuncques M-A, Rezvoy N, et al. Mechanisms underlying resistance to cetuximab in the HNSCC cell line: role of AKT inhibition in bypassing this resistance. Int J Oncol (2011) 38:189–200. doi: 10.3892/ijo_00000838
128. Geiger JL, Grandis JR, Bauman JE. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncol (2016) 56:84–92. doi: 10.1016/j.oraloncology.2015.11.022
129. Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene (2004) 23:7906–9. doi: 10.1038/sj.onc.1208160
130. Maggioni D, Gaini R, Nicolini G, Tredici G, Garavello W. MAPKs activation in head and neck squamous cell carcinomas. Oncol Rev (2011) 5:223–31. doi: 10.1007/s12156-011-0086-z
131. Leelahavanichkul K, Amornphimoltham P, Molinolo AA, Basile JR, Koontongkaew S, Gutkind JS. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol Oncol (2014) 8:105–18. doi: 10.1016/j.molonc.2013.10.003
132. Rampias T, Giagini A, Matsuzaki H, Bartzi V, Siolos S, Vaja E, et al. Genetic alterations in HRAS gene in relation to outcome and response to cetuximab in head and neck squamous cell carcinoma. J Clin Oncol (2012) 30:5574. doi: 10.1200/jco.2012.30.15_suppl.5574
133. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol (2014) 4:64. doi: 10.3389/fonc.2014.00064
134. García-Escudero R, Segrelles C, Dueñas M, Pombo M, Ballestín C, Alonso-Riaño M, et al. Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral Oncol (2018) 79:55–63. doi: 10.1016/j.oraloncology.2018.02.014
135. Wang D, Qian G, Zhang H, Magliocca KR, Nannapaneni S, Amin ARMR, et al. HER3 targeting sensitizes HNSCC to cetuximab by reducing HER3 activity and HER2/HER3 dimerization - evidence from cell line and patient derived xenograft models. Clin Cancer Res (2016) 23:677–86. doi: 10.1158/1078-0432.CCR-16-0558
136. Iida M, Bahrar H, Brand TM, Pearson HE, Coan JP, Orbuch RA, et al. Targeting the HER family with Pan-HER effectively overcomes resistance to cetuximab. Mol Cancer Ther (2016) 15:2175–86. doi: 10.1158/1535-7163.MCT-16-0012
137. Kjær I, Lindsted T, Fröhlich C, Olsen JV, Horak ID, Kragh M, et al. Cetuximab Resistance in Squamous Carcinomas of the Upper Aerodigestive Tract Is Driven by Receptor Tyrosine Kinase Plasticity: Potential for mAb Mixtures. Mol Cancer Ther (2016) 15:1614–26. doi: 10.1158/1535-7163.MCT-15-0565
138. Madoz-Gúrpide J, Zazo S, Chamizo C, Casado V, Caramés C, Gavín E, et al. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J Transl Med (2015) 13:282. doi: 10.1186/s12967-015-0633-7
139. Brand TM, Iida M, Stein AP, Corrigan KL, Braverman C, Luthar N, et al. AXL mediates resistance to cetuximab therapy. Cancer Res (2014) 74:5152–64. doi: 10.1158/0008-5472.CAN-14-0294
140. Quesnelle KM, Grandis JR. Dual kinase inhibition of EGFR and HER2 overcomes resistance to cetuximab in a novel in vivo model of acquired cetuximab resistance. Clin Cancer Res (2011) 17:5935–44. doi: 10.1158/1078-0432.CCR-11-0370
141. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med (2011) 3:99ra86. doi: 10.1126/scitranslmed.3002442
142. Chung CH, Pohlmann PR, Rothenberg ML, Burkey BB, Parker J, Palka K, et al. Insulin-like growth factor-1 receptor inhibitor, AMG-479, in cetuximab-refractory head and neck squamous cell carcinoma. Head Neck (2011) 33:1804–8. doi: 10.1002/hed.21478
143. Zuo Q, Shi M, Li L, Chen J, Luo R. Development of cetuximab-resistant human nasopharyngeal carcinoma cell lines and mechanisms of drug resistance. BioMed Pharmacother (2010) 64:550–8. doi: 10.1016/j.biopha.2010.03.003
144. Barnes CJ, Ohshiro K, Rayala SK, El-Naggar AK, Kumar R. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2007) 13:4291–9. doi: 10.1158/1078-0432.CCR-06-2040
145. McDaniel NK, Iida M, Nickel KP, Rodems TS, Swick AD, Prabakaran PJ, et al. Abstract A128: AXL mediates cetuximab resistance through tyrosine 821 and activation of c-Abl kinase in head and neck cancer. Mol Cancer Ther (2019) 18:A128–8. doi: 10.1158/1535-7163.TARG-19-A128
146. Novoplansky O, Fury M, Prasad M, Yegodayev K, Zorea J, Cohen L, et al. MET activation confers resistance to cetuximab, and prevents HER2 and HER3 upregulation in head and neck cancer. Int J Cancer (2019) 145:748–62. doi: 10.1002/ijc.32170
147. Yang H, Mo C, Xun Y, Liu LG, Li W, Guan J, et al. Combination of cetuximab with met inhibitor in control of cetuximab-resistant oral squamous cell carcinoma. Am J Transl Res (2019) 11:2370–81.
148. Leonard B, Brand TM, O’Keefe RA, Lee ED, Zeng Y, Kemmer JD, et al. BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res (2018) 78:4331–43. doi: 10.1158/0008-5472.CAN-18-0459
149. Deeken JF, Wang H, Marshall J, Subramaniam DS, Hwang JJ, He AR, et al. Final results of a phase I study of lapatinib (LAP) and cetuximab (CET) in patients with CET-sensitive solid tumors. J Clin Oncol (2013) 31:2616. doi: 10.1200/jco.2013.31.15_suppl.2616
150. Deeken JF, Wang H, Subramaniam D, He AR, Hwang J, Marshall JL, et al. A phase 1 study of cetuximab and lapatinib in patients with advanced solid tumor malignancies. Cancer (2015) 121:1645–53. doi: 10.1002/cncr.29224
151. Argiris A, Bauman JE, Ohr J, Gooding WE, Heron DE, Duvvuri U, et al. Phase II randomized trial of radiation therapy, cetuximab, and pemetrexed with or without bevacizumab in patients with locally advanced head and neck cancer. Ann Oncol Off J Eur Soc Med Oncol (2016) 27:1594–600. doi: 10.1093/annonc/mdw204
152. Fury MG, Xiao H, Sherman EJ, Baxi S, Smith-Marrone S, Schupak K, et al. A Phase II Trial of Bevacizumab + Cetuximab + Cisplatin with Concurrent Intensity Modulated Radiation Therapy (IMRT) for Patients with Stage III/IVB Head and Neck Squamous Cell Carcinoma (HNSCC). Head Neck (2016) 38:E566–70. doi: 10.1002/hed.24041
153. Argiris A, Kotsakis AP, Hoang T, Worden FP, Savvides P, Gibson MK, et al. Cetuximab and bevacizumab: preclinical data and phase II trial in recurrent or metastatic squamous cell carcinoma of the head and neck. Ann Oncol Off J Eur Soc Med Oncol (2013) 24:220–5. doi: 10.1093/annonc/mds245
154. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer (2004) 4:361–70. doi: 10.1038/nrc1360
155. Elferink LA, Resto VA. Receptor-Tyrosine-Kinase-Targeted Therapies for Head and Neck Cancer. J Signal Transduct (2011) 2011:1–11. doi: 10.1155/2011/982879
156. McDaniel NK, Iida M, Nickel KP, Longhurst CA, Fischbach SR, Rodems TS, et al. AXL mediates cetuximab and radiation resistance through tyrosine 821 and the c-ABL kinase pathway in head and neck cancer. Clin Cancer Res (2020) 26:4349–59. doi: 10.1158/1078-0432.CCR-19-3142
157. Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res MCR (2007) 5:203–20. doi: 10.1158/1541-7786.MCR-06-0404
158. Forster MD, Dillon MT, Kocsis J, Remenár É, Pajkos G, Rolland F, et al. Patritumab or placebo, with cetuximab plus platinum therapy in recurrent or metastatic squamous cell carcinoma of the head and neck: A randomised phase II study. Eur J Cancer (Oxford Engl 1990) (2019) 123:36–47. doi: 10.1016/j.ejca.2019.08.017
159. Ferrarotto R, William WN, Tseng JE, Marur S, Shin DM, Murphy B, et al. Randomized phase II trial of cixutumumab alone or with cetuximab for refractory recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol (2018) 82:83–90. doi: 10.1016/j.oraloncology.2018.05.014
160. Glisson BS, Tseng J, Marur S, Shin DM, Murphy BA, Cohen EEW, et al. Randomized phase II trial of cixutumumab (CIX) alone or with cetuximab (CET) for refractory recurrent/metastatic squamous cancer of head and neck (R/M-SCCHN). J Clin Oncol (2013) 31:6030. doi: 10.1200/jco.2013.31.15_suppl.6030
161. Lu H, Liang K, Lu Y, Fan Z. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1α. Cancer Lett (2012) 322:78–85. doi: 10.1016/j.canlet.2012.02.012
162. Lu H, Li X, Luo Z, Liu J, Fan Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol Cancer Ther (2013) 12:2187–99. doi: 10.1158/1535-7163.MCT-12-1245
163. Huang S, Benavente S, Armstrong EA, Li C, Wheeler DL, Harari PM. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res (2011) 71:7071–9. doi: 10.1158/0008-5472.CAN-11-0128
164. Boeckx C, Van den Bossche J, De Pauw I, Peeters M, Lardon F, Baay M, et al. The hypoxic tumor microenvironment and drug resistance against EGFR inhibitors: preclinical study in cetuximab-sensitive head and neck squamous cell carcinoma cell lines. BMC Res Notes (2015) 8:203. doi: 10.1186/s13104-015-1197-6
165. Lu H, Lu Y, Xie Y, Qiu S, Li X, Fan Z. Rational combination with PDK1 inhibition overcomes cetuximab resistance in head and neck squamous cell carcinoma. JCI Insight (2019) 4:e131106. doi: 10.1172/jci.insight.131106
166. Zhou G, Liu Z, Myers JN. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J Cell Biochem (2016) 117:2682–92. doi: 10.1002/jcb.25592
167. Nieto MA, Huang RY-J, Jackson RA, Thiery JP, Abell AN, Jordan NV, et al. EMT: 2016. Cell (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
168. Smith A, Teknos TN, Pan Q. Epithelial to Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Oral Oncol (2013) 49:287–92. doi: 10.1016/j.oraloncology.2012.10.009
169. Hsu DS-S, Hwang W-L, Yuh C-H, Chu C-H, Ho Y-H, Chen P-B, et al. Lymphotoxin-β Interacts with Methylated EGFR to Mediate Acquired Resistance to Cetuximab in Head and Neck Cancer. Clin Cancer Res (2017) 23:4388–401. doi: 10.1158/1078-0432.CCR-16-1955
170. Kagohara LT, Zamuner F, Davis-Marcisak EF, Sharma G, Considine M, Allen J, et al. Integrated single-cell and bulk gene expression and ATAC-seq reveals heterogeneity and early changes in pathways associated with resistance to cetuximab in HNSCC-sensitive cell lines. Br J Cancer (2020) 123:101–13. doi: 10.1038/s41416-020-0851-5
171. Stein-O’Brien G, Kagohara LT, Li S, Thakar M, Ranaweera R, Ozawa H, et al. Integrated time course omics analysis distinguishes immediate therapeutic response from acquired resistance. Genome Med (2018) 10:37. doi: 10.1186/s13073-018-0545-2
172. Ozawa H, Ranaweera RS, Izumchenko E, Makarev E, Zhavoronkov A, Fertig EJ, et al. SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin Cancer Res (2017) 23:5162–75. doi: 10.1158/1078-0432.CCR-16-1686
173. Cheng H, Fertig EJ, Ozawa H, Hatakeyama H, Howard JD, Perez J, et al. Decreased SMAD4 expression is associated with induction of epithelial-to-mesenchymal transition and cetuximab resistance in head and neck squamous cell carcinoma. Cancer Biol Ther (2015) 16:1252–8. doi: 10.1080/15384047.2015.1056418
174. La Fleur L, Johansson A-C, Roberg K. A CD44high/EGFRlow Subpopulation within Head and Neck Cancer Cell Lines Shows an Epithelial-Mesenchymal Transition Phenotype and Resistance to Treatment. PloS One (2012) 7:e44071. doi: 10.1371/journal.pone.0044071
175. Keysar SB, Le PN, Anderson RT, Morton JJ, Bowles DW, Paylor JJ, et al. Hedgehog Signaling Alters Reliance on EGF Receptor Signaling and Mediates Anti-EGFR Therapeutic Resistance in Head and Neck Cancer. Cancer Res (2013) 73:3381–92. doi: 10.1158/0008-5472.CAN-12-4047
176. Holz C, Niehr F, Boyko M, Hristozova T, Distel L, Budach V, et al. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol J Eur Soc Ther Radiol Oncol (2011) 101:158–64. doi: 10.1016/j.radonc.2011.05.042
177. Basu D, Nguyen T-TK, Montone KT, Zhang G, Wang L-P, Diehl JA, et al. Evidence for mesenchymal-like subpopulations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene (2010) 29:4170–82. doi: 10.1038/onc.2010.170
178. Boeckx C, Blockx L, de Beeck KO, Limame R, Van Camp G, Peeters M, et al. Establishment and characterization of cetuximab resistant head and neck squamous cell carcinoma cell lines: focus on the contribution of the AP-1 transcription factor. Am J Cancer Res (2015) 5:1921–38. doi: 2156-6976/ajcr0008298
179. Fujiwara T, Eguchi T, Sogawa C, Ono K, Murakami J, Ibaragi S, et al. Anti-EGFR antibody cetuximab is secreted by oral squamous cell carcinoma and alters EGF-driven mesenchymal transition. Biochem Biophys Res Commun (2018) 503:1267–72. doi: 10.1016/j.bbrc.2018.07.035
180. Kagohara LT, Zamuner F, Considine M, Allen J, Yegnasubramanian S, Gaykalova DA, et al. Integrated single cell and bulk multi-omics reveals heterogeneity and early changes in pathways associated with cetuximab resistance in HNSCC sensitive cell lines. bioRxiv (2019), 729384. doi: 10.1101/729384
181. Shimizu R, Ibaragi S, Eguchi T, Kuwajima D, Kodama S, Nishioka T, et al. Nicotine promotes lymph node metastasis and cetuximab resistance in head and neck squamous cell carcinoma. Int J Oncol (2019) 54:283–94. doi: 10.3892/ijo.2018.4631
182. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug Resistance and the Solid Tumor Microenvironment. JNCI J Natl Cancer Inst (2007) 99:1441–54. doi: 10.1093/jnci/djm135
183. Mandal R, Şenbabaoğlu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight (2016) 1:e89829. doi: 10.1172/jci.insight.89829
184. Concha-Benavente F, Ferris RL. Reversing EGFR Mediated Immunoescape by Targeted Monoclonal Antibody Therapy. Front Pharmacol (2017) 8:332. doi: 10.3389/fphar.2017.00332
185. Ferris RL, Saba NF, Gitlitz BJ, Haddad R, Sukari A, Neupane P, et al. Effect of Adding Motolimod to Standard Combination Chemotherapy and Cetuximab Treatment of Patients With Squamous Cell Carcinoma of the Head and Neck: The Active8 Randomized Clinical Trial. JAMA Oncol (2018) 4:1583–8. doi: 10.1001/jamaoncol.2018.1888
186. Chow LQM, Morishima C, Eaton KD, Baik CS, Goulart BH, Anderson LN, et al. Phase Ib Trial of the Toll-like Receptor 8 Agonist, Motolimod (VTX-2337), Combined with Cetuximab in Patients with Recurrent or Metastatic SCCHN. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23:2442–50. doi: 10.1158/1078-0432.CCR-16-1934
187. Shayan G, Kansy BA, Gibson SP, Srivastava RM, Bryan JK, Bauman JE, et al. Phase Ib Study of Immune Biomarker Modulation with Neoadjuvant Cetuximab and TLR8 Stimulation in Head and Neck Cancer to Overcome Suppressive Myeloid Signals. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24:62–72. doi: 10.1158/1078-0432.CCR-17-0357
188. Jie H-B, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4+ Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res (2015) 75:2200–10. doi: 10.1158/0008-5472.CAN-14-2788
189. Stephenson RM, Lim CM, Matthews M, Dietsch G, Hershberg R, Ferris RL. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol Immunother CII (2013) 62:1347–57. doi: 10.1007/s00262-013-1437-3
190. Freiser ME, Serafini P, Weed DT. The immune system and head and neck squamous cell carcinoma: from carcinogenesis to new therapeutic opportunities. Immunol Res (2013) 57:52–69. doi: 10.1007/s12026-013-8462-3
191. Ferris RL. Immunology and Immunotherapy of Head and Neck Cancer. J Clin Oncol (2015) 33:3293–304. doi: 10.1200/JCO.2015.61.1509
192. Saba NF, Chen ZG, Haigentz M, Bossi P, Rinaldo A, Rodrigo JP, et al. Targeting the EGFR and Immune Pathways in Squamous Cell Carcinoma of the Head and Neck (SCCHN): Forging a New Alliance. Mol Cancer Ther (2019) 18:1909–15. doi: 10.1158/1535-7163.MCT-19-0214
193. Bonomo P, Desideri I, Loi M, Mangoni M, Sottili M, Marrazzo L, et al. Anti PD-L1 DUrvalumab combined with Cetuximab and RadiOtherapy in locally advanced squamous cell carcinoma of the head and neck: A phase I/II study (DUCRO). Clin Transl Radiat Oncol (2018) 9:42–7. doi: 10.1016/j.ctro.2018.01.005
194. Tao Y, Auperin A, Sun X, Sire C, Martin L, Coutte A, et al. Avelumab-cetuximab-radiotherapy versus standards of care (SoC) in patients (pts) with locally advanced squamous cell carcinoma of head and neck (LA-SCCHN): Safety phase of randomized trial GORTEC 2017-01 (REACH). Ann Oncol (2019) 30:v454. doi: 10.1093/annonc/mdz252.010
195. Adkins D, Ley J, Neupane P, Worden F, Sacco AG, Palka K, et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: a multicentre, multigroup, phase 2 trial. Lancet Oncol (2019) 20:1295–305. doi: 10.1016/S1470-2045(19)30405-X
196. Faden DL, Concha-Benavente F, Chakka AB, McMichael EL, Chandran U, Ferris RL. Immunogenomic correlates of response to cetuximab monotherapy in head and neck squamous cell carcinoma. Head Neck (2019) 41:2591–601. doi: 10.1002/hed.25726
197. Sacco AG, Chen R, Ghosh D, Worden F, Wong DJ, Adkins D, et al. An open-label, non-randomized, multi-arm, phase II trial evaluating pembrolizumab combined with cetuximab in patients (pts) with recurrent/metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): updated results of cohort 1 analysis. Int J Radiat Oncol • Biol • Phys (2020) 106:1121–2. doi: 10.1016/j.ijrobp.2019.11.376
198. Elbers JBW, Al-Mamgani A, Tesseslaar MET, van den Brekel MWM, Lange CAH, van der Wal JE, et al. Immuno-radiotherapy with cetuximab and avelumab for advanced stage head and neck squamous cell carcinoma: Results from a phase-I trial. Radiother Oncol (2020) 142:79–84. doi: 10.1016/j.radonc.2019.08.007
199. Fayette J, Lefebvre G, Posner MR, Bauman J, Salas S, Even C, et al. Results of a phase II study evaluating monalizumab in combination with cetuximab in previously treated recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). Ann Oncol Off J Eur Soc Med Oncol (2018) 29(Suppl 8):viii374. doi: 10.1093/annonc/mdy287.005
200. Jie H-B, Srivastava RM, Argiris A, Bauman JE, Kane LP, Ferris RL. Increased PD-1+ and TIM-3+ TILs during Cetuximab Therapy Inversely Correlate with Response in Head and Neck Cancer Patients. Cancer Immunol Res (2017) 5:408–16. doi: 10.1158/2326-6066.CIR-16-0333
201. Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S, et al. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin Cancer Res (2017) 23:707–16. doi: 10.1158/1078-0432.CCR-16-0879
202. Bauman J, Ferris RL, Clump DA, Ohr J, Gooding W, Kim S, et al. Phase I trial of cetuximab, intensity modulated radiotherapy (IMRT), and ipilimumab in previously untreated, locally advanced head and neck squamous cell carcinoma (PULA HNSCC). Ann Oncol (2016) 27:vi571. doi: 10.1093/annonc/mdw435.31
203. Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the Cell-Intrinsic and -Extrinsic Pathways Downstream of EGFR and IFNγ That Induce PD-L1 Expression in Head and Neck Cancer. Cancer Res (2016) 76:1031–43. doi: 10.1158/0008-5472.CAN-15-2001
204. Bertino EM, McMichael EL, Mo X, Trikha P, Davis M, Paul B, et al. A Phase I Trial to Evaluate Antibody-Dependent Cellular Cytotoxicity of Cetuximab and Lenalidomide in Advanced Colorectal and Head and Neck Cancer. Mol Cancer Ther (2016) 15:2244–50. doi: 10.1158/1535-7163.MCT-15-0879
205. Ferris RL, Blumenschein GJ, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
206. Forster MD, Sacco JJ, Kong AH, Wheeler G, Forsyth S, Bhat R, et al. EACH: A randomised phase II study evaluating the safety and anti-tumour activity of the combination of avelumab and cetuximab relative to avelumab monotherapy in recurrent/metastatic head and neck squamous cell cancer. J Clin Oncol (2019) 37:TPS6091–TPS6091. doi: 10.1200/JCO.2019.37.15_suppl.TPS6091
207. Hirsch I, Janovec V, Stranska R, Bendriss-Vermare N. Cross Talk between Inhibitory Immunoreceptor Tyrosine-Based Activation Motif-Signaling and Toll-Like Receptor Pathways in Macrophages and Dendritic Cells. Front Immunol (2017) 8:394. doi: 10.3389/fimmu.2017.00394
208. Li J, Yang F, Wei F, Ren X. The role of toll-like receptor 4 in tumor microenvironment. Oncotarget (2017) 8:66656–67. doi: 10.18632/oncotarget.19105
209. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol (2007) 44:2850–9. doi: 10.1016/j.molimm.2007.01.022
210. Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, et al. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene (2009) 28:4353–63. doi: 10.1038/onc.2009.289
211. Liu C-Y, Xu J-Y, Shi X-Y, Huang W, Ruan T-Y, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest (2013) 93:844–54. doi: 10.1038/labinvest.2013.69
212. Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res (2009) 69:3105–13. doi: 10.1158/0008-5472.CAN-08-3838
213. Ju H, Hu Z, Lu Y, Wu Y, Zhang L, Wei D, et al. TLR4 activation leads to anti-EGFR therapy resistance in head and neck squamous cell carcinoma. Am J Cancer Res (2020) 10:454–72. doi: 2156-6976/ajcr0104647
214. Curry JM, Sprandio J, Cognetti D, Luginbuhl A, Bar-ad V, Pribitkin E, et al. Tumor microenvironment in head and neck squamous cell carcinoma. Semin Oncol (2014) 41:217–34. doi: 10.1053/j.seminoncol.2014.03.003
215. Ziani L, Chouaib S, Thiery J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front Immunol (2018) 9:414. doi: 10.3389/fimmu.2018.00414
216. Takahashi H, Sakakura K, Kawabata-Iwakawa R, Rokudai S, Toyoda M, Nishiyama M, et al. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol Immunother (2015) 64:1407–17. doi: 10.1007/s00262-015-1742-0
217. Steinbichler TB, Savic D, Dejaco D, Romani A, Kofler B, Skvortsova II, et al. Pleiotropic Effects of Epithelial Mesenchymal Crosstalk on Head and Neck Cancer: EMT and beyond. Cancer Microenviron (2019) 12:67–76. doi: 10.1007/s12307-019-00228-y
218. Yegodayev KM, Novoplansky O, Golden A, Prasad M, Levin L, Jagadeeshan S, et al. TGF-Beta-Activated Cancer-Associated Fibroblasts Limit Cetuximab Efficacy in Preclinical Models of Head and Neck Cancer. Cancers (Basel) (2020) 12:339. doi: 10.3390/cancers12020339
219. Johansson A-C, Ansell A, Jerhammar F, Lindh MB, Grénman R, Munck-Wikland E, et al. Cancer-Associated Fibroblasts Induce Matrix Metalloproteinase–Mediated Cetuximab Resistance in Head and Neck Squamous Cell Carcinoma Cells. Mol Cancer Res (2012) 10:1158–68. doi: 10.1158/1541-7786.MCR-12-0030
220. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499:214–8. doi: 10.1038/nature12213
221. Zolkind P, Przybylski D, Marjanovic N, Nguyen L, Lin T, Johanns T, et al. Cancer immunogenomic approach to neoantigen discovery in a checkpoint blockade responsive murine model of oral cavity squamous cell carcinoma. Oncotarget (2018) 9:4109–19. doi: 10.18632/oncotarget.23751
222. Hayes DN, Grandis JR, El-Naggar AK. The {Cancer} {Genome} {Atlas}: {Integrated} analysis of genome alterations in squamous cell carcinoma of the head and neck. J Clin Oncol (2013) 31:6009. doi: 10.1200/jco.2013.31.15_suppl.6009
223. Keck MK, Zuo Z, Khattri A, Stricker TP, Brown CD, Imanguli M, et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin Cancer Res Off J Am Assoc Cancer Res (2015) 21:870–81. doi: 10.1158/1078-0432.CCR-14-2481
224. Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria J-C, Postel-Vinay S, et al. Mutational Landscape and Sensitivity to Immune Checkpoint Blockers. Clin Cancer Res (2016) 22:4309–21. doi: 10.1158/1078-0432.CCR-16-0903
225. Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci (2014) 111:15544–9. doi: 10.1073/pnas.1416074111
226. Outh-Gauer S, Le Tourneau C, Broudin C, Scotte F, Roussel H, Hans S, et al. [{Current} events in immunotherapy for upper aerodigestive tract cancer]. Ann Pathol (2017) 37:79–89. doi: 10.1016/j.annpat.2016.12.013
227. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (80-) (2015) 348:56–61. doi: 10.1126/science.aaa8172
228. Zandberg DP, Strome SE. The role of the {PD}-{L1}:{PD}-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol (2014) 50:627–32. doi: 10.1016/j.oraloncology.2014.04.003
229. Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, et al. Evidence for a role of the {PD}-1:{PD}-{L1} pathway in immune resistance of {HPV}-associated head and neck squamous cell carcinoma. Cancer Res (2013) 73:1733–41. doi: 10.1158/0008-5472.CAN-12-2384
230. Ritprajak P, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule {PD}-{L1} in epithelial cells and squamous cell carcinoma. Oral Oncol (2015) 51:221–8. doi: 10.1016/j.oraloncology.2014.11.014
231. Horton JD, Knochelmann HM, Day TA, Paulos CM, Neskey DM. Immune Evasion by Head and Neck Cancer: Foundations for Combination Therapy. Trends Cancer (2019) 5:208–32. doi: 10.1016/j.trecan.2019.02.007
232. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol (2018) 81:45–51. doi: 10.1016/j.oraloncology.2018.04.008
233. Cohen EEW, Soulières D, Le Tourneau C, Dinis J, Licitra L, Ahn M-J, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet (London England) (2019) 393:156–67. doi: 10.1016/S0140-6736(18)31999-8
234. Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet (London England) (2019) 394:1915–28. doi: 10.1016/S0140-6736(19)32591-7
235. Yarchoan M, Hopkins A, Jaffee EM. Tumor {Mutational} {Burden} and {Response} {Rate} to {PD}-1 {Inhibition}. N Engl J Med (2017) 377:2500–1. doi: 10.1056/NEJMc1713444
236. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell (2017) 171:934–49.e16. doi: 10.1016/j.cell.2017.09.028
237. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
238. Pereira C, Gimenez-Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, et al. Genomic {Profiling} of {Patient}-{Derived} {Xenografts} for {Lung} {Cancer} {Identifies} {B2M} {Inactivation} {Impairing} {Immunorecognition}. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23:3203–13. doi: 10.1158/1078-0432.CCR-16-1946
239. Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2006) 12:3890–5. doi: 10.1158/1078-0432.CCR-05-2750
240. Leibowitz MS, Andrade Filho PA, Ferrone S, Ferris RL. Deficiency of activated {STAT1} in head and neck cancer cells mediates {TAP1}-dependent escape from cytotoxic {T} lymphocytes. Cancer Immunol Immunother CII (2011) 60:525–35. doi: 10.1007/s00262-010-0961-7
241. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining {Immunotherapy} and {Radiotherapy} for {Cancer} {Treatment}: {Current} {Challenges} and {Future} {Directions}. Front Pharmacol (2018) 9:185. doi: 10.3389/fphar.2018.00185
242. Demaria S, Golden EB, Formenti SC. Role of {Local} {Radiation} {Therapy} in {Cancer} {Immunotherapy}. JAMA Oncol (2015) 1:1325–32. doi: 10.1001/jamaoncol.2015.2756
243. Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol (2011) 8:151–60. doi: 10.1038/nrclinonc.2010.223
244. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547:217–21. doi: 10.1038/nature22991
245. Guo ZS, Liu Z, Bartlett DL. Oncolytic {Immunotherapy}: {Dying} the {Right} {Way} is a {Key} to {Eliciting} {Potent} {Antitumor} {Immunity}. Front Oncol (2014) 4:74. doi: 10.3389/fonc.2014.00074
246. Dhillon AS, Hagan S, Rath O, Kolch W. {MAP} kinase signalling pathways in cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
247. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
248. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discovery (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
249. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem C, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
250. Ludwig S, Floros T, Theodoraki M-N, Hong C-S, Jackson EK, Lang S, et al. Suppression of {Lymphocyte} {Functions} by {Plasma} {Exosomes} {Correlates} with {Disease} {Activity} in {Patients} with {Head} and {Neck} {Cancer}. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23:4843–54. doi: 10.1158/1078-0432.CCR-16-2819
251. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: {MDSCs}, {TAMs}, and {Tregs} as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol (2016) 58:59–70. doi: 10.1016/j.oraloncology.2016.05.002
252. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma ({ECHO}-301/{KEYNOTE}-252): a phase 3, randomised, double-blind study. Lancet Oncol (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
253. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-{PD1} therapy by {Tim}-3 upregulation is mediated by the {PI3K}-{Akt} pathway in head and neck cancer. Oncoimmunology (2017) 6:e1261779. doi: 10.1080/2162402X.2016.1261779
254. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
255. Yu G-T, Bu L-L, Zhao Y-Y, Mao L, Deng W-W, Wu T-F, et al. {CTLA4} blockade reduces immature myeloid cells in head and neck squamous cell carcinoma. Oncoimmunology (2016) 5:e1151594. doi: 10.1080/2162402X.2016.1151594
256. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. {CTLA}-4 control over {Foxp3}+ regulatory {T} cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
257. Deng W-W, Mao L, Yu G-T, Bu L-L, Ma S-R, Liu B, et al. {LAG}-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology (2016) 5:e1239005. doi: 10.1080/2162402X.2016.1239005
258. Jie H-B, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, et al. Intratumoral regulatory {T} cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer (2013) 109:2629–35. doi: 10.1038/bjc.2013.645
259. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of {Tim}-3 and {PD}-1 identifies a {CD8}+ {T}-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
260. Siu LL, Even C, Mesía R, Remenar E, Daste A, Delord J-P, et al. Safety and {Efficacy} of {Durvalumab} {With} or {Without} {Tremelimumab} in {Patients} {With} {PD}-{L1}-{Low}/{Negative} {Recurrent} or {Metastatic} {HNSCC}: {The} {Phase} 2 {CONDOR} {Randomized} {Clinical} {Trial}. JAMA Oncol (2019) 5:195–203. doi: 10.1001/jamaoncol.2018.4628
261. Ferris RL, Haddad R, Even C, Tahara M, Dvorkin M, Ciuleanu TE, et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: {EAGLE}, a randomized, open-label phase {III} study. Ann Oncol Off J Eur Soc Med Oncol (2020) 31:942–50. doi: 10.1016/j.annonc.2020.04.001
262. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory {T} cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U.S.A. (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
263. Liu J-F, Ma S-R, Mao L, Bu L-L, Yu G-T, Li Y-C, et al. T-cell immunoglobulin mucin 3 blockade drives an antitumor immune response in head and neck cancer. Mol Oncol (2017) 11:235–47. doi: 10.1002/1878-0261.12029
264. Morvan MG, Lanier LL. {NK} cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16:7–19. doi: 10.1038/nrc.2015.5
265. Segal NH, Infante JR, Sanborn RE, Gibney GT, Lawrence DP, Rizvi N, et al. Safety of the natural killer ({NK}) cell-targeted anti-{KIR} antibody, lirilumab (liri), in combination with nivolumab (nivo) or ipilimumab (ipi) in two phase 1 studies in advanced refractory solid tumors. Ann Oncol (2016) 27:vi372. doi: 10.1093/annonc/mdw378.40
266. Bell RB, Leidner RS, Crittenden MR, Curti BD, Feng Z, Montler R, et al. {OX40} signaling in head and neck squamous cell carcinoma: {Overcoming} immunosuppression in the tumor microenvironment. Oral Oncol (2016) 52:1–10. doi: 10.1016/j.oraloncology.2015.11.009
267. Baruah P, Lee M, Odutoye T, Williamson P, Hyde N, Kaski JC, et al. Decreased levels of alternative co-stimulatory receptors {OX40} and 4-{1BB} characterise {T} cells from head and neck cancer patients. Immunobiology (2012) 217:669–75. doi: 10.1016/j.imbio.2011.11.005
268. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi: 10.1038/ni.2703
269. Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young MR. Mechanisms of immune suppression in patients with head and neck cancer: presence of {CD34}(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res Off J Am Assoc Cancer Res (1995) 1:95–103.
270. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol (2013) 91:493–502. doi: 10.1038/icb.2013.29
271. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating {MDSC} correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother CII (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
272. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
273. Costa NL, Valadares MC, Souza PPC, Mendonça EF, Oliveira JC, Silva TA, et al. Tumor-associated macrophages and the profile of inflammatory cytokines in oral squamous cell carcinoma. Oral Oncol (2013) 49:216–23. doi: 10.1016/j.oraloncology.2012.09.012
274. Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med (2013) 5:196ra99. doi: 10.1126/scitranslmed.3005747
275. Ho AL, Hanna GJ, Scholz CR, Gualberto A, Park SH. Preliminary activity of tipifarnib in tumors of the head and neck, salivary gland and urothelial tract with HRAS mutations. J Clin Oncol Am Soc Clin Oncol (ASCO) (2020) 38:6504–4. doi: 10.1200/JCO.2020.38.15_suppl.6504
276. Nathan CAO, Hayes DN, Harismendy O, Flores J, Moore-Medlin T, Gutkind JS, et al. Multi-Institutional Randomized Double-Blind Phase II Trial of Everolimus vs. Placebo as Adjuvant Therapy in Patients with Locally Advanced Squamous Cell Cancer of the Head and Neck (SCCHN). Int J Radiat Oncol Biol Phys (2020) 106:1116. doi: 10.1016/j.ijrobp.2019.11.392
277. Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell (2004) 5:489–500. doi: 10.1016/S1535-6108(04)00112-6
278. Walter V, Yin X, Wilkerson MD, Cabanski CR, Zhao N, Du Y, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PloS One (2013) 8:e56823. doi: 10.1371/journal.pone.0056823
279. Network CGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
280. De Cecco L, Nicolau M, Giannoccaro M, Daidone MG, Bossi P, Locati L, et al. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget (2015) 6:9627–42. doi: 10.18632/oncotarget.3301
281. Foy J-P, Bazire L, Ortiz-Cuaran S, Deneuve S, Kielbassa J, Thomas E, et al. A 13-gene expression-based radioresistance score highlights the heterogeneity in the response to radiation therapy across HPV-negative HNSCC molecular subtypes. BMC Med (2017) 15:165. doi: 10.1186/s12916-017-0929-y
282. Li B, Cui Y, Nambiar DK, Sunwoo JB, Li R. The Immune Subtypes and Landscape of Squamous Cell Carcinoma. Clin Cancer Res (2019) 25:3528–37. doi: 10.1158/1078-0432.CCR-18-4085
283. Ferrarotto R, Mitani Y, McGrail DJ, Li K, Karpinets TV, Bell D, et al. Proteogenomic Analysis of Salivary Adenoid Cystic Carcinomas Defines Molecular Subtypes and Identifies Therapeutic Targets. Clin Cancer Res (2020). doi: 10.1158/1078-0432.CCR-20-1192. clincanres.1192.2020.
284. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell (2017) 171:1611–24.e24. doi: 10.1016/j.cell.2017.10.044
Keywords: head and neck squamous-cell carcinoma, resistance, chemotherapy, cetuximab, immunotherapy, targeted therapy
Citation: Ortiz-Cuaran S, Bouaoud J, Karabajakian A, Fayette J and Saintigny P (2021) Precision Medicine Approaches to Overcome Resistance to Therapy in Head and Neck Cancers. Front. Oncol. 11:614332. doi: 10.3389/fonc.2021.614332
Received: 05 October 2020; Accepted: 08 January 2021;
Published: 25 February 2021.
Edited by:
Floriana Morgillo, Second University of Naples, ItalyReviewed by:
Sandra Schmitz, Catholic University of Louvain, BelgiumEdgar K. Selzer, Medical University of Vienna, Austria
Copyright © 2021 Ortiz-Cuaran, Bouaoud, Karabajakian, Fayette and Saintigny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Ortiz-Cuaran, U2FuZHJhLk9SVElaLUNVQVJBTkBseW9uLnVuaWNhbmNlci5mcg==; Pierre Saintigny, cGllcnJlLnNhaW50aWdueUBseW9uLnVuaWNhbmNlci5mcg==